

Abstract
Calvarial defects are common reconstructive dilemmas secondary to a variety of etiologies including traumatic brain injury, cerebrovascular disease, oncologic resection, and congenital anomalies. Reconstruction of the calvarium is generally undertaken for the purposes of cerebral protection, contour restoration for psychosocial well-being, and normalization of neurological dysfunction frequently found in patients with massive cranial defects. Current methods for reconstruction using autologous grafts, allogeneic grafts, or allo-plastic materials have significant drawbacks that are unique to each material. The combination of wide medical relevance and the need for a better clinical solution render defects of the cranial skeleton an ideal target for development of regenerative strategies focused on calvarial bone. With the improved understanding of the instructive properties of tissue-specific extracellular matrices and the advent of precise nanoscale modulation in materials science, strategies in regenerative medicine have shifted in paradigm. Previously considered to be simple carriers of stem cells and growth factors, increasing evidence exists for differential materials directing lineage specific differentiation of progenitor cells and tissue regeneration. In this work, we review the clinical challenges for calvarial reconstruction, the anatomy and physiology of bone, and extracellular matrix-inspired, collagen-based materials that have been tested for in vivo cranial defect healing.
Keywords: cranial defects, mineralized collagen glycosaminoglycan
1. Introduction
Current investigative strategies for bone tissue engineering integrate three elements: scaffolding material, osteogenic cells, and growth factors. However, this strategy has a few significant limitations. First, the procurement of osteogenic cells usually dictates an additional procedure for harvesting of autologous progenitor cells from the bone marrow, adipose tissue, or other sources. Second, the expansion of progenitor cells is subsequently required in an ex vivo, laboratory setting for an uncertain amount of time until a critical mass of progenitor cells has been proliferated and/or differentiated in preparation for implantation. Third, in order to proliferate and differentiate progenitor cells, growth factors are frequently utilized either in the ex vivo expansion stage or simultaneously with implantation. By definition, any addition of a growth factor is a supraphysiologic dosage which may cause unanticipated consequences such as uncontrolled proliferation. When one truly evaluates this strategy conceptually in an objective manner, it is clear that these deficiencies reduce the practicality of the strategy in clinical translation. Yet, the major argument from proponents of the strategy is that both stem cells and growth factors are absolutely necessary if we are to recapitulate or augment the biological wound healing process.
An alternative to this strategy is to consider that progenitor cells and growth factors are actually quite plentiful in humans and, perhaps, one may be able to recruit and stimulate endogenous progenitor cells to differentiate should we be able to provide the appropriate cues. In physiological differentiation, the appropriate cues are frequently found in the unique combination of molecules that make up extracellular matrices (ECM) of respective tissues. In recent years, an extraordinary expansion of bioinspired, ECM-based materials has been reported for the purposes of skeletal tissue regeneration.[2–13] Similar to other biomaterials fabricated for regeneration, many of these materials have demonstrated early promises in bench research. However, only a small number have progressed to in vivo investigations that accurately mimic a clinical scenario.
As one of the most utilized in vivo models for bone regeneration, critical-sized cranial defects are also one of the most clinically relevant situations as it is encountered in congenital anomalies, trauma, stroke, aneurysms, and cancer. For example, in stroke alone, 4.3 million adults were hospitalized in the United States between 1999 and 2008 with decompressive craniectomies performed in 14.5 patients per 10,000 hospitalizations for elevated intracranial pressures.[14] In a report to the United States Congress in 2015, the Centers for Disease Control and Prevention (CDC) estimated that traumatic brain injuries (TBI) in the civilian population accounted for 283,630 hospitalizations and 52,844 deaths.[15] In addition, the CDC and United States Department of Defense (DoD) reported that 235,046 service members (4.2%) were diagnosed with a TBI between 2000–2011.[15] Within these groups, surgical decompression via craniotomy or craniectomies are performed emergently in between 2–14% of patients depending on the severity of brain injury and, subsequently, delayed, secondary reconstructions may occur following resolution of cerebral edema.[16] In 1 in 2500 children, congenital anomalies of the skull such as craniosynostosis occurs.[17] Additionally, 1 in 700 children are affected by facial clefts, of which the rarest clefts usually affect both the skull and the face. In both pediatric situations, a paucity of bone may require harvest from a distant site and grafting to areas of deficiencies. Undoubtedly, successful technologies targeting cranial defects are likely to benefit a large proportion of the civilian and military population.
In this review, we aim to accomplish three tasks: present the clinical perspectives in treating cranial defects, review the biological organization and components of bone, and discuss the ECM-based materials that have been tested in vivo in cranial defect animal models.
2. Challenges in Cranial Defect Reconstruction
2.1. Indications for Cranial Defect Reconstruction
There are three reasons for reconstruction of a cranial defect: 1) cerebral protection 2) cosmesis 3) prevention or reversal of the “syndrome of the trephined”. Although these three reasons are not always applicable in smaller defects, large defects can clearly be envisaged to lack the ability to withstand even minor head trauma. In addition, the grossly visible deformity of a massive cranial defect is frequently disruptive to psychosocial and vocational well-being.
Beyond empirical cerebral protection and deformity correction, cranial defects 20 cm2 and larger have been reported to be associated with the “syndrome of the trephined” or post-craniotomy syndrome.[18,19] In one study, the prevalence was found to be 24% in patients who had undergone decompressive craniectomy.[20] This syndrome consists of constellation of neurologic symptoms including headache, vertigo, tinnitus, fatigue, lack of concentration, insomnia, memory disturbance, irritability, and mental depression with or without dysphasia, dyspraxia, extremity paresis, and epilepsy.[18] Fodstad and colleagues found that 79% of their patients who suffered from the syndrome of the trephined were relieved of clinical symptoms and another 21% experienced improvement of symptoms after cranioplasty.[18] Using a battery of neurocognitive tests, Corallo et al. evaluated pre-cranioplasty and post-cranioplasty scores at 1 month and 1 year following reconstruction.[21] In all 15 tests, a statistically significant improvement was found between preoperative and postoperative states, however, essentially no statistically significant differences were noted between 1 month and 1 year post reconstruction suggesting that the improvement occurs early following reconstruction. One of the most dramatic accounts of motor recovery from a paralyzed extremity was documented in a case report of a patient with a delayed cranioplasty after gunshot wound to the head.[22] In addition, Suzuki and colleagues showed that their patients not only demonstrated improvement in speech, seizure activity, and cognition, they also demonstrated increased cerebral blood flow based on dynamic computed tomography.[23] Although understanding of the syndrome of the trephined remains incomplete, skeletal reconstruction of the cranial vault is thought to normalize cerebrospinal fluid hydrodynamics and increase cerebral blood flow.[18,23]
2.2. Timing of Cranial Defect Reconstruction
Timing of calvarial reconstruction, or cranioplasty, plays a significant role in the types of materials available for use. In elective, non-emergent neurosurgical procedures, such as tumor resection or treatment of vascular malformations, replacement of the autologous calvarium at the conclusion of the procedure is universally performed provided there are no contraindications such as presence of tumor within the calvarium or concern for elevated intracranial pressures. Thus, a discussion of primary cranioplasties will not be examined here as there is little argument to the treatment. However, in many circumstances, cranioplasty is performed in a delayed fashion.
Secondary cranioplasties occur due to a variety of reasons. The most common reason is the replacement of cranial bone removed for the purposes of relieving elevated intracranial pressures. This scenario occurs frequently in the setting of cerebral edema from traumatic brain injury or stroke. Severe trauma resulting in comminution of cranial bone, resection of cranial bone for malignancy, delayed resorption of bone, infection, radiation, or post-surgical deformities account for the majority of other reasons for secondary cranioplasties. Timing of a secondary cranioplasty depends on the etiology. For example, in the event of an oncologic resection of skin cancer invading the frontal bone, cranioplasty may occur immediately provided that the margins are clear. In the setting of infection, reconstruction of the calvarium is typically delayed for 6–12 months after resolution before definitive repair.
2.3. Clinically Available Materials for Cranial Reconstruction
Clinically available materials used for cranioplasty are categorized as autologous, homologous, or alloplastic (Table 1). For many surgeons, autologous bone graft continues to be the first line method for cranioplasty due to its ability for osteoinduction, osteoconduction, revascularization, and low infection rates.[24,25] Within the category of autologous bone, three subcategories exist: 1) Banked calvarial bone graft 2) Fresh bone graft from split calvarium and other sites 3) Vascularized bone flaps. These three subcategories should be considered separately as all three have their advantages and disadvantages.
Table 1.
Clinically Available Materials for Cranioplasty.
| Source | Type | Vascularity | Defect Fit | Resorption | Extrusion | Infection | Resistance to hostile environments (radiation, previous infection) |
Implant Cost |
Reasons for Failure/ Reoperation |
Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Autologous | Banked Orthotopic Bone Graft (“Bone Flap”) | Requires neo-vascularization | Exact fit | ↑↑↑ | – | 7–10% | ↑↓ | – | Resorption | [26–29] |
| Heterotopic Bone Graft | Requires neo-vascularization | Requires intraoperative shaping | ↑↓ | – | – | ↑↑ | – | Low failure rates, Contour abnormalities secondary to resorption or initial operation | [24,27,31–33] | |
| Heterotopic Vascularized Bone Flap | Vascularized | Requires intraoperative shaping | – | – | – | ↑↑↑↑ | – | Low failure rates, Contour abnormalities secondary to resorption or initial operation | [24,30] | |
| Allogenic | Cadaveric Bone Graft | Requires neo-vascularization | Requires intraoperative shaping | ↑↑↑ | – | 13.2% | ↓↓↓ | ↑↑ | Infection, Resorption | [34,35] |
| Alloplastic | Titanium | – | Either intraoperative shaping or exact fit with 3D printing | – | ↑↑↑ | 8.6– 29% | ↓↓↓ | ↑↑ | Extrusion, Infection | [44,45,48] |
| Hydroxyapatite(Bone Cement) | – | Either intraoperative shaping or exact fit with 3D printing | – | ↑ | 22–62.5% | ↓↓↓ | ↑↑ | Infection | [48] | |
| Methylmethacrylate | – | Either intraoperative shaping or exact fit with 3D printing | – | ↑ | 13–42% | ↓↓↓↓ | ↑↑ | Infection | [140] | |
| Polyetheretherketone (PEEK) | – | Exact fit with 3D printing | – | ? (insufficient time in clinical use) | 7.6–13% | ↓↓↓ | ↑↑↑↑↑ | Infection | [49,50] |
↑ denotes relative increase with multiple arrows denoting higher relative qualities (i.e. increased resorption or cost), ↓ denotes relative decrease with multiple arrows denoting lower relative qualities (i.e. decreased resorption or cost); ↑↓ denote equivocal depending on report; – denotes not applicable.
2.3.1. Autologous Banked Calvarial Bone Grafts (“Bone Flaps”)
Banked calvarial bone graft, termed “bone flaps” in the neurosurgical literature, denotes the removed calvarium that is stored sterilely for planned replacement to the same patient at a later time. Such instances are most common in the setting of traumatic brain injuries where intracranial hypertension requires relief via decompressive craniectomy. Approximately 2–3 months following decompression when the patient’s intracranial pressures are no longer elevated, the patient’s calvarium is replaced as a delayed cranioplasty. This procedure is considered standard of care and should be pursued whenever possible. Although the obvious advantage of this procedure is the orthotopic replacement of the exact bone removed, banked calvarial bone grafts are avascular by definition and the survival of cellular material is likely to be very low considering the long storage times. Thus, failure rates due to resorption tend to be high, particularly in the pediatric patient, where a number of studies have suggested rates between 39% and 50%.[26,27] Adults fare significantly better in comparison to children with reported ranges between 7–20%.[28] Regardless of pediatric or adult, resorption also increases with increasing size of defect in a statistically significant manner. In terms of quantity of resorbed bone, the work by Grant and colleagues noted that, while there were no instances of resorption in defects less than 74 cm2 in size, defects >75 cm2 averaged around 60% resorption of the total bone graft.[26] Beyond resorption and contour defects, few other types of complications exist for this particular modality. Infection rates are generally low, reported between 7–10%.[26–29] When banked calvarial bone grafts fail due to resorption or are removed due to infection, the cranial defect returns and reoperation is necessary with an alternative material for secondary reconstruction.
2.3.2. Autologous Heterotopic Bone Grafts and Flaps
Unlike replacement of banked calvarium, immediate grafting using autologous split cranial bone graft or other sites have a 5% reported rate of significant resorption and the lowest rate of infection based on a systematic review of the literature (Figure 1).[27] However, also unlike banked calvarium, autologous bone grafting has the drawbacks of donor site availability and morbidity, thereby limiting the size of cranial defects that may be approached using this method. While the most common site for bone grafting of cranial defects is the uninvolved calvarium, other common donor sources include the iliac crest and ribs.
Figure 1.

Autologous cranial bone graft reconstruction for traumatic cranial defect. 44 year old male with traumatic cranial defect of the frontal bone measuring 5 × 4 cm. A) Preoperative CT scan with green arrows pointing to defect. B) Donor site defect from harvest of left parietal cranial bone graft, inset shows bone removed for split cranial bone graft. C) Following intraoperative split and shaping of the bone, the reconstructed frontal bone and donor site is secured with titanium plates. D) 1 year postoperative CT scan demonstrating near complete healing of frontal bone defect (green arrows) and donor site defect (blue arrows).
In a hostile environment (radiation, infection, reoperation), non-vascularized bone graft may not fully osseointegrate, resulting in nonunion or osteonecrosis. In such settings, vascularized bone flaps are superior.[24,30] Different from the neurosurgical term of “bone flap”, vascularized bone flaps denote bony tissue harvested with its vascular supply intact for an autologous transplant and re-vascularization to distant vessels. The vascular flap is, thus, transfer of live tissue without requirements of vascular ingrowth. As this procedure necessitates the participation of a skilled microsurgical plastic and reconstructive surgeon, vascularized bone flaps are not widely available as an option and are generally limited to tertiary care academic centers. Typical sites of harvest for cranioplasty include the back where the latissimus dorsi muscle and ribs or scapula are harvested in continuity with the thoracodorsal artery and vein.[24] For both bone grafts and bone flaps, perfect adaptation to the native calvarium is essential to bony union. In addition, precision in generating a convex contour that is harmonious with the rest of the skull is necessary to allow for both appropriate cerebral expansion and a normal appearance. With both of these requirements in mind, reconstruction of the cranial skeleton with autologous bone requires a painstaking intraoperative shaping process using drills, burrs, and saws, which is frequently time-consuming. Although the complications rates vary widely in the literature, the major sequela of significance for using autologous bone graft in the craniofacial skeleton that we and other surgeons face is resorption resulting in contour abnormalities that may require further revision.[24,31–33] Minor wound complications are common and range between 20–30% of all reconstructions. However, such complications are nearly universally treated with local wound care with resolution. Extrusion, removal, and complete replacement of autologous bone are exceedingly rare occurrences. In the largest series of autogenous bone graft harvest in the literature, complications of the donor site ranged from 0.25–0.9% depending on the site.[32] For the purposes of contour and the low donor site morbidity, split cranial bone graft is generally the preferred method for autologous reconstruction.[24,32,33]
2.3.3. Cadaveric Allograft
Cadaveric bone allograft has been sparingly utilized for reconstruction of bony defects as an alternative to autologous bone. Allograft is well known to have inferior abilities for osteoconduction and bony union as well as high infection rates.[34] In addition, the potential for disease transmission is present. Thus, allograft is generally not considered to be an acceptable option for cranioplasty. Recently, however, one Serbian center described a series of cranioplasty patients with greater than 5 years of followup who have been treated with either autografts or allografts.[35] In their work, cadaveric allografts demonstrated an infection rate twice as high as autografts at 13.2% but otherwise had reasonable outcomes.
There are a few dire circumstances in pediatric patients where cadaveric demineralized bone matrix has been reported to have limited success. Children between the ages of 1–4 years represent a subset of cranial defect patients who are particularly challenging. Unlike infants who have a high propensity to heal most cranial defects, older children lose the osteogenic potential yet are severely limited in donor site availability due to the thickness of the calvarium. Unlike adults, alloplastic solutions are less than ideal as the skull is continuing to grow and problems such as intracranial migration of titanium plates may occur. Thus, several groups have presented some potential alternative treatments. In a small series, the Pittsburgh group demonstrated bony union using a combination of resorbable mesh plates composed of 70/30 poly-L/DL-lactide with demineralized bone matrix sandwiched between the plates.[36] Also, in the rare circumstance of craniopagus conjoined twins, Salyer and colleagues described the utility of perforated demineralized allograft bone for staged reconstruction of the massive calvarial deficiencies.[37]
2.3.4. Alloplastic Materials
Alloplastic materials such as titanium alloys, methylmeth-acrylate (MMA), hydroxyapatite (HA) bone cement, and poly-etheretherketone (PEEK) have been applied extensively to cranial defects due to ease of use and the ability to manufacture a customized implant for each defect (Figure 2).[38,39] The major benefits of an alloplastic implant are: 1) “off-the-shelf” placement on a cranial defect, thereby significantly decreasing operative time; 2) ability to have an exact fit with the assistance of 3D printing; 3) ability to reconstruct massive defects that exceed sizes possible with autologous bone due to donor site availability. However, alloplastic materials have the drawbacks of cost, potential extrusion, lack of any re-vascularization, and infection, especially in patients who have undergone radiation treatment or have had previous infections.[24] Despite the excellent results reported by some investigators,[39,40] others have shown failures ranging from 16–62% in the setting of infection or radiated tissues depending on the type of alloplast used.[41–43] In addition, as the majority of studies evaluate cranioplasties of a variety of sizes and etiologies within the first several years after placement, the true long term complication rate is generally difficult to assess from the literature.
Figure 2.
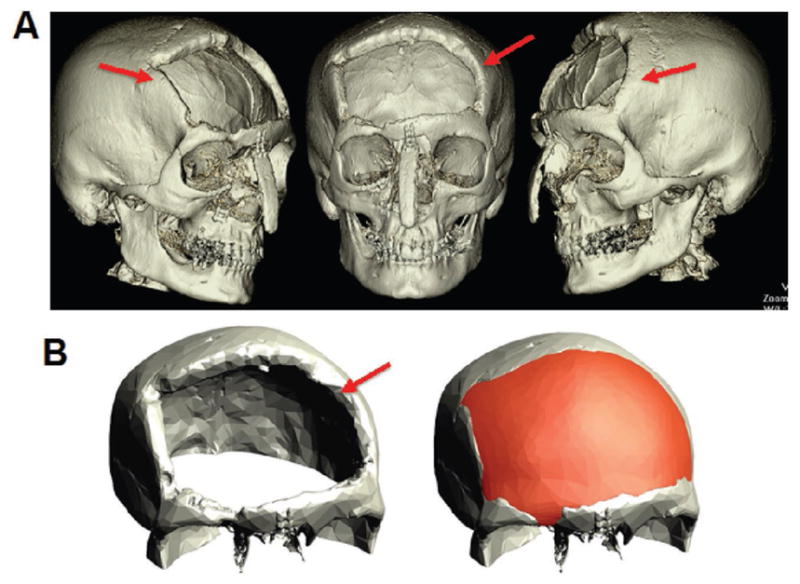
3D printed alloplastic cranioplasty after atypical meningioma resection and radiation. 62 year old female with a 9 × 9 cm cranial defect secondary to resection of atypical meningioma. A) Preoperative CT scan with red arrows pointing to defect. B) Closeup view of cranial defect (left) and virtually planned reconstructed skull with 3D printed alloplastic material.
Titanium is the most widely used alloplastic implant for calvarial reconstruction due to its reasonable biocompatibility, durability, low cost, and ability to be shaped intraoperatively or precisely produced with 3D printing. However, it is radio-opaque and, therefore, can interfere with surveillance imaging following cranioplasty. Similar to other alloplastic materials, titanium has been known to have a range of complication rates. Unlike all other materials, the special feature of titanium is that extrusion is common in large defects long term, particularly when there is soft tissue compromise such as previous infections or radiation. In one Singaporean center, the complication rate of titanium cranioplasty after decompressive craniectomy was found to be 25.0% with a higher percentage of titanium implants extruding in comparison to PEEK implants.[44] One of the largest retrospective studies on titanium cranioplasties, conducted by Mukherjee et al. in London, reported a failure and removal rate of 10.3% and infection rate of 8.6%.[45] When they evaluated reasons for failure, the presence of a previous infection and defect size greater than 100 cm2 were associated with higher complication rates.
Methylmethacrylate (MMA) has been used extensively for its ability to be molded intraoperatively, its excellent tensile strength, low cost, and ability to be custom produced with precision using 3D printing. However, the high rate of infection and fractures in large defects exceeding 42 cm2 has been problematic for broad usage.[26,41] In a long term outcomes study in pediatric patients, Blum et al. noted that 23% of their patients suffered complications within 8 years following MMA cranioplasty which was significantly correlated to radiation therapy, large defects, and involvement of the frontal sinus.[41]
Hydroxyapatite (HA), frequently used as “bone cement”, also has the advantage of allowing for intraoperative molding while being able to be custom produced precisely with 3D printing. Although brittle, HA is biocompatible and osteoconductive. Similar to MMA, HA is prone to high rates of infection.[46] In a small series, Durham and colleagues reported a 22% infection rate requiring reoperation and explantation and suggested that the unacceptably high rate of infection in defects >25 cm2 may limit the utility of HA for reconstruction.[46] Zins and colleagues, in another small series, reported a 62.5% reoperation rate.[43,47] Of the long term complications, 62.5% were attributed to infection and the remainder were attributed to fracture of the reconstruction. Interestingly, a recent randomized controlled trial for large defects demonstrated no significant differences between HA and titanium in terms of reoperation and explantation rates which was 11.5% and 12.5%, respectively.[48]
Polyetheretherketone (PEEK) is the most recent addition to the materials available for alloplastic cranioplasty. Use of PEEK is generally reserved for large defects in which computer aided design (CAD) is used for fabrication of a 3D printed implant based on the patient’s contralateral intact skull or normative data for human skulls. The material is extremely strong, non-porous, radiolucent, and can be re-sterilized for use in the event of a complication requiring explantation. All of these qualities set PEEK apart from MMA and HA as a superior material. However, PEEK is expensive and can cost up to 5–6 times the amount of custom titanium. At this point in time, due to the limited time in use, extrusion of PEEK has not been reported as a long term complication. In two studies, infections with PEEK implants were reported to be comparable to other alloplasts at 7.6–13%.[49,50] In all of the complications experienced in one of the studies, the implants were explanted, resterilized, and reimplanted with success.[49]
2.3.5. Adjunctive BMP-2
In addition to materials for cranioplasty, bone morphogenetic protein-2 (BMP-2) has been anecdotally described as an adjunctive technique for augmenting bone healing.[51,52] Two reports involving three patients received recombinant human BMP-2 (rhBMP-2) in conjunction with autologous bone grafting from either calvarial[52] or femoral[51] sources.
As one of the most common family of growth factors used to induce osteogenesis, the BMP family of growth factors are of clinical significance in that two of their members are available for clinical use.[53] To date, over 15 molecules of the BMP and growth and differentiation factor (GDF) subfamily have been identified and two (BMP-2 and -7) have been approved for use by the United States Food and Drug Administration (FDA) in clinical medicine.[54] Mechanistically, BMPs are first synthesized as precursor proteins that dimerize intracellularly (Figure 3). Upon dimerization, precursor proteins are cleaved at the consensus Arg-x-x-Arg site, yielding carboxy-terminal mature dimers that are secreted. Following secretion from cells, BMP dimers activate intracellular processes by binding to BMP receptor (BMPR) complexes.[55] Both type I and type II BMPR are transmembrane serine threonine kinases.[56] Type I BMPR are generally considered to be the high affinity receptors that determine the specificity of BMP signaling and type II receptors are the constitutively active receptors that activate downstream processes after binding to type I receptors. However, this general rule has been challenged by the binding patterns of certain BMPs such as BMP-9.[57] The mode of BMPR oligomerization at the cell surface is a determinant in downstream signaling pathways.[58] In the BMP-mediated signaling complex (also called the BMP-induced signaling complex or BISC), BMP dimers bind to type I BMPR dimers and recruit type II BMPR dimers to the complex (Figure 4). This complex is internalized in caveolae and activates the non-canonical, Smad-independent BMPR signaling pathway with the activation of ERK, p38 MAPK, JNK1/2, and PI3K pathways without Smad activation. In contrast, type I and type II BMPR can exist in a tetrameric preformed complex. When BMP dimers bind to the tetrameric preformed complex, the canonical pathway is activated with recruitment and phosphorylation of receptor Smads (Smad 1/5/8 or Smad 2/3). Internalization occurs through a clathrin dependent endosomal route. Phosphorylated receptor Smads associate with co-Smad (Smad 4) and translocate to the nucleus to activate Smad-dependent genes such as Id1–3. Crosstalk between the two pathways occurs. Both ERK and p38 MAPK are activated by the Smad independent pathway and both have the capabilities to target receptor Smads for proteasomal degradation.[59] Both canonical and non-canonical pathways can induce osteogenic genes. Despite the abilities for osteogenic stimulation, both cost and complications such ectopic bone formation, resorption, decreased maxillary growth, and potential for malignancy have dampened the enthusiasm for clinically utilizing exogenous, supraphysiologic dosages of BMP.[60]
Figure 3.
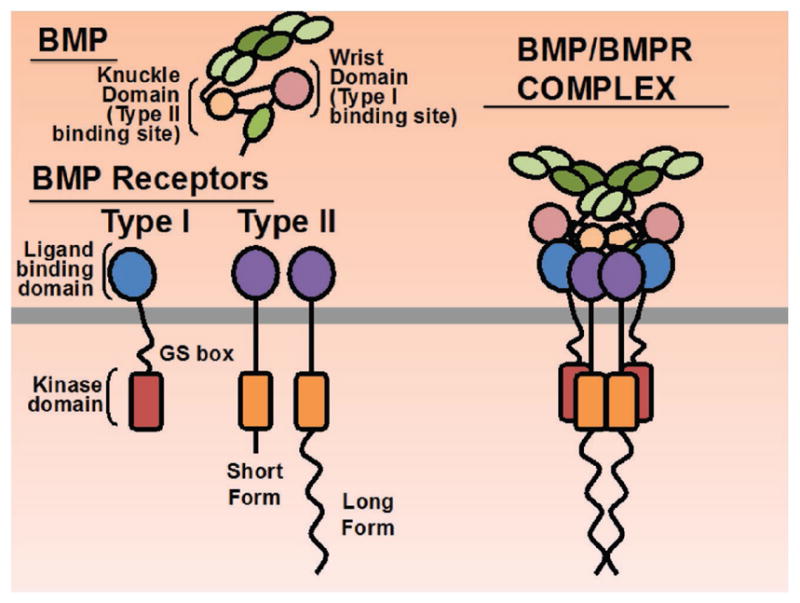
BMP and BMP Receptors. Protein domains of BMP ligands and BMP receptors are shown including binding sites on BMP ligands for receptors. The active intracellular serine threonine kinase domains are depicted as red or orange rectangles on Type I or Type II receptors, respectively, for downstream signaling. Assembled BMP and BMPR complex is shown at the right. Gray line denotes plasma membrane.
Figure 4.

Mode of BMP signaling determines Smad dependent and independent pathways. In the BMP-mediated signaling complex, BMP dimers bind to type I BMPR dimers and recruit type II BMPR dimers to the complex resulting in caveolae-mediated internalization and activation of the non-canonical (Smad independent) pathways including PI3K/Akt, p38 MAPK, ERK1/2, and JNK1/2. While the non-canonical pathways are not as well characterized as the canonical (Smad dependent pathways), BMP stimulation has been found to upregulate both Ras and TAK-1 phosphorylation, the upstream MAPKKKs for the ERK pathway and JNK pathways, respectively.[1] In addition, recent work in our laboratory has demonstrated an interdependence of ERK1/2 and JNK1/2 activation in hMSCs undergoing osteogenic differentiation on collagen glycosaminoglycan scaffolds (JCL, unpublished data). When type I and type II receptors are assembled in a preformed tetrameric complex, Smad dependent canonical pathway is activated via a clathrin-mediated internalization of the receptor complex. Phosphorylated receptor Smads (Smad1/5/8) associate with co-Smad (Smad 4) and translocate to the nucleus to activate Runx2 and other Smad-dependent genes. Crosstalk between the canonical and non-canonical pathways exist at multiple levels. Both p38 MAPK and ERK1/2-mediated phosphorylation has been reported to target Smads for ubiquitination by Smurf1 and proteasomal degradation. Despite the apparent inverse relationship between BMPR-mediated Smad and ERK signaling, both have been reported to contribute to osteogenic differentiation. Adapted with permission.[2] Copyright 2015, Elsevier.
2.3.6. Applying the Requirements of Cranial Defect Reconstruction to Regenerative Strategies
From the vast surgical literature on management of cranial defects, several concepts for successful reconstruction are clear. First, the most stable method of reconstruction with the lowest likelihood of wound complications is autologous, vascularized bone. Second, approximation of the transferred bone graft to the native calvarium is necessary for successful bony union. Thus, despite the risks for complications, one advantage of allo-plastic materials over autologous bone is the ability to generate a precisely shaped implant using 3D printing technologies. Lastly, the utility of exogenous growth factors to achieve bone healing is limited due to the problematic side effect profile. All three concepts serve as guiding principles for a realistic strategy for bone regeneration.
3. Bone Biology: The Basis for Regeneration
Efforts in designing the perfect material for bone regeneration requires a recapitulation of normal bone biology. Bone is a heterogenous and anisotropic material, which consists of a varied arrangement of hierarchical structures that work in concert to perform diverse mechanical, biological and chemical functions, including structural support, protection of vital organs, and regulation of mineral homeostasis (Table 2).[61–63]
Table 2.
The Hierarchical Structural Organization of Bone.
| Level | Structural Components | Size Range |
|---|---|---|
| 1 | Cortical and Cancellous Bone | |
| 2 | Osteons and Trabeculae | 10 to 500 μm |
| 3 | Lamellae | 1–10 μm |
| 4 | Fibrillar Collagen and Embedded Mineral | 250 nm–1 μm |
| 5 | Mineral, Collagen, and Non-collagenous Proteins | <250 nm |
3.1. The Hierarchical Organization of Bone
At the macrostructural level, bone can be separated into cortical (also called compact) and cancellous (also called trabecular) bone (Figure 5). In calvarial bone, cortical bone composes the inner and outer tables separated by the diploie, the cancellous portion. Overall, cortical bone accounts for 80 percent of the body’s bone mass.[62,64] In all bones, the cortical component provides strength and resistance to compression and tension.[62] However, the biomechanical properties of cortical bones can be quite different depending on the anatomical area. While cortical bone in the human femur has been reported to have an average elastic modulus of around 20–25 GPa,[65] cortical calvarial bone has been reported to have an elastic modulus of around 12 GPa.[66] When evaluated in children 1–2 years of age, the elastic modulus of the calvaria was found to be between 1.1–1.3 GPa with frontal bone providing greater stiffness than parietal bone.[67] Cancellous bone makes up the remaining 20 percent of the body’s bone mass.[62,64] Unlike cortical bone, cancellous bone has a relatively porous structure and a significantly larger and lower range of elastic moduli reported anywhere from 1–18 GPa depending on anatomic location of the bone.[68] The elastic modulus of diploie has not been reported in the literature. Although both types of bone are grossly different by porosity or density, the true differentiation comes from histological evaluation of the tissue’s microstructure.[61]
Figure 5.

The Hierarchical Organization of Bone. On the left, the gross anatomical structure of calvarial bone is depicted. Calvarial bone consists of cortical bone (inner and outer tables) separated by the cancellous or trabecular bone (diploe). Cortical bone consists of osteons consisting of osteocytes within lacunae arranged regularly within the lamellae of collagen fibers surrounding the central canal. Cancellous bone consists of a more irregular organization of lacunae, osteocytes, and canaliculi enclosing bone marrow. Collagen fibers that comprise the lamellae are further broken down into collagen proteins arranged with calcium phosphate mineral.
The basic functional unit of cortical bone is the osteon, or Haversian system.[61,62] Each osteon contains osteocytes arranged in regular, concentric layers of mineralized collagen fibers, called lamellae, which surround the long hollow central canal.[61,62] The central canal, or Haversian canal, generally runs parallel to the bone surface and contains neurovascular structures.[61,62,69] Volkmann’s canals are perforating holes or channels that run perpendicular to the central canals and function to interconnect central canals with each other and the periosteum.[62,69] Lacunae are the small spaces between the concentric lamellae where the osteocytes are located.[62,70] As such, the size of the lacunae is related to the original size of the osteoblast from which the osteocyte evolved.[71] The lacunae are linked together by minute channels called canaliculi, which permit the exchange of nutrients and metabolic waste products.[62,70] The small, irregular areas of bone between separate osteons is occupied by interstitial or intercalated lamellae.[62]
In contrast, cancellous bone consists of an irregular lattice of rod and plate like structures called trabeculae.[61,62] Trabeculae contain parallel lamellae, lacunae, osteocytes, and canaliculi.[72] Unlike osteons, trabeculae do not have a central canal with a blood vessel. Rather, the interconnecting framework of trabeculae encloses irregular red bone marrow cavities and hematopoietic stem cells that give rise to platelets, red blood cells and white blood cells.[73] Blood vessels from the periosteum penetrate into the trabeculae lattice, allowing the osteocytes in the trabeculae to receive nourishment from the blood passing through the marrow cavities.[62,73]
3.2. Bone Homeostasis
Bone is a metabolically active tissue which undergoes active homeostasis through the interactions of a number of cell types.[62] At the anatomic level, bone formation and resorption occurs as part of two major mechanisms of bone homeostasis: modeling and remodeling. Bone modeling generates a net positive quantity of bone at specific surfaces separate from the resorptive surfaces. Unlike long bones which model through an endochondral ossification pathway, calvaria and other facial bones ossify directly via the intramembranous pathway. One physiologic example is provided by Sarnat and colleagues who described that mandibular ramal growth occurred at the posterior and inferior borders, while resorption was prominent at the anterior border.[74] In contrast, bone remodeling couples osteoblast and osteoclast activity and is a process common in adult bone. This coupling of bone formation and resorption is spatially enclosed within specialized anatomic structures called basic multicellular units (BMUs).[75] The BMU consists of a cutting cone of osteoclasts followed by a closing cone of osteoblasts. This structure travels across cortical or cancellous bone. For remodeling to occur, BMU activation needs to occur with activation of osteoclasts. This signal has been hypothesized to come from the osteocyte following injury or times of mechanical stress such that a linear correlation exists between apoptotic osteocytes and intracortical remodeling.[76] The proximity of osteoblasts and osteoclasts result in cell-cell interactions that ultimately function to cooperate in bone resorption and formation.[77]
At the cellular level, two major cell lineages are responsible for bone homeostasis: osteoblasts and osteoclasts (Figure 6).[78,79] Mesenchymal stem cells (MSCs) are pluripotent cells that have the ability to differentiate into chondrocytes, adipocytes, osteoblasts, and myoblasts. The osteoblastic lineage begins with the differentiation of MSCs into a fibroblast colony forming unit (CFU-F), which is further differentiated into pre-osteoblasts through the action of a number of signaling pathways that are discussed further below. Central to the signaling pathways is the activation of Runx2, a master transcription factor in osteoblast differentiation. Osteoblasts are responsive to mechanical stimuli and growth factor receptor-mediated signals.[80] Following secretion of bone matrix, a population of osteoblasts undergoes further differentiation to osteocytes and remains embedded within the matrix. Osteocytes, the most abundant cell type in bone, are interconnected via dendritic processes and gap junctions to each other, osteoblasts, and endosteal lining cells. Although the exact purpose of the osteocyte network is controversial, it likely has a role as mechanical sensors for stress and injury.[76,81]
Figure 6.
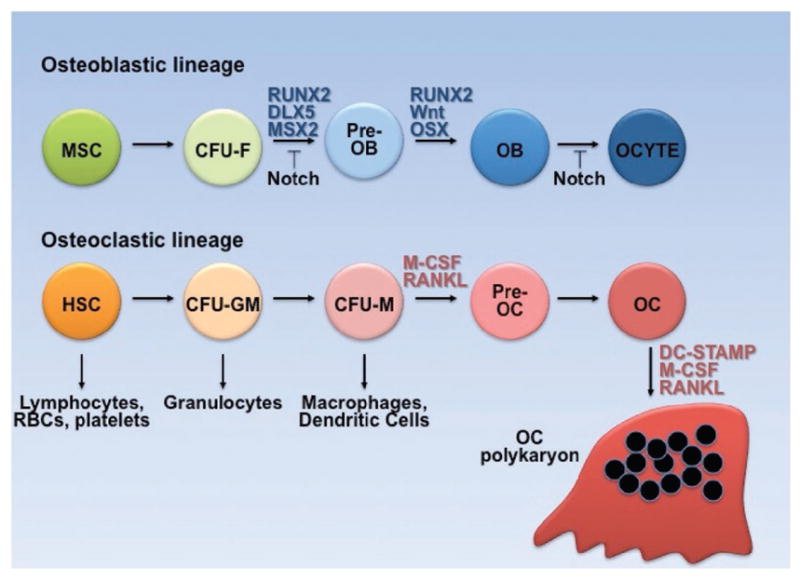
Ontogeny of osteoblast and osteoclast lineage. Colony forming units (CFU), fibroblast (F), granulocyte monocyte (GM), monocyte (M), osteoblast (OB), osteocyte (OCYTE), osteoclast (OC).
Unlike the osteoblast lineage of cells, osteoclasts are multi-nucleated cells differentiated from hematopoietic stem cells (HSCs) that function to resorb and remodel bone.[78] Following differentiation of the HSC to monocyte colony forming units (CFU-M), osteoclastogenesis is stimulated by the action of macrophage colony stimulating factor (M-CSF) and receptor activator of NF-κB (RANK). Osteoclasts exert their function by direct contact with bone, resulting in the formation of resorption pits or tunnels.
Anatomic, cellular, and mechanistic differences exist for long bones versus calvaria. This distinction is not entirely surprising in that the tissues are derived from different embryologic germ layers. In terms of bone resorption, calvarial osteoclasts have been demonstrated to utilize different proteases for matrix degradation in comparison to long bones.[82] When different sources of osteoclasts were investigated, heterogeneity in collagenolysis abilities was found to be present depending on anatomical site which was related to the expression of cathepsin K.[83] Interestingly, further investigation demonstrated that in vitro osteoclast activity was intimately tied to the anatomic source of osteoblasts and that calvarial osteoblasts activated higher numbers of osteoclasts in comparison to long bones.[84]
3.3. The Extracellular Matrix of Bone
Outside of the cells, the extracellular matrix is extraordinarily interesting and has been demonstrated to facilitate a significant amount of biological activity within the bone. The widely separated cells are surrounded by large amounts of calcified matrix, which is composed of approximately 30% organic components, 60% inorganic components, and 10% water.[63] However, the relative proportions of these constituents can vary considerably with age, gender, ethnicity, health status, and bone type.[54] The primary component of the organic matrix is Type I collagen (≈90%), a fibrous protein that provides strength and flexibility.[61,63,70,85] Type I collagen is composed of three polypeptide chains (i.e., two identical alpha-1 chains and one alpha-2 chain) wound together in a triple helix.[54,63] The triple-helical molecules self-assemble into parallel fibrils that are staggered by 67 nm and separated by 35 nm holes between their ends.[54,61] Other proteins, so-called non-collagenous proteins (NCPs), account for a small proportion of the organic matrix (≈10%).[54,61,63,85] The physiologic roles of most of the non-collagenous proteins have not fully been elucidated, but they appear to serve important functions in matrix organization and bone mineralization.[63] Specifically, osteocalcin, osteonectin, osteopontin, and sialoprotein may regulate the size, orientation, and crystallization of the mineral deposits.[61,63]
Additionally, proteoglycans (PGs), consisting of a protein core covalently bound to one or more glycosaminoglycan (GAG) chains, may regulate processes such as adhesion, migration, proliferation, and differentiation due to interactions with diverse proteins, including cytokines, chemokines, growth factors, enzymes, and adhesion molecules.[86,87] Glycosaminoglycans and proteoglycans are thought to be responsible for sequestering soluble factors within the matrix using noncovalent interactions, which is a process that can lead to spatial immobilization and bioactivity. GAGs are long, linear polysaccharides composed of repeating disaccharide units of hexosa-mine and either uronic acid or galactose.[86,87] Less than 1% of the extracellular matrix of bone is composed of GAGs, which consists primarily of chondroitin sulfate, repetitive N-acetylga-lactosamine (GalNac)-glucuronic acid (GlcAc) disaccharide unit polymers that are synthesized intracellularly, sulfated, secreted, and bound to proteoglycans.[87] PGs and their constituent GAG chains also bind to hydroxyapatite (HA) which could also potentially protect these molecules from degradation.[86] PGs and GAGs play a major role in bone morphogenesis, homeostasis and degenerative bone disease.[86] Proteoglycans are also believed to effect the formation, function, and viability of osteoclasts and osteoblasts.[87] Furthermore, small leucine-rich proteoglycans (SLRPs), such as biglycan and decorin, are an important family of proteoglycans that play a major role during all phases of bone formation, including cell proliferation, organic matrix deposition, remodeling, and mineral deposition.[86] Similar to PGs, glycoproteins such as fibronectin are effective at growth factor sequestration and such properties may be harnessed for regenerative materials.[88] Lipoproteins and lipids are also present in the bone matrix and are important for metabolic functions.[54,89] The roles of the supporting organic content, especially the GAGs, are only recently being investigated in bone regeneration.
The inorganic composition of bone consists predominantly of calcium phosphate, however, certain amounts of carbonate, sulfate, sodium, potassium, magnesium, fluoride, zinc, barium, and strontium are also present.[70,85] Within the matrix, phosphate and calcium ions nucleate to form plate-like hydroxyapatite crystals (Ca10(PO4)6(OH)2), which account for the stiffness and hardness of bone. During the early stages of mineralization, the crystals form within the discrete spaces between the collagen fibrils, limiting their possible primary growth and forcing them to be discrete and discontinuous.[54,61,70,85] The primary crystals grow with a specific crystalline orientation with the c-axes of the crystals roughly parallel to the long axes of the collagen fibrils.[54,61] However, as the crystals continue to grow, they penetrate into the overlap zone of the collagen fibrils, compressing the triple-helical collagen molecules to form extended sheets.[54,85]
Significant differences in the extracellular matrix compositions between long bones and calvaria have been reported.[90] Using two-dimensional polyacrylamide electrophoresis, van de Bos et al. evaluated proteomic differences in the matrix of long bones versus calvaria in a murine model. Their work demonstrated that collagen, pigment epithelium derived factor (PEDF), and osteoglycin (a member of the SLRPs) were more abundant in calvaria when compared to long bones. Conversely, the investigators also reported six proteins including chondrocalcin that were more abundant in long bones than in calvaria. The combination of differences in cellular activities, homeostasis, and extracellular matrix compositions between long bones and calvaria suggest that skeletal regenerative technologies may have different requirements depending on the anatomical targets.
4. Challenges in Development of Biomimetic Scaffolds
Due to the limited practicality in stem cell-based and growth factor-based strategies in regenerative medicine and the increasing understanding in the instructive abilities of the extracellular matrix, the impetus to generate ECM-inspired materials has become more and more attractive.[91] Additionally, several challenges have been identified in synthetic polymeric scaffolding material. Polymers made of poly-L-lactic acid (PLLA) and poly-lactide-co-glycolide (PLGA) are clinically available as resorbable surgical implants and, thus, have been commonly tested in laboratories as a carrier of growth factors and/or stem cells for regeneration.[92] However, there are a few significant considerations that have been clinically described. In orthopaedic implants composed of PLGA and ceramic composites, failure due to degradation resulting in screw migration and aseptic cysts have been described in long bones.[93] In an attempt to further understand the reasoning for device failure, Meyer and colleagues demonstrated that high lactic acid and glycolic acid concentrations negatively affected viability, proliferation, and differentiation of osteoprogenitor cells.[92] In addition, the produced acidic metabolites have been reported to induce inflammation via superoxide release from phagocytes.[94] Thus, degradation of the implant and subsequent metabolites generated by the scaffold may be important to consider. Mineralization on PLLA or PLGA does not occur clinically and such materials have no inherent instructive capabilities for osteogenesis. Indeed, osteogenic differentiation of progenitor cells on PLGA is difficult without the support exogenous growth factors.[95] However, as cerebral protection is an important component to reconstruction, there is a clinical need for a structurally stable covering of the skull during regeneration. This is unlikely to be provided by a soft, porous, biocompatible material designed for cellular integration and differentiation. Thus, a protocol for protection prior to complete regeneration would be necessary for future translation which may arrive in the form of a resorbable material.[96]
Recapitulation of the hierarchical structure of bone is difficult prior to implantation as it is composed of both cellular and acellular material. Since the scaffold is a cell free reagent, it is also difficult to predict the organization that will be necessary, how they should be arranged, and which molecules are necessary. An anistropic arrangement of ECM within a scaffold has been reported to be useful for the differentiation of cells into composite materials such as the osteochondral junction or the osteotendinous junction.[5,6] An anisotropic, hierarchical arrangement of porosities and mineralization have also been reported by a number of investigators specific for osteogenic differentiation.[97] However, the biological significance of these arrangements for bone regeneration has not yet been elucidated.
Chemical composition of the scaffold can be based essentially completely off of biological bone. However, there are still some nuances that must be considered. Sources of organic material, such as collagen, proteoglycans, and glycosaminoglycans, are animal in origin. Thus, the immunoreactivity of the proteins may potentially be of concern for implantation. In terms of the minor species of organic content, it is unclear how much and which minor species should be included as the importance of each has not been fully determined. In terms of the inorganic content, the types of calcium phosphate and method of incorporation is also unclear. Although carbonated hydroxyapatite is known to be the major type of calcium phosphate in bone ECM, it is difficult to speculate on whether this is necessary or appropriate for stimulating regeneration as excellent osteogenic differentiation has been reported in scaffolds with brushite as the primary calcium phosphate component.[2,7,9,11,98] Furthermore, the significance of the less abundant inorganic ions such as silicon and magnesium are also incompletely described for regeneration.
Many scaffolds are very similar in composition and even fabrication, however, differences such as crosslinking with either the carbodiimide (EDC/NHS) or the dehydrothermal (DHT) techniques can effect changes in porosity, compression modulus, and degradation rate.[99] All of these attributes have been reported to affect stem cell differentiation.[100] When evaluated in the laboratory, the efficiency of EDC crosslinking of Concanavalin A (ConA) to collagen glycosaminoglycan (CG) scaffolds was significantly increased in the presence of greater concentrations of ConA, a 5:12.5:1 EDC-NHS-COOH ratio, and with the step crosslinking approach versus bulk crosslinking.[8] Pence and Harley demonstrated that the source of collagen can significantly change the immobilization of proteins during crosslinking.[8] Murphy and O’Brien evaluated a range of pore sizes in collagen glycosaminoglycan scaffolds of identical composition, crosslinking, and synthesis methods.[13,101] They demonstrated that wide differences in adhesion, migration, and differentiation in a range of 85–325 μm in pore sizes, with the larger pore size demonstrating the best results. These studies highlight that minute changes can provoke significant differences in the success of regenerative materials. However, this may also point to the underlying power of a materials-based approach to regeneration in that the reagent can be finely tuned to the application at hand. Whereas stem cell and growth factor-based approaches may have difficulties in modular organization at the nanoscale level, materials can be relatively easily fabricated with precise compartments or gradients. In combination with the ability of the extracellular matrix to direct lineage specific differentiation by progenitor cells, it is clear that materials that can mimic tissue-specific extracellular matrix will also direct tissue-specific regeneration.
With respect to the clinical question of regenerating human calvarial defects, the most important piece of evidence in relationship to the potential clinical translation of material is still animal data. Specifically, the critical-size cranial defect is essentially exactly the same defect encountered in the clinical scenario and, thus, successful in vivo healing with supporting mechanistic data would be powerful indicator for clinical utility. Our focus below will be on the current ECM-inspired, collagen-based materials that have been designed and fabricated for bone engineering with in vivo cranial defect testing, thereby representing materials with the highest potential for rapid clinical translation in calvarial reconstruction (Table 3).
Table 3.
Described Collagen Scaffolds and Modifications for Bone Regeneration.
| Scaffold | Mechanism/In Vitro Studies |
In Vivo Model, Defect diameter |
Stem Cells for In vivo |
In vivo Micro-CT data (% BV/TV) |
Biomechanical data |
Special Features | Ref. |
|---|---|---|---|---|---|---|---|
| Collagen Scaffolds | |||||||
| Helistat (commercially available collagen) | None | Rat (Sprague Dawley), 8 mm | Cell Free | None | None | Not quantitative | [117] |
| Collagen with SDF-1 (CXCL12) and BMP-2 | C3H10T1/2 and C2C12 Greatest amount of ALP activity and transwell migration with SDF-1 followed by BMP-2 | Mouse (C57Bl/6 female), 5 mm | Cell free | No percentages but overgrowth seen at 4 wk (5 μg BMP-2) | None | SDF-1 recruitment prior to BMP-2 stimulation | [102] |
| Collagen/anti-BMP-2 (Antibody mediated osseous regeneration, AMOR) | C2C12, stimulates Runx2 and p-Smad1 in presence of BMP-2 | Rat (Sprague Dawley, female) 7 mm | Cell Free | 75% at 8 weeks | None | mAb persisted for 8 weeks | [105] |
| Collagen with human amniotic fluid derived stem cells (hAFSCs) and human dental pulp derived stem cells (hDPSCs) | None | Rat (CD IG5, male), 5 × 8 mm | Primary hAFSCs and primary hDPSCs | None, plain radiographs | None | Noted that human cells seen in regen erated bone and neo-vascularity. Followup study with ferutinin with improved healing | [106,107] |
| Compressed Collagen with rat DPSCs | None | Rat (Wistar), 5 mm | Primary Rat DPSCs | 9% at 5 weeks | No | [108] | |
| Collagen with human alveolar osteoblasts (hAlOBs) | Primary hAlOBs upregulates BMP2, BMP4, OPN, OCN, BSP on 3D collagen sponges but not 2D | Mouse (Scid), 3.5 mm | Primary hAlOBs | None, plain radiographs | None | No growth factors | [109] |
| Collagen with porcine bone marrow stromal cells (pBMSCs) and BMP-2 | None | Miniature swine, 2 × 5 cm | Primary pBMSCs transduced with BMP-2 | No percentage. 2D analysis of CT shows bone fill of 6.32 cm2 of 10 cm2 defect | In Vivo Compressive strength 81 MPa (native bone 86 MPa) | Dura removed | [110] |
| Collagen with murine muscle-derived stem cells (mMDSCs) and BMP-2 | None | Mouse (Scid), 5 mm | Murine muscle derived stem cells trans duced with BMP-2 | No CT scan | None | [111] | |
|
| |||||||
| Collagen with Glycosaminoglycans Scaffolds | |||||||
| Collagen glycosaminoglycan | Primary hMSCs, Primary rabbit MSCs Upregulated ALP, OCN | Rabbit, 14 mm | Cell free and primary rabbit BMSCs | 30–40% at 12 weeks with BMSCs and BMP-2 | Hydrated Elastic Modulus: 208 Pa | Not significantly different than unreconstructed defects | [1–5, 7,8,96,98, 115,141] |
| Collagen hyaluronic acid | Primary rat BMSCs Upregulated VEGF with chondrogenic medium | Rat (Fischer), 7 mm | Primary BMSCs | 20% at 4 weeks; 45% at 8 weeks | None | Only when cultured first in chondrogenic medium with TGF-β, then hypertrophic chondrogenic medium for 35 days before implantation; angiogenesis; endochondral approach to healing calvarium | [116] |
|
| |||||||
| Collagen with Mineral Content Scaffolds | |||||||
| Healos (commercially available collagen coated with hydroxyapatite) | None | Rat (Sprague Dawley), 8 mm | Cell Free | None | None | Not quantitative | [117] |
| Mastergraft (commercially available collagen and biphasic calcium phosphate) | None | Dog (Beagle), 2.5 × 4 cm | Cell Free | None | None | Histomorphometric analysis at 4 months with bone healing equivalent to demineralized bone matrix | [118] |
| Collagen hydroxyapatite (CCP) | Rat primary BMSCs | Rat (Fischer and Wistar), 7 mm | Primary Rat BMSCs and Cell Free | Cell-free: | Initial in vitro compressive modulus: | In osteogenic medium for 28–35 days before implantation. | [12,116] |
| 8% at 4 weeks; 34% at 8 weeks | CCP 10.3 kPa vs. CG 1 kPA | M2: CD163+ at the areas of remodeling | |||||
| With rat MSCS: | M1: CCR7+ only when cultured ahead of time with cells, decreased in cell-free | ||||||
| 5–15% at 4 weeks; 8–15% at 8 weeks | |||||||
| Nanohydroxyapatite (<100 nm) collagen, dispersant aided precipitation | MC3T3-E1 with upregulation ALP, OCN, OPN, mineralization with 5:1 more than 1:1 nHA:collagen | Rat (Wistar), 7 mm | Cell free | 4% at 4 wk, 16% at 8 wk regardless of nHA quantities | Young’s modulus: | No difference in osteogenesis in relationship to nHA ratios | [120] |
| Cell free implantation of scaffolds | Collagen only 0.3 kPa 5:1 nHA: collagen 5.5 kPa 1:1 nHA: collagen 0.4 kPa EDAC crosslinking to generate intermolecular amide bonds between adjacent collagen fibers, led to 38-fold increase in Young’s modulus |
||||||
| Collagen carbonated hydroxyapatite (Col- cHA) in modified simulated body fluid (SBF) | Primary murine calvarial osteoblasts | Mouse (pOBCol3.6tpz irradiated), 3.5 mm | Primary murine calvarial osteoblasts | None, plain radiographs at 4 wk | None | [142] | |
| Murine BMSCs (CD1) | Mouse (NSG immunodeficient), 3.5 mm | Primary murineBMSCs | None | None | [133] | ||
| Primary murine BMSCs with Basement Membrane Extract/Matrigel | Mouse (nod/scid), 3.5 mm | Primary murineBMSCs | None, plain radiographs taken at 3 wk | None | [132] | ||
| Atellocollagen/biphasic calcium phosphate (HA + β-tricalcium phosphate) | hMSCs | Rabbit (New Zealand), 8 mm | Cell free | 35% at 8 wk | None | [121] | |
| ALP activation and expression, Runx2, OCN, Col I expression increase in comparison to BCP alone | |||||||
| Polyphosphate Collagen | Immortalized hPDLSCs and primary human osteoblasts | Mouse (female ICR), 5 mm | Cell free | 8% at 6 weeks | None | Calcium-free scaffolds | [131] |
| Silicified Collagen (SCS) | Primary murine MSCs | Mouse (male C57Bl/6J), 3 mm | Primary murine MSCs | 45% at 4 weeks; 90% at 12 weeks | Compressive strength less than calcified collagen scaffolds | Angiogenic, monocyte immunomodulation, osteoclast inhibition | [134,136,143] |
| Compared to demineralized cancellous allograft bone: upregulate SDF-1, TGF-β, VEGF, PDGF-BB | Tangent modulus: 574.5 kPa; Modulus of toughness: 137.3 kPa | ||||||
| Compared to type I collagen scaffold: Upregulate ALP, Col I, OPN, OPG, mildly upregulate p-ERK1/2; downregulate RANKL | |||||||
|
| |||||||
| Collagen with Glycosaminoglycans and Mineral Content Scaffolds | |||||||
| Mineralized Collagen Glycosaminoglycan | Primary human MSCs, Primary rabbit MSCs Upregulated ALP, OCN, OPN | Rabbit (New Zealand), 14 mm | Cell free and primary rabbit BMSCs | 60% at 12 weeks | Hydrated Elastic Modulus: 4 kPa | [5,8,11,98,138] | |
4.1. Collagen Scaffolds
Materials composed of the major organic component of bone ECM, collagen I, have been present for several decades. However, the majority of the investigations with collagen I have focused on utilizing the material as a carrier of cells and/or growth factors without particular focus on modulating collagen.
Unmodified collagen scaffolds are commercially available (Helistat, Integra Life Sciences Corp, Plainsboro, NJ) and have been tested in the setting of cranial defects. However, collagen lacks structural stability resulting in contraction during in vitro osteogenic differentiation.[7,95] Thus, the usage of collagen scaffolds alone in the in vivo cranial defect literature is limited and universally requires either growth factors or stem cells for any reasonable efficacy.
Hwang et al. provided a growth factor based method for healing cranial defects in murine calvarial defects.[102] Early in wound healing, stromal cell derived factor-1 (SDF-1/CXCL12) binds to its receptor CXCR4 and serves to recruit progenitor cells including mesenchymal and endothelial stem cells to injured tissues.[103] Utilizing commercially available, cell-free collagen scaffolds in a 5 mm murine calvarial defect, their work demonstrated that pretreatment with SDF-1 prior to BMP-2 stimulation resulted in a massive increase in bone formation when compared to treatment with BMP-2 alone or SDF-1 alone. As their work was performed in a cell-free manner, their results support the idea that endogenous progenitor cells can be sufficient for bone regeneration provided that the appropriate signals are present for homing to the defect.[104] The massive overgrowth of bone seen in the defects with supraphysiologic SDF-1 and BMP-2 underscores the dysregulation that may occur with exogenous growth factor supplementation.
An alternative to using growth factors, yet still harnessing growth factor signaling, has been proposed by a few investigators. Antibody-mediated osseous regeneration (AMOR) alludes to a process using monoclonal antibodies to crosslink osteogenic BMPs, thereby activating the BMP receptors without increasing the physiologic quantity of osteogenic BMPs.[105] Although this strategy does not increase the absolute quantities of BMPs, the fact that it results in higher activities of physiologic quantities of BMPs may actually be equivalent to adding BMP-2. However, further work will need to be performed to determine whether this strategy will be fruitful and also decrease the complications induced by exogenous BMP-2.
Stem cell supplementation of various sources have been tested in combination with collagen scaffold carriers in cranial defect healing. De Pol and colleagues compared human amniotic fluid derived (hAFSCs) and human dental pulp derived stem cells (hDPSCs) head to head in rat critical-sized full thickness cranial defects of the parietal region.[106] In the presence of immunosuppression, healing of critical-sized defects was found to be more effective with amniotic fluid-derived stem cells. Their followup work also suggested the utility of ferutinin in further healing as determined by two-dimensional radiographic evidence and histochemical methods.[107] Similarly, Chamieh et al. utilized compressed collagen gel scaffolds in a rat cranial defect model with primary DPSCs.[108] They demonstrated 8–9% of bone volume/total volume (BV/TV) based on micro-computed tomographic data (micro-CT). Using a more differentiated set of progenitor cells, Bartold and colleagues expanded and implanted primary human alveolar osteoblasts cultured on collagen scaffolds into 3.5 mm cranial defects in SCID mice.[109] Taken together, the different investigations demonstrate that osteogenic capabilities of progenitor cells differ by source and degree of differentiation.
A combination of growth factors and stem cell supplementation have been tested by a number of groups. In a swine model, Chang et al. investigated the ability of collagen scaffolds carrying autologous bone marrow stromal cells (BMSCs) transduced with adenoviruses expressing BMP-2 for 3 months.[110] Whereas addition of BMSCs yielded a small amount of bone healing, BMP-2 increased the efficiency of calvarial healing at 3 months. In this work, a two-dimensional analysis of a three-dimensional reconstructed CT scan showed approximately 63% of bone fill. A similar comparison using autologous muscle derived stem cells (MDSCs) seeded on collagen scaffolds demonstrated that healing of murine cranial defects only occurred in the presence of BMP-2 transduction from an adenoviral source.[111]
Ultimately, all of the above studies have concluded that healing of cranial defects using collagen alone as a scaffold does not occur. Supplementation with growth factors or progenitor cells or both are necessary to detect any reasonable amount of bone healing in the presence of collagen only materials.
4.2. Collagen with Glycosaminoglycans
GAGs in the extracellular matrix serve a number of roles, of which cell adhesion and non-covalent interactions with growth factors are likely to be of the greatest significance in regenerative medicine.[112] Although originally evaluated in skin and peripheral nerve regeneration, Farrell et al. in 2006 first demonstrated that addition of GAGs to collagen scaffolds (also called CG or Col-GAG) resulted in a microenvironment with the capabilities of supporting osteogenesis and chondrogenesis of hMSCs.[113] In this initial work, the U0126 small molecule MEK inhibitor was found to inhibit osteogenic differentiation suggestive that the MAP kinase pathway was induced during differentiation. Our group has reported similar results in the osteogenic mechanism of Col-GAG scaffolds with the upregulation of phosphorylated ERK1/2 during osteogenic differentiation.[2,98] Recently, we have also tested the PD98059 small molecule MEK inhibitor on hMSC differentiation on CG scaffolds and noted inhibition of osteogenesis and matrix mineralization (JCL, unpublished observations).
The incorporation of glycosaminoglycans into collagen-based scaffolds has been investigated by several groups due to its capacity to regulate physiology through interactions with growth factors.[13,101,114,115] Termed the “sulfation code”, GAGs may function as an organizer of extracellular signals. Hortensius and Harley evaluated a panel of GAGs including hyaluronic acid, chondroitin sulfate, and heparan sulfate.[115] These investigators found that sulfated GAGs when provided even at low concentrations (collagen:GAG 11.28:1) may provide a depot of growth factors.
To evaluate an alternative route of osteogenic induction via endochondral ossification, Thompson et al. tested a collagen hyaluronic acid (CHyA) scaffold for osteogenic differentiation.[116] When rat bone marrow-derived MSCs were cultured first with TGF-β to induce chondrogenesis followed by osteogenic media, CHyA was found to display less matrix mineralization in comparison to a collagen hydroxyapatite scaffold. Following the sequential culture approach, scaffolds were implanted into 7 mm rat cranial defects and the CHyA scaffolds were seen to result in the maximum amount of bone formation with a BV/TV ratio of around 40%. The interpretation that this may be related to improved vascularization and remodeling remains to be evaluated in greater depth.
4.3. Collagen with Mineral Content
Similar to collagen scaffolds, five commercially available preparations of collagen with mineral content exist and have been approved by the FDA for use as a bone void filler, primarily for spine or orthopaedic surgeries. Of the commercially available composites, two have been evaluated in cranial defect models. Healos (DePuy Spine Inc, Rayham, MA) is a composite of collagen coated with hydroxyapatite and has been approved for use in spinal surgeries in combination with bone marrow. In one comparison between Helistat and Healos in 8 mm rat calvarial defects, Healos did result in improved bone healing in a qualitative evaluation.[117] Similarly, Mastergraft (Medtronic Sofamor Danek, Memphis, TN) has been evaluated in critical sized cranial defects in a canine model.[118] Quantitation at 4 months following implantation was performed using histomorphometric analyses which demonstrated no differences when compared to demineralized bone matrices. Neither of the commercially available products are considered to be standard of care as primary modes of calvarial reconstruction and neither have definitive mechanistic or animal data.
In 2009, O’Brien and colleagues utilized a biphasic immersion process in developing highly porous, collagen scaffolds coated with calcium phosphate (CCP).[119] Their scaffold was characterized with a compressive modulus of 10.3 kPa in comparison to pure collagen scaffolds which have a compressive modulus of 0.28 kPa. When the mineral content was evaluated in detail, hydroxyapatite was found to be the major component of the mineral phase. Subsequently, the same investigators compared the CG to the CCP scaffold.[12] The compressive modulus of CG scaffolds was 1.0 kPa, which was significantly lower that CCP scaffolds but higher than pure collagen scaffolds which increased after seeding with primary rat BMSCs. Eight weeks after implantation into an 8 mm rat cranial defect, micro-CT analysis demonstrated that CCP far exceeded CG in percent BV/TV at 34% and that cell-free implantation performed better that scaffolds containing ex vivo expanded and differentiated BMSCs. Interestingly, the group also noted that one potential explanation for the improved healing in cell-free scaffolds may be related to an increase in M1 subtype macrophages in the presence of MSCs. The technique was later modified to a suspension method with dispersant aided precipitation and also evaluated in vivo demonstrating similar results.[120]
Although the majority of ECM-based materials have generally considered collagen as a base material, one group of investigators have approached scaffold fabrication from the mineral standpoint.[121] Biphasic calcium phosphate (BCP) composed of a 60:40 HA to β-tricalcium phosphate (TCP) ratio coated with a pepsin-cleaved fragment of collagen, termed atelocollagen (AT-Col), was compared to BCP alone in 8 mm rabbit calvarial defects. Their group noted that AT-Col/BCP composite scaffolds demonstrated approximately 34% healing at 8 weeks in comparison to approximately 8% in the BCP only scaffolds. In addition, improved bone healing was correlated to increased osteogenic gene expression, TRAP staining, and CD31 staining suggestive of increased osteogenesis, osteoclastogenesis, and angiogenesis. Taken in total, collagen and mineral content clearly have a synergistic effect on osteogenic differentiation.
One potential explanation for the synergistic effects of mineral content on osteogenic differentiation may be due to the increasing understanding of the signaling capabilities of inorganic ions. The BMP signaling pathway has been shown to have a key role in osteogenesis driven by calcium phosphate ions. Specifically, calcium ion has been found to upregulate gene transcription of BMP-2,[122,123] transcription factors in osteogenic differentiation (Runx2, osterix),[123,124] and late osteogenic genes osteopontin (OPN) and osteocalcin (OCN).[125] However, the interplay between extracellular calcium ion and BMP-2 gene expression is poorly understood although one group has proposed a mechanism involving the Calcium Sensing Receptor (CaSR) and L-type voltage-gated calcium channels (VGCCs).[123] Via a proposed signaling pathway by Barradas et al., extracellular calcium ions are internalized via L-type VGCCs which in turn activates protein kinase C (PKC). PKC phosphorylates the CaSR, diminishing its sensitivity to calcium. Simultaneously, PKC activates the Ras-MAP-kinase signaling pathway. The resulting phosphorylated ERK activates transcription of the c-Fos transcription factor, which binds to c-Jun, forming the AP-1 transcriptional activator, which binds to the AP-1 binding site in the promoter region of the BMP-2 gene, activating BMP-2 transcription.[123]
Similarly, exposure of progenitor cells to phosphate has recently been shown to yield osteogenic effects. Like calcium ions, phosphate ion upregulates osteogenic markers such as osteopontin and osteocalcin.[126,127] Phosphate ion also upregulates BMP-2, Smad 1/5/8, and β-catenin, thereby stimulating osteogenic gene expression and matrix mineralization.[128] A key constituent to phosphate metabolism is the phosphate transporter solute carrier family 20, member 1 (SLC20a1 or PiT-1), a sodium-phosphate symporter that transports extracellular phosphate ions into the cytoplasm, playing a key role in osteoblast and progenitor cell mineralization.[126,129] Via a proposed mechanism by Shih et al., extracellular phosphate enters the cell through SLC20a1 and subsequently enters the mitochondria, serving as a substrate for ATP synthesis. ATP is then secreted and metabolized into adenosine by membrane-bound ectonucleotidases. Adenosine via the A2b adenosine receptor to promote osteogenic differentiation. The key role of exogenous adenosine signaling in osteogenic differentiation is supported with studies of adenosine and A2b adenosine receptor inhibitors and knock-out studies.[126,130] Likely utilizing the phosphate ion-mediated osteogenic pathway, Bae et al. reported on healing of in vivo murine calvarial defects 5 mm in diameter using collagen scaffolds seeded with two polyphosphate compounds.[131] While the amount of increased mineralization was not dramatic, addition of sodium triphosphate demonstrated a 2% increase in BV/TV beyond collagen alone.
Using a co-precipitation approach, one group of investigators utilized a cocktail of salts termed simulated body fluid (SBF) to produce a collagen carbonated hydroxyapatite (Col-cHA).[132,133] In immunodeficient murine models, 3.5 mm calvarial defects demonstrated radiographic bone healing that was comparable to a commercially available collagen hydroxyapatite material (Healos, DePuy). However, unlike the majority of the previous studies, the investigations with the Col-cHA scaffold have been largely performed with defect healing times of 3–4 weeks and evaluated using two-dimensional plain radiographs. Thus, true comparison is difficult to ascertain. In addition, unlike other formulations which have been predominantly bovine, the collagen source in these investigations was rat tail. The significance of xenogeneic source for collagen is yet to be determined. In a followup study, Col-cHA scaffolds with BMSCs were cultured in a basement membrane extract (BME) gel, containing over 40 different growth factors as well as laminin, entactin, and collagen IV.[132] Although the results were somewhat equivocal, the BME gel was thought to potentially improve cell attachment.
Alternative mineralization techniques have also been considered. Silicic acid/silica has been found to stimulate type I collagen synthesis and osteoblast differentiation in human osteoblast like cells in vitro.[134] In addition, a role for silicon in bone development has been reported, thereby suggesting a role for silicon in bone regeneration.[135] Silicified collagen scaffolds (SCS) fabricated with intrafibrillar deposition of amorphous silica nanoparticles within collagen has been fabricated. The particular impetus for evaluating silica was due to the idea that it has the ability to modulate both the anabolic and catabolic aspects of bone metabolism in that it may stimulate osteogenesis as well as inhibit osteoclast differentiation. Using a combination silicified and calcified collagen scaffold, Jiao and colleagues demonstrated in primary murine MSCs both upregulate osteogenic genes including alkaline phosphatase, collagen I, and osteopontin as well as upregulation of osteoprotegerin at the same time as downregulation of RANKL.[136] Interestingly, in a follow up study with the same group, silicified scaffolds fabricated from demineralized bone matrix allograft, demonstrated monocyte regulation and angiogenesis in in vivo 3 mm murine calvarial defects.[134] At 12 weeks following cell-free implantation, micro-CT quantification confirmed a BV/TV ratio of 90%.
4.4. Collagen with Glycosaminoglycans and Mineral Content
With the understanding that CG scaffolds could support osteogenic differentiation, in 2008, Gibson and colleagues developed a mineralized CG (also called MCG or MC-GAG) scaffold using concurrent mapping to mimic the biological components of bone further.[137] Until that point, the majority of methods for mineral content incorporation in collagen scaffold incorporation was focused on extrafibrillar incorporation of minerals through immersion. Such techniques resulted in mineralized scaffolds that were not uniform and did not truly recapitulate bone. This initial work was subsequently expanded by Harley et al. to further evaluate mechanical and compositional properties of a 50 wt% scaffold with a 420 μm mean pore size.[6] In the MCG scaffold, the calcium phosphate phase was characterized to be almost entirely composed of brushite with a minor amount of monetite.
With the ability to modulate essentially every physical and chemical characteristic of the MCG scaffold, biological responses have also been able to be controlled.[4,115] In close collaboration with Harley, our group subsequently evaluated the differences in osteogenesis in primary bone marrow-derived human mesenchymal stem cells (hMSCs).[2,7,10,11,138] Our initial experience in using pure collagen scaffolds (Helistat) was decidedly unimpressive during in vitro osteogenic differentiation.[95] The primary reason was due to the severe structural contraction that occurred during mineralization. Both CG and MCG were found to exhibit significantly improved abilities to withstand contraction. In addition, this was further improved with crosslinking using EDC/NHS. Secondly, both CG and MCG demonstrated improved mineralization with the MCG scaffold far exceeding both CG and collagen.
When the properties of the scaffolds were examined, two differences stood out. First, elution of calcium and phosphate ions were noted in MCG primarily for the first 3 weeks of culture and may potentially contribute to osteogenic signaling through the pathways mentioned above.[11] Secondly, biomechanical testing of the scaffolds demonstrated differences in the stiffness of each material in that the elastic moduli for hydrated, empty CG and MCG scaffolds were 208 Pa and 4 kPa, respectively. Following 8 weeks of culture, the elastic moduli of MCG increased to ≈70 kPa in the presence of hMSCs and osteogenic media. The mechanical differences may be important for cellular adhesion and migration as MCG demonstrated significantly more cellular material in the center of the scaffold when compared to CG scaffolds.[2,7,98] When we further compared osteogenic differentiation on CG and MCG scaffolds in relationship to BMP-2 induction, minimal increases in osteogenic gene expression were found in either hMSCs or rabbit BMSCs.[2,138] However, in vitro mineralization on MCG scaffolds was not improved with BMP-2 whereas a minor increase was seen in CG scaffolds. This finding was also seen in 14 mm rabbit cranial defects 8 weeks following implantation. Surprisingly, similar to the O’Brien study, when we compared cranial defect healing of cell-free scaffolds to both scaffolds seeded with ex vivo expanded BMSCs or scaffolds seeded with BMSCs and treated with BMP-2, MCG cell-free scaffolds demonstrated the highest BV/TV percentage and highest strength and stiffness on microindentation analysis. Further evaluation of the mechanism for elevated osteogenesis revealed that MCG scaffolds stimulated phosphorylation of Smad1/5, the major downstream mediator of the canonical BMPR signaling, even in the absence of BMPs suggesting an autogenous activation of the pathway. This autogenous activation was subsequently confirmed by detection of elevated BMP-2, BMP-4, and BMP-9 expression stimulated by the MCG scaffolds. Both Smad1/5 phosphorylation and transcriptional activation of osteogenic BMPs were not evident in CG scaffolds. The differences in CG and MCG in hMSC differentiation and activation of intracellular signaling molecules suggested that the materials alone instructed differential signaling pathways (Figure 7).
Figure 7.

Proposed Differential Signaling Pathways Activated by CG and MCG Scaffolds. CG (Col-GAG) and MCG (MC-GAG) activate different intracellular signaling pathways in MSCs during osteogenic differentiation. MCG (left) autogenously upregulates BMPR activation as detected by phosphorylation of Smad1/5/8 and gene expression of BMP-2, BMP-9, and BMP-4. This is correlated to both in vitro mineralation of MSCs and in vivo cranial defect healing. Left insert with micro-CT of explanted rabbit calvarium with regenerated bone after 12 weeks of cell-free MCG implantation. CG (right) does not activate Smad1/5/8 phosphorylation but does activate different BMP-4, BMP-7, and BMP-9 expression with ERK1/2 phosphorylation. In vitro MSC mineralization and in vivo rabbit cranial defect healing (right insert with micro-CT of explanted rabbit calvarium after 12 weeks of cell-free CG implantation) is diminished in comparison to MCG albeit detectable. Unlike MCG, CG-mediated osteogenic differentiation is improved with exogenous BMP-2 stimulation.
Approached from a different manner, Zeitouni et al. cultured hMSCs in osteogenic media and isolated a combination of cell and ECM lysate.[139] This lysate, considered to be ECM-enriched, was further dehydrated and utilized as a scaffold for evaluating hMSC directed calvarial regeneration in a 4 mm murine defect. Interestingly, the mineral component in this scaffold was also brushite, indicative that hMSCs undergoing osteogenic differentiation produce brushite during matrix mineralization. In combination with hMSCs treated with an inhibitor of peroxisome proliferator activated receptor-γ (PPAR-γ), this particular biological scaffold was found to be more effective in bone regeneration. However, there are significant unknowns in the exact composition of this scaffold, thus the main driver for mineralization in this situation is not completely clear. Furthermore, as this does not represent completely purified ECM, it is difficult to state whether there are growth factors that are at play in regeneration. This study again underscores the significance of instructive signals for MSC differentiation.
5. Conclusions and Future Directions: Are we almost there yet?
Death Valley in California is a vast expanse of desert for which the vegetation is minimal, illusions are maximal, and the discovery of an oasis may or may not actually occur within one lifetime. It is a metaphor for the great divide between bench science and clinical advancement. Despite the hundreds of publications on bone tissue engineering and the 30 years of work that have been put into investigation, the question of whether or not we have actually achieved a viable strategy to clinical translation remains elusive. However, the increased understanding of the significance of the extracellular matrix and the advances in bio-material fabrication have placed a new optimism on regenerative medicine as we now have an important reagent that can be finely tuned and optimized to control both simple and composite tissue synthesis. Additionally, this understanding is now bringing together the diverse fields of materials science, bone biology, cell biology, potentially cell electro-physiology, and clinicians.
In this new era, the potential for achieving clinical translation ought to be met with optimism but also with a resolute consensus that we must become more systematic in our efforts for fabrication, functional characterization, in vivo testing, and clinical trials. In terms of fabrication, this current review has focused primarily on ECM-inspired, collagen-based materials generated using the major organic and inorganic components of bone with testing in in vivo cranial defect models. Less prominent components of bone including glycoproteins and lipoproteins were not examined. Various other materials that are not naturally found in bone, such as graphene oxide-based composites, were also not examined in this review but may be of potential future interest in bone regeneration.[140] The sheer numbers and combinations of collagen-based scaffolds, even within relatively narrow confines, suggest that a more directed method should be considered with optimization at every step of the fabrication process. That is, we should optimize from the best material or materials reported, rather than to work from materials that have been demonstrated to yield suboptimal results for the same application. For this to occur, we must have quantifiable standardized methods of comparison that can be used from investigator to investigator and material to material with the expectation that results from these measures ought to be part of the literature. For example, while a number of the studies in this review have used micro-CT scanning, several have used plain radiographs and others have used only gross examination. The inability to truly compare these materials is significant as it is difficult to definitively state which method of fabrication is optimal and which components are absolutely necessary.
When one critically evaluates a number of the reports within this review, one also cannot help but be inundated with an immense variety of cell types and cell lines used with variable genetic modifications and growth factor additions. Most cell biologists would agree that immortalized cell lines are, by definition, abnormal. Thus, we must re-evaluate the convenience of utilizing cell lines for in vitro studies as they may not yield comparable results in primary human cells. Furthermore, the continued reliance on growth factors or growth factor cocktails is likely to yield the same conclusions as has been demonstrated in the past: that growth factors will cause unintended complications and ultimately fall out of favor in the clinical realm. Similar to understanding signaling pathways in normal cell physiology or specific pathologies, we should consider regeneration on specific materials as a separate cellular micro-environment. That is, osteogenic differentiation and the interaction between osteoprogenitors and other cell types should be characterized down to the specific signaling pathways activated or inhibited such that fine control can be further accomplished. Lastly, in the selection of in vivo animal models, both the clinical problem at hand and the potential models required for regulatory agencies to approve product development should be kept in mind. In terms of clinical problems, the approach for in vivo model selection should ideally start with some degree of input from clinicians who specifically treat the clinical problem in question such that the in vivo model recapitulates an actual clinical issue. In the setting of calvarial defects, for example, a full thickness calvarial defect (involving both cortices) is a true clinical problem whereas a partial thickness calvarial defect (involving only one cortex) is not. As it is likely that small animal models will only be useful as a portion of preclinical data in preparation for clinical testing, testing in medium or large animal models should be considered as a logical progression following small animal testing.
Acknowledgments
This work was supported by the US Department of Veterans Affairs under award numbers IK2 BX002442-01A2 (JCL), the Bernard G. Sarnat Endowment for Craniofacial Biology (JCL), the Aramont Foundation (JCL), and the Jean Perkins Foundation (JCL).
Biography

Justine C. Lee, MD, PhD, FACS is the Bernard G. Sarnat Endowed Chair in Craniofacial Biology and an Assistant Professor of Surgery in the Division of Plastic and Reconstructive Surgery at the UCLA David Geffen School of Medicine. Dr. Lee received both her MD and PhD in Immunology through the National Institutes of Health Medical Scientist Training Program at the University of Chicago Pritzker School of Medicine. Subsequently, she completed a plastic and reconstructive surgery residency at the University of Chicago and a craniofacial surgery fellowship at UCLA. Clinically, Dr. Lee is a craniofacial reconstructive surgeon who specializes in complex congenital, post-traumatic, and post-oncologic deformities. Her research interests are a combination of basic and clinical science respectively focused on regenerative technologies for skeletal defects independent of exogenous growth factors or ex vivo progenitor cell amplification for rapid translation and improved clinical outcomes in craniofacial surgery.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.a) Kua HY, Liu H, Leong WF, Li L, Jia D, Ma G, Hu Y, Wang X, Chau JF, Chen YG, Mishina Y, Boast S, Yeh J, Xia L, Chen GQ, He L, Goff SP, Li B. Nat Cell Biol. 2012;14:727. doi: 10.1038/ncb2528. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lai CF, Cheng SL. J Biol Chem. 2002;277:15514. doi: 10.1074/jbc.M200794200. [DOI] [PubMed] [Google Scholar]; c) Shirakabe K, Yamaguchi K, Shibuya H, Irie K, Matsuda S, Moriguchi T, Gotoh Y, Matsumoto K, Nishida E. J Biol Chem. 1997;272:8141. doi: 10.1074/jbc.272.13.8141. [DOI] [PubMed] [Google Scholar]
- 2.Ren X, Bischoff D, Weisgerber DW, Lewis MS, Tu V, Yamaguchi DT, Miller TA, Harley BA, Lee JC. Biomaterials. 2015;50:107. doi: 10.1016/j.biomaterials.2015.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Caliari SR, Ramirez MA, Harley BA. Biomaterials. 2011;32:8990. doi: 10.1016/j.biomaterials.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Caliari SR, Harley BA. Biomaterials. 2011;32:5330. doi: 10.1016/j.biomaterials.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Caliari SR, Mozdzen LC, Armitage O, Oyen ML, Harley BA. J Biomed Mater Res A. 2014;102:917. doi: 10.1002/jbm.a.35058. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Caliari SR, Gonnerman EA, Grier WK, Weisgerber DW, Banks JM, Alsop AJ, Lee JS, Bailey RC, Harley BA. Adv Healthcare Mater. 2014;4:58. doi: 10.1002/adhm.201400252. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Caliari SR, Harley BA. Adv Healthcare Mater. 2014;3:1086. doi: 10.1002/adhm.201300646. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Harley BA, Leung JH, Silva EC, Gibson LJ. Acta Biomateralia. 2007;3:463. doi: 10.1016/j.actbio.2006.12.009. [DOI] [PubMed] [Google Scholar]; g) Lynn AK, Best SM, Cameron RE, Harley BA, Yannas IV, Gibson LJ, Bonfield W. J Biomed Mater Res A. 2010;92:1057. doi: 10.1002/jbm.a.32415. [DOI] [PubMed] [Google Scholar]; h) Weisgerber DW, Kelkhoff DO, Caliari SR, Harley BA. J Mech Behav Biomed Mater. 2013;28:26. doi: 10.1016/j.jmbbm.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Keogh MB, O’Brien FJ, Daly JS. Acta Biomateralia. 2010;6:4305. doi: 10.1016/j.actbio.2010.06.001. [DOI] [PubMed] [Google Scholar]; j) Neumann AJ, Alini M, Archer CW, Stoddart MJ. Tissue Eng Part A. 2013;19:1285. doi: 10.1089/ten.TEA.2012.0411. [DOI] [PubMed] [Google Scholar]; k) Raftery RM, Woods B, Marques AL, Moreira-Silva J, Silva TH, Cryan SA, Reis RL, O’Brien FJ. Acta Biomateralia. 2016;43:160. doi: 10.1016/j.actbio.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 4.Caliari SR, Weisgerber DW, Ramirez MA, Kelkhoff DO, Harley BA. J Mech Behav Biomed Mater. 2012;11:27. doi: 10.1016/j.jmbbm.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caliari SR, Harley BA. Tissue Eng Part A. 2014;20:2463. doi: 10.1089/ten.tea.2013.0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harley BA, Lynn AK, Wissner-Gross Z, Bonfield W, Yannas IV, Gibson LJ. J Biomed Mater Res A. 2010;92:1066. doi: 10.1002/jbm.a.32361. [DOI] [PubMed] [Google Scholar]
- 7.Lee JC, Pereira CT, Ren X, Huang W, Bischoff D, Weisgerber DW, Yamaguchi DT, Harley BA, Miller TA. J Craniofac Surg. 2015;26:1992. doi: 10.1097/SCS.0000000000001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pence JC, Gonnerman EA, Bailey RC, Harley BA. Biomater Sci. 2014;2:1296. doi: 10.1039/C4BM00193A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren X, Weisgerber DW, Bischoff D, Lewis MS, Reid RR, He TC, Yamaguchi DT, Miller TA, Harley BA, Lee JC. Adv Healthcare Mater. 2016;5:1821. doi: 10.1002/adhm.201600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weisgerber DW, Caliari SR, Harley BA. Biomater Sci. 2015;3:533. doi: 10.1039/C4BM00397G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisgerber DW, Caliari SR, Harley BA. Biomater Sci. 2015;3:533. doi: 10.1039/C4BM00397G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons FG, Al-Munajjed AA, Kieran SM, Toner ME, Murphy CM, Duffy GP, O’Brien FJ. Biomaterials. 2010;31:9232. doi: 10.1016/j.biomaterials.2010.08.056. [DOI] [PubMed] [Google Scholar]
- 13.Murphy CM, Matsiko A, Haugh MG, Gleeson JP, O’Brien FJ. J Mech Behav Biomed Mater. 2012;11:53. doi: 10.1016/j.jmbbm.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 14.Walcott BP, Kuklina EV, Nahed BV, George MG, Kahle KT, Simard JM, Asaad WF, Coumans JV. PLoS One. 2011;6:e29193. doi: 10.1371/journal.pone.0029193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frieden TR, Houry D, Baldwin G. Centers for Disease Control and Prevention; [accessed April 2015]. https://www.cdc.gov/trau-maticbraininjury/pdf/tbi_report_to_congress_epi_and_rehab-a.pdf. [Google Scholar]
- 16.Lawrence T, Helmy A, Bouamra O, Woodford M, Lecky F, Hutchinson PJ. BMJ Open. 2016;6:e012197. doi: 10.1136/bmjopen-2016-012197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Slack GC, Gruszynski M, Lee JC. In: Craniosynostosis and Rare Craniofacial Clefts. Lee JC, editor. Nova Science Publishers; New York, USA: 2016. p. 67. [Google Scholar]; b) Yuan N, Lee JC. In: Craniosynostosis and Rare Craniofacial Clefts. Lee JC, editor. Nova Science Publishers; New York, USA: 2016. p. 17. [Google Scholar]; c) Willson TD, Lee JC. In: Craniosynostosis and Rare Craniofacial Clefts. Lee JC, editor. Nova Science Publishers; New York, USA: 2016. p. 79. [Google Scholar]
- 18.Fodstad H, Love JA, Ekstedt J, Fridén H, Liliequist B. Acta Neurochir. 1984;70:21. doi: 10.1007/BF01406039. [DOI] [PubMed] [Google Scholar]
- 19.Corallo F, Marra A, Bramanti P, Calabrò RS. Funct Neurol. 2014;29:273. [PMC free article] [PubMed] [Google Scholar]
- 20.Sedney CL, Dillen W, Julien T. J Neurosci Rural Pract. 2015;6:438. doi: 10.4103/0976-3147.158778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corallo F, De Cola MC, Lo Buono V, Marra A, De Luca R, Trinchera A, Bramanti P, Calabrò RS. Int J Neurosci. 2016;127:688. doi: 10.1080/00207454.2016.1235045. [DOI] [PubMed] [Google Scholar]
- 22.Segal DH, Oppenheim JS, Murovic JA. Neurosurgery. 1994;34:729. doi: 10.1227/00006123-199404000-00024. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki N, Suzuki S, Iwabuchi T. Acta Neurochir. 1993;122:49. doi: 10.1007/BF01446986. [DOI] [PubMed] [Google Scholar]
- 24.Lee JC, Kleiber GM, Pelletier AT, Reid RR, Gottlieb LJ. Plast Reconstr Surg. 2013;132:967. doi: 10.1097/PRS.0b013e31829f4b59. [DOI] [PubMed] [Google Scholar]
- 25.Tessier P. Clin Plast Surg. 1982;9:531. [PubMed] [Google Scholar]
- 26.Grant GA, Jolley M, Ellenbogen RG, Roberts TS, Gruss JR, Loeser JD. J Neurosurg. 2004;100:163. doi: 10.3171/ped.2004.100.2.0163. [DOI] [PubMed] [Google Scholar]
- 27.Rocque BG, Amancherla K, Lew SM, Lam S. J Neurosurg Pediatr. 2013;12:120. doi: 10.3171/2013.4.PEDS12605. [DOI] [PubMed] [Google Scholar]
- 28.Corliss B, Gooldy T, Vaziri S, Kubilis P, Murad G, Fargen K. World Neurosurg. 2016;96:510. doi: 10.1016/j.wneu.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 29.Yadla S, Campbell PG, Chitale R, Maltenfort MG, Jabbour P, Sharan AD. Neurosurgery. 2011;68:1124. doi: 10.1227/NEU.0b013e31820a5470. [DOI] [PubMed] [Google Scholar]
- 30.Fong AJ, Lemelman BT, Lam S, Kleiber GM, Reid RR, Gottlieb LJ. J Plast Reconstr Aesthet Surg. 2015;68:1036. doi: 10.1016/j.bjps.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 31.a) Lee HJ, Choi JW, Chung IW. J Craniofac Surg. 2014;25:1337. doi: 10.1097/SCS.0000000000000806. [DOI] [PubMed] [Google Scholar]; b) Fan KL, Federico C, Kawamoto HK, Bradley JP. J Craniofac Surg. 2012;23:2033. doi: 10.1097/SCS.0b013e318262d7fb. [DOI] [PubMed] [Google Scholar]
- 32.Tessier P, Kawamoto H, Posnick J, Raulo Y, Tulasne JF, Wolfe SA. Plast Reconstr Surg. 2005;116:72S. doi: 10.1097/01.prs.0000173841.59063.7e. [DOI] [PubMed] [Google Scholar]
- 33.Tessier P, Kawamoto H, Posnick J, Raulo Y, Tulasne JF, Wolfe SA. Plast Reconstr Surg. 2005;116:54S. doi: 10.1097/01.prs.0000173949.51391.d4. [DOI] [PubMed] [Google Scholar]
- 34.Oklund SA, Prolo DJ, Gutierrez RV, King SE. Clin Orthop Relat Res. 1986;205:269. [PubMed] [Google Scholar]
- 35.Pavlićevicć G, Lepić M, Perić P, Ivetić D, Roganović A, Roganović Z. J Craniomaxillofac Surg. 2017;45:312. doi: 10.1016/j.jcms.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 36.Chao MT, Jiang S, Smith D, DeCesare GE, Cooper GM, Pollack IF, Girotto J, Losee JE. Plast Reconstr Surg. 2009;123:976. doi: 10.1097/PRS.0b013e31819ba46f. [DOI] [PubMed] [Google Scholar]
- 37.Salyer KE, Gendler E, Squier CA. Plast Reconstr Surg. 1997;99:1721. [PubMed] [Google Scholar]
- 38.Marchac D, Greensmith A. J Plast Reconstr Aesthet Surg. 2008;61:744. doi: 10.1016/j.bjps.2007.10.055. [DOI] [PubMed] [Google Scholar]
- 39.O’Reilly EB, Barnett S, Madden C, Welch B, Mickey B, Rozen S. J Plast Reconstr Aesthet Surg. 2015;68:329. doi: 10.1016/j.bjps.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 40.a) Manson PN, Crawley WA, Hoopes JE. Plast Reconstr Surg. 1986;77:888. [PubMed] [Google Scholar]; b) Pennington DG, Stern HS, Lee KK. Plast Reconstr Surg. 1989;83:655. doi: 10.1097/00006534-198904000-00010. [DOI] [PubMed] [Google Scholar]
- 41.Blum KS, Schneider SJ, Rosenthal AD. Pediatr Neurosurg. 1997;26:33. doi: 10.1159/000121158. [DOI] [PubMed] [Google Scholar]
- 42.a) Afifi A, Djohan RS, Hammert W, Papay FA, Barnett AE, Zins JE. J Craniofac Surg. 2010;21:1205. doi: 10.1097/SCS.0b013e3181e17c1e. [DOI] [PubMed] [Google Scholar]; b) Shonka DC, Potash AE, Jameson MJ, Funk GF. Laryngoscope. 2011;121:2305. doi: 10.1002/lary.22191. [DOI] [PubMed] [Google Scholar]; c) Kshettry VR, Hardy S, Weil RJ, Angelov L, Barnett GH. Neurosurgery. 2012;70:8. doi: 10.1227/NEU.0b013e31822fef2c. [DOI] [PubMed] [Google Scholar]
- 43.Zins JE, Moreira-Gonzalez A, Papay FA. Plast Reconstr Surg. 2007;120:1332. doi: 10.1097/01.prs.0000279557.29134.cd. [DOI] [PubMed] [Google Scholar]
- 44.Thien A, King NK, Ang BT, Wang E, Ng I. World Neurosurg. 2015;83:176. doi: 10.1016/j.wneu.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Mukherjee S, Thakur B, Haq I, Hettige S, Martin AJ. Acta Neurochir. 2014;156:989. doi: 10.1007/s00701-014-2024-x. [DOI] [PubMed] [Google Scholar]
- 46.Durham SR, McComb JG, Levy ML. Neurosurgery. 2003;52:842. doi: 10.1227/01.neu.0000054220.01290.8e. [DOI] [PubMed] [Google Scholar]
- 47.Zins JE, Langevin CJ, Nasir S. J Craniofac Surg. 2010;21:1755. doi: 10.1097/SCS.0b013e3181c34675. [DOI] [PubMed] [Google Scholar]
- 48.Lindner D, Schlothofer-Schumann K, Kern BC, Marx O, Müns A, Meixensberger J. J Neurosurg. 2017;126:175. doi: 10.3171/2015.10.JNS151245. [DOI] [PubMed] [Google Scholar]
- 49.Jonkergouw J, van de Vijfeijken SE, Nout E, Theys T, Van de Casteele E, Folkersma H, Depauw PR, Becking AG. J Craniomaxillofac Surg. 2016;44:1266. doi: 10.1016/j.jcms.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Rosenthal G, Ng I, Moscovici S, Lee KK, Lay T, Martin C, Manley GT. Neurosurgery. 2014;75:523. doi: 10.1227/NEU.0000000000000477. [DOI] [PubMed] [Google Scholar]
- 51.Wang F, Hoang D, Medvecky M, Amankulor N, Teng E, Narayan D. J Craniofac Surg. 2012;23:1083. doi: 10.1097/SCS.0b013e31824e69b9. [DOI] [PubMed] [Google Scholar]
- 52.Sung JJ, Mapstone TB, El-Amm CA. J Craniofac Surg. 2011;22:1536. doi: 10.1097/SCS.0b013e31821da32f. [DOI] [PubMed] [Google Scholar]
- 53.a) Kohan E, Roostaeian J, Yuan JT, Fan KL, Federico C, Kawamoto H, Bradley JP. Ann Plast Surg. 2015;74:603. doi: 10.1097/01.sap.0000465206.62522.af. [DOI] [PubMed] [Google Scholar]; b) Ducy P, Karsenty G. Kidney Int. 2000;57:2207. doi: 10.1046/j.1523-1755.2000.00081.x. [DOI] [PubMed] [Google Scholar]
- 54.Mueller TD, Nickel J. FEBS Lett. 2012;586:1846. doi: 10.1016/j.febslet.2012.02.043. [DOI] [PubMed] [Google Scholar]
- 55.Miyazono K, Kamiya Y, Morikawa M. J Biochem. 2010;147:35. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 56.Koenig BB, Cook JS, Wolsing DH, Ting J, Tiesman JP, Correa PE, Olson CA, Pecquet AL, Ventura F, Grant RA. Mol Cell Biol. 1994;14:5961. doi: 10.1128/mcb.14.9.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, Kumar R, Grinberg AV. J Biol Chem. 2012;287:27313. doi: 10.1074/jbc.M112.377960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.a) Gilboa L, Nohe A, Geissendörfer T, Sebald W, Henis YI, Knaus P. Mol Biol Cell. 2000;11:1023. doi: 10.1091/mbc.11.3.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nohe A, Hassel S, Ehrlich M, Neubauer F, Sebald W, Henis YI, Knaus P. J Biol Chem. 2002;277:5330. doi: 10.1074/jbc.M102750200. [DOI] [PubMed] [Google Scholar]
- 59.Lou J, Tu Y, Li S, Manske PR. Biochem Biophys Res Commun. 2000;268:757. doi: 10.1006/bbrc.2000.2210. [DOI] [PubMed] [Google Scholar]
- 60.a) Lewandrowski KU, Nanson C, Calderon R. Spine J. 2007;7:609. doi: 10.1016/j.spinee.2007.01.011. [DOI] [PubMed] [Google Scholar]; b) Smoljanovic T, Bojanic I, Delimar D. Eur Spine J. 2009;18:920. doi: 10.1007/s00586-009-0959-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yee KS, Lee JC, Andrews BT, Bradley JP. Plast Reconstr Surg. 2014;133:63. doi: 10.1097/01.prs.0000445084.23234.03. [DOI] [PubMed] [Google Scholar]
- 61.Rollnick BR, Kaye CI, Nagatoshi K, Hauck W, Martin AO. Am J Med Genet. 1987;26:361. doi: 10.1002/ajmg.1320260215. [DOI] [PubMed] [Google Scholar]
- 62.Grabb WC. Plast Reconstr Surg. 1965;36:485. doi: 10.1097/00006534-196511000-00001. [DOI] [PubMed] [Google Scholar]
- 63.Lewkonia RM, Lowry RB. Am J Med Genet. 1983;14:385. doi: 10.1002/ajmg.1320140220. [DOI] [PubMed] [Google Scholar]
- 64.Fan WS, Mulliken JB, Padwa BL. J Oral Maxillofac Surg. 2005;63:330. doi: 10.1016/j.joms.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 65.Zysset PK, Guo XE, Hoffler CE, Moore KE, Goldstein SA. J Biomech. 1999;32:1005. doi: 10.1016/s0021-9290(99)00111-6. [DOI] [PubMed] [Google Scholar]
- 66.Boruah S, Subit DL, Paskoff GR, Shender BS, Crandall JR, Salzar RS. J Mech Behav Biomed Mater. 2017;65:688. doi: 10.1016/j.jmbbm.2016.09.041. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Zou D, Li Z, Huang P, Li D, Shao Y, Wang H, Chen Y. Med Sci Monit. 2014;20:1808. doi: 10.12659/MSM.892278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.a) Hoffler CE, Moore KE, Kozloff K, Zysset PK, Brown MB, Goldstein SA. Bone. 2000;26:603. doi: 10.1016/s8756-3282(00)00268-4. [DOI] [PubMed] [Google Scholar]; b) Turner CH, Rho J, Takano Y, Tsui TY, Pharr GM. J Biomech. 1999;32:437. doi: 10.1016/s0021-9290(98)00177-8. [DOI] [PubMed] [Google Scholar]
- 69.Werler MM, Starr JR, Cloonan YK, Speltz ML. J Craniofac Surg. 2009;20(Suppl 1):664. doi: 10.1097/SCS.0b013e318193d5d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clauser LC, Tieghi R, Consorti G. J Craniomaxillofac Surg. 2010;38:605. doi: 10.1016/j.jcms.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 71.Sommer A, Gambichler T, Bacharach-Buhles M, von Rothenburg T, Altmeyer P, Kreuter A. J Am Acad Dermatol. 2006;54:227. doi: 10.1016/j.jaad.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 72.Werler MM, Sheehan JE, Hayes C, Padwa BL, Mitchell AA, Mulliken JB. Cleft Palate Craniofac J. 2004;41:494. doi: 10.1597/03-110.1. [DOI] [PubMed] [Google Scholar]
- 73.Pensler JM, Murphy GF, Mulliken JB. Plast Reconstr Surg. 1990;85:669. [PubMed] [Google Scholar]
- 74.Robinson IB, Sarnat BG. Am J Anat. 1955;96:37. doi: 10.1002/aja.1000960103. [DOI] [PubMed] [Google Scholar]
- 75.Parfitt AM. Bone. 2002;30:5. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 76.Hedgecock NL, Hadi T, Chen AA, Curtiss SB, Martin RB, Hazelwood SJ. Bone. 2007;40:627. doi: 10.1016/j.bone.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 77.Matsuo K, Irie N. Arch Biochem Biophys. 2008;473:201. doi: 10.1016/j.abb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 78.Bar-Shavit Z. J Cell Biochem. 2007;102:1130. doi: 10.1002/jcb.21553. [DOI] [PubMed] [Google Scholar]
- 79.Robling AG, Castillo AB, Turner CH. Annu Rev Biomed Eng. 2006;8:455. doi: 10.1146/annurev.bioeng.8.061505.095721. [DOI] [PubMed] [Google Scholar]
- 80.Castro-Malaspina H, Gay RE, Resnick G, Kapoor N, Meyers P, Chiarieri D, McKenzie S, Broxmeyer HE, Moore MA. Blood. 1980;56:289. [PubMed] [Google Scholar]
- 81.Burger EH, Klein-Nulend J. FASEB J. 1999;13(Suppl):S101. [PubMed] [Google Scholar]
- 82.Everts V, Korper W, Hoeben KA, Jansen ID, Bromme D, Cleutjens KB, Heeneman S, Peters C, Reinheckel T, Saftig P, Beertsen W. J Bone Miner Res. 2006;21:1399. doi: 10.1359/jbmr.060614. [DOI] [PubMed] [Google Scholar]
- 83.Merrild DM, Pirapaharan DC, Andreasen CM, Kjærsgaard-Andersen P, Møller AM, Ding M, Delaissé JM, Søe K. Bone Res. 2015;3:15032. doi: 10.1038/boneres.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wan Q, Schoenmaker T, Jansen ID, Bian Z, de Vries TJ, Everts V. Bone. 2016;86:10. doi: 10.1016/j.bone.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 85.Buckwalter JA, Glimcher MJ, Cooper RR, Recker R. Instructional Course Lectures. 1996;45:371. [PubMed] [Google Scholar]
- 86.Cheng H, Jiang W, Phillips FM, Haydon RC, Peng Y, Zhou L, Luu HH, An N, Breyer B, Vanichakarn P, Szatkowski JP, Park JY, He TC. J Bone Joint Surg Am. 2003;85-A:1544. doi: 10.2106/00004623-200308000-00017. [DOI] [PubMed] [Google Scholar]
- 87.Luu HH, Song WX, Luo X, Manning D, Luo J, Deng ZL, Sharff KA, Montag AG, Haydon RC, He TC. J Orthop Res. 2007;25:665. doi: 10.1002/jor.20359. [DOI] [PubMed] [Google Scholar]
- 88.Martino MM, Tortelli F, Mochizuki M, Traub S, Ben-David D, Kuhn GA, Müller R, Livne E, Eming SA, Hubbell JA. Sci Transl Med. 2011;3:100ra89. doi: 10.1126/scitranslmed.3002614. [DOI] [PubMed] [Google Scholar]
- 89.Allam KA, Wan DC, Kawamoto HK, Bradley JP, Sedano HO, Saied S. Plast Reconstr Surg. 2011;127:812. doi: 10.1097/PRS.0b013e318200aa08. [DOI] [PubMed] [Google Scholar]
- 90.van den Bos T, Speijer D, Bank RA, Brömme D, Everts V. Bone. 2008;43:459. doi: 10.1016/j.bone.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 91.Szpalski C, Wetterau M, Barr J, Warren SM. Tissue Eng Part B Rev. 2012;18:246. doi: 10.1089/ten.TEB.2011.0427. [DOI] [PubMed] [Google Scholar]
- 92.Meyer F, Wardale J, Best S, Cameron R, Rushton N, Brooks R. J Orthop Res. 2012;30:864. doi: 10.1002/jor.22019. [DOI] [PubMed] [Google Scholar]
- 93.Dujardin J, Vandenneucker H, Bellemans J. Arthroscopy. 2008;24:238. doi: 10.1016/j.arthro.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 94.a) Jiang WW, Su SH, Eberhart RC, Tang L. J Biomed Mater Res A. 2007;82:492. doi: 10.1002/jbm.a.31175. [DOI] [PubMed] [Google Scholar]; b) Bergsma EJ, Rozema FR, Bos RR, de Bruijn WC. J Oral Maxillofac Surg. 1993;51:666. doi: 10.1016/s0278-2391(10)80267-8. [DOI] [PubMed] [Google Scholar]
- 95.Kruger EA, Im DD, Bischoff DS, Pereira CT, Huang W, Rudkin GH, Yamaguchi DT, Miller TA. Plast Reconstr Surg. 2011;127:2301. doi: 10.1097/PRS.0b013e318213a004. [DOI] [PubMed] [Google Scholar]
- 96.Mozdzen LC, Vucetic A, Harley BA. J Mech Behav Biomed Mater. 2017;66:28. doi: 10.1016/j.jmbbm.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.a) Di Luca A, Longoni A, Criscenti G, Mota C, van Blitterswijk C, Moroni L. Biofabrication. 2016;8:045007. doi: 10.1088/1758-5090/8/4/045007. [DOI] [PubMed] [Google Scholar]; b) Baino F, Fiorilli S, Vitale-Brovarone C. Acta Biomateralia. 2016;42:18. doi: 10.1016/j.actbio.2016.06.033. [DOI] [PubMed] [Google Scholar]; c) Hu C, Zilm M, Wei M. J Biomed Mater Res A. 2016;104:1153. doi: 10.1002/jbm.a.35649. [DOI] [PubMed] [Google Scholar]
- 98.Ren X, Tu V, Bischoff D, Weisgerber DW, Lewis MS, Yamaguchi DT, Miller TA, Harley BA, Lee JC. Biomaterials. 2016;89:67. doi: 10.1016/j.biomaterials.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kozlowska J, Sionkowska A. Int J Biol Macromol. 2015;74:397. doi: 10.1016/j.ijbiomac.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 100.AbdulQader ST, Rahman IA, Thirumulu KP, Ismail H, Mahmood Z. J Biomater Appl. 2016;30:1300. doi: 10.1177/0885328215625759. [DOI] [PubMed] [Google Scholar]
- 101.Murphy CM, Duffy GP, Schindeler A, O’brien FJ. J Biomed Mater Res A. 2016;104:291. doi: 10.1002/jbm.a.35567. [DOI] [PubMed] [Google Scholar]
- 102.Hwang HD, Lee JT, Koh JT, Jung HM, Lee HJ, Kwon TG. Tissue Eng Part A. 2015;21:2125. doi: 10.1089/ten.TEA.2014.0571. [DOI] [PubMed] [Google Scholar]
- 103.a) Kitaori T, Ito H, Schwarz EM, Tsutsumi R, Yoshitomi H, Oishi S, Nakano M, Fujii N, Nagasawa T, Nakamura T. Arthritis Rheum. 2009;60:813. doi: 10.1002/art.24330. [DOI] [PubMed] [Google Scholar]; b) Fujio M, Yamamoto A, Ando Y, Shohara R, Kinoshita K, Kaneko T, Hibi H, Ueda M. Bone. 2011;49:693. doi: 10.1016/j.bone.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 104.Chang JB, Lee JC. Int J Stem Cell Res Ther. 2016;3:26. doi: 10.23937/2469-570x/1410026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ansari S, Moshaverinia A, Pi SH, Han A, Abdelhamid AI, Zadeh HH. Biomaterials. 2013;34:10191. doi: 10.1016/j.biomaterials.2013.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maraldi T, Riccio M, Pisciotta A, Zavatti M, Carnevale G, Beretti F, La Sala GB, Motta A, De Pol A. Stem Cell Res Ther. 2013;4:53. doi: 10.1186/scrt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zavatti M, Bertoni L, Maraldi T, Resca E, Beretti F, Guida M, La Sala GB, De Pol A. Life Sci. 2015;121:174. doi: 10.1016/j.lfs.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 108.Chamieh F, Collignon AM, Coyac BR, Lesieur J, Ribes S, Sadoine J, Llorens A, Nicoletti A, Letourneur D, Colombier ML, Nazhat SN, Bouchard P, Chaussain C, Rochefort GY. Sci Rep. 2016;6:38814. doi: 10.1038/srep38814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao Y, Qian H, Young WG, Bartold PM. Tissue Eng. 2003;9:1167. doi: 10.1089/10763270360728071. [DOI] [PubMed] [Google Scholar]
- 110.Chang SC, Chung HY, Tai CL, Chen PK, Lin TM, Jeng LB. J Biomed Mater Res A. 2010;94:433. doi: 10.1002/jbm.a.32685. [DOI] [PubMed] [Google Scholar]
- 111.Lee JY, Musgrave D, Pelinkovic D, Fukushima K, Cummins J, Usas A, Robbins P, Fu FH, Huard J. J Bone Joint Surg Am. 2001;83-A:1032. doi: 10.2106/00004623-200107000-00008. [DOI] [PubMed] [Google Scholar]
- 112.a) Hudalla GA, Murphy WL. Adv Funct Mater. 2011;21:1754. doi: 10.1002/adfm.201002468. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miguez PA, Terajima M, Nagaoka H, Mochida Y, Yamauchi M. Biochem Biophys Res Commun. 2011;405:262. doi: 10.1016/j.bbrc.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Farrell E, O’Brien FJ, Doyle P, Fischer J, Yannas I, Harley BA, O’Connell B, Prendergast PJ, Campbell VA. Tissue Eng. 2006;12:459. doi: 10.1089/ten.2006.12.459. [DOI] [PubMed] [Google Scholar]
- 114.Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard WA, Nishi A, Hsieh-Wilson LC. Nat Chem Biol. 2006;2:467. doi: 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- 115.Hortensius RA, Harley BA. Biomaterials. 2013;34:7645. doi: 10.1016/j.biomaterials.2013.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thompson EM, Matsiko A, Kelly DJ, Gleeson JP, O’Brien FJ. Tissue Eng Part A. 2016;22:556. doi: 10.1089/ten.TEA.2015.0457. [DOI] [PubMed] [Google Scholar]
- 117.Tcacencu I, Wendel M. J Mater Sci Mater Med. 2008;19:2015. doi: 10.1007/s10856-007-3284-2. [DOI] [PubMed] [Google Scholar]
- 118.He D, Genecov DG, Herbert M, Barcelo R, Elsalanty ME, Weprin BE, Opperman LA. J Neurosurg. 2010;112:319. doi: 10.3171/2009.1.JNS08976. [DOI] [PubMed] [Google Scholar]
- 119.Al-Munajjed AA, O’Brien FJ. J Mech Behav Biomed Mater. 2009;2:138. doi: 10.1016/j.jmbbm.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 120.Cunniffe GM, Curtin CM, Thompson EM, Dickson GR, O’Brien FJ. ACS Appl Mater Interfaces. 2016;8:23477. doi: 10.1021/acsami.6b06596. [DOI] [PubMed] [Google Scholar]
- 121.Kim BS, Yang SS, Lee J. J Biomed Mater Res A. 2017;105:1446. doi: 10.1002/jbm.a.36032. [DOI] [PubMed] [Google Scholar]
- 122.Viti F, Landini M, Mezzelani A, Petecchia L, Milanesi L, Scaglione S. PLoS One. 2016;11:e0148173. doi: 10.1371/journal.pone.0148173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barradas AM, Fernandes HA, Groen N, Chai YC, Schrooten J, van de Peppel J, van Leeuwen JP, van Blitterswijk CA, de Boer J. Biomaterials. 2012;33:3205. doi: 10.1016/j.biomaterials.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 124.Rosen V. Cytokine Growth Factor Rev. 2009;20:475. doi: 10.1016/j.cytogfr.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 125.Dvorak MM, Siddiqua A, Ward DT, Carter DH, Dallas SL, Nemeth EF, Riccardi D. Proc Natl Acad Sci USA. 2004;101:5140. doi: 10.1073/pnas.0306141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shih YR, Hwang Y, Phadke A, Kang H, Hwang NS, Caro EJ, Nguyen S, Siu M, Theodorakis EA, Gianneschi NC, Vecchio KS, Chien S, Lee OK, Varghese S. Proc Natl Acad Sci USA. 2014;111:990. doi: 10.1073/pnas.1321717111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beck GR, Jr, Zerler B, Moran E. Proc Natl Acad Sci USA. 2000;97:8352. doi: 10.1073/pnas.140021997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guerrero F, Herencia C, Almaden Y, Martinez-Moreno JM, Montes de Oca A, Rodriguez-Ortiz ME, Diaz-Tocados JM, Canalejo A, Florio M, Lopez I, Richards WG, Rodriguez M, Aguilera-Tejero E, Munoz-Castaneda JR. PLoS One. 2014;9:e89179. doi: 10.1371/journal.pone.0089179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yoshiko Y, Candeliere GA, Maeda N, Aubin JE. Mol Cell Biol. 2007;27:4465. doi: 10.1128/MCB.00104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Carroll SH, Wigner NA, Kulkarni N, Johnston-Cox H, Gerstenfeld LC, Ravid K. J Biol Chem. 2012;287:15718. doi: 10.1074/jbc.M112.344994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bae WJ, Auh QS, Kim GT, Moon JH, Kim EC. Differentiation. 2016;92:257. doi: 10.1016/j.diff.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 132.Villa MM, Wang L, Rowe DW, Wei M. PLoS One. 2014;9:e109568. doi: 10.1371/journal.pone.0109568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Villa MM, Wang L, Huang J, Rowe DW, Wei M. J Biomed Mater Res B. 2015;103:243. doi: 10.1002/jbm.b.33225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun JL, Jiao K, Niu LN, Jiao Y, Song Q, Shen LJ, Tay FR, Chen JH. Biomaterials. 2017;113:203. doi: 10.1016/j.biomaterials.2016.10.050. [DOI] [PubMed] [Google Scholar]
- 135.Carlisle EM. Calcif Tissue Int. 1981;33:27. doi: 10.1007/BF02409409. [DOI] [PubMed] [Google Scholar]
- 136.Jiao K, Niu LN, Li QH, Chen FM, Zhao W, Li JJ, Chen JH, Cutler CW, Pashley DH, Tay FR. Acta Biomateralia. 2015;19:23. doi: 10.1016/j.actbio.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kanungo BP, Silva E, Van Vliet K, Gibson LJ. Acta Biomateralia. 2008;4:490. doi: 10.1016/j.actbio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 138.Ren X, Tu V, Bischoff D, Weisgerber DW, Lewis MS, Yamaguchi DT, Miller TA, Harley BA, Lee JC. Biomaterials. 2016;89:67. doi: 10.1016/j.biomaterials.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zeitouni S, Krause U, Clough BH, Halderman H, Falster A, Blalock DT, Chaput CD, Sampson HW, Gregory CA. Sci Transl Med. 2012;4:132ra55. doi: 10.1126/scitranslmed.3003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.a) Zhang W, Chang Q, Xu L, Li G, Yang G, Ding X, Wang X, Cui D, Jiang X. Adv Healthcare Mater. 2016;5:1299. doi: 10.1002/adhm.201500824. [DOI] [PubMed] [Google Scholar]; b) Ruan J, Wang X, Yu Z, Wang Z, Zhang D, Huang Y, Zhou H, Bi X, Xiao C, Gu P, Fan X. Adv Funct Mater. 2016;26:1085. [Google Scholar]
- 141.a) Mozdzen LC, Rodgers R, Banks JM, Bailey RC, Harley BA. Acta Biomater. 2016;33:25. doi: 10.1016/j.actbio.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mozdzen LC, Thorpe SD, Screen HR, Harley BA. Adv Healthcare Mater. 2016;5:1731. doi: 10.1002/adhm.201600181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xia Z, Yu X, Jiang X, Brody HD, Rowe DW, Wei M. Acta Biomater. 2013;9:7308. doi: 10.1016/j.actbio.2013.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Niu LN, Jiao K, Ryou H, Yiu CK, Chen JH, Breschi L, Arola DD, Pashley DH, Tay FR. Angew Chem Int Ed. 2013;52:5762. doi: 10.1002/anie.201210259. [DOI] [PMC free article] [PubMed] [Google Scholar]