

Abstract
Bilirubin is a component of the heme catabolic pathway that is essential for liver function and has been shown to reduce hepatic fat accumulation. High plasma bilirubin levels are reflective of liver disease due to an injurious effect to hepatocytes. In a healthy liver, bilirubin is conjugated and excreted to the intestine and converted by microbes to urobilinoids, which is reduced to the predominant pigment in feces, stercobilin, or reabsorbed. The function of urobilinoids in the gut or their physiological relevance of reabsorption is not well understood. In this review, we discuss the relationship of hepatic bilirubin signaling to the intestinal microbiota and their regulation of the liver to gut axis, as well as their capacity to mediate these processes.
Keywords: biliverdin reductase, urobilinoids, bile pigment, fatty liver disease, gut microbiota
Introduction
Symbiotically, a relationship exists between humans and a vast complex of bacteria, viruses, and fungi in the intestine that are collectively referred to as the gut microbiota [1]. The bacteria in the gut, which has been the most studied, have been shown to play a crucial role in maintaining a healthy intestinal function. The contributions of the bacteria include digestion, vitamin synthesis, and catabolism of fats, bile acids, hormones and drugs [2]. A majority of bacteria in the gut are anaerobes mostly due to the intestine being an anaerobic environment [1]. A complex bacterial network develops over a lifetime in animals. Newborns have an immature gut microbiota, partially inherited from their mother, that matures during the first two years of life [3, 4]. It is widely accepted that approximately 1000 different bacterial species inhabit the gut. However, a recent study by Frank et al. estimated that the total number of bacterial species in the gut might be ∼15,000 – 36,000 [5]. There is considerable interest in understanding how the bacterial species reduce bile acids, and more recently the bile pigment, bilirubin, and their role in the hepatic to gut signaling axis. The gut microbiota is modulated by bile acids, which metabolizes them to secondary bile salts. Perturbations in the microbiome affect the bile acid composition. Ultimately, bile acids signal along the liver-gut axis. Bilirubin is also excreted from the liver, and intestinal bacteria catabolize it in the gut to urobilinoids, which can also be reabsorbed through the hepatic portal vein surrounding the intestine. The interactions that microbiota has with bilirubin and metabolic waste, particularly fatty acids and carbohydrates, are crucial to understanding how they relate to several diseases. Urobilinoids also act as antioxidants, similar to bilirubin, and may play a larger part in the hepatic to gut signaling [6, 7]. Herein, we discuss the role of bilirubin in hepatic signaling and catabolism by the gut microbiota and their impact on the intestinal bacterial species in the regulation of this pathway.
Bilirubin and hepatic function
During eryptosis, erythrocytes are broken down in the spleen, liver, and bone marrow by reticuloendothelial cells causing the release of globin and heme. The globin is degraded via proteolytic enzymes into amino acids to be transported and reabsorbed throughout the body including the liver. The heme ring is opened by heme oxygenase (HMOX) releasing iron (Fe), carbon monoxide (CO), and the ring structure is converted to biliverdin (Figure 1) [8, 9]. Subsequently, biliverdin reductase (BVR) reduces biliverdin to bilirubin. BVR exists in two isozymes; BVRA (BLVRA) and BVRB (BLVRB), which form bilirubin IXα and bilirubin IXβ, respectively [8]. BVRA is central to bilirubin production in adults forming bilirubin IXα while bilirubin IXβ is mostly present in the fetal stage of development. Bilirubin IXα is insoluble and binds to albumin in the blood for transport to the liver for conjugation [8]. Bilirubin is eliminated from the body by conjugation to glucuronic acid by the UDP-glucuronosyltransferase (UGT) system in the liver and deposited into feces for removal [9, 10]. For many years, bilirubin was only regarded as the breakdown product of heme and was not known to confer any physiological benefit [11]. The cytoprotective capacity of bilirubin has been shown by its ability to be a potent radical scavenger with its antioxidant function, which reduces reactive oxygen species (ROS) especially in lipid peroxidation [9, 12]. Bilirubin has been shown to have anti-inflammatory effects on T helper type 17 (Th17) immune responses [13] and by inhibiting ROS generated by the toll-like receptor 4 [14]. More recently, bilirubin has been shown to bind to the peroxisome proliferator-activated receptor-α (PPARα), and this interaction may reveal to be key to its protective metabolic functions (Figure 1) [15]. PPARα promotes uptake, utilization, and catabolism of fatty acids by the upregulation of genes involved in fatty acid transport and peroxisomal and mitochondrial fatty acid β-oxidation. The fibrates have a high binding affinity to PPARα and have been shown to reduce inflammation as well as hyperlipidemia and fatty liver disease [16, 17]. The immunosuppressive effects of bilirubin may be related to the antioxidant or PPARα activation properties, which both have been shown to reduce inflammation. More investigations of the role of bilirubin and its molecular signaling properties are needed.
Figure 1. The heme catabolic pathway in liver to gut signaling.
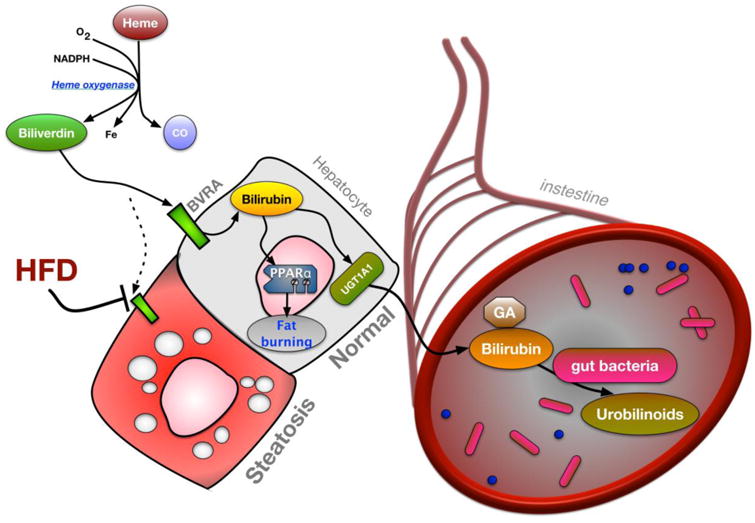
Heme is broken down by heme oxygenase to produce carbon monoxide (CO), iron (Fe), and biliverdin. In normal, healthy hepatocytes biliverdin is converted to bilirubin via biliverdin reductase A (BVRA). Bilirubin prevents hepatic steatosis by elevating the peroxisomal proliferator activated receptor α (PPARα) transcriptional activity to increase fat burning genes. Alterations in bilirubin production by BVRA result in marked steatosis in hepatocytes. Bilirubin is conjugated in hepatocytes by the UGT1A1 UDP-glucuronosyltransferase enzyme and excreted to bile eventually reaching the intestine. Conjugated bilirubin is deconjugated by the gut bacteria and reduced to urobilinoids.
To date, several studies in diverse human patient populations, and experimental investigations in rodents, have demonstrated the protective effects of hyperbilirubinemia in preventing the development of chronic liver disease [15, 18-20], diabetes [11, 21, 22], coronary artery disease [23, 24], and, recently, obesity [8, 15, 20, 25-27]. Multiple investigations have shown a negative correlation between serum bilirubin levels and obesity [25, 28, 29], as well as the prevalence of non-alcoholic fatty liver disease (NAFLD) [18, 19]. Individuals with polymorphisms in the hepatic UDP-glucuronosyltransferase 1-1 (UGT1A1) gene, which is the primary gene responsible for the conjugation of bilirubin and excretion from the liver, have been shown to be protective against adiposity [30-32]. The Gilbert's syndrome (GS) polymorphism, UGT1A1*28, which contains an additional TA repeat in the TATA sequence of the UGT1A1 promoter region has been reported to be associated with a lower risk of cardiovascular disease and NAFLD [30, 33, 34]. There has been more than one gene variant identified in the UGT1A1 locus; the UGT1A1*28 has been the most studied to date. Children with the UGT1A1*6 variant that were shown to have slightly elevated total plasma bilirubin levels at 0.36 mg/dL compared to wild-type genotypes at 0.32 mg/dL, but lower than the UGT1A1*28 at 0.43 mg/dL [34]. Interestingly, the UGT1A1*6 variant was found to be negatively correlated with NAFLD in obese children, even though bilirubin levels were only slightly elevated [34]. These studies suggest that bilirubin may be protective against the progression of liver disease. Patients with unconjugated hyperbilirubinemia were shown to be less likely to progress to nonalcoholic steatohepatitis (NASH) and have a less severe form of hepatic fibrosis with a smaller degree of liver stiffness [35].
Biliverdin, the precursor molecule to bilirubin, has also been shown to protect the liver from ischemia-reperfusion injury (IRI) in pig liver transplants [36]. IRI results from the ischemic insult and inflammation from the transplantation process [36]. Transplanted livers with biliverdin treatment showed a decrease in serum aspartate aminotransferase (AST) compared to control [37]. Elevated serum AST suggests hepatic damage due to the injured hepatocytes [37, 38]. Biliverdin does requires BVRA to protect against acute hepatitis in mice [39]. In addition, biliverdin treated pig livers also had less apoptosis, reduced neutrophil infiltration, and increased cellular proliferation following reperfusion [37]. These observations posit that biliverdin treatment and its conversion via BVRA to bilirubin is most likely protecting the liver against damage and preventing apoptosis of hepatocytes (Figure 1). However, the role(s) of unconjugated or conjugated bilirubin in liver diseases is currently unknown.
Mostly, investigations have measured serum levels of total bilirubin, while some have assayed for unconjugated bilirubin, but not taking into account the level of conjugated bilirubin. Conjugated bilirubin, which is mostly found in the intestine, is also present in the serum at concentrations between 0.06 to 0.48 μM or 3-5% of total bilirubin [40, 41]. Conversely, very high levels of serum total bilirubin >10 mg/dL are indicative of hepatic damage. Patients with hepatic diseases have been shown to have very high concentrations of unconjugated (20 μM) and conjugated (79 μM) bilirubin [41], which is most likely due to significant, irreversible damage to the liver that undermines any potential protective effects of elevations in serum unconjugated bilirubin. There are two genetic causes for increases in conjugated bilirubin (Rotor's and Dubin-Johnson Syndromes) that are not associated with cholestasis or hepatocellular damage. Most of the studies reporting the beneficial actions of moderate hyperbilirubinemia have either reported total or unconjugated bilirubin levels.
Patients with the GS polymorphism have bilirubin levels in the range of 18-58 μM for unconjugated and 0.1-0.6 μM for conjugated, which the latter only makes up 0.9% [41]. Elevated serum conjugated bilirubin has not been shown to prevent cardiovascular or metabolic diseases. The high serum levels of unconjugated bilirubin in GS patients have been negatively correlated with cardiovascular and metabolic protection including lower cholesterol, triglyceride levels, and pro-inflammatory cytokine levels, particularly interleukin-6 (IL-6) [31]. Growing evidence has shown bilirubin modulates lipid metabolism which could contribute to the health benefits associated with GS patients [42]. A recent study used a humanized mouse model for GS containing the human UGT1A1 *28 polymorphism (HuUGT*28) to mimic the hyperbilirubinemia seen in GS patients. The HuUGT*28 mice showed that an elevated unconjugated bilirubin decreased hepatic lipid accumulation on a high-fat diet and increased PPARα activation reducing obesity [20]. A recent study by Molzer et al. in humans with the GS polymorphism reported similar increases in PPARα and pAMPK [43]. However, the relative role of PPARα activation in the anti-steatotic and protective effects of unconjugated bilirubin in the liver has yet to be directly tested [44].
Hepatic bilirubin clearance
Bilirubin IXα is transported into the hepatocyte by the organic anion transporters, OATP1 (SLCO1B1) [45, 46] and OATP2 (SLCO2B1) [47-49], that are present in the lipid bilayer of the cell membrane (Figure 2). Within the cytosol of the hepatocyte, bilirubin is bound to Y-protein (ligandin) and Z-protein, which transports bilirubin to the endoplasmic reticulum for conjugation [50]. In the endoplasmic reticulum, UDP-glucuronosyltransferase (UGT1A1) adds glucuronic acid to the unconjugated bilirubin forming a soluble bilirubin that permits secretion [8]. The newly conjugated bilirubin is secreted by the multidrug resistant protein, MRP2 (ABCC2), through the canalicular membrane into the bile. MRP3 (ABCC3) deposits small amounts of soluble bilirubin back into the blood [50]. Mouse studies by van de Steeg et al. showed that the MRP3 sinusoidal export pump protein also secretes bilirubin conjugates into the blood and that the OATP1B1 and OATP1B3 mediate hepatic reuptake of bilirubin [45].
Figure 2. Hepatic bilirubin elimination.
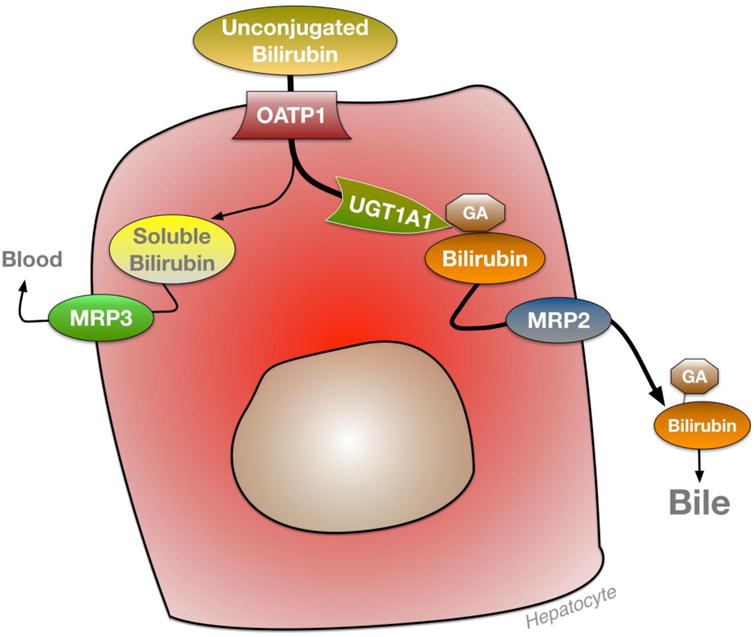
Unconjugated bilirubin enters hepatocytes through the organic anion transporter (OATP) and is conjugated with glucuronic acid (GA) by UDP-glucuronyltransferase UGT1A1 and excreted by multi-drug resistant protein (MRP2) to bile. Soluble bilirubin is secreted back into the blood by MRP3.
Experimental studies in rodents have shown that clearance of bilirubin occurs mostly in the liver and is mainly directed by two nuclear receptors, the constitutive androstane receptor (CAR) and the pregnane X receptor (PXR) [47-49]. CAR and PXR are xenobiotic-sensing transcription factors, which when bound to a ligand mediate gene expression through promoters of target genes [48, 49, 51], including UGT1A1 [48, 52], MRP2 and MRP3 [47, 48], and OATP2 [47-49]. Transactivation of CAR and PXR stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice [47]. CAR knockout mice have elevated total serum bilirubin [49], while PXR knockout mice have substantially lower bilirubin [49]. CAR has been shown to reduce bilirubin and increase the export of bile acids in serum. While CAR has not been shown to increase expression of the bile acid receptor, Farnesoid X Receptor (FXR), it does raise export of bile acids that are known to regulate FXR activity [47]. For these reasons, CAR is most likely an active modulator of bilirubin and bile acid levels. Huang et al. showed that bilirubin treatment in mice increased expression of CAR and induced its gene-regulated activity, but not as a direct agonist [48]. Bilirubin has only been shown to bind directly to PPARα [15], which increases expression of CAR [53, 54]. Most likely bilirubin induction of PPARα transcription activity raises CAR expression, which regulates bile acid and bilirubin turnover, but this has yet to be tested. Overall, there may be a hepatic heme to bile acid conveyer that mediates these processes (see Box 1). It should be noted that these pathways in humans have yet to be shown. More investigations, especially in humans, on bilirubin clearance and factors that regulate its catabolism are needed.
Box 1. Heme synthesis and bile acids.
The heme synthesis pathway in the liver is central to normal hepatocyte function and vitamin and iron homeostasis. Heme plays a critical role in the red blood cell's ability to bind oxygen. In the formation of heme, the rate-limiting enzyme, delta-aminolevulinic acid synthase (ALA synthase), has been studied due to its regulatory effects on heme synthesis. δ-ALAS1 mRNA expression increased significantly upon activation of the farnesoid X receptor (FXR) with chenodeoxycholic acid (CDCA) suggesting that bile acids positively regulate heme synthesis genes (Figure I) [80]. Increasing heme levels activates the heme oxygenase (HO-1) pathway, which produces bilirubin [27, 81]. An increase in bilirubin is known to prevent diabetes and obesity [8, 15, 20, 27]. Bilirubin activates the peroxisome proliferator-activated receptor alpha (PPARα) gene-regulatory activity to increase fatty acid oxidation and decrease lipid accumulation in the liver [27]. The increased levels of PPARα also reduced body weight, insulin sensitivity, and blood glucose. Heme synthesis is thereby central to understanding the signaling between the liver and the gut involving PPARα and bilirubin.
Bilirubin and bile
Very little is known about bilirubin regulation of bile, bile excretion, or on the management of bile acid synthesis or signaling. Primary bile acids are synthesized directly from cholesterol in the liver and excreted into the gall bladder. The bile acids are released from the gall bladder to the intestine during times of feeding to assist in the absorption of fats. Secondary bile acids are produced in the intestine by the gut microbiome [55]. Interestingly, modification of the gut microflora due to a change in diet shifts the bile acid composition which may contribute to inflammatory bowel disease [56]. Bile acids are mostly absorbed in the distal ileum via apical sodium-dependent bile salt transporter and the ileal bile acid transporter to be delivered to the liver for bile acid circulation [57]. The gut to liver reabsorption of bile acids has been shown to be an important mediator of overall health [55]. Studies involving cholestyramine, a bile acid sequestrant, have shown increased levels of total triglycerides in patients, while treatment with bile acids displayed a reduced amount of total triglycerides [58, 59]. Chow-fed mice with impairment of the FXR bile acid receptor, exhibited higher levels of serum triglycerides further illustrating the role of FXR in fat metabolism [60]. These studies suggest that the bacterial composition and reabsorption of bile acids are critical to maintaining metabolic balance. Investigations by Liu et al. and Hinds et al. showed that increasing bilirubin levels in mice lower total cholesterol levels [20, 21] but if bilirubin suppression of cholesterol effects bile acid synthesis remains unknown. These studies imply that the bilirubin bile pigment may impact cholesterol synthesis or secretion, which may also affect bile acid synthesis (see Box 2).
Box 2. Bile acid synthesis and reabsorption.
In the classical pathway, the enzyme, 7-alpha hydroxylase (CYP7A1), acts on cholesterol to form 7α-hydroxycholesterol, and 3β-hydroxy-Δ5-C27-steroid dehydrogenase (HSD3B7) forms 7α-hydroxy-4-cholesten-3-one [82]. After side chain modifications, the primary bile acid, cholic acid (CA) forms. The enzymes bile acid-CoA-synthase (BACS) and bile acid-amino acid transferase (BAT) conjugate primary bile acids with taurine or glycine forming secondary bile acids. Primary bile acid conjugation can be induced by the farnesoid X receptor (FXR), which binds directly to bile acids, and increases the expression of genes encoding for BACS and BATS [83]. Conjugation of the primary bile acids results in increased solubility and their release to the gallbladder. The gut microbiota achieves this by the dehydroxylation of the primary bile acids CA and CDCA via bacterial hydroxysteroid dehydrogenases [57, 71]. The newly formed secondary bile acids are recirculated to the liver where they are re-conjugated to glycine or taurine. The apical sodium-dependent bile acid transporter (ASBT) has been established as the bile acid transporter delivering bile acids from the epithelium to the intestinal lumen [84, 85]. Bile acid reabsorption and signaling pathways have been widely studied, and the sodium-taurocholate co-transporting polypeptide (NTCP) on hepatocytes, has been identified as the transporter that delivers bile acids to the liver from the portal circulation. The bile salt export pump (BSEP) secretes bile salts out of hepatocytes through the canalicular membrane and into the bile ducts. The processes that bilirubin uses for reabsorption in the intestine remain to be determined.
Bilirubin excretion to the gut and microbiota catabolism
Bilirubin secreted from the liver into the bile joins the biliary system and flows to the gut for excretion which undergoes microbial catabolism to urobilinoids in the intestinal lumen (Figure 2). Bilirubin reductase is currently a putative unknown enzyme but is most likely present in bacterial cells allowing them to reduce bilirubin to urobilinoids. The bacterial species that catabolize bilirubin to urobilinoids may be essential for normal gut function or could play a part in the development of intestinal diseases. Indeed, serum bilirubin levels have been shown to be negatively correlated with colorectal cancer [61]. However, there is very little known about the physiological role of urobilinoids, especially in their role in cancer, diabetes, or cardiovascular diseases. Of the bacteria within the gut microbiota, only four have been identified that are known to reduce bilirubin to urobilinoids, which include: Clostridium ramosum, Clostridium perfringens, Clostridium difficile, and Bacteroides fragilis [62-64]. Bilirubin catabolism to urobilinoids by the intestinal bacteria, mainly stercobilin, serves as the principal pigment in feces.
The most predominant bacteria in the gut are Bacteroidetes and Firmicutes, which comprises almost 90% of the phyla in the intestinal lumen [1]. The expression of bacteria in the gut varies with age. Newborns lack gut bacteria, and the feces comprises bilirubin in the intestine until the fifth day of life, in which the microbiota are more developed and capable of catabolizing bilirubin to urobilinoids [65]. Older infants and adults have more developed microbiota and can produce higher levels of urobilinoids [65]. The Firmicutes/Bacteroidetes ratio is high during adulthood but roughly a tenth of that during infancy and in adults older than 70. The total number and diversity of microbiota are reduced in the elderly as compared to adults, and thus the composition of the gut microbiota is frequently changing [66]. Bacteroidetes is a gram-negative anaerobic non-spore forming bacteria, while Firmicutes is a gram-positive anaerobic spore-forming bacteria that ferment sugars [3]. Bacteroides fragilis is the most common Bacteroides species and has very potent virulence factors. Bacteroides fragilis also has a unique ability to use oxygen which allows for it to occupy the mucosal surface, and has a capsule that produces an abscess formation [67]. These bacteria collectively produce urobilinoids from conjugated bilirubin and allow for pigmentation of excrement and reabsorption of urobilinogen into the portal circulation (Figure 3) [68]. Conjugated bilirubin delivered to the gut from the biliary system is deconjugated by β-glucuronidase and subsequently reduced by intestinal microbiota in the intestinal lumen to bilirubin metabolic compounds including urobilinogen, urobilin, dihydrobilirubin, mesobilirubin, dihyrdomesobilirubin, urobilinogen, half-stercobilinogen, and stercobilinogen which is oxidized to form the feces pigment stercobilin [68]. Clostridium perfringens has been found to reduce mesobilirubin, bilirubin glucuronides, amides, alkyl esters, taurates, and urobilin to the terminal bilirubin reduced product, stercobilin [68].
Figure 3. Urobilinoid excretion and reabsorption.
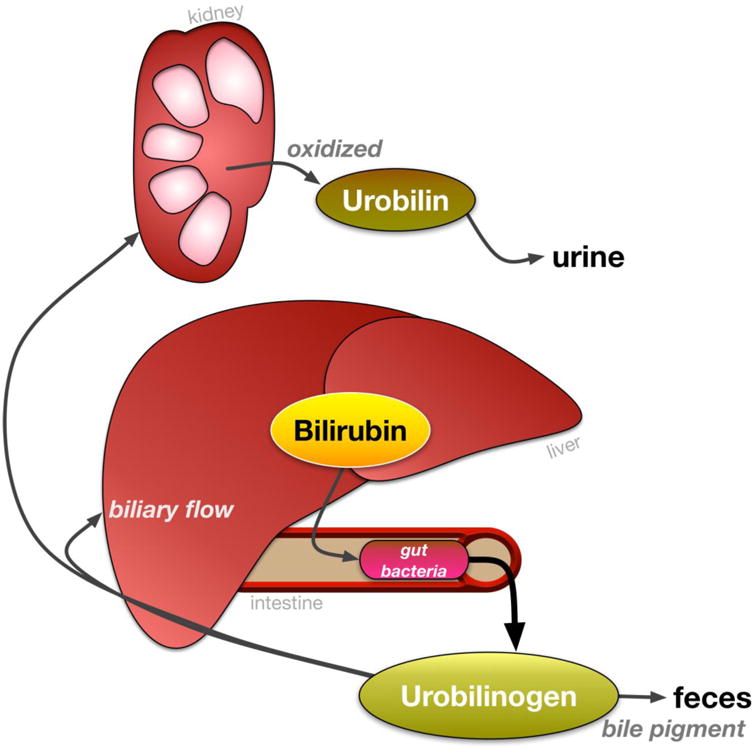
Bilirubin is excreted into the intestines and reduced to urobilinoids/urobilinogens by gut bacteria. Then, urobilinogens are either deposited into feces as the bile pigment or reabsorbed into hepatic portal circulation. Urobilinogens are taken up by the kidney and oxidized to urobilin and excreted into urine.
The regulation of bilirubin catabolism to urobilinoids by the gut microbiota has been shown in studies involving germfree rats fed a synthetic diet and the subsequent examination of their fecal matter. This was accomplished by examining the fecal content of fourth generation germfree rats. The rats with conventional intestinal microbiota produced a mean of 3.4 micromoles of stercobilinogen per day per 1 Kg body weight while germfree rats produced zero micromoles of stercobilinogen [69]. Clostridium ramosum was isolated and shown to increase the amount of urobilins in feces [69, 70]. This early study showed the central role of the gut microbiota in the formation of bilirubin derivatives and bacterial imbalance leading to variable urobilinoid levels. This dysbiosis has also helped elucidate the relationship between the liver and the gut via the portal vein through studies involving variable bacterial composition induced by antibiotic treatment. There is nothing known about why the intestinal bacteria have adapted to reduce bilirubin. However, the bacterial 7α dehydroxylation transformation of primary bile acids is necessary for normal gut and liver function (reviewed in detail in [55]), which may be found to be analogous to bilirubin reduction.
A wide array of bacteria possess bile salt hydrolases (BSH) which deconjugate bile salts and a smaller amount of bacteria contain bacterial hydroxysteroid dehydrogenases which dehydrogenate bile salts [71]. Elevated levels of secondary bile acids, lithocholic acid (LCA) and ursodeoxycholic acid (UDCA), act as effective antimicrobials reducing the levels of a broad array of bacteria in the gut, including Clostridium difficile [71]. The changes in bile acid and gut microbiota composition imply a similar variation in FXR. Mice fed a high-fat diet were administered an FXR antagonist and displayed a decrease in the Firmicutes/Bacteroidetes ratio [72]. The composition of bile acids regulates the formation of the gut microbiota. This relationship involving FXR relates to lipid metabolism directly, by increasing hepatic PPARα, and indirectly by affecting the gut microbiota, which affects bilirubin reduction and reabsorption. Bilirubin activates transcriptional activity of PPARα, which induces lipid metabolism in liver and adipose tissue by increasing carnitine palmitoyltransferase (CPT1) and fibroblast growth factor 21 (FGF21) expression [15]. PPARα is a sensor for fatty acids that improves mitochondrial function and β-oxidation. However, the impact that bilirubin catabolism or the selection of bacteria with bilirubin reductase has on these processes is unknown.
Bilirubin and urobilinoid regulation by the diet
Bilirubin and urobilinogen are antioxidants which can prevent lipid peroxidation and low-density lipoprotein (LDL) accumulation [7, 73]. The similarities between bilirubin and its derivatives and their function in the gut or liver are unknown. A study by Walker et al. showed distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 mouse strains on a high-fat diet [6]. Interestingly, they also found that urobilinoids were much more prominent in the cecal and liver meta-metabolomes of C57N obese mice. The investigation also found that the higher body weight in the C57N mice had more Bacteroidetes and fewer Firmicutes in cecal bacterial profiles, as well as higher Proteobacteria and fewer Defferribacteres. Several studies have shown in obese humans that plasma bilirubin levels are decreased [8, 20, 21, 25-28, 34, 74]. Bilirubin may be protective against obesity both in its ability to activate lipid metabolism and its function as an antioxidant. If bilirubin levels decrease in the obese due to changes in the gut microbiota is unknown. We can postulate that the shift in the gut microbiota is due to the increased nutrients and bile acids entering the intestinal environment permitting the growth of specific bacterial species. The change in gut microbiota may also be responsible for the increased urobilinoids that have been previously reported. Although the exact function of these urobilinoids is not known; it is possible they may have similar features to bilirubin. However, more work is needed to elucidate this process in humans and rodents. It is unclear whether or not urobilinoids play a direct role in any signaling pathway between the liver and the gut. More studies examining the relationship between bilirubin, urobilinoids, and the gut microbiota as they relate to hepatic steatosis and obesity are needed to further understand this pathway.
Bilirubin reabsorption
Enterohepatic circulation of bilirubin occurs exclusively from the bacterial de-conjugated form because conjugated bilirubin is bulky, highly polar, and soluble. Enterocytes prevent the passive transport of bulky amphiphiles into the portal circulation. Unconjugated bilirubin has been shown to be reabsorbed from the large and small intestines [75, 76]. There have been no specific transporters that have been shown to be involved in the reabsorption of bilirubin or bilirubin metabolites. However, transporters are known to exist in the intestine for bile acids, and specific transporters for bilirubin may be present. The bacterial populations vary in different regions of the gut making up a microbiota biogeography [77], which will affect the catabolism of products and metabolites produced and reabsorbed. Urobilinoids are formed from unconjugated bilirubin in the terminal ileum and the large intestine due to the gut microbiota. The Firmicutes bacterial phylum, including Clostridium that has been shown to reduce bilirubin, is found in both the small and large intestine. However, urobilinoid production may be more efficient in the colon since there are higher levels of total bacterial load compared to the small intestine [78]. Urobilinogen is reabsorbed into the portal circulation and delivered to the liver where it is recycled back into the biliary flow (Figure 3) [76]. Small amounts of urobilinogens are also deposited in the kidneys which oxidize them to urobilin, a yellow pigment seen in urine [79]. The physiological role of urobilinogen or other bilirubin metabolites and their reabsorption is unknown.
Conclusions
The gut microbiota act as a point of convergence for varying pathways that contribute to lipid metabolism and disease states. The catabolism of bilirubin to urobilinoids may regulate a phylum or subspecies of bacteria in the gut that helps maintain homeostasis. Determining the gut bacteria that express bilirubin reductase to produce urobilinoids may be critical to understanding intestinal diseases and overall health. Increasing plasma bilirubin decreases cholesterol levels and may regulate bile acid production, but the impact that this has on human diseases remains unknown. The role of hepatic BVRA in the regulation of bilirubin levels and their impact on the gut microbiome is an area that needs more investigation. Additionally, studies that measure the roles of unconjugated or conjugated bilirubin in the regulation of hepatic fat accumulation and obesity are essential to further understanding liver diseases. A clearer role of urobilinoid production and reabsorption and their physiological signaling will further advance our understanding of how bile pigments maintain metabolic balance.
Figure I. Theoretical heme and bile acid hepatic conveyor.

The bile acid receptor, Farnesoid X Receptor (FXR), is a transcription factor that increases heme synthesis genes and lipid-reducing genes. Heme is broken down by heme oxygenase (HO-1) to biliverdin and is further reduced to bilirubin by biliverdin reductase. Bilirubin is excreted into the intestine and further processed into urobilinoids. Unconjugated bilirubin can bind to the peroxisomal proliferator activated receptor α (PPARα) to increase fat burning genes which also activates constitutive androstane receptor (CAR) that regulates bile acid secretion. Bile acids are catabolized by gut bacteria.
Trends.
Bilirubin has been shown to reduce adiposity and fatty liver in the obese. Recent data show that bilirubin signals through nuclear receptors transcription factors and has the capacity to function as an antioxidant.
The gut microbiota reduces bilirubin to urobilinogens in the intestine. Emerging data have shown that urobilinogens are reabsorbed in the intestine of the obese. However, their function remains elusive.
Bilirubin derivatives contribute to the pigmentation in urine and feces, and may have a major role in the gut to liver axis.
Increasing unconjugated bilirubin levels in the plasma is a promising therapeutic for obesity and type II diabetes mellitus.
Outstanding Questions.
What is the physiological role of the gut microbiota in the catabolism of bilirubin? Do specific bacterial populations reduce bilirubin levels as a consequence of obesity or adiposity?
Does bilirubin and its byproducts regulate specific bacterial phyla? Does high bilirubin in the intestine cause selection of distinct bacterial species that may cause or reduce hepatic inflammation?
What is the function of bilirubin in the regulation of cholesterol levels and does this impact bile acid synthesis?
Are urobilinogens protective against obesity and diabetes? What is the role of urobilinogens in hepatic reabsorption?
Does reabsorption of urobilinogens and stercobilinogens have a positive or negative physiological impact?
Footnotes
Disclosure Statement: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sekirov I, et al. Gut microbiota in Health and Disease. Physiological Reviews. 2010;90(3):859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 2.Clarke G, et al. Minireview: Gut microbiota: The neglected Endocrine Organ. Molecular endocrinology. 2014;28(8):1221–1238. doi: 10.1210/me.2014-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans J, Morris L, Marchesi J. The gut microbiome: the role of a virtual organ in the host endocrinology. Journal of endocrinology. 2013;218(3):R37–R47. doi: 10.1530/JOE-13-0131. [DOI] [PubMed] [Google Scholar]
- 4.Kundu P, et al. Our Gut Microbiome: The Evolving Inner Self. Cell. 2017;171(7):1481–1493. doi: 10.1016/j.cell.2017.11.024. [DOI] [PubMed] [Google Scholar]
- 5.Frank D, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker A, et al. Distinct signatures of host-microbial meta-metabolome and gut microbiome in two C57BL/6 strains under high-fat diet. ISME J. 2014;8(12):2380–96. doi: 10.1038/ismej.2014.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura T, et al. Urobilinogen, as a bile pigment metabolite, has an antioxidant function. Journal of Oleo Science. 2006;55(4):191–197. [Google Scholar]
- 8.O'Brien L, et al. Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab. 2015;26(4):212–20. doi: 10.1016/j.tem.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomaro ML, Batlle AM. Bilirubin: its role in cytoprotection against oxidative stress. Int J Biochem Cell Biol. 2002;34(3):216–20. doi: 10.1016/s1357-2725(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 10.Sundararaghavan VL, Sindhwani P, Hinds TD., Jr Glucuronidation and UGT isozymes in bladder: new targets for the treatment of uroepithelial carcinomas? Oncotarget. 2016 doi: 10.18632/oncotarget.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitek L. The role of bilirubin in diabetes, metabolic syndrome, and cardiovascular diseases. Front Pharmacol. 2012;3:55. doi: 10.3389/fphar.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sedlak TW, et al. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci U S A. 2009;106(13):5171–6. doi: 10.1073/pnas.0813132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longhi MS, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. 2017;2(9) doi: 10.1172/jci.insight.92791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Idelman G, Smith DL, Zucker SD. Bilirubin inhibits the up-regulation of inducible nitric oxide synthase by scavenging reactive oxygen species generated by the toll-like receptor 4-dependent activation of NADPH oxidase. Redox Biol. 2015;5:398–408. doi: 10.1016/j.redox.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stec DE, et al. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS One. 2016;11(4):e0153427. doi: 10.1371/journal.pone.0153427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo YS, et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J Gastroenterol Hepatol. 2008;23(1):102–9. doi: 10.1111/j.1440-1746.2006.04819.x. [DOI] [PubMed] [Google Scholar]
- 17.Harano Y, et al. Fenofibrate, a peroxisome proliferator-activated receptor alpha agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26(5):613–20. doi: 10.1111/j.1478-3231.2006.01265.x. [DOI] [PubMed] [Google Scholar]
- 18.Jang BK. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2012;18(4):357–9. doi: 10.3350/cmh.2012.18.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kwak MS, et al. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. 2012;18(4):383–90. doi: 10.3350/cmh.2012.18.4.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hinds TD, Jr, et al. Mice with hyperbilirubinemia due to Gilbert's Syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARalpha. Am J Physiol Endocrinol Metab. 2017:ajpendo 00396 2016. doi: 10.1152/ajpendo.00396.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, et al. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism, Adipokines and PPARgamma Levels. Sci Rep. 2015;5:9886. doi: 10.1038/srep09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitek L, et al. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis. 2002;160(2):449–56. doi: 10.1016/s0021-9150(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 23.Mayer M. Association of serum bilirubin concentration with risk of coronary artery disease. Clin Chem. 2000;46(11):1723–7. [PubMed] [Google Scholar]
- 24.Zhang ZY, et al. Inverse relation of total serum bilirubin to coronary artery calcification score detected by multidetector computed tomography in males. Clin Cardiol. 2012;35(5):301–6. doi: 10.1002/clc.21964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Belo L, et al. Body Fat Percentage Is a Major Determinant of Total Bilirubin Independently of UGT1A1*28 Polymorphism in Young Obese. PLoS One. 2014;9(6):e98467. doi: 10.1371/journal.pone.0098467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinds TD, Jr, et al. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J Biol Chem. 2016 doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds TD, Jr, et al. Increased HO-1 levels ameliorate fatty liver development through a reduction of heme and recruitment of FGF21. Obesity (Silver Spring) 2014;22(3):705–12. doi: 10.1002/oby.20559. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Dong H, et al. Bilirubin increases insulin sensitivity in leptin-receptor deficient and diet-induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology. 2014;155(3):818–28. doi: 10.1210/en.2013-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, et al. Bilirubin Increases Insulin Sensitivity by Regulating Cholesterol Metabolism. Adipokines and PPARγ Levels. 2015;5:9886. doi: 10.1038/srep09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin JP, et al. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation. 2006;114(14):1476–1481. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 31.Wallner M, et al. Protection from age-related increase in lipid biomarkers and inflammation contributes to cardiovascular protection in Gilbert's syndrome. Clin Sci (Lond) 2013;125(5):257–264. doi: 10.1042/CS20120661. [DOI] [PubMed] [Google Scholar]
- 32.Bulmer AC, Verkade HJ, Wagner KH. Bilirubin and beyond: a review of lipid status in Gilbert's syndrome and its relevance to cardiovascular disease protection. Prog Lipid Res. 2013;52(2):193–205. doi: 10.1016/j.plipres.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 33.Schwertner HA, Vitek L. Gilbert syndrome, UGT1A1*28 allele, and cardiovascular disease risk: possible protective effects and therapeutic applications of bilirubin. Atherosclerosis. 2008;198(1):1–11. doi: 10.1016/j.atherosclerosis.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Lin YC, et al. Variants in the UGT1A1 gene and the risk of pediatric nonalcoholic fatty liver disease. Pediatrics. 2009;124(6):e1221–7. doi: 10.1542/peds.2008-3087. [DOI] [PubMed] [Google Scholar]
- 35.Kumar R, et al. Unconjugated hyperbilirubinemia in patients with non-alcoholic fatty liver disease: a favorable endogenous response. Clin Biochem. 2012;45(3):272–4. doi: 10.1016/j.clinbiochem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Zhai Y, et al. Ischaemia-reperfusion injury in liver transplantation--from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andria B, et al. Biliverdin protects against liver ischemia reperfusion injury in swine. PLoS One. 2013;8(7):e69972. doi: 10.1371/journal.pone.0069972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giannini EG, Testa R, Savarino V. Liver enzyme alteration: a guide for clinicians. CMAJ. 2005;172(3):367–79. doi: 10.1503/cmaj.1040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wegiel B, et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem. 2009;284(32):21369–78. doi: 10.1074/jbc.M109.027433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fevery J, et al. Plasma bilirubin pigments in health and disease. Mol Aspects Med. 1987;9(5):391–404. doi: 10.1016/0098-2997(87)90005-7. [DOI] [PubMed] [Google Scholar]
- 41.Fevery J, Vanstapel F, Blanckaert N. Bile pigment metabolism. Baillieres Clin Gastroenterol. 1989;3(2):283–312. doi: 10.1016/0950-3528(89)90002-x. [DOI] [PubMed] [Google Scholar]
- 42.Bulmer AC, et al. Improved resistance to serum oxidation in Gilbert's syndrome: a mechanism for cardiovascular protection. Atherosclerosis. 2008;199(2):390–6. doi: 10.1016/j.atherosclerosis.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 43.Molzer C, et al. Features of an altered AMPK metabolic pathway in Gilbert's Syndrome, and its role in metabolic health. Sci Rep. 2016;6:30051. doi: 10.1038/srep30051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinds TD, Jr, et al. Does bilirubin prevent hepatic steatosis through activation of the PPARalpha nuclear receptor? Med Hypotheses. 2016;95:54–57. doi: 10.1016/j.mehy.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van de Steeg E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122(2):519–28. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Steeg E, et al. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest. 2010;120(8):2942–52. doi: 10.1172/JCI42168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wagner M, et al. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42(2):420–30. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- 48.Huang W, et al. Induction of bilirubin clearance by the constitutive androstane receptor (CAR) Proc Natl Acad Sci U S A. 2003;100(7):4156–61. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saini SP, et al. Dual role of orphan nuclear receptor pregnane X receptor in bilirubin detoxification in mice. Hepatology. 2005;41(3):497–505. doi: 10.1002/hep.20570. [DOI] [PubMed] [Google Scholar]
- 50.Roberts M, et al. Enterohepatic Circulation. Clinical Pharmacokinetics. 2002;41(10):751–790. doi: 10.2165/00003088-200241100-00005. [DOI] [PubMed] [Google Scholar]
- 51.Yan J, Xie W. A brief history of the discovery of PXR and CAR as xenobiotic receptors. Acta Pharm Sin B. 2016;6(5):450–452. doi: 10.1016/j.apsb.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sugatani J, et al. Regulation of pregnane X receptor (PXR) function and UGT1A1 gene expression by posttranslational modification of PXR protein. Drug Metab Dispos. 2012;40(10):2031–40. doi: 10.1124/dmd.112.046748. [DOI] [PubMed] [Google Scholar]
- 53.Wieneke N, et al. PPARalpha-dependent induction of the energy homeostasis-regulating nuclear receptor NR1i3 (CAR) in rat hepatocytes: potential role in starvation adaptation. FEBS Lett. 2007;581(29):5617–26. doi: 10.1016/j.febslet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 54.Saito K, et al. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonists induce constitutive androstane receptor (CAR) and cytochrome P450 2B in rat primary hepatocytes. Drug Metab Pharmacokinet. 2010;25(1):108–11. doi: 10.2133/dmpk.25.108. [DOI] [PubMed] [Google Scholar]
- 55.Ridlon JM, et al. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. doi: 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Devkota S, Chang E. Interactions between diet, bile acid metabolism, gut microbiota, and inflammatory bowel diseases. Dig Dis. 2015;33(3):351–356. doi: 10.1159/000371687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Staley C, et al. Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Applied microbiology and biotechnology. 2016;100(613):1–18. doi: 10.1007/s00253-016-8006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bateson M, et al. Chenodeoxycholic acid therapy for hypertriglyceridaemia in men. Br J Clin Pharmacol. 1978;5:249–254. doi: 10.1111/j.1365-2125.1978.tb01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones R, Dobrilovic L. Lipoprotein lipid alterations with cholestyramine administration. J Lab Clin Med. 1970;75:953–966. [PubMed] [Google Scholar]
- 60.Sinal C, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 61.Zucker S, Horn P, Sherman K. Serum bilirubin levels in the U.S. population-gender effect and inverse correlation with colorectal cancer. Hepatology. 2004;40(4):827–835. doi: 10.1002/hep.20407. [DOI] [PubMed] [Google Scholar]
- 62.Fahmy K, Gray CH, Nicholson DC. The reduction of bile pigments by faecal and intestinal bacteria. Biochim Biophys Acta. 1972;264(1):85–97. doi: 10.1016/0304-4165(72)90119-5. [DOI] [PubMed] [Google Scholar]
- 63.Vitek L, et al. The impact of intestinal microflora on serum bilirubin levels. Journal of hepatology. 2005;42(2):238–243. doi: 10.1016/j.jhep.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 64.Konickova R, et al. Reduction of bilirubin ditaurate by the intestinal bacterium Clostridium perfringens. The Journal of the Polish Biochemical Society and of the Committee of Biochemistry and Biophysics Polish Academy of Sciences. 2012;59(2):289–291. [PubMed] [Google Scholar]
- 65.Vitek L, et al. Intestinal colonization leading to fecal urobilinoid excretion may play a role in the pathogenesis of neonatal jaundice. J Pediatr Gastroenterol Nutr. 2000;30(3):294–8. doi: 10.1097/00005176-200003000-00015. [DOI] [PubMed] [Google Scholar]
- 66.Mariat D, et al. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiology. 2009;9(1):123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wexler H. Bacteroides: the good, the bad, the nitty-gritty. Clinical Microbiology Reviews. 2007;20(4):593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vitek L, et al. Identification of bilirubin reduction products formed by Clostridium perfringens isolated from human neonatal fecal flora. Journal of Chromatography. 2006;833(2):149–157. doi: 10.1016/j.jchromb.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 69.Gustafsson B, Lanke L. Bilirubin and urobilins in germfree, ex-germfree, and conventional rats. Journal of experimental medicine. 1960;1(112):975–981. doi: 10.1084/jem.112.6.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saxerholt H, et al. Influence of antibiotics on the faecal excretion of bile pigments in healthy subjects. Scandinavian Journal of Endocrinology. 1986;21(8):991–996. doi: 10.3109/00365528608996410. [DOI] [PubMed] [Google Scholar]
- 71.Nie Y, Hu J, Yan X. cross-talk between bile acids and intestinal microbiota in host metabolism and health. Journal of Zhejiang university science B. 2015;16(6):436–446. doi: 10.1631/jzus.B1400327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L, et al. Farnesoid X Receptor signaling shapes the Gut Microbiota and Controls Hepatic Lipid Metabolism. American Society for microbiology. 2016;1(5):e00070–16. doi: 10.1128/mSystems.00070-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dudnik LB, et al. Effect of bilirubin on lipid peroxidation, sphingomyelinase activity, and apoptosis induced by sphingosine and UV irradiation. Biochemistry (Mosc) 2001;66(9):1019–27. doi: 10.1023/a:1012329911696. [DOI] [PubMed] [Google Scholar]
- 74.Andersson C, et al. Acute effect of weight loss on levels of total bilirubin in obese, cardiovascular high-risk patients: an analysis from the lead-in period of the Sibutramine Cardiovascular Outcome trial. Metabolism. 2009;58(8):1109–15. doi: 10.1016/j.metabol.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 75.Vitek L. Intestinal metabolism of bilirubin in the pathogenesis of neonatal jaundice. J Pediatr. 2003;143(6):810. doi: 10.1067/S0022-3476(03)00542-0. author reply 811-2. [DOI] [PubMed] [Google Scholar]
- 76.Lester R, Schmid R. Intestinal Absorption of Bile Pigments. 3. The Enterohepatic Circulation of Urobilinogen in the Rat. J Clin Invest. 1965;44:722–30. doi: 10.1172/JCI105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14(1):20–32. doi: 10.1038/nrmicro3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jandhyala SM, et al. Role of the normal gut microbiota. World journal of Gastroenterology. 2015;21(29):8787–8803. doi: 10.3748/wjg.v21.i29.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rupe CO, Fetter MC. Urinary urobilinogen determined by a mercuric chloride procedure. Clin Chem. 1981;27(8):1385–7. [PubMed] [Google Scholar]
- 80.Peyer AK, et al. Regulation of human liver delta-aminolevulinic acid synthase by bile acids. Hepatology. 2007;46(6):1960–1970. doi: 10.1002/hep.21879. [DOI] [PubMed] [Google Scholar]
- 81.Vanella L, et al. Increased heme-oxygenase 1 expression in mesenchymal stem cell-derived adipocytes decreases differentiation and lipid accumulation via upregulation of the canonical Wnt signaling cascade. Stem Cell Res Ther. 2013;4(2):28. doi: 10.1186/scrt176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chiang J. Bile acids: regulation of synthesis. The Journal of Lipid Research. 2009;50(10):1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reshetnyak V. Physiological and molecular biochemical mechanisms of bile formation. World journal of Gastroenterology. 2013;19(42):7341–7360. doi: 10.3748/wjg.v19.i42.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alrefai W, Gill R. Bile Acid Transporters: Structure, Function, Regulation and Pathophysiological Implications. Pharmaceutical Research. 2007;24(10):1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 85.Kosters A, Karpen S. Bile acid transporters in health and disease. Xenobiotica. 2008;38(7-8):1043. doi: 10.1080/00498250802040584. [DOI] [PMC free article] [PubMed] [Google Scholar]