

Abstract
Objective
Reverse cholesterol transport (RCT) comprises cholesterol efflux from ABCA1-expressing macrophages to apo AI giving nascent high density lipoprotein (nHDL), esterification of nHDL-free cholesterol (FC), selective hepatic extraction of HDL-lipids, and hepatic conversion of HDL-cholesterol to bile salts, which are excreted. We tested this model by identifying the fates of nHDL-[3H]FC, [14C]PL, and [125I]apo AI in serum in vitro and in vivo.
Approach and Results
During in vitro incubation of human serum, nHDL-[3H]FC and [14C]PL rapidly transfer to HDL and low density lipoproteins (LDL; t1/2 = 2–7 min) while nHDL-[125I]apo AI transfers solely to HDL (t1/2<10 min) and to the lipid-free form (t1/2 >480 min). Following injection into mice nHDL-[3H]FC and [14C]PL rapidly transfer to liver (t1/2 ~ 2–3 min) whereas apo AI clears with t1/2 = ~460 min. The plasma nHDL-[3H]FC esterification rate is slow (0.46%/h) compared to hepatic uptake. Phospholipid transfer protein enhances nHDL-[14C]PL but not nHDL-[3H]FC transfer to cultured Huh7 hepatocytes.
Conclusions
nHDL-FC, PL, and apo AI enter different pathways in vivo. Most nHDL-[3H]FC and [14C]PL are rapidly extracted by the liver via scavenger receptor class B member 1 and spontaneous transfer; hepatic PL uptake is promoted by phospholipid transfer protein. nHDL-[125I]apo AI transfers to HDL and to the lipid-free form that can be recycled to nHDL formation. Cholesterol esterification by LCAT is a minor process in nHDL metabolism. These findings could guide the design of therapies that better mobilize peripheral tissue-FC to hepatic disposal.
Keywords: reverse cholesterol transport, nascent high density lipoprotein biogenesis, atherogenesis, ATP-binding cassette transporter A1, lipoprotein receptors
Subject Codes: Lipids and cholesterol, metabolism, atherosclerosis, cell biology/structural biology
Graphic Abstract
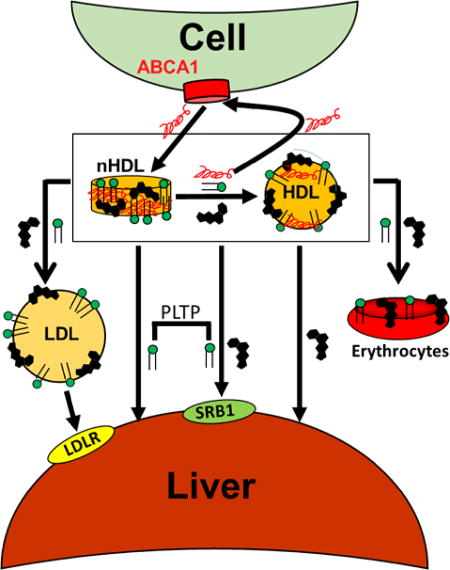
Visual Overview – An online visual overview is available for this article.
Atherosclerosis and its major sequela, cardiovascular disease, are major causes of mortality and morbidity in the United States.1 Atherosclerosis, which is characterized by the accumulation of cholesterol in macrophages within the subendothelial space of the arterial wall, is suppressed by reverse cholesterol transport (RCT).2–4 According to one model,3–5 RCT is a three-step process whereby arterial macrophage-cholesterol transfers to the liver for disposal. The initial RCT step is the formation of nascent high density lipoproteins (nHDL) via the interaction of apo AI with the ABCA1 lipid transporter; the nHDL so-formed contains mainly apo AI, free cholesterol (FC), and phospholipid (PL).6,7 The second step has both molecular and macromolecular consequences, respectively, esterification of nHDL-FC to cholesteryl ester (CE) by plasma lecithin:cholesterol acyltransferase (LCAT), and concurrent conversion of nHDL from a disc to a sphere with a central CE core. In the third step—cholesterol disposal—hepatic scavenger receptor class B member 1 (SR-B1) selectively extracts lipids from HDL via a nibbling mechanism in which apo AI and apo AII are excluded from uptake and HDL is reduced to a remnant while releasing lipid-free apo AI.8 Although cited to near-consensus,4 this RCT model has never been rigorously validated. Herein we provide data showing that the major components of ABCA1-derived HDL—FC, PL, and apo AI—segregate in serum in vitro and enter different pathways for clearance in vivo. Importantly, our results reveal that a major portion of the rapid hepatic nHDL-FC and PL clearance is independent of the reaction catalyzed by LCAT. These data are important in the context of the current segue from “HDL-C is the good cholesterol” to “HDL functionality is at least equally important.”4,9–13
Materials and Methods
Materials and Methods are available in the online-only Data Supplement
Results
SEC Analysis of nHDL
nHDL was produced by incubation of apo AI with ABCA1-expressing BHK cells with and without methyl β-cyclodextrin (CDX)-mediated FC-loading. The media from each incubation were collected, concentrated, and analyzed. After the cells were FC-loaded with 0, 0.06, and 0.26 mM FC (0, 2.5, and 5 mM CDX respectively), the resulting cellular FC contents were 29 ± 2, 42 ± 6, and 52 ± 6 µg/mg cell protein. The size exclusion chromatography (SEC) profiles of the media (Figure 1) revealed two well-resolved peaks for protein absorbance corresponding to medium and small nHDL, nHDL-M and nHDL-S, respectively, which were collected and analyzed by SEC (Figure 1 A–C, chromatograms b, c). Based on past in vitro and in vivo studies6,14 showing a large nHDL is formed from FC-loaded cells, we collected a third larger (L), less prominent, and earlier-eluting nHDL-L (Figure 1 A–C, chromatogram a). SEC analysis of the pooled fractions for nHDL-L, -M and -S showed that in the absence of CDX-mediated FC-loading, only two nHDL were formed (Figure 1 A, chromatograms b, c). However, the SEC profile of nHDL from CDX-mediated FC-loaded cells revealed three fractions (Figure 1 B, C, chromatograms a–c). The first two, nHDL-M and S, were similar to those found without CDX-mediated FC-loading; the third, larger nHDL-L, was unique to nHDL formed from CDX/FC-loaded cells. Based on SEC and comparison with the elution volumes of standard proteins, the respective diameters of nHDL-L, M, and S were 14.5 nm, 12.2 nm and 8.8 nm; the corresponding molecular weights were 510, 262, and 96 kDa. According to non-denaturing PAGE (Figure 1 D), the molecular weights were 482, 298 and 91 kDa respectively. Of note, while not detected by SEC, a minor amount of nHDL-L, detected by non-denaturing PAGE, was formed in cells without FC loading (Lane L, No CDX). As expected and shown in the SDS-PAGE (Figure 1 E), apo AI is the dominant protein in all three nHDL fractions but minor bands in the 60 – 200 kD range are also detected. The remainder of our studies were performed with the most abundant fraction, nHDL-M, which for brevity will from hereon be called nHDL. nHDL used in our study is, according to SEC, stable; repeat analytical SEC runs 1 and 3 months apart were identical.
Figure 1.

SEC Isolation of nHDL. nHDL was collected from the media after apo AI incubation of BHK cells over expressing ABCA1 with no FC-loading (A, 0 CDX) and with FC-loading with 2.5 mM (B) and 5.0 mM CDX (C) and analyzed by SEC. Grey-filled plots are the A280 SEC of the media; red bars denote the fractions from media that were collected and pooled as large (L), medium (M) and small (S) nHDL. Analytical A280 SEC of the pooled fractions L, M and S are shown in the chromatograms labeled a, b and c respectively. Non-denaturing PAGE (D) and SDS-PAGE (E) of the L, M and S pooled fractions from each of the three cell cholesterol loading conditions as labeled. Molecular weight standards are in the left lanes, and apo AI and BSA are as labelled.
nHDL Composition
The trends in nHDL composition as a function of particle size were similar for all three FC-loading conditions. PL/apo AI, FC/apo AI and FC/PL molar ratios and mol% FC in each particle increased with nHDL particle size (Table 1). The PC/SM ratios were 4.3 ± 0.2, 5.6 ± 0.2, and 7.0 ± 0.9 for the large, medium, and small nHDL respectively; the PC/SM ratio for the BHK-ABCA1 cells from which the nHDL were formed was 7.4 ± 0.2. The PC/SM ratios of the large and medium nHDL were significantly less than those of the small nHDL and the cells, p< 0.005 for all of these comparisons.
Table 1.
Nascent HDL Molar Compositions
| CDX* | nHDL† | PL/apo AI mol/mol |
FC/apo AI mol/mol |
FC / PL mol/mol |
mol% FC |
mol% PL |
|---|---|---|---|---|---|---|
| 0.0 | Large | 43.2 ± 1.8 | 76.7 ± 2.5 | 1.77 ± 0.05 | 64.0 | 36.0 |
| 0.0 | Medium | 34.2 ± 1.5 | 54.6 ± 2.3 | 1.60 ± 0.03 | 61.5 | 38.5 |
| 0.0 | Small | 17.9 ± 0.6 | 16.1 ± 0.5 | 0.90 ± 0.03 | 47.3 | 52.7 |
| 2.5 | Large | 39.4 ± 1.7 | 66.1 ± 1.0 | 1.68 ± 0.07 | 62.7 | 37.3 |
| 2.5 | Medium | 33.7 ± 1.4 | 49.6 ± 2.0 | 1.47 ± 0.00 | 59.5 | 40.5 |
| 2.5 | Small | 17.7 ± 0.4 | 13.7 ± 0.4 | 0.77 ± 0.02 | 43.6 | 56.4 |
| 5.0 | Large | 48.6 ± 0.8 | 83.8 ± 1.4 | 1.72 ± 0.01 | 63.3 | 36.7 |
| 5.0 | Medium | 37.6 ± 1.0 | 56.3 ± 1.6 | 1.50 ± 0.02 | 60.0 | 40.0 |
| 5.0 | Small | 16.2 ± 1.0 | 13.3 ± 1.0 | 0.82 ± 0.02 | 45.0 | 55.0 |
Concentration, mM
nHDL sizes are from Figure 1.
nHDL-[3H]FC Rapidly Transfers to HDL and Low Density Lipoproteins (LDL) In Vitro
nHDL-[3H]FC was incubated with serum under several conditions of time and temperature, and analyzed by SEC (Figure 2). nHDL-[3H]FC elutes as a single SEC peak (Figure 2 A). When added to HDL, nHDL-[3H]FC rapidly transfers to HDL. After rapid mixing at 4 °C followed by immediate injection into the FPLC, SEC shows all the [3H]FC is already associated with HDL (Figure 2 B). When incubated with whole serum, nHDL-[3H]FC transfers to all lipoproteins. Initially, most is associated with HDL, but lesser amounts co-elute with HDL and more are associated with the apo B-containing lipoproteins with increasing incubation time at 37° C. (Figure 2 C). The kinetics of transfer of nHDL-[3H]FC to other lipoproteins were too fast to quantify by SEC. [3H]FC accumulated in LDL with a t1/2 = 15 min (Figure 2D). Parallel studies of the transfer of native human HDL-[3H]FC to serum gave remarkably similar results with the majority of HDL-[3H]FC transferring to LDL within 2 h (Supplementary Figure I). Given time limitations of SEC, the kinetics of nHDL-[3H]FC transfer to LDL were determined by heparin-Mn+2 precipitation of LDL15,16 after its incubation with nHDL-[3H]FC at 37° C for various times; the halftime for this process was 2.1 ± 0.5 min (Figure 2E). Notably, transfer of nHDL- and HDL-FC to LDL occurred over a time frame in which less than 5% of nHDL- and HDL-[3H]FC are converted to CE by mouse plasma LCAT activity (Supplementary Figure II).
Figure 2.
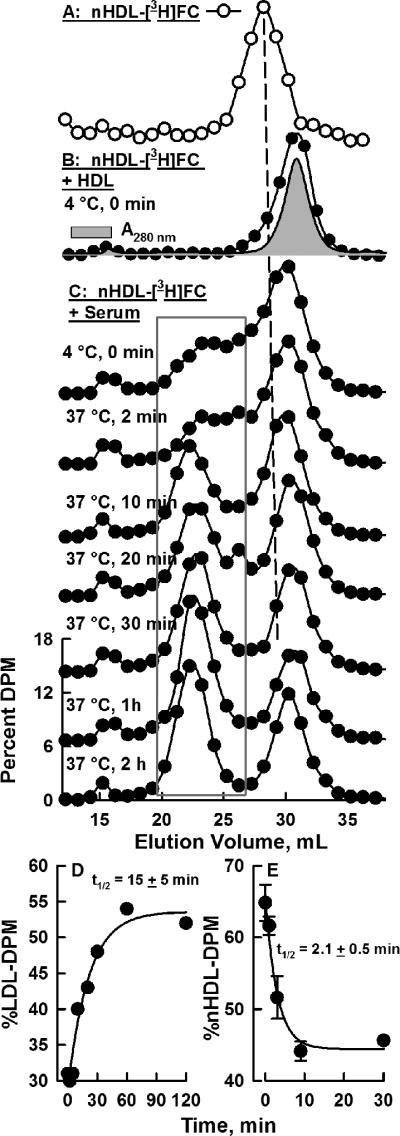
Transfer of nHDL-[3H]FC to Serum Lipoproteins. nHDL-[3H]FC was prepared from BHK-ABCA1 cells labeled with [3H]FC as in Figure 1. nHDL-[3H]FC were tested under several conditions and analyzed by SEC, which was monitored by β-counting of the collected fractions. A) nHDL-[3H]FC; B) nHDL-[3H]FC incubated with HDL at 4 °C and immediately analyzed by SEC (0 min). C) nHDL-[3H]FC incubated with serum at 4 °C or 37oC for various times as labeled and analyzed by SEC. D) Kinetics of [3H]FC accumulation in LDL (fractions encompassed by grey box in Panel C). E) The kinetics of nHDL-[3H]FC transfer to isolated LDL determined by heparin-Mn+2 precipitation of LDL.
nHDL-[14C]PL Transfers to HDL and LDL In Vitro
Experiments similar to those of Figure 2 were conducted with nHDL-[14C]PL. Like nHDL-[3H]FC, the elution volume of nHDL-[14C]PL was distinct from those of the major native serum lipoproteins (Figure 3 A). SEC analysis following incubation of nHDL-[14C]PL with serum revealed a rapid transfer of [14C]PL to LDL (peak elution volume 22 – 23 mL) but not to very low density lipoproteins (VLDL; Figure 3, peak elution volume 16 mL); the former had a transfer halftime = ~6.5 min and reached ~33% of total DPM at t ≥ 30 min (Figure 3 C). In whole serum, the halftime for nHDL-[14C]PL transfer to HDL appeared to be on a time scale similar to that for transfer to LDL but could not be quantified because the elution volumes of nHDL-[14C]PL and HDL overlap (Figure 3). The halftime for HDL-[14C]PL transfer to serum lipoproteins was slightly faster, t1/2 ≤5 min (data not shown). Halftimes for nHDL-[14C]PL transfer to lipoproteins in the absence of non-lipidated proteins were slower: for nHDL-[14C]PL transfer to LDL in total lipoproteins, to isolated LDL, and to HDL respectively t1/2 = 3.4, 4.1, and < 0.5 h. (Supplementary Figures III–V). Transfer of nHDL-[14C]PL to total lipoproteins, which do not contain lipid transfer factors, was enhanced by the addition of PLTP (Supplementary Figure III, Panel A, right insert).
Figure 3.

Transfer of nHDL-[14C]PL to Serum Lipoproteins: A) Control nHDL-[14C]PL, no serum (O). B) nHDL-[14C]PL was incubated with serum at 37 °C for various times (as labeled) and separated by SEC after which collected aliquots were β-counted (●). The grey rectangle circumscribes the regions of LDL-[14C]PL that were summed to calculate kinetics of nHDL-[14C]PL transfer to LDL, which is shown in Panel C.
nHDL-[125I]Apo AI Transfers to HDL In Vitro
Experiments similar to those in Figures 2 and 3 were also conducted with nHDL-[125I]apo AI. The SEC elution profile of nHDL-[125I]apo AI was distinct from those of the native serum lipoproteins (Figure 4 A) and from that of lipid-poor [125I]apo AI (Figure 4 B). According to SEC analysis, nHDL-[125I]apo AI rapidly transferred to HDL, as seen after only brief incubation with serum at 4 °C (Figure 4C). Changes in the nHDL-[125I]apo AI distribution occurred in two time frames. At early times (0.5 – 2 h), nearly all nHDL-[125I]apo AI coeluted with HDL (Figure 4 D–F). At later times, ≥ 4 h, some [125I]apo AI co-eluted with lipid-poor apo AI (Compare Figures 4 B and I–K). A trace of [125I]apo AI was detected in the LDL and none appeared in the VLDL fractions. A direct comparison of the SEC distribution of nHDL-[3H]FC, [14C]PL, and [125I]apo AI before and after short incubations with HDL or serum is shown in Supplementary Figure IX.
Figure 4.

Transfer of nHDL-[125I]Apo AI to Serum Lipoproteins. A) SEC Profile of nHDL-[125I]apo AI (O). B) SEC of [125I]Apo AI(●) and HDLA280 nm (grey fill). C–K) SEC profiles of [125I]apo AI after incubation with serum at various temperatures and times as labeled.
Serum Factors Segregate nHDL-FC, PL, and Apo AI
Figure 5 compares the distribution of nHDL-FC, PL, and apo AI at the end of 2 and 24 h incubations with serum (from the data of Figures 2, 3, and 4). These data show that each component of nHDL has a different fate. After a 2 h incubation, nHDL-FC distributes as LDL > HDL >> VLDL; nHDL-PL distributes as HDL > LDL > lipid-poor (LP) apo AI > VLDL; nHDL-apo AI distributes as HDL > LP apo AI >> LDL. The distribution of nHDL analytes is only slightly different after a 24 h incubation with serum.
Figure 5.

Distributions of nHDL-[3H]FC, [14C]PL, and [125I]apo AI into Serum Lipoproteins and Lipid-Poor Apo AI. Bars represent the percent distribution of each radio-analyte from Figures 2–4 at 2 h. The 24 h values for [125I]apo AI are shown as a red line above the 2 h bar.
In vivo nHDL Metabolism
The plasma clearance of the constituents of nHDL and their appearance in major organs was followed in groups of mice after injection of nHDL-[3H]FC, [14C]PL, and [125I]apo AI. The respective halftimes for disappearance of nHDL-[3H]FC and nHDL-[14C]PL from plasma were fast, t1/2 = 5.2 ± 0.07 and 2.0 ± 0.4 min (Figure 6 A, B). The in vivo kinetics for nHDL-[125I]apo AI were profoundly different from those for FC and PL (Figure 6 C). These data best fit a bi-exponential curve having fast and slow components with half times of 2.1 ± 1.0 min and 7.7 ± 1.3 h; the respective pre-exponential factors, which reflect the relative contributions of the two components to the total decay, were 0.68 and 1.9, i.e., 25 and 75% of the total. In the same experiment, we collected tissues at various times and determined their associated [3H]FC, [14C]PL and [125I]apo AI. The majority of the tissue [3H]FC- and [14C]PL-uptake was hepatic (Figure 6 D—F; Supplementary Figure VI); the corresponding halftimes for hepatic uptake were 2.6 ± 0.7 and 1.4 ± 0.1 min (Figure 6 D, E inserts). These data complemented the plasma kinetic data by showing the rapid disappearance of [3H]FC and [14C]PL coincided with rapid hepatic uptake. Similarly, the much slower disappearance of plasma [125I]apo AI coincided with little hepatic uptake, which declined with time (4.5 → 0.5 % over 24 h); the initial rapid apo AI clearance has been attributed to transfer from plasma to extravascular spaces.17–19 At later times, hepatic [3H]FC and [14C]PL declined while accumulating in other tissue sites, especially in erythrocytes, which respectively contained 13 and 7% of injected [3H]FC and [14C]PL at 30 min and 8% and 10% at 24 h (Supplementary Figures VI and VII). In a second set of in vivo experiments we compared the plasma kinetics and organ uptake of nHDL- vs. HDL-[3H]FC and [14C]PL. Rates of plasma clearance of FC and PL from the two lipoproteins were similarly rapid (t1/2 <3 min; Supplementary Figure VIII) and organ uptake of nHDL- and HDL-FC and PL was mostly hepatic (data not shown). Kinetic constants for the plasma clearance and liver accretion of nHDL-FC, PL, and apo AI are summarized in Table 2.
Figure 6.

nHDL-FC, PL and Apo AI Kinetics for Plasma Clearance and Hepatic Uptake in Mice. Plasma and tissue were collected and analyzed at various times following the injection of nHDL-[3H]FC, -[14C]PL, and -[125I]Apo AI; details are in Material and Methods. A – C) Plasma decay plots for each analyte. Data are plotted as smoothed curves with the ordinates adjusted to the same scale for comparison. For C, data were fitted to a four-parameter bi-exponential equation having the form, Weight-Normalized Plasma DPM = ae-k1t + be-k2t where t is time, k1 and k2 are rate constant, and a and b are their respective pre-exponential constants that reflect injected dose. D – E) Hepatic uptake plots for each analyte. Inserts in D and E show the kinetics for the initial (0 – 30 min) rapid hepatic uptake of [3H]FC and [14C]PL.
Table 2.
Kinetic Constants for the Plasma Clearance and Liver Accretion of nHDL-FC, PL, and Apo AI
| Plasma Decay Kinetics* | Liver Uptake, | |||
|---|---|---|---|---|
|
| ||||
| Plasma Analyte | k, min−1 | t1/2, min | k, min−1; uptake max, (%) | t1/2, min |
| HDL-[3H]FC | 0.68 ± 0.09 | 1.0 ± 0.09 | 0.91 ± 0.7 (25 ± 6) | 0.77 ± 0.33 |
| HDL-[14C]PL | 0.46 ± 0.16 | 1.5 ± 0.4 | 0.46 ± 0.23 (12 ± 2) | 1.5 ± 0.5 |
| nHDL-[3H]FC | 0.22 ± 0.12 | 3.2 ± 2.1 | 0.17 ± 0.06 (18 ± 3) | 4.1 ± 1.1 |
| nHDL-[14C]PL | 0.52 ± 0.23 | 1.3 ± 0.7 | 0.71 ± 0.2 (11 ± 2) | 1.4 ± 0.05 |
| nHDL-[125I]Apo AI† | 0.34 ± 0.29 | 2.1 ± 1.1 | --- | --- |
| (1.5 ± 0.32) × 10−3 | 460 ± 80 | |||
From Figure 6 and Supplementary Figure VIII.
apoAI kinetics were conducted on a longer time scale (Supplementary Figure VI) from which short and long components of the kinetics were calculated.
PLTP Increases Hepatocyte nHDL-PL Uptake
Given the rapid removal of nHDL-[14C]PL by liver in vivo (Figure 6), its faster in vitro transfer to lipoproteins in serum (Figure 3) compared to TLP (Supplementary Figure III), and the PLTP-mediated increase in nHDL-[14C]PL transfer to TLP (Supplementary Figure III), we tested the effects of PLTP on nHDL- and HDL-FC and PL uptake by cultured human hepatocyte Huh7 cells. Our data revealed similar rates of nHDL- and HDL-[3H]FC uptake, which were not affected by addition of PLTP to the incubation media (Figure 7 A and C). The rates of nHDL- and HDL-[14C]PL uptake were also similar. However, addition of PLTP to the incubation media increased nHDL-[14C]PL uptake (+85%; p = 0.0016) but not that of HDL-[14C]PL (Figure 7 B and D). When uptake was calculated as percent of input, FC uptake from both nHDL and HDL was 30% at 180 min, while PL uptake, in this case PC uptake, in the absence of PLTP was 5%, and increased to 10% from nHDL in the presence of PLTP. Greater uptake of FC vs PC has also been reported for fibroblasts.20
Figure 7.

Comparison of the rates of Hepatic nHDL- and HDL-[14C]PL Uptake In Vitro. nHDL- and HDL-[14C]PL (20 µg/mL protein) were incubated with Huh7 cells in the presence or absence of PLTP (10 µg/mL) and the cell-associated radioactivity determined at various times as shown. Additional details are provided in the Materials and Methods. Slopes in panel B were different, p = 0.0016. Slopes in the other panels were not significantly different.
Discussion
Formulation of the RCT Hypothesis
Discovery of the role of LCAT in HDL metabolism provoked the hypothesis that cholesterol esterification is part of a broader cholesterol transport process that prevents accumulation of cytotoxic FC levels in peripheral tissue.21–23 This process, now known as RCT, was hypothesized decades before the transporters involved in the first step, nHDL production mainly by ABCA1,24,25 and the selective hepatic lipid-uptake receptor, SR-B1,26 involved in the final disposal step, were identified. In the context of atheroprotection by HDL, RCT evolved into a three-step process—cholesterol efflux from ABCA1-expressing macrophages in the subendothelial space of the arterial wall to apo AI or preβ HDL giving rise to nHDL, esterification of nHDL-FC by plasma LCAT producing more mature, spherical HDL, and selective hepatic extraction of HDL-CE by SR-B1, which “nibbles” a few CE from many HDL.8 In the liver, CE is converted to FC, some of which is converted to bile salts, both of which enter the bile and then transfer to the feces for excretion.27
nHDL Composition, Size and Structure
ABCA1-derived nHDL is thought to be a precursor to the mature forms of HDL that occur in plasma. The composition, size and structure of ABCA1-derived nHDL6,7 as well as the metabolism of nHDL-protein derived from several cell types, including macrophage foam cells, have been reported and are similar.18,28,29 Cell-ABCA1-derived nHDL is both similar and different from human plasma pre-β HDL. Both contain FC, PL and apo AI. However, the human plasma pre-β HDL is smaller and is ~90% protein, mostly apo AI,30 and has been proposed to be a terminal nascent HDL product,18 a conclusion that is consistent with the likely rapid removal of lipids from nascent HDL soon after its production as reported here and by others.19,31 This difference may limit the transferability of findings with ABCA1-derived nHDL to human pre-β HDL metabolism.
nHDL-lipid metabolism has not been reported so that we have advanced the state of knowledge of the role of nHDL-analytes in HDL metabolism and are the first to study the plasma clearance and tissue uptake of nHDL-FC, PL, and apo AI derived from the same cell system and using the same mouse model, C57BL/6J mice. The major novelty of our contribution is the observed rapid, quantitative in vivo metabolism of the nHDL-FC and PL in a background of slower apo AI metabolism in which LCAT plays a minor role in the nHDL-FC clearance and tissue uptake. These findings should guide the design of therapies that promote the transfer of peripheral tissue-FC to the liver for disposal.
Our findings are consistent with previous nHDL studies: Interaction of apo AI with the cellular ABCA1 transporter produces multiple nHDL species with dimensions that are a function of cellular cholesterol content6,7,32,33 and cell type34 with lower ratios of FC/PL in nHDL of J774 macrophage cells and human fibroblasts. A high FC/PL molar ratio, with FC > PL (Table 1) has been reported previously for large and medium nHDL formed by BHK-ABCA1 cells6,32 as well as HEK293-ABCA1 cells.7 Our data confirm that nHDL-SM content increases with increasing nHDL size35 and that erythrocytes are FC and PL reservoirs (Supplementary Figures VI and VII).36
Most variously-sized nHDL are discoidal,37 and according to electron microscopy studies, both 9 nm and 12 nm diameter nHDL from J774 macrophage cells are discoidal.34 Our preliminary electron cryo-microscopic studies of nHDL from BHK-ABCA1 cells also show a discoidal structure (J. Michael Bell, et al., unpublished). In contrast, nHDL from HEK293-ABCA1 cells were reportedly mostly spherical.7 We confirmed6,7 that the nHDL from BHK-ABCA1 cells is FC-rich and contains PL and apo AI and then studied the in vitro and in vivo fates of [3H]FC, [14C]PL and [125I]apo AI of the major nHDL-M species.
In Vitro Fates of nHDL-FC, PL, and Apo AI
The rate-limiting step for spontaneous lipid transfer from particle surfaces is desorption,38,39 which follows Kelvin’s law stating that desorption halftimes are a positive function of particle radius.40,41 Differences in particle shape—HDL is a sphere and nHDL is a disc, and composition—nHDL and HDL are 60 (Table 1) and 10 mol% FC,42 play unknown roles. However, with these caveats we conclude that their similar Stokes radii would dictate similar halftimes for FC desorption from nHDL and HDL, i.e., ~2–4 min.15 Consistent with this, the halftimes for nHDL-[3H]FC transfer to LDL is 2.1 ± 0.5 min (Figure 2 E). Given that FC desorption rates are independent of the identity of the acceptor,15 the rate of nHDL-[3H]FC transfer to HDL is the same as its transfer to LDL, ~2–4 min. In whole serum the number of HDL particles is ten times that of all other lipoprotein particles combined43 so that based on collision theory the first encounter of desorbing nHDL-[3H]FC is expected to be HDL, the same as that found for plasma acceptors of macrophage FC efflux.44 nHDL- and HDL-[3H]FC are esterified to [3H]CE by LCAT.21 However, the FC esterification rate is slow compared to the rate of transfer such that >90% of the [3H] activity transferred to LDL in the initial 10 min is [3H]FC, not [3H]CE (Supplementary Figure II). Slow FC esterification vs. rapid hepatic uptake in mice has also been observed for HDL-FC.19
The in vitro transfer halftime of dipalmitoylphosphatidylcholine (DPPC) from HDL3 to LDL is longer than that of FC, i.e., 5 h,15 which is similar to what we observed for nHDL-PL transfer to total lipoproteins (3.4 h), LDL (4.1 h), and HDL (2.1 h) (Supplementary Figures III–V). In contrast, nHDL-[14C]PL transfer to lipoproteins in whole serum is faster (t1/2 = 6.5 min; Figure 3C). We attribute the faster rate of PL transfer in whole serum to PLTP activity45 for several reasons as discussed below. nHDL-apo AI transfers to HDL within 10 min in vitro (Figure 4) and between 2 and 24 h, some HDL-associated apo AI transfers to the lipid-free or lipid-poor form, an effect that is likely a function of HDL instability;46,47 HDL is disrupted by many HDL-modifying factors–LCAT, CE and PL transfer proteins, and endothelial and hepatic lipases.48–52 The distribution of nHDL-[3H]FC, [14C]PL, and [125I]apo AI into lipoproteins in serum at 2 h (Figure 5), which is near equilibrium, is similar to the natural distribution of FC, PL, and apo AI among serum lipoproteins.42,53 Most FC is in LDL, while more PL is in HDL than LDL, and nearly all apo AI is in HDL.53
Metabolic Fates of nHDL-FC, PL, and Apo AI In Vivo
The disappearance halftime for plasma nHDL-[3H]FC in vivo (t1/2 = 5.2 min; Figure 6 A) is comparable to its in vitro transfer to plasma lipoproteins, primarily HDL (t1/2 = 2.1 min; Figure 2 E); the longer t1/2 observed by SEC (Figure 2 D) is due to the intrinsic limitation of this method to t1/2 >15 min. Rapid plasma clearance of nHDL-[3H]FC coincided with that of [3H]FC accretion by the liver, initially the main tissue site of uptake (Figure 6 D). nHDL-[3H]FC is likely cleared by multiple pathways. In one, nHDL-[3H]FC transfers directly to liver cells. In the other, nHDL-[3H]FC transfers to HDL from whence [3H]FC transfers to the liver. Two likely mechanisms underlie these pathways. One is mediated by SR-B1, which plays a role in reversible HDL-FC flux.54–56 SR-B1 overexpression in CHO cells increases their uptake of HDL-FC,57 and compared to WT mice, hepatic removal of HDL-FC is faster in mice overexpressing SR-B1.19 Given the size and compositional similarities of nHDL and HDL, we propose that some nHDL-[3H]FC is also cleared by hepatic SR-B1. The rates of desorption of both nHDL- and HDL-FC are rapid so that direct receptor-independent, spontaneous transfer to the plasma membranes of liver cells is a likely second mechanism underlying the accretion of [3H]FC by the liver and other major tissue sites at later times post nHDL-[3H]FC injection (Supplementary Figures VI and VII). Both mechanisms could support the role of HDL as the shuttle between the liver and extrahepatic tissues. The halftime for nHDL-FC transfer to the liver is 2.6 min during which mouse plasma only esterifies 2.6×0.46% FC/min =~1% of nHDL-FC (Supplementary Figure II). Thus, most hepatic nHDL-FC uptake is independent of LCAT activity; a similar finding has been reported for HDL-FC in mice19 and humans.58
Similarly, t1/2 for the in vivo disappearance of nHDL-[14C]PL coincided with its rapid appearance in liver (Figure 6 B, E). However, unlike nHDL-[3H]FC, plasma clearance and hepatic accretion of nHDL-[14C]PL was faster—t1/2 = 2.0 and 1.4 min respectively (Figure 6 B, E)—than its in vitro desorption to isolated lipoproteins (t1/2 = 2– 4 h; Supplementary Figure III – V) but comparable to nHDL-[14C]PL transfer to lipoproteins in whole serum, t1/2 = 6.5 min (Figure 3). Thus, transfer of nHDL-[14C]PL from plasma to liver must involve a plasma factor or a hepatic receptor. This factor is likely PLTP, a conclusion supported by four observations: nHDL is a PLTP target;18,28 injection of PLTP into rat doubles the rate of clearance of an rHDL-PC ether;59 transfer of nHDL-[14C]PL to LDL in serum is faster than transfer in total lipoproteins in the absence of the lipoprotein-deficient fractions (6.5 min vs. 3.4 h; Figure 3, Supplementary Figure III); addition of PLTP increases transfer to LDL in a dose-dependent manner (Supplementary Figure IIIA, right insert); and PLTP increases the rate of transfer of nHDL-[14C]PL to Huh7 hepatocytes (Figure 7). nHDL-[14C]PL could, like nHDL- and HDL-FC,19,57 also be cleared by hepatic SR-B1 although the much slower rate of selective uptake compared to FC (12%),60 could make this mechanism quantitatively less important. Hydrolysis could also play a role but only at much longer times. For example, in rat, the plasma t1/2 for the disappearance of [3H]PC is ~1 h. This process is mediated, in part, by LCAT and hepatic lipase-mediated hydrolysis, which account for ≥90% of PC clearance.61 In contrast to the nHDL-lipids, the plasma t1/2 for nHDL-[125I]apo AI is longer, 460 min, and after fusion with HDL and subsequent its release as lipid-free or poor, apo AI is available for additional efflux cycles.
Relevance to Human Pathophysiology
It is interesting to compare our data with similar human kinetic studies of plasma HDL-[3H]FC. If one considers that human metabolism is slower than that of mouse, there are striking similarities. Among human volunteers, only a small fraction of intravenously injected HDL-[3H]FC is converted to HDL- and LDL-[3H]CE; the majority of FC is rapidly cleared from plasma into tissue without esterification with t1/2 ~7– 9 min.31,58 Moreover, as with our observed rapid in vivo hepatic nHDL-[3H]FC uptake data, human HDL-[3H]FC rapidly appears in bile reaching a maximum within 40 min.62
A Model for nHDL Metabolism
nHDL, with its high FC content, and the transient nature of its lipid components, appears to be a key modulator of in vivo cholesterol homeostasis and phospholipid exchange among lipoproteins and tissues. Our data support a model of hepatic nHDL metabolism as follows (Figure 8, Steps 1 – 8): Apo AI interaction with cellular ABCA1 forms nHDL containing FC, PL, and apo AI. nHDL-FC rapidly transfers to the liver either directly or via initial transfer to HDL, followed by transfer to the liver; this occurs via both SR-B1 and spontaneous transfer. nHDL-PL transfer to liver occurs directly or via HDL and although our cell data implicate PLTP in this process, it is not clear whether PLTP is a direct mediator or that PLTP simply modifies nHDL in a way that accelerates PL uptake; the latter hypothesis is supported by the observation that PLTP is essential to nHDL remodeling.18 Ultimately, FC and PL equilibrate among all lipoproteins giving steady-state plasma concentrations that are the targets of enzymes and transfer proteins. Although our model also shows hepatic LDL-lipid uptake via the LDLR, this process is not as important during the early rapid hepatic uptake but contributes to hepatic removal at longer times when lipoprotein concentrations have equilibrated to a stationary state. Notably, these fast lipid transfer processes are independent of LCAT activity, which acts on a slower time scale. Over longer time intervals, erythrocytes accrue ~10% of nHDL-FC and PL deposition.
Figure 8.

Model of nHDL Remodeling and Metabolism In Vivo. Cells extrude FC and PL via the interaction of apo AI with ABCA1 giving nHDL (1). nHDL-FC, PL, and apo AI rapidly transfer to HDL, t1/2 < 2 min (2); concurrently nHDL-FC and PL transfer to LDL (3). Over the same time frame, nHDL- and HDL-FC and PL transfer mainly to the liver (4, 5, 6) while some nHDL-apo AI is recycled to ABCA1 (7). Over time, FC and PL equilibrate among erythrocytes (8), extrahepatic tissues, and lipoproteins, which achieve a steady state concentration that is the target of enzymes, transfer proteins, and hepatic receptors. nHDL- and HDL-FC and PL accretion in the liver occurs via spontaneous transfer and SR-B1 (4, 5, 6), with the latter being promoted by PLTP (4). Symbols for FC, PL, and apo AI are labeled as shown in the legend at the top of the figure.
In vitro, nHDL-apo AI associates with HDL with some appearing lipid-free at longer incubation times with serum. In vivo, nHDL-apo AI also transfers to HDL where it remains with a halftime of 460 min (Figure 6 C). No lipid-free apo AI was detected in vivo suggesting that soon after plasma factors convert nHDL-apo AI to the lipid-free form, it immediately reassociates with HDL or initiates another cycle of tissue cholesterol-efflux. Clearly, nHDL structure is disrupted by plasma factors in vitro, and in vivo its components—FC, PL, and apo AI—are cleared by independent mechanisms that do not involve holo-particle uptake. nHDL-apo AI persists in plasma longer than nHDL-FC and PL, which rapidly move through the plasma compartment while maintaining a stationary state of HDL-FC, PL, and apo AI. Given the similarities between HDL and nHDL metabolism, we conclude by paraphrasing from Schwartz et al. who studied human HDL-FC metabolism, i.e., “nHDL-FC and PL constantly exchange between HDL and tissue-membranes, while maintaining a steady state HDL composition.”63
Supplementary Material
Highlights.
Nascent (n) HDL-FC, PL and apo AI have distinct destinations and serum kinetics in vitro and enter different pathways for clearance in vivo.
HDL is an early acceptor of nHDL-FC, which later transfers to LDL but mainly to the liver.
Hepatic nHDL-FC uptake is rapid and faster than its esterification by LCAT.
The terminal RCT step for most FC and PL respectively occurs by spontaneous and protein-mediated transfer to tissue, mainly liver.
nHDL, with its high FC content and labile lipids, is a key modulator of in vivo cholesterol homeostasis and phospholipid exchange among lipoproteins and tissues.
Acknowledgments
The authors thank Dedipya Yelamanchili for her excellent help with the mouse experiments.
Sources of Funding
Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number HL056865 (CR and HJP), the Bass Foundation (AMG) and Houston Methodist Foundation funding from Charif Souki, Patrick Studdert and the Jerold B. Katz family. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health
Nonstandard Abbreviations and Acronyms
- Apo
apolipoprotein
- CDX
methyl β-cyclodextrin
- CE
cholesteryl ester
- CPM
counts per minute
- DPM
disintegrations per minute
- FC
free cholesterol
- HDL
high density lipoprotein
- LCAT
lecithin:cholesterol acyltransferase
- LDL
low density lipoprotein
- nHDL
nascent HDL
- S-, M-, and L-nHDL
small, medium, and large nascent high density lipoprotein
- PC
phosphatidylcholine
- PL
phospholipid
- PLTP
phospholipid transfer protein
- RCT
reverse cholesterol transport
- SEC
size exclusion chromatography
- SM
sphingomyelin
- SR-B1
scavenger receptor class B member 1
- TLP
total lipoprotein
- VLDL
very low density lipoproteins
Footnotes
Disclosures
None.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacko AG, Pritchard PH. International Symposium on Reverse Cholesterol Transport. Report on a meeting. J Lipid Res. 1990;31:2295–2299. [PubMed] [Google Scholar]
- 3.Rosenson RS, Brewer HB, Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth PP, Barter PJ, Rosenson RS, et al. High-density lipoproteins: a consensus statement from the National Lipid Association. Journal of clinical lipidology. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Rosenson RS, Brewer HB, Jr, Ansell BJ, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nature reviews Cardiology. 2016;13:48–60. doi: 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyssenko NN, Nickel M, Tang C, Phillips MC. Factors controlling nascent high-density lipoprotein particle heterogeneity: ATP-binding cassette transporter A1 activity and cell lipid and apolipoprotein AI availability. FASEB J. 2013;27:2880–2892. doi: 10.1096/fj.12-216564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sorci-Thomas MG, Owen JS, Fulp B, et al. Nascent high density lipoproteins formed by ABCA1 resemble lipid rafts and are structurally organized by three apoA-I monomers. J Lipid Res. 2012;53:1890–1909. doi: 10.1194/jlr.M026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillard BK, Bassett GR, Gotto AM, Jr, Rosales C, Pownall HJ. Scavenger receptor B1 (SR-B1) profoundly excludes high density lipoprotein (HDL) apolipoprotein AII as it nibbles HDL-cholesteryl ester. J Biol Chem. 2017;292:8864–8873. doi: 10.1074/jbc.M117.781963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz GG, Olsson AG, Abt M, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 10.Cao G, Beyer TP, Zhang Y, et al. Evacetrapib is a novel, potent, and selective inhibitor of cholesteryl ester transfer protein that elevates HDL cholesterol without inducing aldosterone or increasing blood pressure. J Lipid Res. 2011;52:2169–2176. doi: 10.1194/jlr.M018069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohatgi A, Khera A, Berry JD, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;384:618–625. doi: 10.1016/S0140-6736(14)61217-4. [DOI] [PubMed] [Google Scholar]
- 13.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 14.Massey JB, Pownall HJ. Cholesterol is a determinant of the structures of discoidal high density lipoproteins formed by the solubilization of phospholipid membranes by apolipoprotein A-I. Biochim Biophys Acta. 2008;1781:245–253. doi: 10.1016/j.bbalip.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lund-Katz S, Hammerschlag B, Phillips MC. Kinetics and mechanism of free cholesterol exchange between human serum high- and low-density lipoproteins. Biochemistry. 1982;21:2964–2969. doi: 10.1021/bi00541a025. [DOI] [PubMed] [Google Scholar]
- 16.Davidson WS, Heink A, Sexmith H, et al. The effects of apolipoprotein B depletion on HDL subspecies composition and function. J Lipid Res. 2016;57:674–686. doi: 10.1194/jlr.M066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glass C, Pittman RC, Weinstein DB, Steinberg D. Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A. 1983;80:5435–5439. doi: 10.1073/pnas.80.17.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulya A, Lee JY, Gebre AK, et al. Initial interaction of apoA-I with ABCA1 impacts in vivo metabolic fate of nascent HDL. J Lipid Res. 2008;49:2390–2401. doi: 10.1194/jlr.M800241-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji Y, Wang N, Ramakrishnan R, et al. Hepatic scavenger receptor BI promotes rapid clearance of high density lipoprotein free cholesterol and its transport into bile. J Biol Chem. 1999;274:33398–33402. doi: 10.1074/jbc.274.47.33398. [DOI] [PubMed] [Google Scholar]
- 20.Picardo M, Massey JB, Kuhn DE, Gotto AM, Jr, Gianturco SH, Pownall HJ. Partially reassembled high density lipoproteins. Effects on cholesterol flux, synthesis, and esterification in normal human skin fibroblasts. Arteriosclerosis. 1986;6:434–441. doi: 10.1161/01.atv.6.4.434. [DOI] [PubMed] [Google Scholar]
- 21.Glomset JA. The plasma lecithins:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 22.Ross R, Glomset JA. Atherosclerosis and the arterial smooth muscle cell: Proliferation of smooth muscle is a key event in the genesis of the lesions of atherosclerosis. Science. 1973;180:1332–1339. doi: 10.1126/science.180.4093.1332. [DOI] [PubMed] [Google Scholar]
- 23.Glomset JA, Wright JL. Some Properties of a Cholesterol Esterifying Enzyme in Human Plasma. Biochim Biophys Acta. 1964;89:266–276. doi: 10.1016/0926-6569(64)90215-9. [DOI] [PubMed] [Google Scholar]
- 24.Brooks-Wilson A, Marcil M, Clee SM, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet. 1999;22:336–345. doi: 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- 25.Bodzioch M, Orso E, Klucken J, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–351. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- 26.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 28.Ji A, Wroblewski JM, Webb NR, van der Westhuyzen DR. Impact of phospholipid transfer protein on nascent high-density lipoprotein formation and remodeling. Arterioscler Thromb Vasc Biol. 2014;34:1910–1916. doi: 10.1161/ATVBAHA.114.303533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bielicki JK, McCall MR, Forte TM. Apolipoprotein A-I promotes cholesterol release and apolipoprotein E recruitment from THP-1 macrophage-like foam cells. J Lipid Res. 1999;40:85–92. [PubMed] [Google Scholar]
- 30.Kunitake ST, La Sala KJ, Kane JP. Apolipoprotein A-I-containing lipoproteins with pre-beta electrophoretic mobility. J Lipid Res. 1985;26:549–555. [PubMed] [Google Scholar]
- 31.Schwartz CC, Berman M, Vlahcevic ZR, Swell L. Multicompartmental analysis of cholesterol metabolism in man. Quantitative kinetic evaluation of precursor sources and turnover of high density lipoprotein cholesterol esters. J Clin Invest. 1982;70:863–876. doi: 10.1172/JCI110683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lund-Katz S, Lyssenko NN, Nickel M, et al. Mechanisms responsible for the compositional heterogeneity of nascent high density lipoprotein. J Biol Chem. 2013;288:23150–23160. doi: 10.1074/jbc.M113.495523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oram JF, Yokoyama S. Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res. 1996;37:2473–2491. [PubMed] [Google Scholar]
- 34.Duong PT, Collins HL, Nickel M, Lund-Katz S, Rothblat GH, Phillips MC. Characterization of nascent HDL particles and microparticles formed by ABCA1-mediated efflux of cellular lipids to apoA-I. J Lipid Res. 2006;47:832–843. doi: 10.1194/jlr.M500531-JLR200. [DOI] [PubMed] [Google Scholar]
- 35.Liu L, Bortnick AE, Nickel M, et al. Effects of apolipoprotein A-I on ATP-binding cassette transporter A1-mediated efflux of macrophage phospholipid and cholesterol: formation of nascent high density lipoprotein particles. J Biol Chem. 2003;278:42976–42984. doi: 10.1074/jbc.M308420200. [DOI] [PubMed] [Google Scholar]
- 36.Li XM, Tang WH, Mosior MK, et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler Thromb Vasc Biol. 2013;33:1696–1705. doi: 10.1161/ATVBAHA.113.301373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doody MC, Pownall HJ, Kao YJ, Smith LC. Mechanism and kinetics of transfer of a fluorescent fatty acid between single-walled phosphatidylcholine vesicles. Biochemistry. 1980;19:108–116. doi: 10.1021/bi00542a017. [DOI] [PubMed] [Google Scholar]
- 39.Massey JB, Gotto AM, Jr, Pownall HJ. Kinetics and mechanism of the spontaneous transfer of fluorescent phosphatidylcholines between apolipoprotein-phospholipid recombinants. Biochemistry. 1982;21:3630–3636. doi: 10.1021/bi00258a016. [DOI] [PubMed] [Google Scholar]
- 40.Thomson SW. On the equilibrium of vapour at curved surface of a liquid. Philosophical Magazine. 1871;42:5. [Google Scholar]
- 41.Moore WJ. Physical Chemistry. 4. Englewood Cliffs, NJ: Prentice Hall; 1962. [Google Scholar]
- 42.Havel RJ, Goldstein JL, Brown MS. Lipoproteins and Lipid Transport. In: Bondy PE, Rosenberg LE, editors. Metabolic Control of Disease. Philadelphia: Saunders Publishing; 1980. pp. 393–493. [Google Scholar]
- 43.Smith LC, Massey JB, Sparrow JT, Gotto AM, Jr, Pownall HJ. Structure and dynamics of human plasma lipoproteins. In: Pifat G, Herak JN, editors. Supramolecular Structure and Function. 1983. pp. 205–243. [Google Scholar]
- 44.Vasudevan M, Tchoua U, Gillard BK, Jones PH, Ballantyne CM, Pownall HJ. Modest diet-induced weight loss reduces macrophage cholesterol efflux to plasma of patients with metabolic syndrome. Journal of clinical lipidology. 2013;7:661–670. doi: 10.1016/j.jacl.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brewster ME, Ihm J, Brainard JR, Harmony JA. Transfer of phosphatidylcholine facilitated by a component of human plasma. Biochim Biophys Acta. 1978;529:147–159. doi: 10.1016/0005-2760(78)90113-3. [DOI] [PubMed] [Google Scholar]
- 46.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328:183–192. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 47.Pownall HJ, Hosken BD, Gillard BK, Higgins CL, Lin HY, Massey JB. Speciation of human plasma high-density lipoprotein (HDL): HDL stability and apolipoprotein A-I partitioning. Biochemistry. 2007;46:7449–7459. doi: 10.1021/bi700496w. [DOI] [PubMed] [Google Scholar]
- 48.Clay MA, Pyle DH, Rye KA, Barter PJ. Formation of spherical, reconstituted high density lipoproteins containing both apolipoproteins A-I and A-II is mediated by lecithin:cholesterol acyltransferase. J Biol Chem. 2000;275:9019–9025. doi: 10.1074/jbc.275.12.9019. [DOI] [PubMed] [Google Scholar]
- 49.Rye KA, Hime NJ, Barter PJ. Evidence that cholesteryl ester transfer protein-mediated reductions in reconstituted high density lipoprotein size involve particle fusion. J Biol Chem. 1997;272:3953–3960. doi: 10.1074/jbc.272.7.3953. [DOI] [PubMed] [Google Scholar]
- 50.Settasatian N, Duong M, Curtiss LK, et al. The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J Biol Chem. 2001;276:26898–26905. doi: 10.1074/jbc.M010708200. [DOI] [PubMed] [Google Scholar]
- 51.Liang HQ, Rye KA, Barter PJ. Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J Lipid Res. 1996;37:1962–1970. [PubMed] [Google Scholar]
- 52.Kee P, Rye KA, Taylor JL, Barrett PH, Barter PJ. Metabolism of apoA-I as lipid-free protein or as component of discoidal and spherical reconstituted HDLs: studies in wild-type and hepatic lipase transgenic rabbits. Arterioscler Thromb Vasc Biol. 2002;22:1912–1917. doi: 10.1161/01.atv.0000038485.94020.7f. [DOI] [PubMed] [Google Scholar]
- 53.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yancey PG, de la Llera-Moya M, Swarnakar S, et al. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275:36596–36604. doi: 10.1074/jbc.M006924200. [DOI] [PubMed] [Google Scholar]
- 55.Yancey PG, Bortnick AE, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Rothblat GH. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–719. doi: 10.1161/01.ATV.0000057572.97137.DD. [DOI] [PubMed] [Google Scholar]
- 56.Jian B, de la Llera-Moya M, Ji Y, et al. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 57.Ji Y, Jian B, Wang N, et al. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz CC, VandenBroek JM, Cooper PS. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J Lipid Res. 2004;45:1594–1607. doi: 10.1194/jlr.M300511-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Pownall HJ, Hickson-Bick D, Massey JB. Effects of hydrophobicity on turnover of plasma high density lipoproteins labeled with phosphatidylcholine ethers in the rat. J Lipid Res. 1991;32:793–800. [PubMed] [Google Scholar]
- 60.Thuahnai ST, Lund-Katz S, Williams DL, Phillips MC. Scavenger receptor class B, type I-mediated uptake of various lipids into cells. Influence of the nature of the donor particle interaction with the receptor. J Biol Chem. 2001;276:43801–43808. doi: 10.1074/jbc.M106695200. [DOI] [PubMed] [Google Scholar]
- 61.Shamburek RD, Zech LA, Cooper PS, Vandenbroek JM, Schwartz CC. Disappearance of two major phosphatidylcholines from plasma is predominantly via LCAT and hepatic lipase. Am J Physiol. 1996;271:E1073–1082. doi: 10.1152/ajpendo.1996.271.6.E1073. [DOI] [PubMed] [Google Scholar]
- 62.Schwartz CC, Halloran LG, Vlahcevic ZR, Gregory DH, Swell L. Preferential utilization of free cholesterol from high-density lipoproteins for biliary cholesterol secretion in man. Science. 1978;200:62–64. doi: 10.1126/science.204996. [DOI] [PubMed] [Google Scholar]
- 63.Schwartz CC, Vlahcevic ZR, Berman M, Meadows JG, Nisman RM, Swell L. Central role of high density lipoprotein in plasma free cholesterol metabolism. J Clin Invest. 1982;70:105–116. doi: 10.1172/JCI110582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.