

Abstract
Many cobalamin (Cbl)-dependent radical S-adenosyl-L-methionine (SAM) methyltransferases have been identified through sequence alignment and/or genetic analysis; however, few have been studied in vitro. GenK is one such enzyme that catalyzes methylation of the 6′-carbon of gentamicin X2 (GenX2) to produce G418 during the biosynthesis of gentamicins. Reported herein, several alternative substrates and fluorinated substrate analogs were prepared to investigate the mechanism of methyl transfer from Cbl to the substrate as well as the substrate specificity of GenK. Experiments with deuterated substrates are also shown here to demonstrate that the 6′-pro-R-hydrogen atom of GenX2 is stereoselectively abstracted by the 5′-dAdo· radical and that methylation occurs with retention of configuration at C6′. Based on these observations, a model of GenK catalysis is proposed wherein free rotation of the radical-bearing carbon is prevented and the radical SAM machinery sits adjacent rather than opposite to the Me-Cbl cofactor with respect to the substrate in the enzyme active site.
A typical biological methylation reaction proceeds via an SN2-like mechanism that involves direct attack of a nucleophilic functional group at the electrophilic methyl of the sulfonium moiety in S-adenosyl-L-methionine (SAM, 1) releasing S-adenosyl-L-homocysteine (SAH, 2) as the leaving group.1,2 However, a group of methyltransferases has been recently discovered that belong to the radical SAM superfamily of enzymes. Distinct from the classic paradigm for biochemical methylation,3 the mechanisms of these enzymes involve radical chemistry with methyl transfer to unactivated (i.e., non-nucleophilic) sp2- or sp3-hybridized carbons or phosphinate centers.4
Radical SAM methyltransferases (RSMTs) can be divided into four groups based on protein architecture, predicted reaction mechanism, and cofactor dependence.4 The subgroup comprising the cobalamin (Cbl)-dependent RSMTs has the most known members and the greatest recognized diversity of reactions catalyzed. One such enzyme is GenK, which catalyzes the methylation of gentamicin X2 (GenX2, 4) at the C6′ position to yield G418 (5)5 during the biosynthesis of gentamicin C1 (6) and C2 (Scheme 1).5 The GenK-catalyzed reaction is important for the production of this class of clinically useful aminoglycoside antibiotics.6
Scheme 1.

C6′ Methylation Reaction Catalyzed by GenK in the Gentamicin Biosynthetic Pathway
To probe the ability of GenK to accept different substrates, paromamine (7), neamine (8), and five pseudotrisaccharides including gentamicin A (3), A2 (9) and B (10), along with JI-20A (11) and 2′-deamino-2′-hydroxy-GenX2 (12) were synthesized as described in the Supporting Information and tested for their competence as GenK substrates (Figure 1). These compounds were separately incubated with GenK in 50 mM Tris·HCl buffer (pH 8.0) in the presence of 10 mM DTT, 4 mM SAM, 1 mM methyl viologen, 4 mM NADPH, and 1 mM methylcobalamin for 18 h.5 Reactions were then characterized by mass spectrometry and HPLC with UV detection at 254 nm. The assay was also performed with GenX2 (4) as a reference. HPLC analysis of the reactions mixtures showed 5′-dAdo (13) formation when GenK was incubated with the pseudotrisaccharides 3 and 9–12 but not the pseudodisaccharides 7 and 8. Likewise, mass spectroscopic analysis of the product mixtures demonstrated that all pseudotrisaccharides tested were converted to their methylated products (see Figure S1) albeit with reduced efficiency compared to GenX2 (4) as measured in terms of 5′-dAdo production (see Table S1). Therefore, GenK can recognize and process alternative substrates; however, the biologically relevant substrate GenX2 appears to be preferred.
Figure 1.
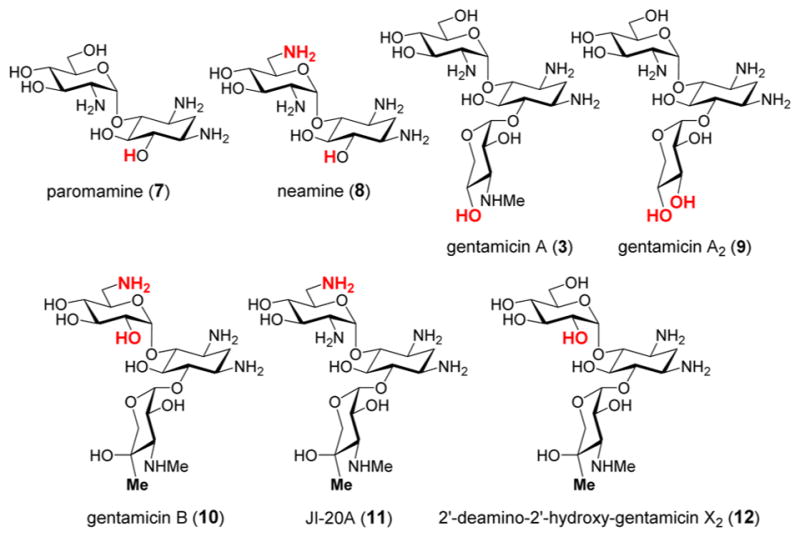
Compounds tested as substrates for GenK. The structural variations from GenX2 (4) are highlighted in red.
With regards to the mechanism of GenK-catalyzed methylation, three hypotheses have been proposed (see Scheme 2).5 Similar to other radical SAM enzymes,7 the reaction is initiated by reductive homolysis of SAM via single electron transfer from the reduced [4Fe-4S]1+ cluster in the active site to produce methionine (14) and a 5′-deoxyadenosyl radical (5′-dAdo·, 15) that abstracts a hydrogen atom from C6′ of GenX2 (4). In the first mechanism (see Scheme 2A), the resulting substrate radical (16) then abstracts the methyl group from Me-Cbl to give G418 (5) and Cbl(II). Single electron reduction of Cbl(II) followed by methyl transfer from SAM regenerates Me-Cbl after each turnover. Analogous catalytic cycles have been proposed for most other Cbl-dependent RSMTs.8–12 However, due to the decreased pKa of the 5′ α-hydroxyalkyl radical in 16, this intermediate may be deprotonated to form a ketyl radical anion (17/18) as proposed in the reactions catalyzed by DesII13–17 and (R)-2-hydroxyacetyl-CoA dehydratase.18,19 Subsequent nucleophilic attack by the ketyl intermediate 18 at Me-Cbl could result in an alkoxy radical (19) and a Cbl(I) intermediate. The alkoxy radical could then be reduced either by an external electron followed by protonation (mechanism B in Scheme 2) or via hydrogen atom transfer from 5′-dAdo (13) (mechanism C in Scheme 2). In the latter scenario, the SAM so produced could serve to regenerate Me-Cbl such that SAH is the sole reaction byproduct and 5′-dAdo is not produced. However, the 1:1:1 stoichiometry of G418 (5), SAH (2) and 5′-dAdo (13) formation observed in early experiments casts doubt on mechanism C.5
Scheme 2. Possible Mechanisms of Catalysis by GenKa.

a(A) Methylation occurs at C6′ of 16 by radical-mediated methyl transfer. (B) Methylation occurs at C6′ via SN2-type chemistry involving a ketyl radical anion intermediate 17/18 as the nucleophile. (C) Same as mechanism B, except the product radical 19 is quenched by reclaiming a hydrogen atom from 5′-dAdo (13) to regenerate SAM (1).
To distinguish among the mechanisms in Scheme 2, three mechanistic probes, [6′-F]-GenX2 (20), [6′-OMe]-GenX2 (21) and [5′-F]-GenX2 (22), were prepared (see Figure 2) as described in the Supporting Information. If GenK can accept [6′-F]-GenX2 (20) as a substrate, then mechanisms B and C would be ruled out, because the α-fluoroalkyl radical intermediate would be electron-deficient at C6′ and is thus not expected to be a Lewis base. The same mechanistic prediction also holds for [6′-OMe]-GenX2 (21). As for [5′-F]-GenX2 (22), if mechanism A is operative, the turnover product should still retain the fluoro group at C5′. In contrast, elimination of the 5′-fluoride is possible if the GenK reaction proceeds via mechanisms B or C due to predicted carbanionic character of C6′ in the ketyl intermediate.
Figure 2.
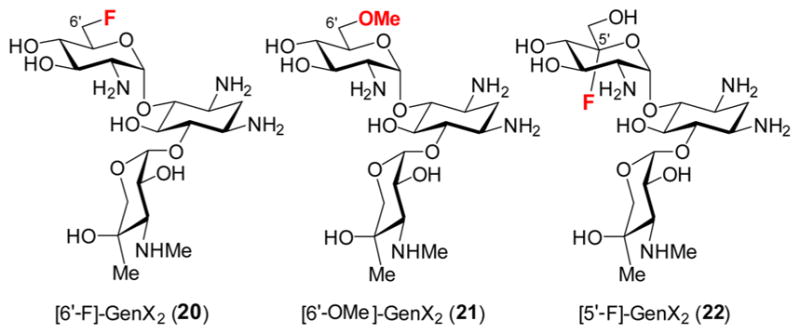
Analogs used to probe the GenK mechanism.
These substrate analogs were separately incubated with GenK, with GenX2 (4) used as a positive control. However, none of these analogs showed any evidence of reaction with GenK (Figure S2). Instead, [6′-F]-GenX2 (20) was found to be a weak inhibitor for GenK via end point assays conducted in the presence of 1 mM GenX2 with the concentration of [6′-F]-GenX2 varied from 0.01 to 20 mM (see Figure S3). These results imply that the 6′-F analog does bind into the GenK active site and that the 6′-OH group is likely important to properly orient the substrate to facilitate initial hydrogen atom abstraction.
To probe the orienting role of the 6′-OH group in substrate binding and to gain more insight into the catalytic cycle of GenK, the stereochemical course of the conversion of GenX2 (4) to G418 (5) was also investigated. This was accomplished by synthesizing the deuterated substrate isotopologs [6′S-2H]-GenX2 (23) and [6′R-2H]-GenX2 (24) (see Supporting Information). These isotopologs (23 and 24, 1 mM each) were separately incubated with GenK as described above. The 5′-dAdo (13) produced was collected by HPLC and deuterium incorporation was analyzed by mass spectroscopy (Figures S3 and S4). When [6′S-2H]-GenX2 (23) was used in the GenK reaction, the isotopic envelope of the isolated 5′-dAdo was not found to differ significantly from that produced in a control reaction with unlabeled GenX2 (4) indicating natural abundance labeling (Figure 3a vs 3b). However, when [6′R-2H]-GenX2 (24) was employed, the peak corresponding to incorporation of a single deuterium was found to be dominant (Figure 3c) indicating deuterium transfer from 24 to 5′-dAdo· (15) to produce deuterated 5′-dAdo.
Figure 3.
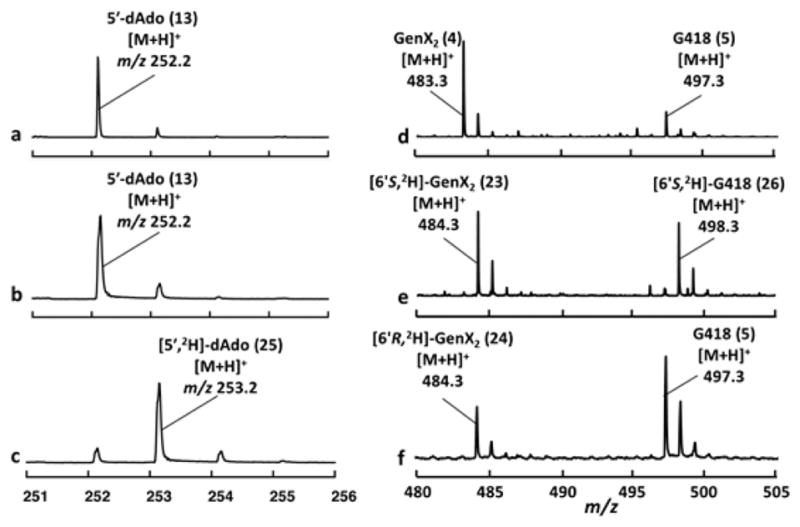
Electrospray ionization (ESI) mass data of (a–c) 5′-dAdo (13, [M + H]+ = 252 m/z) and (d–f) G418 (5, [M + H]+ = 497.28 m/z) generated during incubation of GenK with (a and d), GenX2 (4, [M + H]+ = 483.28 m/z), (b and e) [6′S-2H]-GenX2 (23), and (c and f) [6′R-2H]-GenX2 (24).
The G418 product (5) formed during incubation of GenK with the isotopologs 23 and 24 was also analyzed by mass spectrometry. When [6′S-2H]-GenX2 (23) was utilized as the substrate, the deuterium label was found to be retained in the G418 consistent with the observation of natural abundance labeling in the 5′-dAdo produced (see Figure 3e). In contrast, when [6′R-2H]-GenX2 (24) was used, the majority of the Gen418 produced was unlabeled (Figure 3f). Nevertheless, the isotopic envelope in this reaction deviated significantly from that expected for natural abundance labeling (compare Figure 3d vs 3f) implying partial retention (i.e., ca. 20%) of the deuterium label in G418 when [6′R-2H]-GenX2 is the substrate. Similar observations were made when the assay mixtures were treated with 1-fluoro-2,4-dinitrobenzene at the end of the incubation and the resulting dinitrophenyl (DNP)-derivatized G418 was isolated by HPLC and analyzed by mass spectrometry (see Figure S5).
These results reveal that H atom abstraction from C6′ of GenX2 is stereoselective for the pro-R C6′-H; however, the primary deuterium kinetic isotope effect (KIE) on [2H]-atom abstraction from [6′R-2H]-GenX2 can lead to increased abstraction of the otherwise disfavored pro-S H atom. Consequently a mixture of labeled and unlabeled Gen418 are observed with this isotopolog. Because G418 (5) is known to have the R-configuration at C6′,20 these results demonstrate that the majority (i.e., at least 95% assuming a primary deuterium KIE greater than 5) of the reaction flux proceeds with retention of configuration as the pro-R H atom is abstracted and the methyl group is transferred from Me-Cbl to the GenX2 substrate radical.
In contrast to the 5′-dAdo and Gen418 reaction products, no deuterium incorporation was detected in either the SAH (2) produced during the reaction nor the residual SAM (1) regardless of which isotopolog (i.e., 23 or 24) was included as the substrate (Figures S6 and S7). These results are inconsistent with mechanism C where either SAH or the regenerated SAM is expected to be at least partially labeled with deuterium when [6′R-2H]-GenX2 (24) serves as the substrate. This provides additional evidence arguing against the plausibility of mechanism C.
The stereochemical course of only a handful of reactions catalyzed by Cbl-dependent RSMTs have been reported.21 For GenD1 (3 → 4)11 and MoeK5,22 this information can be directly deduced from the structures of the substrate and product as methylation occurs at a stereogenic center in both cases (e.g., C4″ of 3 for GenD1). In contrast, the stereo-specificity of the β-methylation reactions in the biosynthesis of bottromycin23 and the β-methylation step of 2-hydroxyethyl-phosphonate (HEP) catalyzed by Fom3 during fosfomycin biosynthesis9,24 were determined experimentally by feeding studies with labeled precursors. In all of these cases, the reaction occurs with inversion of configuration at the carbon center where methylation takes place.21 These findings led to the suggestion that Me-Cbl and the radical SAM machinery are situated on opposite sides of the center to be methylated once bound in the active site of these enzymes.9,11,21 This hypothesis is consistent with the only known crystal structure of a Cbl-dependent radical SAM enzyme, OxsB.25 Though not a RSMT, the substrate binding site of OxsB is postulated to be interposed equidistant between the C5′ of SAM and the Co of the Cbl cofactor.
The generality of this hypothesis has, however, been brought into question by recent studies on Fom3 where the substrate was found to be cytidylyl-HEP and the methylation reaction was shown to be nonstereoselective.10 The present results with GenK provide further evidence that reactions catalyzed by this class of enzymes do not necessarily proceed via a mechanism that is stereochemically uniform over all cases. Though rotation of the carbon center bearing the unpaired electron may occur in the case of the Fom3 substrate radical intermediate, such free rotation is not applicable to the mechanism of GenK catalysis given the stereoselectivity of the reaction. Thus, the C5′ hydroxymethyl functionality of GenX2 is predicted to be constrained in a single orientation during the pro-R H atom abstraction event to generate a transient α-hydroxyalkyl or ketyl radical intermediate. As the results with the 6′-fluoro (20), 6′-OMe (21) and 6′-amino (11) derivative suggest, this configuration may be crucial for H atom abstraction from the substrate during turnover. Finally, this constraint is maintained during the subsequent radical or nucleophilic attack on the Me-Cbl donor that likely sits adjacent rather than opposite to the radical SAM machinery in the GenK Michaelis complex, thereby leading to retention of configuration during H atom abstraction and methyl addition.
In summary, the Cbl-dependent RSMTs represent the largest and most diverse collection of RSMTs presently known. Based on prior in vitro investigations of eight of these enzymes (TsrM,26–28 Fom3,8–10 GenD1,11 ThnK,12 PhpK,29–31 PoyC,32 CysS33 and GenK5), a model of substrate binding wherein the substrate is sandwiched between the Cbl cofactor and the radical SAM machinery has been suggested. However, the present results with GenK suggest that this may not necessarily be a general property of these enzymes. Instead, the GenK Michaelis complex is predicted to involve a front-side juxtaposition of both cofactors with respect to the substrate. Hence, from just this small number of Cbl-dependent RSMTs studied to date, the chemistry and structural features of these enzymes are proving to be quite varied, and they are expected to be rich source of new discoveries for future investigations.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM035906) and the Welch Foundation (F-1511).
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b09890.
Details regarding GenK assays along with HPLC protocols, and chemical syntheses of related compounds (PDF)
References
- 1.Tehlivets O, Malanovic N, Visram M, Pavkov-Keller T, Keller W. Biochim Biophys Acta, Mol Basis Dis. 2013;1832:204–215. doi: 10.1016/j.bbadis.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grillo MA, Colombatto S. Amino Acids. 2008;34:187–193. doi: 10.1007/s00726-007-0500-9. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Q, van der Donk WA, Liu W. Acc Chem Res. 2012;45:555–564. doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauerle MR, Schwalm EL, Booker SJ. J Biol Chem. 2015;290:3995–4002. doi: 10.1074/jbc.R114.607044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim HJ, McCarty RM, Ogasawara Y, Liu Y-n, Mansoorabadi SO, LeVieux J, Liu H-w. J Am Chem Soc. 2013;135:8093–8096. doi: 10.1021/ja312641f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinstein MJ, Luedemann GM, Oden EM, Wagman GH, Rosselet JP, Marquez JA, Coniglio CT, Charney W, Herzog HL, Black J. J Med Chem. 1963;6:463–464. doi: 10.1021/jm00340a034. [DOI] [PubMed] [Google Scholar]
- 7.Frey PA, Hegeman AD, Ruzicka FJ. Crit Rev Biochem Mol Biol. 2008;43:63–88. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- 8.Allen KD, Wang SC. Arch Biochem Biophys. 2014;543:67–73. doi: 10.1016/j.abb.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodyer RD, Li G, Zhao H, van der Donk WA. Chem Commun. 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 10.Sato S, Kudo F, Kim SY, Kuzuyama T, Eguchi T. Biochemistry. 2017;56:3519–3522. doi: 10.1021/acs.biochem.7b00472. [DOI] [PubMed] [Google Scholar]
- 11.Huang C, Huang F, Moison E, Guo J, Jian X, Duan X, Deng Z, Leadlay PF, Sun Y. Chem Biol. 2015;22:251–261. doi: 10.1016/j.chembiol.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marous DR, Lloyd EP, Buller AR, Moshos KA, Grove TL, Blaszczyk AJ, Booker SJ, Townsend CA. Proc Natl Acad Sci U S A. 2015;112:10354–10358. doi: 10.1073/pnas.1508615112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruszczycky MW, Choi S-h, Liu H-w. J Am Chem Soc. 2010;132:2359–2369. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruszczycky MW, Choi S-h, Mansoorabadi SO, Liu H-w. J Am Chem Soc. 2011;133:7292–7295. doi: 10.1021/ja201212f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruszczycky MW, Choi S-h, Liu H-w. Proc Natl Acad Sci U S A. 2013;110:2088–2098. doi: 10.1073/pnas.1209446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin G-M, Choi S-h, Ruszczycky MW, Liu H-w. J Am Chem Soc. 2015;137:4964–4967. doi: 10.1021/jacs.5b02545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruszczycky MW, Liu H-w. Isr J Chem. 2015;55:315–324. doi: 10.1002/ijch.201400130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Darley DJ, Buckel W, Pierik AJ. Nature. 2008;452:239–242. doi: 10.1038/nature06637. [DOI] [PubMed] [Google Scholar]
- 19.Buckel W. Angew Chem, Int Ed. 2009;48:6779–6787. doi: 10.1002/anie.200900771. [DOI] [PubMed] [Google Scholar]
- 20.Shalev M, Kondo J, Kopelyanskiy D, Jaffe CL, Adir N, Baasov T. Proc Natl Acad Sci U S A. 2013;110:13333–13338. doi: 10.1073/pnas.1307365110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou S, Alkhalaf LM, de los Santos ELC, Challis GL. Curr Opin Chem Biol. 2016;35:73–79. doi: 10.1016/j.cbpa.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Ostash B, Doud EH, Lin C, Ostash I, Perlstein DL, Fuse S, Wolpert M, Kahne D, Walker S. Biochemistry. 2009;48:8830–8841. doi: 10.1021/bi901018q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kellenberger JL. PhD thesis. ETH; Zürich: 1997. [Google Scholar]
- 24.Hammerschmidt F, Kaehlig H. J Org Chem. 1991;56:2364–2370. [Google Scholar]
- 25.Bridwell-Rabb J, Zhong A, Sun HG, Drennan CL, Liu H-w. Nature. 2017;544:322–326. doi: 10.1038/nature21689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierre S, Guillot A, Benjdia A, Sandström C, Langella P, Berteau O. Nat Chem Biol. 2012;8:957–959. doi: 10.1038/nchembio.1091. [DOI] [PubMed] [Google Scholar]
- 27.Benjdia A, Pierre S, Gherasim C, Guillot A, Carmona M, Amara P, Banerjee R, Berteau O. Nat Commun. 2015;6:8377. doi: 10.1038/ncomms9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Blaszczyk AJ, Silakov A, Zhang B, Maiocco SJ, Lanz ND, Kelly WL, Elliott SJ, Krebs C, Booker SJ. J Am Chem Soc. 2016;138:3416–3426. doi: 10.1021/jacs.5b12592. [DOI] [PubMed] [Google Scholar]; (b) Blaszczyk AJ, Wang B, Silakov A, Ho JV, Booker SJ. J Biol Chem. 2017;292:15456–15467. doi: 10.1074/jbc.M117.778548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werner WJ, Allen KD, Hu K, Helms GL, Chen BS, Wang SC. Biochemistry. 2011;50:8986–8988. doi: 10.1021/bi201220r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen KD, Wang SC. Biochim Biophys Acta, Proteins Proteomics. 2014;1844:2135–2144. doi: 10.1016/j.bbapap.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu K, Werner WJ, Allen KD, Wang SC. Magn Reson Chem. 2015;53:267–272. doi: 10.1002/mrc.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parent A, Guillot A, Benjdia A, Chartier G, Leprince J, Berteau O. J Am Chem Soc. 2016;138:15515–15518. doi: 10.1021/jacs.6b06697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Schnell B, Baumann S, Müller R, Begley TP. J Am Chem Soc. 2017;139:1742–1745. doi: 10.1021/jacs.6b10901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.