

Abstract
Traumatic brain injury (TBI) is a prevalent disorder, but no effective therapies currently exist. An underlying pathophysiology of TBI includes the pathological elevation of autophagy. β-Catenin, a downstream mediator of the canonical Wnt pathway, is a repressor of autophagy. The Wnt/β-catenin pathway plays a crucial role in cell proliferation and neuronal plasticity/repair in the adult brain. We hypothesized that activation of this pathway could promote neuroprotection and neural regeneration following TBI. In the controlled cortical impact (CCI) model of TBI in C57BL/6 mice (total n = 160), we examined intranasal application of recombinant Wnt3a (2 μg/kg) in a short-term (1 dose/day for 2 days) and long-term (1 dose/day for 7 days) regimen. Immunohistochemistry was performed at 1 to 14 days post-TBI to assess cell death and neurovascular regeneration. Western blotting measured canonical Wnt3a activity, expression of growth factors, and cell death markers. Longitudinal behavior assays evaluated functional recovery. In short-term experiments, Wnt3a treatment with a 60-min delay post-TBI suppressed TBI-induced autophagic activity in neurons (44.3 ± 6.98 and 4.25 ± 2.53 LC3+/NeuN+ double positive cells in TBI+Saline and TBI+Wnt3a mice, respectively; p < 0.0001, n = 5/group), reduced autophagic markers light chain 3 (LC3)-II and Beclin-1, as well as injury markers caspase-3 and matrix metalloproteinase 9 (MMP-9). The Wnt3a treatment reduced cell death and contusion volume (0.72 ± 0.07 mm2 and 0.26 ± 0.04 mm2 in TBI+Saline and TBI+Wnt3a mice, respectively; p < 0.001, n = 5/group). The 7-day Wnt3a treatment increased levels of β-catenin and growth factors glial-derived growth factor (GDNF) and vascular endothelial growth factor (VEGF). This chronic Wnt3a therapy augmented neurogenesis (0.52 ± 0.09 and 1.25 ± 0.13 BrdU+/NeuN+ co-labeled cells in TBI+Saline mice and TBI+Wnt3a mice, respectively; p < 0.01, n = 6/group) and angiogenesis (0.26 ± 0.07 and 0.74 ± 0.13 BrdU+/GLUT1+ co-labeled cells in TBI+Saline and TBI+Wnt3a mice, respectively; p = 0.014, n = 6/group). The treatment improved performance in the rotarod test and adhesive removal test. Targeting the Wnt pathway implements a unique combination of protective and regenerative approaches after TBI.
Keywords: : apoptosis, autophagy, β-catenin, functional recovery, regeneration, traumatic brain injury, Wnt
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity among children and young adults.1 Although neuroprotective strategies are being investigated, there currently exist no effective treatments, making innovation of new therapeutic approaches for TBI an unmet clinical need.2 TBI results in diverse pathophysiology, including the initial physical deformation and secondary effects, such as excitotoxicity, increased reactive oxygen species (ROS) production, and inflammation during the sub-acute phase of TBI (hours to days post-injury) that cause further cell death.3 The natural regenerative capacity of the central nervous system (CNS) is inadequate in replacing the lost neurons and restoring original connectivity and function, which warrants the need for exogenous interventions. Given the complexity of TBI and the need for a multi-factorial therapeutic approach, we evaluated a novel pharmacological therapy for TBI that is translationally applicable and can provide both neuroprotective and pro-regenerative effects to bolster recovery in the sub-acute phase of TBI and beyond.
The canonical Wnt pathway (also known as the Wnt/β-catenin pathway given its reliance upon β-catenin), is critical for proper neurogenesis during development and after injury, including neural stem cell division and differentiation.4–7 Wnt activation stabilizes intracellular levels of β-catenin, which then acts on its intranuclear target TCF/LEF (T-cell-specific transcription factor/lymphoid enhancer-binding factor 1) family of transcriptional transactivation factors to drive the expression of Wnt-dependent genes that promote cell survival, proliferation, and plasticity.8 The Wnt/β-catenin pathway also regulates autophagy9 and other adaptive responses to metabolic stress.10,11 Autophagy is a conserved adaptive response to stress, such as nutrient deprivation and hypoxia, through which proteins and organelles are degraded in autophagosomes and recycled.12,13 β-catenin and autophagy constitute a mutually regulated homeostatic duality. β-catenin negatively regulates autophagy via direct inhibition of autophagosome formation, as well as repression of the scaffold protein p62/SQSTM1 on a transcriptional level.9,14 Conversely, the microtubule-associated protein light chain 3 (LC3), a key autophagosomal component upregulated during autophagy, forms complexes with β-catenin for autolysosomal degradation.9 Normally, autophagic activity is suppressed by multiple factors, including β-catenin and mechanistic target of rapamycin (mTOR).15 However, pathological events such as nutrient deprivation that inhibit mTOR trigger the induction of autophagy,16 which then depletes β-catenin levels and disinhibits autophagosome formation to activate the cascade of events involved with autophagy.9,14,17
Because of this interplay between the Wnt/β-catenin pathway and the autophagy pathway, it allows for a novel approach of modulating autophagy activity through driving Wnt/β-catenin activity. Furthermore, TBI results in dysregulated autophagy, which manifests as lysosomal dysfunction, impaired clearance of autophagosomes, and increased autophagic cell death.18,19 To this end, we aimed to explore the translational potential of exogenous activation of the Wnt/β-catenin pathway to upregulate β-catenin levels as a therapeutic strategy to mitigate autophagic dysregulation and promote neuronal survival after TBI. Indeed, blockade of autophagy using phosphoinositide 3-kinase (PI3K) inhibitors has been shown to improve neuronal viability and functional outcomes following TBI,18 suggesting that suppression of autophagy by elevating β-catenin could confer similar neuroprotective benefits.
In this novel paradigm, we utilized the recombinant form of the canonical Wnt3a protein because of multiple considerations, including its facility to be delivered as a pharmacological agent and its multi-factorial benefits, which makes it more suitable to combat complex intractable disorders such as TBI. Aside from stabilization of autophagic activity, activation of the Wnt/β-catenin pathway results in other pro-recovery effects, including neuronal survival and neural regeneration by enhancing the division and differentiation of neural stem cells (NSCs)4,5,20 Importantly, Wnt signaling has been shown to not only be critical for NSC proliferation during development21-–23 but also serves as a key reactive pathway to promote neural plasticity and repair after brain injuries.22 Recently, the Wnt pathway has been identified as a therapeutic candidate, with Wnt3a overexpression by lentiviral injection successfully providing regenerative and functional benefits after ischemic stroke.6 In the present investigation, we aimed to upregulate Wnt3a via a pharmacological strategy, thus providing a greater translational applicability. The brain delivery of the recombinant Wnt protein was performed through the non-invasive intranasal route, which allows for crossing of small molecules, proteins, and even cells into the CNS.24–28 In a mild controlled cortical impact (CCI) TBI model of the mouse, we demonstrate the efficacy of intranasal Wnt therapy for suppressing autophagic and apoptotic cell death and promoting neurovascular repair, which together ultimately resulted in improved functional outcomes after the TBI insult.
Methods
Primary cortical neuron cultures and siRNA transfection
Primary cortical neurons were obtained from the cortex of gestational day 18 C57BL/6 mouse embryos (E18) according to published protocols.29 Dissected cells were plated on 6- or 24-well plates (five hemispheres/plate, ∼2.5 × 105 cells/plate) pre-coated with poly-D-lysine (Sigma, St. Louis, MO) and laminin (Sigma). The cells were maintained as a monolayer in 2 mL Neurobasal media with B-27 serum-free culture supplement and L-glutamine (Invitrogen, ThermoFisher, NY) until time of experiments. Cytosine arabinoside (ARA-C, 5 μM) was added on days 5–6 in vitro (DIV) to halt proliferation of glial cells for a nearly pure neuronal population.
Small interfering RNA (siRNA) transfection was carried out according to the Lipofectamine 2000 (Invitrogen) user manual. The required amount of β-catenin siRNA (1 μM; Cell Signaling, Danvers, MA) and Lipofectamine 2000 was diluted and mixed together in Opti-MEM I (Gibco, ThermoFisher, NY) for 10 min to allow for liposome formation. Cortical neurons (DIV 10–11) were then incubated with liposomes for 40 h. A control group was incubated with liposomes containing non-targeting siRNA (NT-siRNA, Invitrogen). After transfection, cells were washed with complete neuronal media, followed by exposure to oxygen-glucose deprivation (OGD).
Oxygen-glucose deprivation (OGD) and cell death assessment
OGD, an in vitro model well-established to induce autophagy, was performed in DIV 12–13 cortical cultures.30,31 In the OGD groups, media was exchanged for a physiological buffer solution lacking glucose (120 mM NaCl, 25 mM Tris-HCl, 5.4 mM KCl, 1.8 mM CaCl2, pH to 7.4 with NaOH). Cells were then incubated in a calibrated hypoxia chamber perfused with 5% CO2 and balanced nitrogen for a final ambient oxygen level of 0.2% for 3.5 h. Oxygen level was established, maintained, and monitored by the ProOx 360 sensor (Biospherix, NY). After 3.5 h, cells were returned to the normal oxygen condition (20% O2), and the existing OGD media was diluted by half with oxygenated complete neuronal culture media. During OGD, cortical neurons were incubated with either recombinant Wnt3a (100 ng/mL, R&D Systems, Minneapolis, MN), or Wnt3a plus XAV-939 (10 μM, Sigma). After 40 h, release of lactate dehydrogenase (LDH) into the medium was detected using an LDH-based cytotoxicity detection kit (Roche, Nutley, NJ), according to the user manual. Briefly, 50 μL medium and 50 μL mixture of reagent A and B were co-incubated for 30 min and then absorbance (492 nm) was detected using a spectrophotometer (Sunrise Instruments, LLC, Hebron, NH). Full kill neuronal cultures were treated with 0.5% Triton X-100 to generate the upper limit for normalization (normalized to be 100% absorbance).
Controlled cortical impact (CCI) TBI model of the mouse
CCI was induced in mice as previously described.32 C57BL/6 mice (8–12 week old, 160 total) were housed in standard cages with a 12-h light/dark cycle and had access to food and water ad libitum. The animal protocol was approved by the Emory University Institutional Animal Care and Use Committee (IACUC), in compliance with the National Institutes of Health (NIH) guidelines. For TBI surgery, mice were anesthetized with isoflurane (3% induction, 1.5% maintenance) and placed on a stereotaxic frame. After a midline skin incision, a 3.5-mm circular craniotomy was performed midway between the lambda and bregma, 2.0 mm to the right of the central suture using an electric microdrill (Dremel, Mount Prospect, IL). During the craniotomy, mice were excluded from the study if the dura mater was breached. CCI was induced with an electric impact device using a steel cylindrical flat impact tip. In this study, we used a PCI3000 precision cortical impactor (Hatteras Instruments, Cary, NC) and a 2.8-mm diameter impact tip (velocity = 3.0 m/sec, depth = 0.5 mm, and contact duration = 150 msec). Body temperature was maintained during surgery by a feedback-controlled homeothermic blanket (Harvard Apparatus, Holliston, MA). After the injury, the skin was sealed using Vetbond (3M, St. Paul, MN), and mice were allowed to recover in a humidity- and temperature-controlled incubator (Thermocare, Incline Village, NV). Sham animals received the craniotomy, but no impact was applied.
Intranasal drug administration
Intranasal administration is a well-established non-invasive route for drug administration to the brain and allows for permeation of proteins and even cells across the blood–brain barrier (BBB).26–28 Recombinant Wnt3a protein (R&D Systems) was reconstituted at 10 ng/μL in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin (BSA). Prior to the injection, the solution was diluted to 2 ng/μL with 0.9% saline. All injections were delivered intranasally. Starting at 1 h following the injury, injections were administered daily for 7 days. For each injection, the mouse was scruffed by the ears and inverted to expose the nostrils. A 25 μL Hamilton syringe was used to administer 5 μL of either Wnt3a (2 ng/μL) or 0.9% saline into alternating nostrils, starting with the left, and repeating for 5 injections, for a total of 25 μL injections/day (total of 50 ng Wnt3a injected/day).
Immunohistochemistry
Immunohistochemistry procedures were carried out to measure proliferation as previously described.32 Mice were euthanized by CO2 (3 L/min) and sacrificed by decapitation. Brains were removed and frozen using Tissue-Tek optimal cutting temperature compound (VWR International, Radnor, PA) and kept overnight in a −80°C freezer. Brain tissue was then sliced into 10 μm-thick coronal sections using a cryostat vibratome (Leica CM 1950; Leica Microsystems, Buffalo Grove, IL). Sections were dried on a slide warmer for 30 min, fixed with 10% formalin buffer, and followed by a methanol fix for 15 min. The sections were then denatured using 2 N hydrochloric acid in 37°C, and washed with 0.1-M borate buffer (11:9 ratio of 0.2 M boric acid and 0.05 M Borax) and permeabilized with 0.2% Triton-X 100 solution for 5 min. All slides were washed 3 times with PBS (5 min each) after each step. Tissue sections were blocked with 1% fish gelatin (Sigma) in PBS for 1 h at room temperature, then incubated with the primary antibody against Beclin-1 (1:200, BD, East Rutherford, NJ), LC3-II (1:200, Cell Signaling), BrdU (1:400; ABD Serotec, Raleigh, NC), glucose transporter 1 (GLUT1; 1:200; Millipore, Billerica, MA), and/or neuronal nuclear antigen (NeuN, 1:300; Millipore) overnight at 4°C. The next day, the slides were washed 3 times with PBS for 5 min, then reacted with the secondary antibodies Alexa Fluor®488 goat anti-mouse (1:300; Life Technologies, Grand Island, NY) and Cy3-conjugated donkey anti-rat (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA) or Cy5-conjugated donkey anti-rabbit (1:400; Jackson ImmunoResearch Laboratories) for 90 min at room temperature. After 3 washes with PBS, nuclei were stained with Hoechst 33342 (1:20,000; Molecular Probes) for 5 min as a counterstain, then mounted with Vectashield fluorescent mounting medium (Vector Laboratory, Burlingame, CA), and coverslipped for microscopy and image analysis.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
A TUNEL assay kit was used to examine cell death by detecting fragmented DNA in 10-μM-thick coronal fresh frozen sections as described previously.33 After fixation (10% buffered formalin for 10 min and then ethanol:acetic acid (2:1) solution for 5 min) and permeabilization (0.2% Triton X-100 solution), brain sections were incubated in the equilibration buffer for 10 min. Recombinant terminal deoxynucleotidyl transferase (rTdT) and nucleotide mixture were then added on the slide at 37°C for 60 min in the dark. Reactions were terminated by 2X SSC solution for 15 min. Nuclei were counterstained with Hoechst 33342 (1:20,000; Molecular Probes, Eugene, OR) for 5 min.
Nissl staining and contusion volume quantification
Contusion volume was quantified using volumetric Nissl staining analysis as previously described.32 The brains were collected at 2 days post-injury, and 10-μm-thick coronal sections were taken using the cryostat vibratome, with 200-μm separation between each section. The sections were then treated with a 1:1 mixture of 10% formalin and acetic acid, followed by staining with the working solution comprised of a buffer solution (94:6 ratio of sodium acetate and acetic acid) and cresyl violet at a ratio of 5:1. Sections were scanned and imported into ImageJ for area and total volume quantification. For each section, the contusion area was demarcated and normalized using an imported image of a standard ruler. Volumetric analysis was carried out using the Cavalieri method:
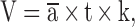 |
where 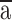 is the average contusion area for each animal, t is the section thickness (200 μm), and k is the number of sections.34 To account for edema and hemorrhage, hemispheric tissue loss was calculated by the following equation:
is the average contusion area for each animal, t is the section thickness (200 μm), and k is the number of sections.34 To account for edema and hemorrhage, hemispheric tissue loss was calculated by the following equation:
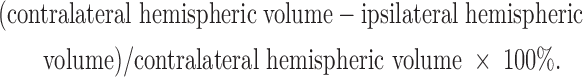 |
Western blot analysis
Western blot analysis was used to detect the activation of the canonical Wnt pathway, as well as for factors involved in apoptosis, BBB disruption, and neurovascular regeneration. Western blot was carried out using the protocols as outlined previously.33 Animals were sacrificed at day 2 or day 14 after TBI. The peri-injury cortical tissue was isolated and lysed in a buffer containing 0.02 M Na4P2O7, 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton, 1 mM EGTA, 2 mM Na3VO4, and a protease inhibitor cocktail (Sigma). The supernatant was collected after centrifugation at 15,000g for 10 min at 4°C. Protein concentration was determined with a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Equivalent amounts of total protein were separated by molecular weight on an SDS-polyacrylamide gradient gel, and then transferred to a PVDF membrane. The blot was incubated in 5% BSA for 1 h and then treated with primary antibodies at 4°C overnight.
The primary antibodies used and the dilutions for each were rat anti-Wnt3a antibody (R&D Systems) at 1:400, mouse anti-β-catenin antibody (R&D Systems) at 1:1000, rabbit anti-cleaved caspase 3 antibody (Cell Signaling) at 1:400, rabbit anti-Bcl-2 (Cell Signaling) at 1:1000, rabbit anti-matrix metalloproteinases 9 (MMP-9) antibody (Millipore) at 1:2500, rabbit anti-MMP-2 antibody (Millipore) at 1:2500, rabbit anti-glial-derived growth factor (GDNF) antibody (Santa Cruz Biotechnology) at 1:1000, and anti-vascular endothelial growth factor (VEGF) antibody (Sigma) at 1:5000. After washing with TBST, membranes were incubated with AP-conjugated or HRP-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ) for 2 h at room temperature. After final washing with TBST, the signals were detected with bromochloroidolyl-phosphate/nitroblue tetrazolium (BCIP/NBP) solution (Sigma) or film. Signal intensity was measured by ImageJ (NIH) and normalized to the actin signal intensity.
Adhesive removal test for sensorimotor functions
The adhesive removal test is a sensitive assay for sensorimotor deficits32,35 Prior to the injury, mice were trained to remove a quarter-circle adhesive tape (Genesee Scientific, San Diego, CA) from alternating forelimb paws. The training was repeated 3 times for each paw for 3 days. Two latencies were recorded: 1) latency to contact, which indicated the time the mouse took to recognize and demonstrate the conviction to remove the adhesive, and 2) latency to removal. The maximum recording latency was 180 sec. Mice that were unable to rapidly remove the adhesive (<10 sec) after the training period were excluded from behavior testing. Following the injury, the adhesive removal task was performed at 3, 7, 10, and 14 days post-injury. The latencies were scored with the experimenter blinded to the treatment conditions. Animals were randomly assigned to each group prior to the injury, with an n = 5 for the sham group, n = 8 for the TBI + saline group, and n = 8 for the TBI + Wnt3a group.
Rotarod test for locomotor function
The rotarod test is sensitive for deficits in gross motor skills and motor coordination.36 Mice are placed onto a spinning cylindrical beam (Ugo Basile, Italy) that accelerated from 4 rpm to 40 rpm over the course of 5 min. Prior to the injury, mice were trained on the rotarod, and animals that were unable to remain on the beam for 5 min after the training period were excluded from future testing. Following the injury, the rotarod test was performed at 1, 2, 3, 7, and 10 days post-injury. Animals were randomly assigned to a group prior to the injury, with n = 6 for sham, n = 12 for TBI + saline, and n = 11 for TBI + Wnt3a.
Statistical analysis
GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for statistical analysis and graphic presentation. Student's two-tailed t test was used for comparison of two experimental groups. For multiple comparisons, a one-way analysis of variance (ANOVA) was used, followed by Bonferroni's correction. For repeated measurements, two-way ANOVA followed by Bonferroni's correction was used. Significance was defined to be a p value ≤0.05. All data are presented as mean ± standard error of the mean (SEM).
Results
Intranasal Wnt3a treatment decreased autophagy in the post-TBI brain
To understand the role of the canonical Wnt/β-catenin system in autophagy of CNS neurons, we tested the effect of recombinant Wnt3a on autophagic activity in a CCI-induced TBI model of mice.37,38 Immunofluorescence imaging of the peri-contusion region at 24 h after TBI revealed an upregulation of LC3 in NeuN-positive (NeuN+) neurons (Fig. 1A,E). At high magnifications, distinct accumulation of LC3 puncta predominantly within the cell soma was evident (Fig. 1D). Sham surgery did not result in similar accumulation of the LC3 signal (Fig. 1C). Treatment with recombinant Wnt3a protein (10 ng/uL, 25 μL injection volume of intranasal delivery) at 60 min post-TBI significantly reduced the numbers of LC3+/NeuN+ cells (Fig. 1E), suggesting that Wnt/β-catenin signaling downregulated autophagic activity. At 48 h after TBI, the numbers of LC3+/NeuN+ cells in the TBI + saline group declined to levels that were similar to those of the Wnt3a-treated group (Fig. 1B,E). This suggests that autophagic activity in neurons peaks within the first 24 h after injury, and Wnt3a treatment within this period provides a robust anti-autophagic effect. We also performed western blotting to corroborate the immunohistochemical findings. In the peri-contusion region at 1 day post-TBI, we confirmed that the Wnt3a treatment increased canonical Wnt activity by probing for the expression level of Wnt3a and β-catenin. TBI resulted in lower levels of β-catenin, possibly as a result from LC3-associated lysosomal destruction of β-catenin or due to fewer viable cells in the peri-contusion tissue (Fig. 1F,H). TBI did not significantly affect Wnt3a levels (Fig. 1F,G), which suggests that the reduction in β-catenin is Wnt-independent, corroborating the possibility of autolysosomal destruction. The treatment with recombinant Wnt3a resulted in significant upregulation of both Wnt3a and β-catenin levels as compared with the saline control group (Fig. 1F–H). Further, Wnt3a suppressed autophagy proteins, Beclin-1 and LC3-II; both were elevated after TBI (Fig. 1F,I,J).
FIG. 1.

Activation of the Wnt/β-catenin pathway reduces autophagic activity following traumataicbrain injury (TBI) in mice. TBI was induced in a mouse model, followed by intranasal Wnt3a (2 μg/kg) delivery 60 min after the TBI insult. (A and B) Representative immunohistochemical images of LC3 (red) and NeuN (blue) fluorescence and co-localization (arrowheads) following TBI at 24 h and 48 h post-injury in the peri-contusion region. (C) Sham injury did not result in noticeable accumulation of LC3. (D) High magnification (100 × ) images of LC3 puncta (arrowheads) in brain sections of the TBI+Saline group at 24 h post-injury. (E) Quantified numbers of LC3+/NeuN+ cells at 24 h post-injury, and at 48 h post-injury. Only cells containing LC3 signal in the soma was considered to be LC3+. *p < 0.05, **p < 0.01, *** p < 0.001, ****p < 0.0001. A total of 600–700 images were quantified. n = 3 for Sham group, n = 5 for both TBI+Saline and TBI+Wnt3a groups for both time-points (24 h and 48 h post-TBI). (F–J) Western blotting was performed on peri-contusion tissue at 24 h post-TBI to evaluate the canonical Wnt signaling and autophagic activity. (F) Representative immunoblots of markers for Wnt signaling and autophagy. All immunoblots were normalized to the β-actin control, and then to sham within group. (G) Quantification of normalized immunoblot intensity of Wnt3a. (H) Quantification of normalized immunoblot intensity of β-catenin. (I) Quantification of normalized immunoblot intensity of Beclin-1. (J) Quantification of normalized immunoblot intensity of LC3-II. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. n = 3 for Sham group, n = 5 for both TBI+Saline and TBI+Wnt3a groups. Color image is available online at www.liebertpub.com/neu
Intranasal Wnt3a treatment endorsed neuroprotection after TBI through activation of the Wnt/β-catenin pathway
Next, we sought to investigate whether Wnt-associated repression of autophagy would ameliorate cell death in the mouse TBI model. At 2 days post-TBI, cell death was quantified by immunostaining of Beclin-1 and TUNEL, markers of autophagy and cell death, respectively (Fig. 2A). The TBI insult caused significant autophagic activity, as indicated by the increased numbers of Beclin-1+ cells (Fig. 2B). The intranasal administration of Wnt3a significantly mitigated the magnitude of autophagic activity (Fig. 2B). There was also a trend in the Wnt-associated reduction in autophagic cell death (Beclin+/TUNEL+ cells; Fig. 2C), although the difference between TBI control and TBI plus Wnt3a was not statistically significant (n = 7 per group).
FIG. 2.

Neuroprotective effects of Wnt3a against autophagic cell death and brain damage following mild traumatic brain injury (TBI) (A–C) The presence of autophagic cell death was visualized by immunohistochemistry at 2 days post-TBI. (A) Representative images of immunofluorescence (red: Beclin-1, green: TUNEL, blue: DAPI). (B) Quantification of Beclin-1+ cells per 40 × field. (C) Quantification of Beclin-1+/TUNEL+ co-labeled cells per 40 × field. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. A total of 600–700 images were quantified. n = 4 for Sham group, n = 7 for both TBI+Saline and TBI+Wnt3a groups. (D) Representative immunofluorescence images are shown (green: TUNEL, red: NeuN, blue: DAPI) revealed significantly greater levels of TUNEL+ cells, as well as TUNEL+/NeuN+ co-labeled cells (arrows). (E) Quantification of total number of TUNEL+ cells. (F) Quantification of total number of NeuN+ cells. (G) Quantification of total number of TUNEL+/NeuN+ colocalized cells. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. A total of 500–600 images were quantified. n = 5 per group. (H) Representative images of Nissl-stained sections for post-TBI animals receiving either intranasal vehicle saline injection or Wnt3a injection at 1 h post-injury and 1 day post-injury. Tissue loss can be observed in the ipslateral hemisphere (bordered by dashed line). (I) Volumetric analysis by the Cavalieri method was used to calculate total injury volumes from cumulative summation of injury areas of adjacent sections spanning the length of the contusion. (J) Hemispheric tissue loss was calculated by comparing the difference in volume between the ipsilateral hemisphere and the contralateral hemisphere. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. n = 5 per group. Color image is available online at www.liebertpub.com/neu
Because TBI results in cell death in multiple cell types, we next investigated the effects of Wnt therapy on both overall cell death and exclusively neuronal cell death. At 2 days post-TBI, immunofluorescence staining of TUNEL and the mature neuron marker NeuN, was performed (Fig. 2D). TBI caused significant total cell death, as indicated by TUNEL+ cells, and neuronal death, as indicated by NeuN+/TUNEL+ cells (Fig. 2E,G). The Wnt3a intranasal treatment attenuated both the overall cell death and neuronal cell death (Fig. 2E,G). Although TBI resulted in a significant decline in the numbers of neurons within the peri-contusion region, there was not a significant change in Wnt3a-treated animals, as compared with the sham group (Fig. 2F).
To examine the manifestations of cell death on a tissue level, brain sections were stained with cresyl violet to visualize tissue loss in the ipsilateral hemisphere (Fig. 2H). TBI animals that received Wnt3a treatment (1 h and 24 h after TBI, 2 μg/kg, intranasal [i.n.]) had significantly smaller contusion volumes, as compared with saline controls (Fig. 2I). To account for parenchymal swelling due to edema or hemorrhage, the ipsilateral tissue loss was compared as a percentage against the contralateral hemisphere. Again, Wnt3a-treated animals possessed much lower ratios of hemispheric loss, as compared with saline controls (Fig. 2J).
To elucidate and corroborate the mechanisms behind the Wnt3a-associated neuroprotection observed after TBI, we carried out an in vitro model of neuronal injury with OGD in primary cortical neurons of 12–13 DIV. To identify the neuroprotective effects associated to the Wnt3a/β-catenin pathway, we performed loss-of-function studies with the Wnt3a inhibitor, XAV939, as well as knockdown studies with β-catenin siRNA. To validate the successful knockdown of β-catenin levels, we probed the levels of β-catenin following the addition of either NT-siRNA or β-catenin siRNA concurrently with OGD (Fig. 3A). OGD itself (with NT-siRNA) reduced β-catenin levels, an effect similar to the result in TBI animals, and is likely a result of LC3-associated lysosomal degradation of β-catenin (Fig. 3B). β-catenin siRNA further suppressed levels of β-catenin to around 78% of that as OGD + NT-siRNA (Fig 3B). Finally, application of Wnt3a rescued the β-catenin levels by stabilizing levels of cytoplasmic β-catenin. We next performed a cell death assay by measuring LDH release. Concomitant Wnt3a administration with OGD mitigated cell death (lower LDH release) as compared with the OGD control cultures; this effect was abolished by co-applied XAV939. Further, β-catenin knockdown with β-catenin siRNA exacerbated OGD-induced cell death, supporting the idea that β-catenin plays a protective role. This conclusion was corroborated by Wnt3a administration together with β-catenin siRNA, which elevated β-catenin expression and reduced the magnitude of cell death to similar levels as OGD alone (Fig. 3C).
FIG. 3.

Anti-apoptotic and blood–brain barrier (BBB) protective effects of Wnt3a/β-catenin signaling in vitro and after TBI in vivo. (A) Primary neuronal cultures were dissected from E16 mice and then plated until day 12 in vitro (12 DIV). Groups exposed to oxygen-glucose deprivation (OGD), concurrently with either Wnt3a administration or phosphate-buffered saline (PBS) vehicle control administration. Western blot evaluation of salient protein levels were performed. (B) Quantification of normalized intensity of β-catenin. (C) Levels of cell death as measured by LDH release after OGD, and cultures were treated with either just Wnt3a, Wnt3a + Wnt pathway inhibitor, XAV939, non-targeting (NT)-siRNA, β-catenin siRNA, or β-catenin + Wnt3a. *p < 0.05, compared with Control group. p < 0.05, compared with β-catenin siRNA group. Δp < 0.05, compared with Wnt3a-only group. (D-H) Mice were sacrificed at 2 days post-TBI after having been given two intranasal injections of either saline or Wnt. (D) Representative immunoblots for markers related to cell death and BBB disruption. All immunoblots were normalized to the β-actin control, and then to Sham within group. (E) Quantification of the normalized intensity of cleaved caspase-3. (F) Quantification of the normalized intensity of Bcl-2. (G) Quantification of the normalized intensity of matrix metalloproteinases 9 (MMP-9). (H) Quantification of the normalized intensity of MMP-2. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. n = 3 for Sham group, n = 7 for both TBI+Saline and TBI+Wnt3a groups.
Western blotting at the same time-point of 2 days post-TBI explored additional mechanisms aside from autophagy that may be implicated in the Wnt3a action. The intranasal Wnt3a treatment decreased cleaved caspase-3 levels and increased Bcl-2 level in the peri-contusion region, suggesting anti-apoptotic activities enhanced by Wnt3a (Fig. 3D,E). In addition, the level of MMP-9, a primary mediator for BBB disruption,39 was significantly reduced in animals that received Wnt3a treatment, although MMP-2 levels were relatively unchanged (Fig. 3G,H). These results suggest that Wnt3a conferred an anti-apoptotic effect and promoted the stabilization of the BBB.
Wnt3a increased neurotrophins and regenerative activities in the post-TBI brain
Western blotting was performed during the chronic phase of TBI to investigate regenerative effects associated with the Wnt/catenrin pathway (Fig. 4A). At 14 days post-TBI, higher levels of Wnt3a and β-catenin persisted in the peri-contusion region of Wnt3a-treated animals, suggesting sustained elevation of canonical Wnt activity (Fig. 4B,C). Saline-treated animals did not show significant differences in levels of Wnt3a and β-catenin as compared with sham controls (Fig. 4B,C). To characterize downstream neurovascular effects of Wnt stimulation, we investigated growth factors specific to neurogenesis and angiogenesis, including GDNF and VEGF. Western blotting showed that levels of both growth factors significantly increased at this delayed time-point after the Wnt3a treatment (Fig. 4D,E).
FIG. 4.

Regenerative effects of Wnt3a/β-catenin signaling during the chronic phase after traumatic brain injury (TBI). (A-E) At 14 days post-TBI, western blot analysis was performed using peri-contusion tissue from animals given a 7-day treatment regimen. (B) Quantification of the normalized intensity of Wnt3a. (C) Quantification of the normalized intensity of β-catenin. (D) Quantification of the normalized intensity of glial-derived growth factor (GDNF). (E) Quantification of the normalized intensity of vascular endothelial growth factor (VEGF). *p < 0.05. n = 3 for Sham group, n = 9 for both TBI+Saline and TBI+Wnt3a groups. (F-I) Mice received intraperitoneal (i.p.) injections of BrdU starting at 3 days post-TBI and continuing until sacrifice at 14 days post-TBI. (F) Immunohistochemical examination (green: NeuN, red: BrdU:, blue: GLUT1) of the cortical region surrounding the injury site shows that Wnt3a treatment resulted in higher levels of total BrdU+ cells, BrdU+/NeuN+ co-labeled cells (arrow), and BrdU+/GLUT1+ co-labeled cells (arrowheads). (G-I) Distinct subtypes of proliferating cells were quantified and analyzed. Wnt3a treatment resulted in significantly greater levels of overall proliferation (total BrdU+ cells), neurogenesis (BrdU+/NeuN+ co-labeled cells), and angiogenesis (BrdU+/GLUT1+ co-labeled cells) following TBI. *p < 0.05, **p < 0.01. n = 3 for Sham group, n = 6 for both TBI+Saline and TBI+Wnt3a groups. Color image is available online at www.liebertpub.com/neu
To evaluate the magnitude of regeneration within the peri-contusion region, immunofluorescence assays were carried out 14 days after TBI using antibodies against NeuN, BrdU, and glucose transporter 1 (GLUT1), markers for mature neurons, newly divided cells, and blood vessels, respectively. Neurogenesis was quantified by co-localization of NeuN and BrdU, whereas angiogenesis was quantified by co-localization of GLUT1 and BrdU (Fig. 4F). Although TBI itself triggered neurovascular regeneration via spontaneous endogenous recovery, Wnt3a administration significantly augmented levels of overall proliferation, as well as levels of neurogenesis and angiogenesis (Fig.4G–I).
Wnt3a treatment preserves sensorimotor function after TBI
To assess for functional preservation and recovery, two behavioral tests were performed, the adhesive removal test and the rotarod test, both of which are sensitive for sensorimotor competency.35,36 The TBI insult resulted in quantifiable deficits in adhesive removal at 3 days post-TBI, as indicated by the greater latency to removal of the adhesive irritant (Fig. 5A). Wnt3a treatment was successful in eliminating this functional deficit, with the TBI-Wnt3a cohort performing at a similar competency as the sham cohort. A similar effect was evident in the rotarod test, with the TBI resulting in deficits in maintaining balance on the accelerating rotational beam, whereas Wnt3a improved the performance (Fig. 5B). At later time-points (14 days post-TBI), no significant difference was seen between groups mainly due to spontaneous functional recovery in this mild TBI model (Fig. 5B).
FIG. 5.

Intranasally delivered Wnt3a promoted sensorimotor functional recovery following traumatic brain injury (TBI). (A) All animals were pre-trained in the adhesive removal task prior to the injury to ensure competent removal. Baseline latencies (day 0) were measured to ensure no significant differences across groups. After injury, Wnt3a preserved the mouse forelimb sensorimotor function, as indicated by the significantly shorter latency to removal in the contralateral forepaw of the Wnt3a-treated animals. (B) Mice were pre-screened and pre-trained on the rotarod task to ensure adequate performance of ≥90% maximum running time (i.e., ≥270 sec, day 0). Wnt3a-treated animals were able to remain on the accelerating beam significantly longer than the Saline animals, whereas there were no significant differences between the Wnt3a-treated animals and the Sham animals. *p < 0.05, compared with Sham. #p < 0.05, compared with TBI+Saline. n = 6 for Sham group, n = 12 for TBI+Saline group, and = 11 for TBI+Wnt3a groups.
Discussion
Wnt3a is endogenous to the brain and is predominantly active during neurodevelopment.20,40 Traditionally, Wnt proteins have been placed in the context of cell renewal and differentiation.41,42 We now report the novel finding that Wnt3a is also a crucial regulator of cell death and regeneration after brain injuries such as TBI. Although β-catenin has been shown to play a central role in directing cell fate toward cell survival or cell death, few investigations have looked at a potential neuroprotective action of Wnt3a following TBI.9–11 TBI is a multi-factorial condition encompassing multiple deleterious secondary effects such as excitotoxicity, inflammation, increased apoptosis, and dysregulation of autophagy.3,19,43 In particular, autophagic flux is impaired following brain injury, resulting in greater accumulation of autophagosomes and is correlated with greater neuronal cell death.18,19 The current study explored whether modulation of the Wnt/β-catenin pathway could attenuate apoptosis as well as autophagic death in vitro and in the post-TBI brain. In the animal model of TBI, we found that intranasal delivery of Wnt3a after TBI enhanced canonical Wnt signaling and subsequent growth factor expression, attenuated autophagic and apoptotic cell death, and improved neurovascular repair and functional outcomes following TBI. Given the complexity and multi-variate nature of the pathophysiology of brain injury, Wnt proteins provide a unique cytoprotective and regenerative strategy to promote functional recovery by global transcriptional modulation along the acute and chronic phases after TBI.
Our study highlights the interconnectivity between three distinct systems: Wnt/β-catenin signaling, autophagy, and TBI pathophysiology (Fig. 6). The Wnt/β-catenin pathway operates by the canonical Wnt protein binding to Frizzled (FZD) receptor, which forms a complex with the co-receptor low-density lipoprotein receptor-related protein (LRP) to inactivate the β-catenin destruction complex consisting of Axin, adenomatous polyposis coli protein (APC), and glycogen synthase kinase 3β (GSK3β).41 This allows for the accumulation of β-catenin in the cytoplasm and nucleus. Aside from regulating pro-mitogen expression, β-catenin has been shown to be a key regulator in autophagy progression in colorectal cells by downregulating autophagic activity.9 However, this homeostatic duality is perturbed by metabolic stress, in which cells respond by upregulating LC3,44,45 which then targets β-catenin for non-proteasomal degradation, resulting in higher levels of autophagic induction.9 We sought to investigate whether this is also the case in the CNS and whether modulation of the Wnt/β-catenin pathway could affect cell fate in pathological conditions such as TBI.
FIG. 6.

Concerted interactivity among three distinct systems: Wnt/β-catenin pathway (green), autophagy (gray), and traumatic brain injury (TBI) pathophysiology (red). Under normal conditions, cytoplasmic β-catenin and autophagic activity are mutually regulated. β-catenin inhibits the initiation and elongation of the phagophore, whereas LC3, which is upregulated upon induction of autophagy, will target β-catenin for autolysosomal degradation. TBI is a pathological event that results in hypoxia and oxidative stress that trigger autophagy induction. Further, due to unknown mechanisms, TBI also results in lysosomal impairment, preventing the degradation of autophagosomes. The accumulation of autophagosomes then contributes to greater levels of neuronal death. Wnt3a can stabilize cytoplasmic β-catenin through its downstream inhibition of the Axin-APC-GSK3β complex, which would otherwise ubiquitinate β-catenin for degradation. Upregulation of β-catenin results in suppression of autophagic induction and amelioration of cell death. Color image is available online at www.liebertpub.com/neu
Using a mild TBI model, we demonstrated multiple key findings. First, the focal cortical injury in mice resulted in reduced levels of β-catenin, possibly due to autolysosomal destruction of β-catenin in cells undergoing increased autophagic activity.9 We also observed this molecular effect with our in vitro study using primary cortical neuronal cultures, in which OGD resulted in decreased levels of β-catenin. This was further supported by the fluctuation in β-catenin in the sub-acute phase of TBI (2 days post-injury) as compared with the chronic phase of TBI (14 days post-injury). By 14 days post-injury, the levels of β-catenin had returned to their original levels, even in the saline control group, and this time course aligns with the time course of autophagy, which peaks around 24 to 48 h post-TBI before waning and returning to basal levels by 7 days post-TBI.18,19 Second, the intranasal Wnt3a administration was able to partially rescue the levels of β-catenin by inhibiting its proteasomal degradation. Higher β-catenin levels re-established levels of suppression of autophagic activity similar to those of the unstressed state, even concomitant with a stressor such as TBI. The lower magnitude of autophagy was instantiated by decreased levels of both Beclin-1 and LC3-II. Finally, greater autophagic induction was also correlated with elevated neuronal cell death, and suppression of autophagy similarly reduced cell death. Autophagy and cell death are evident in multiple cell types following TBI, including neurons, astrocytes, and oligodendrocytes.19,46 We demonstrated Wnt3a-associated neuroprotection by the suppression of apoptosis and autophagy, leading to decreased neuronal cell death, reduced contusion volumes, and enhanced functional recovery. Although this study focused exclusively on neuronal death in TBI, Wnt therapy may have bolstered glial viability as well, as suggested by the greater overall cell viability and benefits observed on the tissue and functional levels.
Multiple reports that implemented the CCI model have consistently demonstrated the deleterious effects of autophagic dysregulation after TBI18,19 To our knowledge, we are the first to investigate a translational approach to stabilize this dysregulation to improve neuronal resilience. An important consideration is the unforeseen consequences associated with this particular pharmacological approach, including potential disproportionate sparing of glial/neuronal subtypes or tumorigenic effects of Wnt3a.47,48 Although we did observe prolonged Wnt3a upregulation, there were no histological or behavioral phenotypes to suggest any adverse effects of Wnt3a therapy. However, more studies are warranted in the future both for long-term monitoring of potential side-effects and for determining the therapeutic index of the drug and its optimal dosing and regimen after TBI.
Canonical Wnt proteins hold great neural regenerative potential because they regulate cell division, proliferation, and cell fate specification.8,41,42 Within the nervous system, in addition to its anti-autophagic and anti-apoptotic effects, Wnt3a promotes neurogenesis within native neural stem cell niches found in the sub-ventricular zone (SVZ) and the hippocampal sub-granular zone (SGZ).20,49,50 Following focal cortical injury, Mash1+ transit amplifying cells in the SVZ are stimulated to proliferate, differentiate, and migrate to the injury region51 along the stromal derived factor 1α (SDF1α)-dependent chemotactic gradient.52 Whereas this was the case following TBI, we also observed even greater numbers of newly formed neuronal and endothelial cells in the peri-contusion region following Wnt3a treatment. These findings indicate that the recombinant Wnt3a protein is likely capable of augmenting proliferation and migration of neuroprogenitor cells from the SVZ into the lesion site. Further, Wnt proteins are involved in regulating the expression and paracrine activity of a variety of neurotrophins.53,54 In particular, we observed that Wnt therapy resulted in a robust upregulation of GDNF, a factor that promotes both survival55 and axonal outgrowth56 of surrounding neurons, both characteristics that are pivotal for tissue repair after injury. Wnt3a also increased levels of VEGF, a potent inducer of angiogenesis.57,58 Although Wnt signaling may be directly mediating VEGF expression, there is not a clear link between Wnt and VEGF expression. Thus, it is more likely that the heightened VEGF levels are due to neurovascular coupling of angiogenesis with Wnt-associated neurogenesis.59
Our report provides evidence that intranasal administration of recombinant Wnt3a provides multiple beneficial effects following brain injury, including amelioration of cell death, increased trophic factors, and importantly, improved functional outcomes. One of the challenges facing neuroprotective strategies is that brain injury acts through multiple pathophysiological mechanisms, including inflammation, oxidative damage, BBB disruption, and hypoxia.3 The multi-factorial nature of this disorder makes it difficult for a single therapeutic strategy to be effective. Thus, it may be more clinically viable to pursue either combination therapy options or a treatment that targets multiple phenotypes. In this regard, Wnt therapy encompasses a comprehensive therapeutic approach. Indeed, along with the neuroprotective effects of Wnt3a, we observed greater stabilization of the BBB after TBI with Wnt3a therapy. This finding warrants further investigation into the underlying mechanisms by which Wnt3a may impact BBB integrity, possibly through recruitment of pericytes.60 Moreover, Wnt therapy stimulates robust regenerative benefits to repair the damaged neurovascular unit, which provides a viable and perhaps more realistic long-term treatment for restoring function and reducing symptomatology after acute brain damage. In concert with the neuroprotective and neural regenerative benefits associated with Wnt3a, we further demonstrate that Wnt3a improved functional outcomes after TBI.
A final important consideration is the intranasal route of administration chosen for drug delivery. This is a non-invasive route of administration that confers greater clinical feasibility and provides direct access across the BBB into the CNS.24,26,28 The intranasal route is a well-established non-invasive delivery for drugs,61 molecules,24 and cells24,25,62 to the brain because perforations within the olfactory epithelium allow the reagents to cross the BBB. After permeating the olfactory epithelium, there are multiple ways through which the agents can enter into the CNS, including through the perineural spaces surrounding the cranial nerves and then disseminating through the cerebrospinal fluid (CSF) to more distal sites.63 Alternatively, the agents can bypass the CSF and enter the CNS directly through intracellular axonal transport along cranial nerves, such as the olfactory and trigeminal nerves.64,65 Although the CNS bioavailability of intranasally administered substances is still unclear, the cortical uptake of drugs by the intranasal route is 6 times greater than that of intraperitoneal injections.66 We were able to demonstrate that intranasal delivery of Wnt3a successfully resulted in efficient CNS permeation of the Wnt3a protein, which then upregulated canonical Wnt activity and conferred multiple beneficial effects in the injured brain.
To our knowledge, this is the first study that evaluated the interplay between the Wnt/β-catenin pathway and the autophagy pathways and leveraging this connection to counteract the dysregulated autophagic activity of TBI. Although the focus of this study was on TBI, this highly translatable treatment could be applied to other disorders as well. Wnt3a is a versatile treatment approach that provides multi-factorial benefits, including autophagy and cell death reduction, gene regulation to improve trophic support, and enhanced neurovascular regeneration. These elements provide a unique treatment paradigm that can be applied to other disorders as well, such as stroke and neurodegenerative disorders. Although the long-term beneficial and adverse effects have yet to be fully elucidated, the current findings warrant further examination of targeting the Wnt/β-catenin pathway for prospective treatments, especially in the realm of dysregulated autophagy. In conclusion, our study has presented compelling evidence that the clinically applicable intranasal delivery of the recombinant Wnt3a protein provides significant therapeutic benefits toward both neuroprotection and neural regeneration after focal brain injury, ultimately culminating in enhanced functional outcomes.
Author Disclosure Statement.
No competing financial interests exist.
References
- 1.Andelic N. The epidemiology of traumatic brain injury. (2013). Lancet Neurol. 12, 28–29 [DOI] [PubMed] [Google Scholar]
- 2.Loane D.J., and Faden A.I. (2010). Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 31, 596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Werner C., and Engelhard K. (2007). Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9 [DOI] [PubMed] [Google Scholar]
- 4.Muroyama Y., Kondoh H., and Takada S. (2004). Wnt proteins promote neuronal differentiation in neural stem cell culture. Biochem. Biophys. Res. Commun. 313, 915–921 [DOI] [PubMed] [Google Scholar]
- 5.Kalani M,Y., Cheshier SH, Cord B.J., Bababeygy S.R., Vogel H., Weissman I.L., Palmer T.D., and Nusse R. (2008).Wnt-mediated self-renewal of neural stem/progenitor cells. Proc. Natl. Acad. Sci. U S A 105, 16970–16975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shruster A., Ben-Zur T., Melamed E., Offen D. (2012). Wnt signaling enhances neurogenesis and improves neurological function after focal ischemic injury. PloS One 7, e40843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whyte J.L., Smith A.A., and Helms J.A. (2012). Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 4, a008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wodarz A., and Nusse R. (1998). Mechanisms of Wnt signaling in development. Ann. Rev. Cell Dev. Biol. 14, 59–88 [DOI] [PubMed] [Google Scholar]
- 9.Petherick K.J., Williams A.C., Lane J.D., Ordóñez-Morán P., Huelsken J., Collard T.J., Smartt H.J., Batson J., Malik K., Paraskeva C., and Greenhough A. (2013). Autolysosomal β‐catenin degradation regulates Wnt‐autophagy‐p62 crosstalk. EMBO J. 1903–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaidi A., Williams A.C., and Paraskeva C. (2007). Interaction between β-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat. Cell Biol. 9, 210–217 [DOI] [PubMed] [Google Scholar]
- 11.Korswagen H. (2005). Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 12.Kang R., Zeh H.J., Lotze M.T., and Tang D. (2005). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baehrecke E.H. (2005). Autophagy: dual roles in life and death? Nat. R Mol. Cell Biol. 6, 505–510 [DOI] [PubMed] [Google Scholar]
- 14.Kühn K., Cott C., Bohler S., Aigal S., Zheng S., Villringer S., Imberty A., Claudinon J., and Römer W. (2015). The interplay of autophagy and β-Catenin signaling regulates differentiation in acute myeloid leukemia. Cell Death Discov., 1, 15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shang L., and Wang X. (2011). AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 7, 924–926 [DOI] [PubMed] [Google Scholar]
- 16.Kroemer G., Mariño G., and Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell 40, 280–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia Z., Wang J., Wang W., Tian Y., XiangWei W., Chen P., Ma K., and Zhou C. (2014). Autophagy eliminates cytoplasmic β-catenin and NICD to promote the cardiac differentiation of P19CL6 cells. Cell. Signal. 26, 2299–2305 [DOI] [PubMed] [Google Scholar]
- 18.Luo C.-L., Li B.X., Li Q.Q., Chen X.P., Sun Y.X., Bao H.J., Dai D.K., Shen Y.W., Xu H.F., Ni H., Wan L., Qin Z.H., Tao L.Y., and Zhao Z.Q. (2011). Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184, 54–63 [DOI] [PubMed] [Google Scholar]
- 19.Sarkar C., Zhao Z., Aungst S., Sabirzhanov B., Faden A.I., and Lipinski M.M. (2014). Impaired autophagy flux is associated with neuronal cell death after traumatic brain injury. Autophagy 10, 2208–2222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshinaga Y., Kagawa T., Shimizu T., Inoue T., Takada S., Kuratsu J., and Taga T. (2010). Wnt3a promotes hippocampal neurogenesis by shortening cell cycle duration of neural progenitor cells. Cell. Mol. Neurobiol.. 30, 1049–1058 [DOI] [PubMed] [Google Scholar]
- 21.Inoue T., Kagawa T., Fukushima M., Shimizu T., Yoshinaga Y., Takada S., Tanihara H, and Taga T. (2006). Activation of canonical Wnt pathway promotes proliferation of retinal stem cells derived from adult mouse ciliary margin. Stem Cells 24, 95–104 [DOI] [PubMed] [Google Scholar]
- 22.Piccin D., and Morshead C.M. (2011). Wnt signaling regulates symmetry of division of neural stem cells in the adult brain and in response to injury. Stem Cells 29, 528–538 [DOI] [PubMed] [Google Scholar]
- 23.Wang Y.Z., Yamagami T., Gan Q., Wang Y., Zhao T., Hamad S., Lott P., Schnittke N., Schwob J.E., and Zhou C.J. (2011). Canonical Wnt signaling promotes the proliferation and neurogenesis of peripheral olfactory stem cells during postnatal development and adult regeneration. J. Cell Sci. 124, 1553–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lochhead J.J., and Thorne R.G. (2012). Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 64, 614–628 [DOI] [PubMed] [Google Scholar]
- 25.Wei N., Yu S.P., Gu X., Taylor T.M., Song D., Liu X.F., and Wei L. (2013). Delayed intranasal delivery of hypoxic-preconditioned bone marrow mesenchymal stem cells enhanced cell homing and therapeutic benefits after ischemic stroke in mice. Cell Transplant. 22, 977–991 [DOI] [PubMed] [Google Scholar]
- 26.Danielyan L., L., Schäfer R., von Ameln-Mayerhofer A., Buadze M., Geisler J., Klopfer T., Burkhardt U., Proksch B., Verleysdonk S., Ayturan M., Buniatian G.H., Gleiter C.H., Frey W.H., 2nd (2009). Intranasal delivery of cells to the brain. Eur. J. Cell Biol. 88, 315–324 [DOI] [PubMed] [Google Scholar]
- 27.Donega V., van Velthoven C.T., Nijboer C.H., van Bel F., Kas M.J., Kavelaars A., and Heijnen C.J. (2013). Intranasal mesenchymal stem cell treatment for neonatal brain damage: long-term cognitive and sensorimotor improvement. PloS One 8, e51253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koch S.B., van Zuiden M., Nawijn L., Frijling J.L., Veltman D.J., Olff M. (2014). Intranasal oxytocin as strategy for medication-enhanced psychotherapy of PTSD: salience processing and fear inhibition processes. Psychoneuroendocrinology 40, 242–256 [DOI] [PubMed] [Google Scholar]
- 29.Ogle M.E., Gu X., Espinera A.R., and Wei L. (2012). Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1α. Neurobiol. Dis. 45, 733–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin A.-P., Liu C.F., Qin Y.Y., Hong L.Z., Xu M., Yang L., Liu J., Qin Z.H., and Zhang H.L. (2010). Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 6, 738–753 [DOI] [PubMed] [Google Scholar]
- 31.Sheng R., Liu X.Q., Zhang L.S., Gao B., Han R., Wu Y.Q., Zhang X.Y., and Qin Z.H. (2012). Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy 8, 310–325 [DOI] [PubMed] [Google Scholar]
- 32.Lee J.H., Wei L., Gu X., Wei Z., Dix T.A., and Yu S.P. (2014). Therapeutic effects of pharmacologically induced hypothermia against traumatic brain injury in mice. J. Neurotrauma 31, 1417–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi K.-E., Hall C.L., Sun J.M., Wei L., Mohamad O., Dix T.A., and Yu S.P. A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. The FASEB J. 26, 2799–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coggeshall R.E. (1992). A consideration of neural counting methods. Trends Neurosci. 15, 9–13 [DOI] [PubMed] [Google Scholar]
- 35.Bouet V., Boulouard M., Toutain J., Divoux D., Bernaudin M., Schumann-Bard P., and Freret T. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. 4, 1560–1564 [DOI] [PubMed] [Google Scholar]
- 36.Hamm R.J., Pike B.R., O'Dell D.M., Lyeth B.G., and Jenkins L.W. The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 11, 187–196 [DOI] [PubMed] [Google Scholar]
- 37.Lighthall J.W. (1988). Controlled cortical impact: a new experimental brain injury model. J. Neurotrauma 5, 1–15 [DOI] [PubMed] [Google Scholar]
- 38.Hall E.D., Sullivan P.G., Gibson T.R., Pavel K.M., Thompson B.M., and Scheff S.W. (2005). Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J. Neurotrauma 22, 252–265 [DOI] [PubMed] [Google Scholar]
- 39.Lucivero V., Prontera M., Mezzapesa D.M., Petruzzellis M., Sancilio M., Tinelli A., Di Noia D., Ruggieri M., and Federico F. (2007). Different roles of matrix metalloproteinases-2 and-9 after human ischaemic stroke. Neurol. Sci. 28, 165–170 [DOI] [PubMed] [Google Scholar]
- 40.Wang Y.-Z., Yamagami T., Gan Q., Wang Y., Zhao T., Hamad S., Lott P., Schnittke N., Schwob J.E., and Zhou C.J. (2011). Canonical Wnt signaling promotes the proliferation and neurogenesis of peripheral olfactory stem cells during postnatal development and adult regeneration. J. Cell Sci. 124, 1553–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nusse R., Fuerer C., Ching W., Harnish K., Logan C., Zeng A., ten Berge D., and Kalani Y. (2008). Wnt signaling and stem cell control. in Cold Spring Harb. Symp. Quant. Biol. 73, 59–66 [DOI] [PubMed] [Google Scholar]
- 42.Salinas P.C., and Zou Y. (2008). Wnt signaling in neural circuit assembly. Annu. Rev. Neurosci. 31, 339–358 [DOI] [PubMed] [Google Scholar]
- 43.Liu C.L., Chen S., Dietrich D., and Hu B.R. (2008). Changes in autophagy after traumatic brain injury. J. Cereb. Blood Flow Metab. 28, 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young J.E., Martinez R.A., and La Spada A.R. (2009). Nutrient deprivation induces neuronal autophagy and implicates reduced insulin signaling in neuroprotective autophagy activation. J. Biol. Chem. 284, 2363–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deretic V. (2008). Autophagosome and Phagosome. Springer; [DOI] [PubMed] [Google Scholar]
- 46.Raghupathi R. (2004). Cell death mechanisms following traumatic brain injury. Brain Pathol. 14 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaur N., Chettiar S., Rathod S., Rath P., Muzumdar D., Shaikh M.L., and Shiras A. (2013). Wnt3a mediated activation of Wnt/β-catenin signaling promotes tumor progression in glioblastoma. Mol. Cell Neurosci. 54, 44–57 [DOI] [PubMed] [Google Scholar]
- 48.Qi L., Sun B., Liu Z., Cheng R., Li Y., and Zhao X. (2014). Wnt3a expression is associated with epithelial-mesenchymal transition and promotes colon cancer progression. J. Exp. Clin. Cancer Res. 33, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu J.M., Kim J.H., Song G.S., and Jung J.S. (2006). Increase in proliferation and differentiation of neural progenitor cells isolated from postnatal and adult mice brain by Wnt-3a and Wnt-5a. Mol. Cell Biochem. 288, 17–28 [DOI] [PubMed] [Google Scholar]
- 50.Munji R.N., et al. , Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci, 2011. 31(5): p. 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomsen G.M., Choe Y., Li G., Siegenthaler J.A., and Pleasure S.J. (2014). Traumatic brain injury reveals novel cell lineage relationships within the subventricular zone. Stem Cell Res. 13, 48–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Imitola J., Raddassi K., Park K.I., Mueller F.J., Nieto M., Teng Y.D., Frenkel D., Li J., Sidman R.L., Walsh C.A., Snyder E.Y., and Khoury S.J. (2004). Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1α/CXC chemokine receptor 4 pathway. Proc. Natl. Acad. Sci. 101, 18117–18122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yi H., Hu J., Qian J., and Hackam A.S. (2012). Expression of brain-derived neurotrophic factor (BDNF) is regulated by the Wnt signaling pathway. Neuroreport 23, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Majumdar A., Vainio S., Kispert A., McMahon J., and McMahon A.P. (2003). Wnt11 and Ret/Gdnf pathways cooperate in regulating ureteric branching during metanephric kidney development. Development 130, 3175–3185 [DOI] [PubMed] [Google Scholar]
- 55.Clarkson E.D., Zawada W.M., and Freed C.R. (1997). GDNF improves survival and reduces apoptosis in human embryonic dopaminergic neurons in vitro. Cell Tissue Res. 289, 207–210 [DOI] [PubMed] [Google Scholar]
- 56.Wood M.D., Kim H., Bilbily A., Kemp S.W., Lafontaine C., Gordon T., Shoichet M.S., Borschel G.H. GDNF released from microspheres enhances nerve regeneration after delayed repair. Muscle Nerve 46, 122–124 [DOI] [PubMed] [Google Scholar]
- 57.Chen J., and Chopp M. (2012). Angiogenesis and arteriogenesis as stroke targets, in Translational Stroke Research: From Target Selection to Clinical Trials. Lapchak Paul A. and Zhang John H. (eds). Springer, pps. 231–249 [Google Scholar]
- 58.Dzietko M., Derugin N., Wendland M.F., Vexler Z.S., Ferriero D.M. )2013). Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl.l Stroke Res. 4, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohab J.J., Fleming S., Blesch A., and Carmichael S.T. (2006). A neurovascular niche for neurogenesis after stroke. J. Neurosci. 26, 13007–13016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu S., Agalliu D., Yu C., and Fisher M. (2012). The role of pericytes in blood-brain barrier function and stroke. Curr. Pharm. Des. 18, 3653–3662 [DOI] [PubMed] [Google Scholar]
- 61.Chen D., Lee J., Gu X., Wei L., and Yu S.P. Intranasal delivery of Apelin-13 is neuroprotective and promotes angiogenesis after ischemic stroke in mice. ASN Neuro 7,pii: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wei Z.Z., Gu X., Ferdinand A., Lee J.H., Ji X., Ji X.M., Yu S.P., and Wei L. (2015). Intranasal delivery of bone marrow mesenchymal stem cells improved neurovascular regeneration and rescued neuropsychiatric deficits after neonatal stroke in rats. Cell Transpl. 24, 391–402 [DOI] [PubMed] [Google Scholar]
- 63.Bradbury M., and Cserr H. (1985). Drainage of cerebral interstitial fluid and of cerebrospinal fluid into lymphatics. Exp. Biol. Lymph. Circ. 9, 355–394 [Google Scholar]
- 64.Thorne R., Hanson L.R., Ross T.M., Tung D., Frey W.H., 2nd (2008). Delivery of interferon-β to the monkey nervous system following intranasal administration. Neurosci. 152, 785–797 [DOI] [PubMed] [Google Scholar]
- 65.Thorne R., Pronk G.J., Padmanabhan V., Frey W.H., 2nd (2004). Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neurosci. 127, 481–496 [DOI] [PubMed] [Google Scholar]
- 66.Chauhan M.B., and Chauhan N.B. Brain uptake of neurotherapeutics after intranasal versus intraperitoneal delivery in mice. J. Neurol. Neurosurg. 2, pii: [PMC free article] [PubMed] [Google Scholar]