

SUMMARY
Type I interferon is produced when host sensors detect foreign nucleic acids, but how sensors differentiate self from nonself nucleic acids, such as double-stranded RNA (dsRNA), is incompletely understood. Mutations in ADAR1, an adenosine-to-inosine editing enzyme of dsRNA, cause Aicardi-Goutieres syndrome, an autoinflammatory disorder associated with spontaneous interferon production and neurologic sequelae. We generated ADAR1 knockout human cells to explore ADAR1 substrates and function. ADAR1 primarily edited Alu elements in RNA polymerase II (pol II) transcribed mRNAs, but not putative pol III transcribed Alus. During the IFN response, ADAR1 blocked translational shutdown by inhibiting hyperactivation of PKR, a dsRNA sensor. ADAR1 dsRNA binding and catalytic activities were required to fully prevent endogenous RNA from activating PKR. Remarkably, ADAR1 knockout neuronal progenitor cells exhibited MDA5 (dsRNA sensor)-dependent spontaneous interferon production, PKR activation, and cell death. Thus, human ADAR1 regulates sensing of self versus nonself RNA, allowing pathogen detection while avoiding autoinflammation.
IN BRIEF
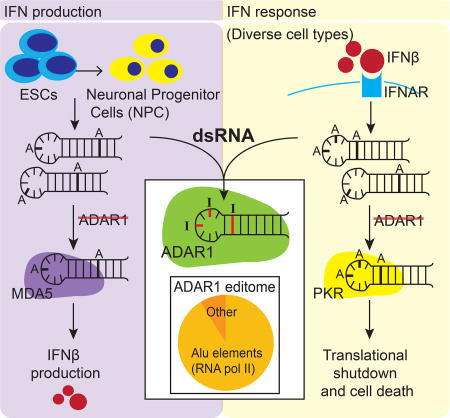
The human RNA editing enzyme ADAR1 prevents endogenous RNA from activating innate immune sensors (PKR, MDA5), which allows efficient translation during the IFN response.
INTRODUCTION
Type I interferon (IFN) is an essential component of innate immunity that protects against virus infection. Type I IFN is produced by almost all cell types when pattern recognition receptors (PRRs) sense pathogen associated molecular patterns (PAMPs). For example, cytosolic PRRs such as MDA5 and RIG-I detect double-stranded RNA (dsRNA) commonly produced during many viral infections, leading to type I IFN production and induction of hundreds of IFN-stimulated genes (ISGs) with antiviral activity (Schneider et al., 2014). However, this seemingly efficient mechanism poses a challenge to the host cell. How do cytosolic PRRs selectively respond to viral dsRNA in the milieu of host-derived dsRNA structures also found in the cytoplasm?
Direct modification of endogenous dsRNA may play a role in shielding endogenous dsRNA away from PRRs. Interestingly, mutations in the RNA editing enzyme ADAR1 (adenosine deaminase acting on RNA 1) cause Aicardi-Goutieres syndrome (AGS), an autoinflammatory disorder associated with spontaneous IFN production in the absence of virus infection (Rice et al., 2012). ADAR1 deaminates adenosine (A) to inosine (I) in cellular dsRNA substrates (Bass and Weintraub, 1988), thereby catalyzing the most common type of RNA editing found in humans. Among the different ADAR proteins – ADAR1, -2, and -3 – ADAR1 is ubiquitously expressed and exists in two different isoforms: a constitutively expressed 110 kDa protein, ADAR1p110, and the IFN-inducible 150 kDa protein, ADAR1p150 (George and Samuel, 1999). Since inosine is read as guanosine by the translational machinery, RNA editing by ADAR1 can result in non-synonymous coding changes (Yeo et al., 2010). Additionally, ADAR1 can also function as a dsRNA binding protein independent of its editing activity. For instance, ADARs can modulate microRNA pathways without editing (Ota et al., 2013). Thus, ADAR1 is a multifunctional protein with editing-dependent and -independent activities.
Due to a lack of human model systems, how ADAR1 deficiencies lead to AGS remains unknown. In AGS patients, elevated type I IFN is detected in body fluids such as plasma and cerebrospinal fluid (CSF), but the mechanism by which IFN is produced and the cell type that produces IFN has not been determined (Rodero et al., 2017). In addition, it is not clear how chronic exposure to IFN leads to disease. Intriguingly, AGS patients with mutations in ADAR1 are associated with clinically distinct neurological phenotypes including microcephaly, but it is not known why symptoms tend to manifest in the brain (Rice et al., 2017).
Mouse models have been invaluable for understanding ADAR1 functions in mammals. Similar to humans, ADAR1 deficiency has been linked to spontaneous type I IFN signaling in mice. ADAR1 deficient murine embryos display an elevated ISG signature and rapid induction of apoptosis in hematopoietic cells leading to embryonic lethality (Hartner et al., 2009). However, concurrent deletion of MDA5 or MAVS (a downstream adaptor for MDA5 and RIG-I) can rescue the elevated ISG signature and embryonic lethality of ADAR1 deficient mice (Liddicoat et al., 2015; Mannion et al., 2014; Pestal et al., 2015). These observations led to a model in which ADAR1 A-to-I editing is hypothesized to destabilize host-derived dsRNA structures resulting in evasion from MDA5 detection and downstream IFN production.
However, there are fundamental differences in ADAR biology between humans and mice. In humans, while some A-to-I editing takes place in protein coding regions, the majority of A-to-I editing takes place in primate-specific Alu elements – a class of repetitive SINE (short interspersed elements) retroelements (Ramaswami and Li, 2013). Although the mouse genome contains B1 and B2 SINEs, it lacks Alu elements. Consequently, conserved A-to-I editing sites only minimally overlap between humans and other mammals (Pinto et al., 2014; Tan et al., 2017). Moreover, A-to-I editing is vastly more prevalent in the human transcriptome than in mice (at least 100 fold) (Ramaswami and Li, 2013). In addition, it is not known why AGS patients with ADAR1 mutations display neurologic symptoms (Rice et al., 2017) whereas mice lacking ADAR1 are most severely affected in hematopoietic compartments (Hartner et al., 2004). Therefore, to understand how dysfunctional ADAR1 causes disease, it is imperative to explore human model systems to identify ADAR1 specific substrates and function.
In this study, we generated ADAR1- and ADAR1p150 knockout (KO) human cells. We determined ADAR1-specific substrates during IFN signaling, and further examined ADAR1 regulation of IFN production and IFN response. Our findings collectively suggest that human ADAR1 RNA editing plays a key role in preventing autoinflammatory reactions against endogenous RNA.
RESULTS
Generation of ADAR1 deficient cell lines
A-to-I RNA editing sites have been identified by high throughput sequencing techniques, typically by detecting nucleotide mismatches between the transcriptome and genome (Ramaswami and Li, 2013). Although this approach globally maps RNA editing sites, it does not link specific editing sites to a particular RNA editing enzyme. Using CRISPR/Cas9 gene-editing technology, we generated ADAR1 KO 293T cell lines – lacking both ADAR1p110 and ADAR1p150 – to robustly map the ADAR1-specific editome (Table S1 and S2). To differentiate ADAR1p150-specific edited sites from total ADAR1 edited sites, we also generated ADAR1p150 KO cell lines (Figure 1A, 1B) (George and Samuel, 1999). In parallel, we derived control wild100 type (WT) clones, which maintained constitutive expression of ADAR1p110 and IFN-inducible expression of ADAR1p150 as seen in parental 293T cells (Figure 1B).
Figure 1. RNA-Seq analysis pipeline to determine the ADAR1 editome.

(A) Two isoforms of ADAR1 are generated by usage of different promoters and alternative splicing. Pc, constitutively active promoter that initiates transcription of exon 1B. Pi, interferon inducible promoter that initiates transcription of exon 1A. Exon 1B or exon 1A splices to exon 2 leading to ADAR1p110 or ADAR1p150 expression, respectively. ATG designates the translational start site for ADAR1p110 (orange) or ADAR1p150 (green). Arrows indicate gRNAs targeting expression of ADAR1 (blue), ADAR1p150 (red), and ADAR1p110 (purple) (see Tables S1 and S2 for more detail).
(B) Western blot of WT, ADAR1- and ADAR1p150 KO 293T cell clones after mock or IFNβ treatment. Par, parental 293T cell line.
(C, D) Schematic of comparative RNA-Seq analysis pipeline for the identification of ADAR1 and ADAR1p150-dependent mismatches, with examples of per sample nucleotide counts for both mismatch types (n = 3 independent clones for each genotype) (C). Pol II and pol III Alu elements refer to putative RNA polymerase II transcribed Alu elements, or RNA polymerase III transcribed Alu elements, respectively. Total mismatch counts associated with ADAR1 (red) and ADAR1p150 (blue) (D). All mismatch sites identified are listed in Tables S3 and S4.
ADAR1 KO and ADAR1p150 KO cell clones exhibited normal morphology and growth rates comparable to WT clones (Figure S1A). We did attempt to generate ADAR1p110 KO cell lines by deleting the ADAR1p110-specific exon 1B and its constitutively active promoter (Figure 1A, Table S1 and S2). However, despite successful deletion of exon 1B and its promoter, ADAR1p110 protein expression was only partially reduced (Figure S1B). Lastly, we also derived ADAR2 KO 293T cells to identify ADAR2 edited sites that may be shared with ADAR1 or unique to ADAR2 (Figure S1C, Table S1 and S2).
RNA-Seq analysis pipeline to determine the ADAR1 editome
Despite the critical role ADAR1 plays in innate immunity, the human ADAR1 editome during the IFN response has not been well characterized. To address this deficit, we implemented a comparative RNA-Seq screen (Rosenberg et al., 2011) on WT, ADAR1 KO, and ADAR1p150 KO cells with or without exposure to IFNβ - a type I IFN. A novel analysis pipeline was developed to identify sites edited by ADAR1 (ADAR1p110 and ADAR1p150) or ADAR1p150 only. In parallel, differential gene expression analysis was performed to examine ADAR1 regulation of global gene expression (Figure 1C). We identified a total of 9420 ADAR1 associated read:reference mismatches (Table S3 and S4). Among the twelve possible nucleotide changes, 99.7% of identified mismatches were A-to-G (A-to-I), validating our analysis strategy for robustly identifying ADAR1-mediated editing events with minimal background (Figure 1D).
ADAR1p110 and ADAR1p150 edit pol II Alu elements in predicted loop regions
When we analyzed the distribution of ADAR1 edit sites in the genome with respect to repetitive elements, we found that ADAR1 primarily edits Alu elements (91.2%), which is consistent with previous reports showing that most A-to-I editing occurs in Alu elements (Ramaswami and Li, 2013). ADAR1-edited Alu elements were mainly found in introns (48.4%) and 3´UTRs (37.4 %) (Figure 2A). Interestingly, editing events dependent upon the IFN-inducible isoform ADAR1p150 were also concentrated in Alu elements (87.4%), many of which were found in 3´UTRs (46.0%) and introns (36.9%) (Figure 2B). In contrast, very few (less than 0.1%) of the ADAR1 and ADAR1p150 edits were found in open reading frames (ORFs; Table S5). Furthermore, gene ontology (GO) analysis of genes edited by ADAR1 or ADAR1p150 alone demonstrated no significant enrichment of any GO terms. Interestingly, a substantial portion of non-Alu ADAR1- and ADAR1p150-dependent editing sites were detected in other transposable elements, including endogenous retroviruses (ERVs) and LINEs such as L1s (Figure 2A, 2B).
Figure 2. Identification and characterization of the ADAR1 editome during IFN response.

(A–B) Distribution of ADAR1- (A) and ADAR1p150- (B) edit sites with respect to repetitive elements. Edit sites within Alu, L1 (LINE1), and ERV1 (Endogenous retroviral sequence 1) elements are indicated. Remaining edit sites within another class of repetitive element or within a non-repetitive element are shown as "Other repeat" and "No repeat", respectively. Spoke axis indicates the percentage of edit sites located within each repetitive element class. Size and color of wedges indicate the proportion of edits found within different transcript components. (C) Fraction of A-to-G read:reference mismatches (in red), and all other read:reference mismatches (in gray) identified in pol II Alu elements (‘transcript-embedded Alus’) and pol III Alu elements (‘independently transcribed Alus’). n = 3 clones for each genotype.
(D) ADAR1-associated edits identified within Alu sequences enumerated and shown on the predicted secondary structure of the Alu consensus sequence. The color bar represents the number of ADAR1 edits identified per position in the Alu consensus sequence.
(E–F) Editing frequencies at ADAR1- (black) and ADAR1p150- (red) associated edit sites are shown for mock (x-axis) versus IFN-treated (y-axis) cells in WT (E) and ADAR1p150 KO cells (F).
(G–H) Gene expression (Log2 read counts per million, CPM) in mock treated WT versus ADAR1 KO (G), and WT versus ADAR1p150 KO cells (H). The median value from n = 3 cell clones is shown per gene.
(I–J) Fold change gene expression (moderated fold-change values, limma analysis) upon IFN treatment are shown for WT versus ADAR1 KO (I), and WT versus ADAR1p150 KO cells (J).
(G–J) Colored dots indicate genes with edit sites. Color bar represents the number of ADAR1- or ADAR1p150- associated edit sites identified per gene. See also Figure S2 and Table S6.
Two types of Alu elements have been characterized in the human cell: Alus transcribed by RNA polymerase II (pol II) or RNA polymerase III (pol III). Pol II transcribed Alus are embedded in mRNAs and comprise the vast majority of Alu RNAs expressed in human cells. In contrast, pol III transcribed Alus are transcribed as independent units, present at low levels, and can retrotranspose (Conti et al., 2015). We implemented an analysis pipeline to classify putative pol II Alu elements (‘transcript-embedded Alus’) and pol III Alu elements (‘independently transcribed Alus’) based on RNA-Seq read coverage patterns (Figure 1C) (Conti et al., 2015). ADAR1 signature A-to-G mismatches were found in many pol II Alu elements (Figure 2C), consistent with our finding of many editing sites in mRNA 3´UTRs and introns (Figures 2A and 2B). Although we were able to identify 214 independently expressed putative pol III Alu elements, surprisingly, we observed no evidence of ADAR1 editing in these transcripts (Figure 2C).
In mice, ADAR1 has been shown to edit long dsRNA structures in transcript 3´UTRs. Such modifications are predicted to destabilize these dsRNA structures and possibly shield them from MDA5 detection (Liddicoat et al., 2015). Therefore, we examined our dataset to see if human ADAR1 preferentially edited long dsRNA regions within Alu elements. When sites of ADAR1 editing along the Alu consensus sequence were superimposed on a predicted Alu RNA secondary structure model, we found that ADAR1- (Figure 2D) and ADAR1p150-dependent (Figure S2A) edit sites were frequently detected in predicted single-stranded loops rather than dsRNA forming stems. However, individual Alu elements can fold differently from the consensus sequence. Moreover, two adjacent Alu elements in reverse orientation in the same RNA transcript can form an extended intramolecular dsRNA structure (Athanasiadis et al., 2004). Therefore, we used secondary structure predictions for each edited Alu and for multiple inverted Alu elements in the same transcript to further assess editing sites. For both types of secondary structures, editing sites were found less frequently in double-stranded regions than other positions within the Alu sequence (Figure S2B and S2C).
In sharp contrast to the ADAR1 editome, we did not detect any ADAR2-associated editing when our RNA-Seq analysis pipeline was applied to ADAR2 KO versus WT control cell lines (data not shown). This was not due to the absence of ADAR2 protein in WT 293T cells (Figure S1C). Thus, ADAR1, rather than ADAR2, is the active A-to-I editor of Alu elements in human 293T cells. Consistent with these results, an extensive analysis of A-to-I editing in human tissue also suggested that ADAR1, rather than ADAR2, is the primary editor of repetitive sites (Tan et al., 2017).
ADAR1p150 is required to enhance overall A-to-I editing during the IFN response
To investigate how ADAR1 shapes genome-wide A-to-I editing during the human IFN response, we compared the editing frequency at each edited site between mock and IFN treated cells. When we treated WT cells with IFN, overall editing frequency increased at both ADAR1- and ADAR1p150-dependent sites (Figure 2E). In ADAR1p150 KO cells, editing was observed at some sites – likely via ADAR1p110 – but IFN treatment did not increase editing frequency (Figure 2F). Similarly, A-to-G mismatch frequencies from ADAR1 editing in pol II Alus did increase in WT cells upon IFN treatment, but did not increase in ADAR1p150 KO cells upon IFN treatment (Figure 2C). Thus, ADAR1p150 is required to enhance the overall A-to-I editing during the human IFN response. This has also been observed in mouse models, where ADAR1p150 was required for enhanced A-to-I editing in murine embryonic fibroblasts (MEFs) exposed to IFN (George et al., 2016).
A-to-I editing in Alu elements does not affect transcript abundance
We also performed differential gene expression analysis on the WT and ADAR1 deficient cells to determine if ADAR1 might regulate transcript abundance (Figure 1C). In mock treated 293T cells, we detected few differentially expressed genes between WT and ADAR1- or ADAR1p150 KO cell clones (Figure 2G and 2H). In addition, we did not detect significant differential enrichment of any gene sets (MSigDB) between mock treated WT and ADAR1-deficient cells (Table S6). Thus, significant spontaneous upregulation of type I IFN or ISG expression does not occur in ADAR1-deficient 293T cells, highlighting that ADAR1-mediated negative regulation of type I IFN production may occur in a cell type specific manner.
Inosine-containing synthetic dsRNA substrates can be cleaved by certain cellular RNases (Morita et al., 2013; Scadden, 2005). Thus it has been proposed that ADAR1 edited transcripts may undergo enhanced degradation. However, genes carrying ADAR1 editing sites were expressed at similar levels between WT and ADAR1-deficient cell lines (scatter plots: Figure 2G and 2H, volcano plots: Figure S2D and S2E), indicating that ADAR1 editing does not appreciably regulate abundance of edited transcripts in these cells.
Next, we examined differential gene expression during response to IFN. Upon IFN treatment, ISG transcripts, including those containing Alu elements (Figure S2F and S2G), were efficiently induced in both WT and ADAR1-deficient cells (Figure 2I and 2J). Similarly, no gene sets (MSigDB) were differentially enriched in the IFN response by WT versus ADAR1- or WT versus ADAR1p150-deficient cells (Table S6). Most edited sites associated with ADAR1 and ADAR1p150 were detected in non-ISGs during the IFN response (Figure 2I and 2J). Taken together, although ISGs are markedly induced by IFN, we did not observe an increase in ADAR1 editing sites or editing frequency in ISGs as compared to non-ISGs.
Lastly, we examined the abundance of pol III Alu elements during IFN treatment. Interestingly, pol III Alu expression levels were modestly increased upon IFN treatment in most ADAR1 KO and ADAR1p150 KO clones, but not in WT clones (Figures S2H). However, as mentioned above we detected no ADAR1-mediated editing in these pol III Alus (Figure 2C).
In summary, in 293T cells, ADAR1-mediated A-to-I editing – the majority of which is found in Alu elements within introns and 3´UTRs of pol II transcripts – does not affect transcript abundance of edited genes. Surprisingly, the lack of ADAR1 protein and genome-wide A-to-I editing does not appear to have a major effect on overall gene expression at the mRNA level.
ADAR1 prevents shutdown of translation and cell growth arrest during the human IFN response
Next, we investigated whether ADAR1 regulates gene expression at the protein level. Strikingly, despite comparable levels of ISG transcript induction upon IFN treatment (Figure 3A, 2I, and 2J), protein induction of several ISGs was significantly lower in ADAR1 KO and ADAR1p150 KO cells compared to WT cells. Protein expression of cell surface ISGs such as β2M – a component of MHC class I – as well as the intracellular ISG STAT1 was reduced in ADAR1 KO and ADAR1p150 KO cells (Figure 3B and 3C). Although mRNA expression of IFN-γ receptor 1 (IFNγR1, an ISG) remained consistent during IFN treatment (Figure 3A), we still observed a decrease in IFNγR1 protein levels in ADAR1-deficient cells (Figure 3C). In contrast, we did not observe reduced protein expression of the cell surface ISG CD9 in IFN treated ADAR1-deficient cells (Figure 3C). In mock treated cells, similar basal levels of ISG transcripts and proteins were detected among different genotypes (Figure S3A and S3B), indicating that the difference in protein expression between WT and ADAR1-deficient cells depends on IFN treatment.
Figure 3. ADAR1 prevents global translational shutdown and cell growth arrest during IFN response.

(A–D) WT or ADAR1-deficient 293T cells were treated with IFNβ (24 hours). ISG mRNA fold change upon IFN treatment measued by qRT-PCR (A). β2M protein expression (B) and mean fluorescence intensity (MFI) fold change measured by flow cytometery upon IFN treatment (C). Polysome gradients (D). RNP, ribonucleoprotein. 40S, 60S and 80S denote the corresponding ribosomal subunits and monosome, respectively. Polysome, translating ribosomes bound to transcript. Data shown as mean ± SEM. n = 3 cell clones for each genotype. One-way ANOVA and Tukey’s post hoc test, *P < 0.05, **P < 0.01.
(E) Cell density during IFN treatment. Data shown as mean ± SEM (n = 3 experimental replicates). Student’s t-test, **P < 0.01, ***P < 0.001.
See also Figures S3 and S4.
To assess if reduced protein expression in IFN treated ADAR1-deficient cells was caused by down-regulation of translation, we examined polysome profiles to assess global translation efficiency. In WT cells, polysome profiles of mock and IFN treated cells were closely aligned, indicating that translation efficiency is not affected by IFN treatment. However, IFN treatment of ADAR1 KO and ADAR1p150 KO cells resulted in a dramatic shift from heavy polysomes to ribosomal subunits (40S, 60S) (Figure 3D). Therefore, ADAR1-deficient cells undergo translational shutdown during the IFN response.
In addition to a defect in mRNA translation, we also observed a profound defect in cell growth in ADAR1-deficient cells starting 48 hours after IFN treatment (Figure 3E). We observed a more severe cell growth defect in ADAR1 KO cells compared to ADAR1p150 KO cells, suggesting that both isoforms of ADAR1 are required for maintaining optimal cell growth during the IFN response. In stark contrast, there was no significant difference in the growth kinetics between mock and IFN treated WT cells (Figure 3E).
We examined whether cell death or an effect on cell proliferation was responsible for the growth defect in ADAR1 deficient cells. At 48 hours of IFN treatment, we did not observe an increase in apoptosis or cell death (Figure S4A). Next, we monitored cell proliferation by CFSE dilution. CFSE intensity was markedly higher in IFN treated ADAR1 KO and ADAR1p150 KO cells compared to their mock treated cells, indicating a proliferation defect in ADAR1-deficient cells (Figure S4B). Moreover, when we performed dual labeling for CFSE and β2M, the β2MlowCFSEhigh population was distinct in IFN treated ADAR1 KO and ADAR1p150 KO cell lines but not in WT cells (Figure S4C and S4D).
In summary, in addition to previous reports that ADAR1 negatively regulates IFN production (Rice et al., 2012; Rodero et al., 2017), our findings add another layer of complexity to the role of ADAR1. We show that ADAR1 is in fact required to maintain efficient translation and cell proliferation during the IFN response.
ADAR1p110 and ADAR1p150 suppress PKR activation, allowing efficient gene translation during the IFN response
Phosphorylation of eukaryotic initiation factor 2 (eIF2α) results in global repression of translation by reducing its ability to deliver the initiator Met-tRNAi to ribosomes (Sonenberg and Hinnebusch, 2009). In ADAR1 KO and ADAR1p150 KO cells, but not in WT cells, IFN treatment gave rise to a distinct population of cells that were p-eIF2α+ (phosphorylated eIF2α) and β2Mlow (Figure 4A and 4B). Thus exposure to IFN increases eIF2α phosphorylation in cells lacking ADAR1. mRNA translation initiation is also heavily dependent on the cap binding protein eIF4E. The abundance of total eIF4E remained relatively consistent during IFN treatment. However, phosphorylated eIF4E levels were specifically reduced in ADAR1-deficient cells undergoing IFN response (Figure S5A). Prior studies have correlated eIF4E phosphorylation to tumorigenesis and have shown that eIF4E phosphorylation may stimulate the translation of a subset of mRNAs (Furic et al., 2010).
Figure 4. ADAR1 enables efficient cellular translation by preventing PKR activation.

(A–C) WT or ADAR1-deficient 293T cells were mock or IFNβ treated (24 hours).
(A, B) Flow cytometry analysis of phosphorylated eIF2α (p-eIF2α) and β2M. Representative dot plots of IFN treated samples only (A). Bar graph showing the frequency (%) of p-eIF2α+ cells (B). Data shown as mean ± SEM (n = 3 experimental replicates). Student’s t-test, ***P < 0.001.
(C) Western blot of phosphorylated PKR (p-PKR), total PKR, and β-actin control.
(D) β2M and IFNγR1 mean fluorescence intensity (MFI) fold change upon IFNβ treatment (24 hours) in PKR knock-down cells. PKR shRNA #1 and #2 are two independent shRNAs targeting different regions of PKR. CTRL, control. Data shown as mean ± SEM (n = 3 experimental replicates). One-way ANOVA and Tukey’s post hoc test, *P < 0.05, **P < 0.01.
(A–D) Par, parental 293T cell line.
See also Figure S5.
Protein Kinase R (PKR) is one of several mammalian kinases that can phosphorylate eIF2α. PKR is constitutively expressed but also an antiviral ISG that functions as a cytoplasmic PRR. When activated by dsRNA, PKR dimerizes and autophosphorylates providing a site for eIF2α binding and phosphorylation (Garcia et al., 2006). This leads to inhibition of viral protein synthesis and reduced virus replication. Prior studies have shown that ADAR1 can inhibit viral RNA mediated PKR activation (Gelinas et al., 2011). Furthermore, virus infection or IFN treatment of ADAR1 knockdown (KD) Hela cells led to an increase in stress granule formation, which was dependent on PKR activation (John and Samuel, 2014). Therefore we tested if PKR activation was increased by IFN in ADAR1-deficient cells. In mock treated cells, phosphorylated PKR (p-PKR) was not increased in ADAR1-deficient cells. However, we observed markedly higher levels of p-PKR in the IFN treated ADAR1 KO and ADAR1p150 KO cells compared to WT cells (Figure 4C). Thus, PKR activation can occur in the absence of foreign viral RNA in ADAR1-deficient cells after IFN treatment. In addition, p-PKR levels were higher in IFN treated ADAR1 KO cells than in ADAR1p150 KO cells (Figure 4C). Thus, ADAR1p110 and ADAR1p150 are both required to fully block PKR activation.
To determine if enhanced PKR activation is indeed causing down-regulation of protein expression in ADAR1-deficient cells, we knocked-down PKR expression with short hairpin RNA (shRNA) (Figure S5B). In ADAR1-deficient cells transduced with PKR shRNAs, β2M and IFNγR1 expression increased to higher levels during IFN signaling compared to cells transduced with the control shRNA (Figure 4D). In conclusion, ADAR1p110 and ADAR1p150 enable efficient translation during response to IFN by preventing PKR activation.
PKR activation depends on newly transcribed endogenous RNA in ADAR1-deficient cells
Potentially, an endogenous RNA or protein could activate PKR. Since PKR is a PRR ubiquitously expressed in the cytoplasm and binds dsRNA without sequence specificity (Garcia et al., 2006), ADAR1 may have evolved to prevent unwanted PKR activation by endogenous RNA species present in the cytoplasm. Alternatively, ADAR1 could prevent host proteins from activating PKR, since proteins such as PRKRA (Protein Activator Of Interferon Induced Protein Kinase EIF2AK2) or ISG15 can trigger PKR activation in the absence of virus infection (Garcia et al., 2006; Okumura et al., 2013).
To examine if PKR activation depends on a host protein or host RNA in ADAR1-deficient cells, we treated cells with cycloheximide (CHX) or actinomycin-D (ActD), which suppress de novo translation and transcription, respectively. In brief, cells were mock or IFN treated for 24 hours in total; after a 12 hour pre-treatment with IFN, CHX or ActD were added to cells for the remaining 12 hours (Figure S6A). A pre-treatment period with IFN allows induction of ISG mRNAs, which serve as a source for ISG translation after addition of drugs. This allows ActD to prevent additional ISG transcription without significantly affecting ISG protein levels. As expected for these conditions, ActD treatment did not suppress ISG protein synthesis – as measured by PKR expression – but it effectively suppressed ISG transcript induction – as measured by IFIT1 mRNA, a highly inducible ISG. In contrast, CHX treatment suppressed ISG protein synthesis but did not change ISG transcript induction (Figure S6B and S6C). Strikingly, in IFN treated ADAR1 KO and ADAR1p150 KO cells, p-PKR levels were significantly reduced upon incubation with ActD, but not CHX (Figure 5A and 5B). In addition, when cells were treated with α-amanitin, a potent inhibitor of pol II and a weak inhibitor of pol III (Bensaude, 2011), p-PKR levels dramatically decreased in ADAR1 KO cells (Figure 5C and S6C). Thus, in the absence of ADAR1, PKR activation depends heavily on IFN-induced de novo transcription rather than translation.
Figure 5. PKR activation depends on newly transcribed endogenous RNA, and is suppressed by the dsRNA binding and catalytic activities of ADAR1.

(A–C) ADAR1 KO or ADAR1p150 KO 293T cells were mock or IFNβ treated for 24 hours in total. After the initial 12 hours of IFN treatment, cells were co-incubated with cycloheximide (CHX, 30ug/ml), actinomycin-D (ActD, 10ug/ml), or α-amanitin (25ug/ml) for the latter 12 hours (Figure S6A). Western blot of phosphorylated PKR (p-PKR) (A, C). p-PKR band intensity was normalized to that of total PKR (n = 2~3 experimental replicates) (B).
(D) Doxycycline-inducible expression of PKR in ADAR1 KO cells. As a positive control for PKR induction, ADAR1 KO cells were treated with IFNβ. p-PKR and total PKR was measured by western blot.
(E–F) ADAR1 KO cells were transduced with puromycin-selectable lentiviruses containing the following constructs; p110, WT ADAR1p110; ADAR1, WT ADAR1p110 and ADAR1p150; EAA, ADAR1 (both isoforms) dsRBD mutant; QA, ADAR1 (both isoforms) deaminase domain mutant; Empty, empty vector negative control. Stable cell lines were mock or IFNβ treated for 24 (E) or 48 hours (F). Representative western blots (left panels) and bar graphs summarizing p-PKR band intensities normalized to that of total PKR (right panels, n= 3~5 experimental replicates). (B, E, F) Data shown as mean ± SEM. Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
See also Figure S6.
Since PKR requires dimerization for its activation, it was possible that merely up-regulating PKR expression rendered it more susceptible to activation by endogenous transcripts. We therefore tested whether up-regulating PKR protein expression, in the absence of IFN, might activate PKR in ADAR1-deficient cells. Interestingly, doxycycline (Dox) inducible expression of PKR in ADAR1 KO cells did not lead to PKR activation (Figure 5D). Taken together, these results suggest that ADAR1 prevents IFN regulated endogenous transcripts from mediating PKR activation.
We also examined if ADAR1 could inhibit PKR activation mediated by exogenous RNA stimulants such as viral RNA or the potent adjuvant poly(I:C), a synthetic dsRNA. Upon infection with Sindbis virus (SINV), a positive-strand RNA virus, we observed only a moderate increase in PKR activation in ADAR1 KO cells compared to WT cells. No difference in PKR activation was observed between ADAR1p150 KO and WT cells (Figure S6D and S6E). Additionally, at all doses of poly(I:C) tested, we did not observe greater PKR activation in ADAR1 KO cells compared to WT cells (Figure S6F). Thus, ADAR1 may be more effective at inhibiting PKR activation stimulated by endogenous RNA than exogenous RNA directly introduced into the cytoplasm.
ADAR1 dsRNA binding and catalytic activities are both required to suppress PKR activation
Next we examined which functional domains of ADAR1 are required for regulating PKR activation. ADAR1 is composed of an N-terminal Z-DNA binding domain, three double stranded RNA binding domains (dsRBD), and a C-terminal deaminase domain (Nishikura, 2010). Both the dsRBDs and the deaminase domain are required for optimal ADAR1 RNA editing activity. Introducing two point mutations in the deaminase domain – H910Q and E912A – results in an editing deficient ADAR1 still intact in its dsRNA binding activity (ADAR1-QA) (Liu and Samuel, 1996). Mutating the KKXXK motif to EAXXA in all three dsRBDs generates an ADAR1 mutant defective in binding to dsRNA (ADAR1-EAA); as a consequence ADAR1-EAA is also defective in A-to-I editing (Valente and Nishikura, 2007). We exogenously expressed WT or mutant forms of ADAR1 in ADAR1 KO cells followed by IFN treatment. As in WT cells, co-expression of the two WT ADAR1 isoforms efficiency suppressed p-PKR after IFN treatment. WT ADAR1p110 alone cannot completely suppress PKR activation (Figure 5E and 5F), reinforcing our observation that both ADAR1p110 and ADAR1p150 are required to fully inhibit PKR activation.
Interestingly, both the ADAR1-EAA and ADAR1-QA mutants inhibited PKR phosphorylation after 24 hours of IFN treatment (Figure 5E). However, by 48 hours both mutants had lost effectiveness (Figure 5F). Thus, the dsRBD and deaminase activities are both required to fully suppress PKR activation. These results suggest that ADAR1 editing of an endogenous RNA plays a role in preventing PKR activation. In addition, since the editing-defective mutant could still exert transient suppression of p-PKR (Figure 5E), ADAR1 may also function through dsRBD-dependent, editing-independent mechanisms.
Generation and characterization of ADAR1 KO human embryonic stem cells (hESCs)
To further evaluate the biological significance of ADAR1-regulated PKR activation in other human cell types, we derived ADAR1 KO human embryonic stem cells (hESCs) following the iCRISPR protocol (Gonzalez et al., 2014) (Table S1 and S2). WT and ADAR1 KO hESC clones expressed hESC markers such as Oct-3/4 and SSEA4 (Figure 6A) and ADAR1 deficiency did not compromise hESC cell growth (Figure 6B). Similar to 293T cells, there was no difference in IFNβ and ISG mRNA levels between WT and ADAR1 KO hESCs (Figure 6C). When hESCs were exogenously treated with IFNβ, p-PKR increased in ADAR1 KO but not in WT hESCs (Figure 6D). Therefore, ADAR1-mediated negative regulation of PKR activation is not limited to 293T cells, but also applies to other human cell types such as hESCs.
Figure 6. Generation and characterization of ADAR1 KO hESCs.

(A) Immunofluorescence staining of WT and ADAR1 KO hESCs. SSEA4 (green), Oct-3/4 (red), and DAPI (blue). Scale bar, 100um.
(B) hESC growth rate/day measured by the CellTiter-Glo assay.
(C) qRT-PCR of IFNβ and ISGs. Data shown as mean ± SEM (n = 3~6 experimental replicates).
(D) Western blot of mock and IFNβ treated (24 hours) hESCs. p-PKR, PKR phosphorylation.
(A–D) Par., parental hESC clone. WT, CRISPR/Cas9 derived WT hESC cell clone. ADAR1 KO #1 and #2 are two independent CRISPR/Cas9 derived ADAR1 KO hESC clones (see Table S2).
Loss of ADAR1 in neuronal progenitor cells leads to MDA5-mediated IFN production, PKR activation, and apoptotic cell death
As mentioned, ADAR1 is one of several human genes associated with AGS and severe neuropathology (Rice et al., 2017). However, such neurological symptoms have not been observed in ADAR1−/− mice (Hartner et al., 2004). To evaluate ADAR1 function in human neuronal lineage cells, we differentiated WT and ADAR1 KO hESCs to neuronal progenitor cells (NPCs) using adherent culture conditions (Chambers et al., 2009) (Figure S7A). We observed no difference in neural induction efficiency between WT and ADAR1 KO hESCs as assessed by NPC markers such as PAX6 and Nestin (Figure S7B). However, in contrast to WT hESCs, ADAR1 KO hESCs spontaneously up-regulated IFNβ and ISG mRNA expression upon differentiation into NPCs (Figure 7A).
Figure 7. Differentiation of ADAR1 KO hESCs to NPCs leads to spontaneous IFNβ production, PKR activation, and apoptotic cell death.

(A–D) WT and two independent ADAR1 KO hESC clones were differentiated into monolayer NPCs (Figure S7A). qRT-PCR of IFNβ and two ISGs (STAT1, ISG15) (n = 3 experimental replicates) (A). Western blot of p-PKR in NPCs (B). Bright field images of NPC monolayer at 20 days post differentiation (C). Western blot of full length and cleaved forms of PARP1 and Caspase-3, markers of apoptosis (D). As a positive control for apoptosis, WT NPCs were treated with 1uM of Staurosporine (STA).
(E-F) RIG-I, MDA5, or PKR were knocked down in ADAR1 KO NPCs with lentiviral shRNAs (Figure S7F). Representative western blot (uppler panel) and band intensities normalized to β- actin (lower panels, n = 2~5 experimental replicates) (E). qRT-PCR of IFNβ mRNA (n = 4~5 experimental replicates) (F).
(A, E, F) Data shown as mean ± SEM. Student’s t-test, *P < 0.05, **P < 0.01, ***P < 0.001, ns = not significant.
See also Figure S7.
Spontaneous IFNβ production in ADAR1 KO NPCs was accompanied by increased p-PKR (Figure 7B). Additionally, in contrast to the dense monolayer observed with WT NPCs, we observed apparent cell death in ADAR1 KO NPCs (Figure 7C). PKR activation has been shown to trigger apoptosis (Garcia et al., 2006). Accordingly, in ADAR1 KO NPCs, but not in WT NPCs, we detected distinct signs of apoptosis including cleavage of caspase-3 and PARP-1 (Figure 7D).
We also differentiated hESCs to hepatocyte-like cells (HLCs). Both WT and ADAR1 KO HLCs efficiently expressed hepatocyte markers alpha-fetoprotein (AFP) and albumin (Figure S7C). In ADAR1 KO HLCs, we did not observe elevated IFNβ expression compared to WT HLCs (Figure S7D). When HLCs were treated with IFNβ, we observed markedly higher p-PKR in ADAR1 KO HLCs compared to WT HLCs (Figure S7E). Findings with HLCs emphasize that ADAR1 suppresses spontaneous IFN production in only certain cell types such as NPCs. Nonetheless, once IFN signaling initiates, ADAR1 can suppress spontaneous PKR activation in a broad range of cell types, including 293T cells, hESCs, and HLCs.
MDA5 and RIG-I are the major cytoplasmic dsRNA sensors known to induce type I IFN production (Schneider et al., 2014). In some instances, PKR activation can also induce IFN production (Pham et al., 2016). We examined if these dsRNA sensors played a role in inducing IFN production in ADAR1 KO NPCs. To do so, we used shRNAs to KD MDA5, RIG-I, and PKR expression in ADAR1 KO NPCs (Figure 7E and S7F), and subsequently measured IFNβ mRNA expression (Figure 7F). Strikingly, MDA5 KD led to a remarkable reduction in IFNβ mRNA levels (Figure 7F) and suppression of downstream ISG protein expression (e.g. RIG-I and PKR in Figure 7E). In contrast, cells reduced in PKR expression maintained high IFNβ mRNA expression (Figure 7F) and high ISG protein expression (e.g. MDA5 and RIG-I in Figure 7E). Interestingly, suppressing RIG-I expression modestly reduced IFNβ mRNA expression (Figure 7F), but high ISG protein expression persisted in these cells (e.g. MDA5 and PKR in Figure 7E).
MDA5 shRNAs were extremely effective at depleting MDA5 expression (Figure 7E). It is likely that as a result of MDA5 KD, less IFN expression leads to less induction of MDA5 mRNAs, allowing MDA5 shRNAs to more effectively deplete MDA5 expression. Taken together, our data support a model in which MDA5 – rather than PKR and RIG-I – is the major dsRNA sensor mediating IFN induction and downstream ISG expression in ADAR1 KO NPCs.
In summary, loss of ADAR1 in NPCs leads to MDA5-mediated IFNβ production, ISG induction, PKR activation, and apoptotic cell death.
DISCUSSION
In addition to previously known functions of ADAR1 as a negative regulator of ‘IFN production’ (Rodero et al., 2017), we now show that ADAR1 also regulates the ‘response to IFN.’ During the response to IFN, ADAR1p110 and the IFN-inducible ADAR1p150 prevented global translational shutdown and cell death by preventing PKR activation. Our findings suggest that ADAR1p150 evolved as an ISG to ensure an effective response to IFN in establishing an antiviral state.
By examining ADAR1 function in different human cell types we show that ADAR1 is a key player in preventing the spontaneous activation of multiple dsRNA sensing PRRs such as PKR, MDA5, and potentially RIG-I. This is consistent with a recent study in human A549 cells demonstrating that ADAR1 could also suppress the OAS (oligoadenylate synthetases)/RNase L (Ribonuclease L) system, another dsRNA sensing PRR pathway in human cells (Li et al., 2017). Our findings also highlight similarities and uncover differences in ADAR1 function between human and mouse. In mice, ADAR1−/− embryonic lethality was rescued by loss of MDA5 but not PKR (Liddicoat et al., 2015; Pestal et al., 2015; Wang et al., 2004). Thus, while ADAR1 suppression of MDA5 activation appears to be essential in both species, ADAR1 may play a more critical role in preventing PKR activation in humans.
We also characterized ADAR1 substrates during the IFN response. In human 293T cells, we show that the overall frequency of A-to-I editing increases during response to IFN. Interestingly, although ISGs are robustly induced during IFN response, we did not observe an increase in ADAR1 editing sites or editing frequency in ISGs as compared to non-ISGs. ADAR1 primarily edited pol II Alu elements embedded in mRNA, but not independently transcribed putative pol III Alus. This new finding raises interesting questions as to whether pol III Alus – that can potentially retrotranspose – actively evade ADAR1 editing, or whether ADAR1 evolved to specifically target pol II Alus. In addition, ADAR1 editing of pol II Alus does not appear to play a major role in regulating transcript abundance of edited genes. Rather, ADAR1 editing may play a key role in decreasing the innate immunostimulatory potential of endogenous transcripts preventing activation of PRRs such as PKR and MDA5.
The endogenous RNA species that mediates activation of PRRs remains to be identified. Since ADAR1 predominantly edited pol II Alu elements, ADAR1 editing or binding of pol II Alu elements may prevent them from activating PKR. In support of this hypothesis, one study proposed that Alu elements in mRNA could activate PKR during mitosis (Kim et al., 2014). If unedited pol II Alu elements are activating PKR in ADAR1 deficient cells during IFN treatment, it is not clear why PKR activation does not occur in mock treated ADAR1 deficient 293T cells given the high abundance of unedited Alu elements present in the transcriptome. IFN induces rapid transcription, which could perturb the equilibrium between RNA to RNA binding proteins in the cell. As a result, IFN may transiently increase the abundance of “naked” Alu elements – Alus not bound to RNA binding proteins – available for PKR binding and activation. Alternatively, it is possible that an IFN-induced, non-Alu RNA species is activating PKR in ADAR1 deficient cells.
Another important question is how A-to-I editing might prevent PKR activation. In our analysis, editing was frequently detected in Alu element loops rather than stems. Thus, destabilization of dsRNA structures by A-to-I editing may not be the major function of human ADAR1. Alternatively, inosine binding proteins could modulate dsRNA detection by PKR or dsRNA elements containing inosines might bind PKR and inhibit activation. These and other possible mechanisms merit further investigation when specific PKR-activating species are identified
We propose that ADAR1 plays a ‘dual’ protective role against autoinflammatory disease by regulating ‘IFN production’ and the ‘response to IFN.’ First, ADAR1 safeguards cells from unwanted MDA5-mediated ‘IFN production’ in specific cell types such as neuronal lineage cells. This may explain the severe neuropathology found in AGS patients. Second, during the ‘response to IFN’, ADAR1 safeguards cells from translational shutdown and cell death by preventing PKR activation. Remarkably, ADAR1 prevented PKR activation in all human cell types we tested. In follow up studies, it will be important to investigate PKR and MDA5 activation directly in AGS patients.
When intrinsic MDA5 transcript expression was analyzed during the course of hESC to NPC differentiation, we did not observe an increase in MDA5 expression during differentiation (Figure S7G). Furthermore, MDA5 protein expression was consistently below our detection limit in WT NPCs (Figure 7E). Therefore, the spontaneous IFN production in ADAR1 KO NPCs may not be due to an intrinsic increase in MDA5 expression during NPC differentiation. Instead, there may be an NPC specific RNA species that initiates MDA5-mediated IFN production.
Recent studies have suggested ADAR1 may also be involved in promoting tumorigenesis (Han et al., 2015; Paz-Yaacov et al., 2015). Elevated IFN and ADAR1 expression are observed in many cancer types, and ADAR1 KD in breast cancer cells reduced cell proliferation (Fumagalli et al., 2015). This has led to the idea that increased editing in tumors may lead to coding changes in proteins that drive tumorigenesis. We propose an alternative albeit not mutually exclusive hypothesis: Increased ADAR1 expression in cancer cells prevents PKR activation allowing continued proliferation in the face of IFN. In support of this hypothesis, PKR activation has been shown to inhibit tumor growth (Meurs et al., 1993). Historically, IFN cancer therapies have been hampered by tumor resistance to its anti-proliferative effects (Parker et al., 2016). Inhibiting ADAR1 during IFN therapy could lead to PKR activation and subsequent cell growth arrest thereby enhancing the anti-proliferative effects and efficacy of IFN therapy.
Throughout evolution A-to-I editing has been adapted by different species for different key physiological roles. Strikingly, a recent study reported that organisms in other taxa such as cephalopods use extensive A-to-I editing to diversify their neural proteome (Liscovitch-Brauer et al., 2017). Humans are on the opposite end of the spectrum with millions of editing events found predominantly in non-coding regions. In this study, we propose that in humans A-to-I editing evolved to discriminate self from nonself RNA. This function of ADAR1 regulates the delicate balance between pathogen detection and protection versus autoinflammation and disease.
STAR ★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles M. Rice (ricec@rockefeller.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
293T cells
ADAR1 KO and ADAR1p150 KO HEK 293T cell lines (human; sex: female) were generated via CRISPR-Cas9 genome editing. gRNAs were cloned into pSpCas9 (BB)-2A-GFP (pX458) (Ran et al., 2013). All gRNA sequences are listed in Table S1. Briefly, ADAR1 KO cells were generated with gRNAs against exon 2, which is shared by both ADAR1 isoforms (Figure 1A). We generated ADAR1p150 KO cell lines by deleting the ADAR1p150-specific exon 1A and its IFN-inducible promoter (Figure 1A) (George and Samuel, 1999). HEK 293T cells were seeded into 24 well plates (200,000 cells/well) and transfected with 500ng of pX458 using Lipofectamine 2000 (ThermoFisher Scientific). After 2 days, GFP+ single cells were isolated by flow cytometry and plated into a 96 well plate. Expanded clones were screened for ADAR1 deficiency by western blot and disruption of the ADAR1 genomic locus was confirmed by Sanger sequencing using PCR®8/GW/TOPO® TA Cloning Kit (Thermo Fisher Scientific).
hESCs
To generate ADAR1 KO hESCs, gRNAs targeting ADAR1 (Table S1 and S2) were transfected into HUES8 (human; sex: male; NIHhESC-09-0021)-iCas9 cells, which upon treatment with Dox (2ug/ml) express Cas9 (Gonzalez et al., 2014). HUES8-iCas9 cells were treated with Dox for 48 hrs prior to transfection and for 24 hrs post transfection. Transfected cells were seeded in clonal limiting dilution and grown on irradiated mouse embryonic fibroblast in cloning medium (DMEM/F12, 20% KOSR, 1% GlutaMax, 1% NEAA, 0.1% β-mercaptoethanol, and 10ng/ml bFGF). Medium was replenished daily. Clones were expanded and screened by western blot and homozygous ADAR1 knockout clones were selected for further characterization.
METHOD DETAILS
Cell culture reagents
For IFN-treated conditions, one 1nM IFNβ (PBL Assay Science) was used. 293T cells were cultured in DMEM/10%FBS and ActD (10ug/ml), CHX (30ug/ml), or α-amanitin (25ug/ml) was added to the media as shown in Figure S6A.
The following lentiviruses were produced from the following vectors; pSCRPSY (Dittmann et al., 2015), vector expressing wildtype or mutant forms of ADAR1; pTRIPZ-PKR, doxycycline inducible PKR expression (modified pTRIPZ vector (Dharmacon); GIPZ-PKR, -RIG-I, or -MDA5 shRNAs (Dharmacon). ADAR1 KO 293T cells were transduced with the respective lentivirus followed by selection with puromycin (3ug/ml) to establish stable cell lines. hESCs were cultured in mTESR1 with supplement (Stem cell Technologies) and split with ReLeSR™ (Stem Cell Technologies).
NPC differentiation
hESCs were differentiated into NPCs using adherent culture conditions (Chambers et al., 2009). We followed the monolayer culture protocol from STEMdiff™ Neural System (Stem cell technologies). Briefly, hESC monolayers were plated onto matrigel coated plates, and cells were incubated with neural induction medium for 13 days, followed by incubation with neural progenitor medium to maintain and expand NPCs (Figure S7A). Gene expression of MDA5, RIG-I, and PKR was knocked-down in NPCs as shown in Figure S7F. Briefly, a monolayer of hESCs were plated onto matrigel coated plates and incubated with neural induction medium. After 8 days, cells were split (50% confluency) into matrigel coated plates. The next day, cells were transduced with lentiviruses produced from pGIPZ shRNA lentiviral vectors (Dharmacon) – targeting MDA5, RIG-I, and PKR - and were further incubated with neural induction medium for 5 days before harvest.
HLC differentiation
hESCs were differentiated to definitive endoderm (DE) by harvesting with gentle cell dissociation reagent (Stemcell Technologies) and plating into Matrigel-coated culture dishes (Corning, New York, NY) in mTeSR1 medium (Stemcell Technologies). The next day, DE differentiation was initiated by using the STEMdiff Definitive Endoderm Kit (Stemcell Technologies). To induce hepatic differentiation, definitive endoderm cells were re-seeded in Matrigel-coated plates and cultured in the presence of medium A (BM, 100ng/ml hepatocyte growth factor [HGF]), 1% DMSO) for eight days followed by incubation in medium B (BM, 100ng/ml HGF, 1% DMSO, 40ng/ml Dexamethasone) for three more days. BM (basal media) consists of CTS KnockOut DMEM/F12, 10% KOSR, 0.5% GlutaMAX supplement, and 0.5% non-essential amino acids (all from ThermoFisher Scientific). Cells were further matured in Hepatocyte Culture Medium (HCM, Lonza) supplemented with 20ng/ml oncostatin-M for five to seven days. HGF was purchased from Peprotech, dexamethasone from Sigma, and oncostatin- M from R&D Systems.
Flow cytometry
Live cell surface staining was conducted with antibodies to detect β2M (Biolegend), IFNγR1 (Biolegend), and CD9 (Biolegend). BD Cytofix/Cytoperm™ Fixation/Permeabilization kit used to stain for STAT1 (EPR4407, abcam), PKR (EPR19374, abcam), and p-eIF2α (phospho S51, abcam). BD Pharmingen™ Factor Buffer Set used to stain for Nestin (Stem Cell Technologies) and Pax6 (BD). Secondary antibodies used were Alexa fluor 488, 594, 647 goat-anti rabbit (Thermo Fisher Scientific). Antibodies used for flow cytometry are listed in the Key Resources Table.
qRT-PCR
RNA was extracted using the Direct-zol™ RNA MiniPrep (Zymo Research). RNA was converted to cDNA using the SuperScript™ III First-Strand Synthesis System (Thermo Fisher Scientific). qRT-PCR was performed with SYBR Green on the LightCycler® 480 instrument. Unless stated otherwise, the housekeeping gene RPS11 was used for normalization. All primer sequences are listed in Table S7.
Western Blot
Cells were directly lysed in NuPAGE® LDS Sample Buffer (Thermo Fisher Scientific) with 400mM DTT (Sigma Aldrich). Lysates were passed through 26G needle and boiled for 10min before loading on NuPAGE™ 4–12% Bis-Tris Protein Gels (Thermo Fisher Scientific). Protein was blotted to nitrocellulose membrane and probed for ADAR1 (D-8, Santa Cruz Biotechnology), ADAR2 (Sigma-Aldrich), p-PKR (phospho T451, abcam), PKR (abcam), β-actin, Caspase-3 (abcam), PARP1 (Santa Cruz Biotechnology), eIF4E and p-eIF4E (Cell Signaling Technology), eIF2α (Thermo Fisher Scientific), p-eIF2α (abcam), MDA5 and RIG-1 (Cell Signaling Technology). Blots were developed using Fluorescent (LI-COR, Odyssey CLx) or Chemiluminescent (film) detection. Band intensity was quantified with Image J. Antibodies used for western blot are listed in the Key Resources Table.
Cell growth assays
Viable cells were identified by Trypan Blue (Thermo Fisher Scientific) exclusion and counted with a hemocytometer. The CellTiter-Glo® Luminescent Cell Viability Assay (Promega) was also used to assess viable cells over time.
Polysome profiling
Polysome profiling was conducted as described by Su et al. (Su et al., 2015). WT, ADAR1 KO, and ADAR1p150 KO 293T cells were treated with IFNβ (1nM) for 24 hours and then treated with CHX (100 µg/ml) for 2 min. Cells were then washed with cold PBS/CHX (100 µg/ml), lysed in polysome lysis buffer (20 mM Tris, pH 8.0, 140 mM NaCl, 15 mM MgCl2, 100 µg/ml CHX, 0.5% Triton X-100) and protease inhibitor cocktail (Roche). Lysates were passed ten times through a 26-gauge needle, and laid onto a 10%–50% sucrose-density gradient to isolate polysome fractions. Gradients were centrifuged in an SW41 ultracentrifuge rotor at 38,000 r.p.m. for 2 h, and OD260 was measured.
RNA extraction and library preparation
WT, ADAR1p150 KO and ADAR1 KO 293T cells (n = 3 clones for each genotype) were mock or IFNβ (1nM) treated for 24 hours. RNA was extracted with the Direct-zol™ RNA MiniPrep (Zymo Research). ERCC RNA Spike-In Mix (Thermo Fisher Scientific) was added to each RNA sample to achieve a standard measure for gene expression. Libraries were prepared with the TruSeq Stranded Total RNA with Ribozero kit (Illumina) using 1ug input RNA/library. Two modifications were applied to the standard kit protocol in order to generate libraries with long fragments. First, RNA was fragmented at 94°C for 2 minutes. Second, a fter ligating adaptors, the volume of AMPure XP Beads (Beckman Coulter) used was reduced to 60% of the suggested volume in the protocol. The 18 libraries were pooled and sequenced on the Illumina Nextseq platform (High output, 2 runs) with read length of 150 nt in paired end configuration.
Virus infection and poly(I:C)
WT- or ADAR1- deficient 293T cells were plated in a 24 well (60,000 cells/well) and infected with a reporter SINV (TE/5′2J strain), which carries a blue fluorescent protein (BFP) fused to nsp3. Alternatively, cells were transfected with poly(I:C) (Invivogen) using lipofectamine 2000 (ThermoFisher Scientific).
RNA sequencing data analysis
Read mapping and filtering
Paired end reads were mapped with Tophat2 (version 2.0.14). Read pair insert size statistics were determined using the CollectInsertSizeMetrics module from Picard (version 1.130) on 5 million randomly selected read pairs. Mean insert sizes and standard deviations for the different dataset/lane combinations obtained in this analysis ranged from 352 to 438 (median = 403) and 169 to 259 (median = 193), respectively. Dataset specific insert size statistics per run were used in subsequent read mapping steps, after which the alignments from the separate runs were combined for each sample.
Read mapping and alignment filtering workflows included several features intended to facilitate identification of RNA editing sites. RNA-Seq read pairs were mapped to human reference genome sequence (GRCh37) supplemented with splice-junction information from Ensembl (version 75) transcript annotations. Relatively permissive read:reference mismatch parameters (--read-mismatches 15; --read-edit-dist 15) were selected to enable mapping of reads potentially derived from transcripts with multiple editing sites. Only mapped, concordant read pairs were included in downstream RNA editing analyses. Read pairs were defined as concordant when both mates mapped to an exonic sequence of the same transcript, or when spanning a genomic sequence less than 5000 nucleotides. Concordant read pair alignments with a minimum total mate pair read:reference distance from the reference sequence were identified, and selected only if a unique alignment was found.
ADAR1- and ADAR1p150- associated mismatch identification
In order to focus the editing analysis to relevant transcripts, we used Stringtie (version 1.2.3) to generate sample specific transcript annotations from RNA-Seq read alignments. Annotations were constructed from each dataset independently, and then combined using the merge functionality in Stringtie.
In order to evaluate potential RNA editing sites, read alignment files were processed for coverage and nucleotide content at single nucleotide resolution. First, regions of overlapping mate pairs were collapsed using clipOverlap (bamUtil version 1.0.13) to prevent redundant counting. Read pairs aligned with insertions or deletions were discarded. The first and last three nucleotide positions of each read were not included in quantification. Furthermore, all read positions with Q-value less than 20 were excluded from quantification. Next, for each genomic position in the Stringtie-derived transcript annotations, strand-specific read nucleotide frequencies were quantified using Mpileup (samtools version 0.1.18).
Genomic positions with coverage of at least 5 reads in all biological replicates (e.g. all mock treated samples or all IFN treated samples) were selected for editing site analysis. Positions on ERCC spike-in sequences or those annotated as genomic SNPs (Common dbSNP version 142) were excluded from subsequent analyses. For each sample at each position, read:reference mismatches were called as present if at least 2 reads and more than 5% of all reads contained the same read:reference mismatch. For each sample at each position, read:reference mismatches were called as absent if less than 1% of the reads contained a read:reference mismatch, or if maximally 1 read contained the read:reference mismatch representing less than 5% of all reads.
For each read:reference mismatch, we evaluated if the mismatch was associated with ADAR1 and/or ADAR1p150 genotype status based on the following criteria, using the samples that received similar treatment, i.e. mock or IFN treatment. First, the read:reference mismatch was required to be present in all WT samples and absent in all ADAR1 KO samples that received similar treatment. Second, the read:reference mismatch should not be present in any of the ADAR1 KO samples from any treatment group. Third, a mismatch presence in any of the ADAR1p150 KO samples was required for assignment as an ADAR1-associated mismatch, and absence of mismatches in all of the ADAR1p150 KO samples was required for assignment as an ADAR1p150-associated mismatch.
If a mismatch was assigned as ADAR1p150-associated in one treatment condition but ADAR1-associated in the other treatment condition, the site was designated as ADAR1-associated. Note, as ADAR1p150 is an isoform of ADAR1, the ADAR1-associated sites in all analyses include all identified ADAR1p150-associated mismatches.
Gene expression analysis
Gene-level read counts were quantified using htseq-count (version 0.6.1) based on Ensembl (version 75) reference transcriptome annotation. Differential gene expression analysis was performed with voom-limma (Law et al., 2014) using a linear model incorporating ADAR1 genotype, treatment and batch factors. Gene set enrichment testing was conducted with the CAMERA (Wu and Smyth, 2012) tool on select MSigDB (Subramanian et al., 2005) gene sets; Categories H (hallmark), C5 (GO gene sets), C6 (Oncogenic gene sets), and C2 (canonical pathways). For GO term analysis of ADAR1 or ADAR1p150 edited genes, genes with ADAR1 or ADAR1p150 edits were compared to expression-matched genes that do not contain edits using the Gene Ontology enRIchment anaLysis and visuaLizAtion tool (GOrilla) (Eden et al., 2009). Expression-matched genes were found by comparison of TPM expression values obtained using the Salmon gene expression quantification software (Patro et al., 2017).
Mismatch site mapping to Alu consensus
To evaluate which sites of the Alu sequence are preferentially edited, ADAR1- or ADAR1p150-associated mismatches were overlaid on the Alu consensus sequence (Bao et al., 2015). First, Alus that contained mismatches were aligned to the Alu consensus sequence using the local pairwise alignment tool implemented in the Biopython module (version 1.65). Next, the location of the mismatch position in the Alu consensus sequence was derived from the alignment. Using this approach, 8492 of 8566 (99%) of the mismatches could be mapped to the Alu consensus sequence.
Alu secondary structure prediction
Alu RNA secondary structure prediction was performed for all Alu sequences containing an ADAR1- or ADAR1p150- associated mismatch. For “Intra Alu” (single Alu) secondary structure prediction, RNAfold (version 2.1.9) was used to predict separately the structures of the left and right arm, and resulting structures were then joined. The left and right arm comprise 45% and 55%, respectively, of the Alu consensus sequence, and were annotated in each Alu element according to this 45/55 ratio. For “Inter Alu” (two Alu elements with opposite orientation within the same transcript) secondary structure prediction, RNAcofold (version 2.1.9) was used to identify the “co-fold” with maximal binding free energy. In order to ensure that single Alu elements were not incorrectly considered for potential inter Alu secondary structures, transcript isoforms from genes containing multiple Alu elements were included in co-fold analyses only if the Alu elements examined are present in all annotated transcript isoforms. Predicted base pairs were counted for the ADAR1- or ADAR1p150-associated mismatch sites, as well as for all (mismatch and non-mismatch) sites, and compared. These comparisons were made using Intra Alu folds or Inter Alu folds, ADAR1 or ADAR1p150 associated mismatch, and for different selections of Alus.
Mismatch site annotation
As we generated a custom transcript annotation (constructed with Stringtie based on read mappings), not all ADAR1- and ADAR1p150- associated mismatches correspond to reference annotated transcripts. Therefore, annotations from Stringtie output and Ensembl were combined to classify sites as within 5´UTR, ORF, 3´UTR, Intronic, or Other. First, positions annotated as intronic by both Stringtie and Ensembl were classified as Intronic. Second, if a Stringtie gene was associated with an Ensembl transcript annotation with non-zero expression, it was annotated according to the Ensembl reference (transcript isoform with the highest expression value). In the absence of a corresponding Ensembl gene annotation, remaining Stringtie positions were annotated as Other. Based on the Ensembl transcript annotations of protein coding sequences, exonic positions were classified as 5´UTR, ORF or 3´UTR. Positions in exonic sequences without translation frame annotations, e.g. for non-protein coding genes, were classified as Other.
Repeatmasker annotations for the human genome were downloaded from UCSC in April 2016. Specific classifications were made for Alu, ERV1 and L1 elements; additional repetitive elements were classified as OtherRM/Other Repeat. Positions with no Repeatmasker annotation were marked as noRM/No Repeat.
Classification of independently expressed Alu transcripts (putative pol III Alu elements) and transcript-embedded Alu elements (putative pol II Alu elements) in RNA-Seq data
Alu elements can be transcribed as part of a larger transcript, often within intronic or 3’UTR segments of an RNA polymerase II (pol II) transcribed mRNA. Alternatively, they can be expressed as independent RNA species transcribed by RNA polymerase III (pol III). Herein, we refer to these distinct modes of Alu expression as transcript-embedded Alus (pol II Alu elements) and independently expressed Alus (pol III Alu elements), respectively. In order to distinguish between transcript-embedded Alus (putative pol II Alu elements) and independently expressed Alus (putative pol III Alu elements) in genome-mapped RNA-Seq data, we compared read coverage of each Alu element with the read coverage of flanking genomic sequences, based on a strategy described by Conti et al. (Conti et al., 2015). Alu elements annotated in the repeatMasker database (downloaded via UCSC in April 2016) were selected for analysis if the Alu interval and flanking regions (1000 nucleotides up and downstream) share the same transcript classification (exonic, intronic, non-genic, as per Ensembl reference). For each sample, strand specific read coverage was determined for the Alu element interval and flanking regions (1000 nucleotides up and downstream, each in 10 concurrent 100 nucleotide intervals). Alu elements with an average read coverage of less than 5 reads in all samples were excluded from further analysis.
In order to identify independently expressed Alus, Alu sequences annotated as non-genic (from Ensembl transcript annotation) were further assessed for read coverage patterns. The search image for an independently expressed Alu is an Alu sequence with continuous read coverage from the start of the Alu interval (defined by repeatmasker) until some unknown point 3’ of the Alu element terminus. First, read coverage in the 10 upstream (5’) intervals was compared to the read coverage within the Alu element. If, in all samples, read coverage in the 10 concurrent 100nt upstream intervals was at least 10-fold lower than in the Alu interval, the Alu element was selected for further assessment of downstream (3’) read coverage. For consideration as an independently expressed Alu element, downstream read coverage was required to decrease at some interval downstream of the Alu interval. Therefore, read coverage in downstream (3’) intervals was assessed for instances in which coverage in an Alu-proximal interval was at least 10-fold higher than coverage in subsequent Alu-distal intervals. If these conditions were met, an Alu element was annotated as an independently expressed Alu (pol III Alu element). Using this approach, we identified 214 independently expressed Alus putatively transcribed by RNA pol III. All of the independent Alus identified were detected on the strand corresponding to the "sense" orientation of the Alu consensus sequence.
In order to identify transcript-embedded Alus (pol II Alu elements), Alu sequences annotated as intronic or exonic were further assessed for read coverage patterns. The search image for a transcript-embedded Alu is continuous read coverage at the Alu sequence as well as over the downstream (3’) and upstream (5’) regions adjacent to the ALU interval. Therefore, for each sample, if at least 80% of the downstream and upstream 100 nucleotide intervals demonstrated read coverage within 3-fold of read coverage over the Alu interval, the Alu was annotated as a transcript-embedded Alu (pol II Alu element). Using this approach, we identified 1991 transcript-embedded Alus, of which 903 (45%) were in "sense" orientation of the Alu consensus sequence, as expected for Alu elements that can have either "sense" or "antisense" orientations within a larger RNA pol II transcript.
Independent Alu expression analysis
Annotations for independently expressed (putative pol III) Alu transcripts (as defined using the Alu transcript type differentiation scheme above) were appended to reference transcriptome annotation (Ensembl version 75). Read counts per gene or pol III Alu element were quantified by htseq-count (version 0.6.1). Gene- and Alu- level read counts were normalized with voom-limma (Law et al., 2014). Per sample Z-scores were calculated on log2 transformed TMM normalized (Robinson et al., 2010) expression values for each independent Alu transcript.
Alu read:reference mismatch analysis
For each set of Alu elements (either independently expressed Alus or transcript-embedded Alus), mapped reads were assessed for read:reference mismatches. Using Mpileup (samtools version 0.1.18), single base resolution coverage was assessed for each strand-specific position in the Alu elements. Positions annotated as genomic SNPs (Common dbSNP version 142) were excluded from this analysis. The observed (read) bases were compared to the expected (reference) base, and counts for each read:reference combination were summed per sample. Per sample, for each read:reference mismatch, the read:reference mismatch count was normalized to the summed number of read:reference counts with the same reference nucleotide. Thus, results are presented as the frequency of mismatches relative to the total number of read bases mapped to a certain reference nucleotide.
QUANTIFICATION AND STATISTICAL ANALYSIS
For qRT-PCR, mRNA expression was determined using the ΔΔCt method with normalization to the housekeeping gene RPS11. All statistical tests, exact value of n, and what n represents (e.g. number of cell clones or experimental replicate) can be found in figure legends. Statistical significance is shown as * (p<0.05), ** (p<0.01), *** (p<0.001), and ns (not significant). Results are presented as means ± standard error of mean (SEM) in graphs. Comparisons between groups were made using the Student’s t test, One-way ANOVA/Tukey’s post host test, or Mann Whitney U test (Bonferroni corrected). Statistical analyses were performed in Graph Pad PRISM 4.
DATA AND SOFTWARE AVAILABILITY
All RNA-Seq data have been deposited under the GEO accession number GSE99249. ADAR1 and ADAR1p150 edit sites identified in this study are listed in Table S3, S4, and S5. Gene expression data are included in Table S6.
Supplementary Material
(A) Growth rate/day measured by the CellTiter-Glo assay.
(B) Cell clones with reduced ADAR1p110 expression. Western blot of ADAR1 expression after generating cell clones deleted of the constitutively active promoter (Pc) and exon 1B using CRISPR/Cas9 genome editing (Figure 1A and Table S1). Deletion of Pc and exon 1B was confirmed by DNA sequencing for clone #1 and #3 (Table S2).
(C) Western blot of CRISPR/Cas9-derived ADAR2 KO cell clones. Blots were probed with antibodies detecting ADAR2 and β-actin control. Disruption of the ADAR2 genomic locus was confirmed by DNA sequencing (Table S1 and S2).
(A–C) Par, parental 293T cell line. WT, CRISPR/Cas9 derived WT 293T cell clone.
(A) Number of ADAR1p150-associated edit sites enumerated and shown on the predicted secondary structure of the Alu consensus sequence. The color bar represents the number of ADAR1p150-associated edit sites identified per position in the Alu consensus sequence.
(B, C) The secondary structures of individual Alu elements (Intra Alu fold) or multiple Alu elements within the same transcript (Inter Alu fold) were predicted as described (see Methods). The fraction of edited sites (Edits) and all sites (All) predicted to be base paired (double stranded) in the predicted Alu secondary structure are plotted. Edit sites were analyzed in groups composed of those in all Alu sequences (All Alus), those in Alus sequences within transcripts without additional reverse complementary Alu sequences in cis (Mono Alus), or those in Alu sequences within transcripts containing an additional reverse complementary Alu sequence in cis (Multi Alus).
(D, E) Volcano plots of moderated log2 gene expression fold change for ADAR1 KO versus WT (D) or ADAR1p150 KO versus WT (E) in mock treated conditions (see Methods). Each dot represents a gene; colored dots indicate genes with edit sites, where the color hue indicates the number of ADAR1- or ADAR1p150- associated edits.
(F-G) Gene expression was analyzed as in Figures 2I and 2J. All genes containing Alu elements within any of their exonic parts are colored red. WT versus ADAR1 KO (F), and WT versus ADAR1p150 KO cells (G).
(H) Z-scaled expression values for 211 of 214 putative pol III Alus (‘independently transcribed Alus’), grouped by cell clone. Each point represents an individual Alu transcript and overlaid boxplot summarizes the distribution per sample. Significant differences of the IFN treated Z-scaled expression values relative to those values in the corresponding Mock treated sample are indicated (values compared by Mann Whitney U test, Bonferroni corrected for multiple testing; *p<0.05; **p<0.01;***p<0.001.). TMM, trimmed mean of M-values (Robinson et al., 2010)
(A) qRT-PCR analysis of relative mRNA levels, normalized to RPS11.
(B) ISG protein expression measured by flow cytometry. MFI, mean fluorescence intensity.
(A) Cell death/apoptosis assay. 293T cells were mock or IFNβ treated (48 hours) and harvested for flow cytometry analysis. Cells were stained with Annexin V and Propidium Iodide (PI) to detect the frequency (%) of different types of cell death. Early apoptotic cells, Annexin V+PI−. Late apoptotic cells/Dead cells, Annexin V+PI+. As a positive control, 293T cells were treated with 2uM staurosporine (STA) for 4 hours to induce apoptosis/cell death. DMSO, Dimethyl sulfoxide. Data are shown as mean ± SEM from n = 2~3 experimental replicates.
(B–D) Cell proliferation assay. WT or ADAR1 deficient cells were labeled with CFSE, equal numbers plated into a 24 well plate, and mock or IFNβ treated. CFSE intensity measured by flow cytometry over time (B). Representative flow cytometry plots of CFSE and β2M expression in mock and IFN treated cells (48 hours) (C). Frequency of β2MlowCFSEhigh summarized in bar graph (D). Data are shown as mean ± SEM from n = 3 experimental replicates. Student’s t-test, ***P < 0.001.
(A) WT, ADAR1 KO, and ADAR1p150 KO 293T cells were treated with IFNβ for 24 hours. Representative western blots labeled for total or phosphorylated forms of eIF2α or eIF4E.
(B) Western blot assessing the efficiency of shRNA-mediated PKR knock-down. WT or ADAR1-deficient 293T cells were transduced with lentiviruses expressing pGIPZ-PKR shRNA or pGIPZ-control shRNA (CTRL), followed by selection with puromycin to establish stable cell lines. Lysates were harvested for western blot analysis, and blots were probed with antibodies detecting PKR and β-actin control. Par, parental 293T cell line.
(A–C) ADAR1 KO 293T cells were mock or IFNβ treated for in total 24 hours. After the initial 12 hours of IFN treatment, cells were co-incubated with cycloheximide (CHX, 30ug/ml), actinomycin-D (ActD, 10ug/ml), or α-amanitin (25ug/ml) for the latter 12 hours (A). Cell pellets were harvested for western blot (B) or qRT-PCR (C). Western blots were probed with an anti- PKR and anti-β-actin antibody. Relative PKR band intensity (normalized to β-actin control) was measured by image J (B). Levels of IFIT1 mRNA, an RNA polymerase II derived ISG, was normalized to the housekeeping gene 18S rRNA, a relatively stable RNA polymerase I derived transcript (C). Data are shown as mean ± SEM from n = 3 experimental replicates. Student’s t-test, *P < 0.05, **P <0.01, ***P < 0.001, ns = not significant.
(D–E) 293T cells were infected with a blue fluorescent protein (BFP)- expressing Sindbis virus (SINV) at MOI 1 or MOI 4. Cells were harvested at 24 hours post-infection and analyzed by western blot to measure PKR phosphorylation (p-PKR) (D). Percent infection of cells was measured by flow cytometry (E).
(F) 293T cells were transfected with increasing doses of poly(I:C), and cell lysates were harvested at 5 hours post-transfection. PKR phosphorylation (p-PKR) was measured by western blot.
(A–B) WT and ADAR1 KO hESCs (two independent clones) were differentiated into NPCs as shown in the schematic (A). The frequency of NPC markers (Nestin, Pax6) was assessed by flow cytometry at 13 days post differentiation (n = 4 experimental replicates) (B).
(C–E) Par. (parental), WT, and ADAR1 KO hESCs (two independent clones) were differentiated into hepatocyte-like cells (HLCs). The hepatocyte markers Albumin and AFP (alpha-fetoprotein)were measured by qRT-PCR and the values were normalized to RPS11 (n = 2~4 experimental replicates) (C). WT NPCs were used as a negative control for Albumin and AFP levels. qRT-PCR of IFNβ mRNA levels that were normalized to RPS11 (n = 6 experimental replicates) (D). HLCs were treated with IFNβ for 24 hours and harvested for western blot to measure PKR phosphorylation (p-PKR) (E).
(F) Schematic of gene knock-down (KD) protocol in NPCs. ADAR1 KO NPCs were transduced with lentiviruses derived from GIPZ lentiviral shRNA vectors (Dharmacon) – targeting MDA5, RIG-I, and PKR - at 9 days post differentiation. Cells were harvested at 14 days post differentiation for western blot and qRT-PCR analysis.
(G) We examined gene expression of MDA-5, RIG-I, and PKR using an RNA-Seq data set from a recent study that examined transcript expression during the course of hESC to NPC differentiation (hESC → ectoderm (ECT) → NPC) (Wu et al. 2018). TPM, transcript per million. (B, C, D, G) Data are shown as mean ± SEM. Student’s t-test, ns = not significant.
Column A (genomicPosition) lists the strand-specific genomic position of the mismatch. Column B (mutationType) lists the type of mismatch, where X>Y indicates the reference (X) and read (Y) nucleotide of the mismatch. Column C (editType) indicates whether the mismatch is ADAR1- or ADAR1p150- associated. Columns D (StringtieGenes) and E (StringtieAssociatedEnsembleGeneIDs) indicate which Stringtie-determined gene was annotated at the genomic position, and which Ensembl genes were associated with the Stringtie determined gene, respectively. Column G (EnsemblAssociatedGeneIDs) indicates which Ensembl genes are present at the genomic position. The gene names from the Ensembl gene IDs in columns E and G are given in columns F (StringtieAssociatedEnsembleGenenames) and H (EnsemblAssociatedGenenames), respectively. Column I (RepeatMaskerElements) indicates any repeat sequence that was present at the genomic position. Columns J-AA list for each sample the number and fraction of total reads that are mismatched; e.g. if 13 As, 10 Gs and 2 Cs are found at a position in a specific sample, the A>G edit frequency is 10/25(0.4).
For each Stringtie-annotated gene (column A, StringtieGene), the number of ADAR1- and ADAR1p150- associated read:reference mismatch sites are listed in columns D (ADAR1- associated mismatch count) and E (ADAR1p150-associated mismatch count), respectively. Ensembl gene IDs that were associated by Stringtie to the Stringtie annotated gene are listed in column B (StringtieAssociatedEnsembleGeneIDs). Gene names from the Ensembl gene IDs in column B are listed in column C (StringtieAssociatedEnsembleGenenames).
All read:reference that map to the ORF of a transcript in the Ensembl annotations are listed here. Column A (Mismatch in highest expressed transcript isoform) indicates if the mismatch is in a transcript isoform that was most highly expressed in comparison to the other transcript isoforms of the same gene. The chromosome, position, and strand of the mismatch are listed in columns B (Chromosome), C (position), and D (strand), respectively. The Ensembl transcript ID and gene name are listed in columns F (transcript IDs) and E (gene), respectively. Column G (Alu sequence) indicates whether an Alu sequence was present at the mismatch position. Column H (mismatch association) indicates whether the mismatch was ADAR1- or ADAR1p150- associated.
For different comparisons as described in column A (comparison), differentially enriched (p-value<0.05 and FDR<0.1) gene sets are listed. Column B (gene set name) lists the name of the gene set. Column C (gene set size) lists the number of genes in the set. Column D (Expression up/down) indicates whether the set gene expression was up or down regulated in the comparison. The p-value and FDR of the test for up/down regulation, as determined using CAMERA, are listed in columns E (p-value) and F (FDR), respectively.
HIGHLIGHTS.
-
-
ADAR1 edits are found in putative RNA polymerase II (but not III) derived Alu repeats
-
-
ADAR1 prevents translational shutdown during the type I IFN response
-
-
ADAR1 dsRNA binding and catalytic activities are required to block PKR activation
-
-
ADAR1 KO NPCs exhibit MDA5-dependent IFN production, PKR activation, and cell death
Acknowledgments
We would like to thank M. MacDonald and W. Schneider for helpful comments on experimental design and the manuscript. We also thank: Laboratory for Investigative Dermatology (J. Gonzalez), Genomics Resource Center, and the Flow Cytometry Resource Center at The Rockefeller University, D. Huangfu for HUES8-iCas9 cells, and L. Marraffini for CRISPR reagents. This work was supported by NIAID/NIH grants R01-AI091707 (C.M.R., H.C., B.R.R., J.J.A.C.), F32AI114211 (H.C.), the John C. Whitehead Presidential Fellowship (B.R.R.), and the Netherlands Organization for Scientific Research (NWO Rubicon)(J.J.A.C.). V.L.D.T. was supported by a Helmsley postdoctoral fellowship at The Rockefeller University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
H.C., C.M.R. and B.R.R. designed the project. H.C. wrote the manuscript. H.C., C.M.R., J.J.A.C., B.R.R. designed experiments. H.C., X.W., T.S., Y.Y., S.S., A.R.S., V.L.D.T., and H.-H.H. performed experimental work. J.J.A.C. and B.R.R. performed RNA-Seq data analysis. Editorial input from all co-authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alucontaining mRNAs in the human transcriptome. PLoS biology. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–1098. doi: 10.1016/0092-8674(88)90253-x. [DOI] [PubMed] [Google Scholar]
- Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription. 2011;2:103–108. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti A, Carnevali D, Bollati V, Fustinoni S, Pellegrini M, Dieci G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015;43:817–835. doi: 10.1093/nar/gku1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann M, Hoffmann HH, Scull MA, Gilmore RH, Bell KL, Ciancanelli M, Wilson SJ, Crotta S, Yu Y, Flatley B, et al. A serpin shapes the extracellular environment to prevent influenza A virus maturation. Cell. 2015;160:631–643. doi: 10.1016/j.cell.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell reports. 2015;13:277–289. doi: 10.1016/j.celrep.2015.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, Petroulakis E, Robichaud N, Pollak M, Gaboury LA, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14134–14139. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiology and molecular biology reviews : MMBR. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JF, Clerzius G, Shaw E, Gatignol A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J Virol. 2011;85:8460–8466. doi: 10.1128/JVI.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Ramaswami G, Li JB, Samuel CE. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. J Biol Chem. 2016;291:6158–6168. doi: 10.1074/jbc.M115.709014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell stem cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John L, Samuel CE. Induction of stress granules by interferon and downregulation by the cellular RNA adenosine deaminase ADAR1. Virology. 2014;454–455:299–310. doi: 10.1016/j.virol.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lee JH, Park JE, Cho J, Yi H, Kim VN. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes & development. 2014;28:1310–1322. doi: 10.1101/gad.242644.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, Weiss SR. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife. 2017;6 doi: 10.7554/eLife.25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015 doi: 10.1126/science.aac7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJ, Eisenberg E. Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell. 2017;169:191–202 e111. doi: 10.1016/j.cell.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel CE. Mechanism of interferon action: functionally distinct RNA-binding and catalytic domains in the interferon-inducible, double-stranded RNA-specific adenosine deaminase. J Virol. 1996;70:1961–1968. doi: 10.1128/jvi.70.3.1961-1968.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell reports. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:232–236. doi: 10.1073/pnas.90.1.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita Y, Shibutani T, Nakanishi N, Nishikura K, Iwai S, Kuraoka I. Human endonuclease V is a ribonuclease specific for inosine-containing RNA. Nature communications. 2013;4:2273. doi: 10.1038/ncomms3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura F, Okumura AJ, Uematsu K, Hatakeyama S, Zhang DE, Kamura T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J Biol Chem. 2013;288:2839–2847. doi: 10.1074/jbc.M112.401851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, Ariyoshi K, Iizasa H, Davuluri RV, Nishikura K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nature reviews Cancer. 2016;16:131–144. doi: 10.1038/nrc.2016.14. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell reports. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43:933–944. doi: 10.1016/j.immuni.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marie IJ, Levy DE. PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLoS Pathog. 2016;12:e1005489. doi: 10.1371/journal.ppat.1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome biology. 2014;15:R5. doi: 10.1186/gb-2014-15-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Li JB. RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kitabayashi N, Barth M, Briggs TA, Burton AC, Carpanelli ML, Cerisola AM, Colson C, Dale RC, Danti FR, et al. Genetic, Phenotypic, and Interferon Biomarker Status in ADAR1-Related Neurological Disease. Neuropediatrics. 2017 doi: 10.1055/s-0037-1601449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, McGlasson SL, Alyanakian MA, Bader-Meunier B, Barnerias C, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. The Journal of experimental medicine. 2017 doi: 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg BR, Hamilton CE, Mwangi MM, Dewell S, Papavasiliou FN. Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3' UTRs. Nat Struct Mol Biol. 2011;18:230–236. doi: 10.1038/nsmb.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden AD. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12:489–496. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annual review of immunology. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, Cross JR, Ratsch G, Rice CM, Ivashkiv LB. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. Dynamic landscape and regulation of RNA editing in mammals. Nature. 2017;550:249–254. doi: 10.1038/nature24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente L, Nishikura K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J Biol Chem. 2007;282:16054–16061. doi: 10.1074/jbc.M611392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- Wu D, Smyth GK. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 2012;40:e133. doi: 10.1093/nar/gks461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, et al. Intrinsic immunity shapes viral resistance of stem cells, Cell. 2018 doi: 10.1016/j.cell.2017.11.018. https://doi.org/10.1016/j.cell.2017.11.018. [DOI] [PMC free article] [PubMed]
- Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Growth rate/day measured by the CellTiter-Glo assay.
(B) Cell clones with reduced ADAR1p110 expression. Western blot of ADAR1 expression after generating cell clones deleted of the constitutively active promoter (Pc) and exon 1B using CRISPR/Cas9 genome editing (Figure 1A and Table S1). Deletion of Pc and exon 1B was confirmed by DNA sequencing for clone #1 and #3 (Table S2).
(C) Western blot of CRISPR/Cas9-derived ADAR2 KO cell clones. Blots were probed with antibodies detecting ADAR2 and β-actin control. Disruption of the ADAR2 genomic locus was confirmed by DNA sequencing (Table S1 and S2).
(A–C) Par, parental 293T cell line. WT, CRISPR/Cas9 derived WT 293T cell clone.
(A) Number of ADAR1p150-associated edit sites enumerated and shown on the predicted secondary structure of the Alu consensus sequence. The color bar represents the number of ADAR1p150-associated edit sites identified per position in the Alu consensus sequence.
(B, C) The secondary structures of individual Alu elements (Intra Alu fold) or multiple Alu elements within the same transcript (Inter Alu fold) were predicted as described (see Methods). The fraction of edited sites (Edits) and all sites (All) predicted to be base paired (double stranded) in the predicted Alu secondary structure are plotted. Edit sites were analyzed in groups composed of those in all Alu sequences (All Alus), those in Alus sequences within transcripts without additional reverse complementary Alu sequences in cis (Mono Alus), or those in Alu sequences within transcripts containing an additional reverse complementary Alu sequence in cis (Multi Alus).
(D, E) Volcano plots of moderated log2 gene expression fold change for ADAR1 KO versus WT (D) or ADAR1p150 KO versus WT (E) in mock treated conditions (see Methods). Each dot represents a gene; colored dots indicate genes with edit sites, where the color hue indicates the number of ADAR1- or ADAR1p150- associated edits.
(F-G) Gene expression was analyzed as in Figures 2I and 2J. All genes containing Alu elements within any of their exonic parts are colored red. WT versus ADAR1 KO (F), and WT versus ADAR1p150 KO cells (G).
(H) Z-scaled expression values for 211 of 214 putative pol III Alus (‘independently transcribed Alus’), grouped by cell clone. Each point represents an individual Alu transcript and overlaid boxplot summarizes the distribution per sample. Significant differences of the IFN treated Z-scaled expression values relative to those values in the corresponding Mock treated sample are indicated (values compared by Mann Whitney U test, Bonferroni corrected for multiple testing; *p<0.05; **p<0.01;***p<0.001.). TMM, trimmed mean of M-values (Robinson et al., 2010)
(A) qRT-PCR analysis of relative mRNA levels, normalized to RPS11.
(B) ISG protein expression measured by flow cytometry. MFI, mean fluorescence intensity.
(A) Cell death/apoptosis assay. 293T cells were mock or IFNβ treated (48 hours) and harvested for flow cytometry analysis. Cells were stained with Annexin V and Propidium Iodide (PI) to detect the frequency (%) of different types of cell death. Early apoptotic cells, Annexin V+PI−. Late apoptotic cells/Dead cells, Annexin V+PI+. As a positive control, 293T cells were treated with 2uM staurosporine (STA) for 4 hours to induce apoptosis/cell death. DMSO, Dimethyl sulfoxide. Data are shown as mean ± SEM from n = 2~3 experimental replicates.
(B–D) Cell proliferation assay. WT or ADAR1 deficient cells were labeled with CFSE, equal numbers plated into a 24 well plate, and mock or IFNβ treated. CFSE intensity measured by flow cytometry over time (B). Representative flow cytometry plots of CFSE and β2M expression in mock and IFN treated cells (48 hours) (C). Frequency of β2MlowCFSEhigh summarized in bar graph (D). Data are shown as mean ± SEM from n = 3 experimental replicates. Student’s t-test, ***P < 0.001.
(A) WT, ADAR1 KO, and ADAR1p150 KO 293T cells were treated with IFNβ for 24 hours. Representative western blots labeled for total or phosphorylated forms of eIF2α or eIF4E.
(B) Western blot assessing the efficiency of shRNA-mediated PKR knock-down. WT or ADAR1-deficient 293T cells were transduced with lentiviruses expressing pGIPZ-PKR shRNA or pGIPZ-control shRNA (CTRL), followed by selection with puromycin to establish stable cell lines. Lysates were harvested for western blot analysis, and blots were probed with antibodies detecting PKR and β-actin control. Par, parental 293T cell line.
(A–C) ADAR1 KO 293T cells were mock or IFNβ treated for in total 24 hours. After the initial 12 hours of IFN treatment, cells were co-incubated with cycloheximide (CHX, 30ug/ml), actinomycin-D (ActD, 10ug/ml), or α-amanitin (25ug/ml) for the latter 12 hours (A). Cell pellets were harvested for western blot (B) or qRT-PCR (C). Western blots were probed with an anti- PKR and anti-β-actin antibody. Relative PKR band intensity (normalized to β-actin control) was measured by image J (B). Levels of IFIT1 mRNA, an RNA polymerase II derived ISG, was normalized to the housekeeping gene 18S rRNA, a relatively stable RNA polymerase I derived transcript (C). Data are shown as mean ± SEM from n = 3 experimental replicates. Student’s t-test, *P < 0.05, **P <0.01, ***P < 0.001, ns = not significant.
(D–E) 293T cells were infected with a blue fluorescent protein (BFP)- expressing Sindbis virus (SINV) at MOI 1 or MOI 4. Cells were harvested at 24 hours post-infection and analyzed by western blot to measure PKR phosphorylation (p-PKR) (D). Percent infection of cells was measured by flow cytometry (E).
(F) 293T cells were transfected with increasing doses of poly(I:C), and cell lysates were harvested at 5 hours post-transfection. PKR phosphorylation (p-PKR) was measured by western blot.
(A–B) WT and ADAR1 KO hESCs (two independent clones) were differentiated into NPCs as shown in the schematic (A). The frequency of NPC markers (Nestin, Pax6) was assessed by flow cytometry at 13 days post differentiation (n = 4 experimental replicates) (B).
(C–E) Par. (parental), WT, and ADAR1 KO hESCs (two independent clones) were differentiated into hepatocyte-like cells (HLCs). The hepatocyte markers Albumin and AFP (alpha-fetoprotein)were measured by qRT-PCR and the values were normalized to RPS11 (n = 2~4 experimental replicates) (C). WT NPCs were used as a negative control for Albumin and AFP levels. qRT-PCR of IFNβ mRNA levels that were normalized to RPS11 (n = 6 experimental replicates) (D). HLCs were treated with IFNβ for 24 hours and harvested for western blot to measure PKR phosphorylation (p-PKR) (E).
(F) Schematic of gene knock-down (KD) protocol in NPCs. ADAR1 KO NPCs were transduced with lentiviruses derived from GIPZ lentiviral shRNA vectors (Dharmacon) – targeting MDA5, RIG-I, and PKR - at 9 days post differentiation. Cells were harvested at 14 days post differentiation for western blot and qRT-PCR analysis.
(G) We examined gene expression of MDA-5, RIG-I, and PKR using an RNA-Seq data set from a recent study that examined transcript expression during the course of hESC to NPC differentiation (hESC → ectoderm (ECT) → NPC) (Wu et al. 2018). TPM, transcript per million. (B, C, D, G) Data are shown as mean ± SEM. Student’s t-test, ns = not significant.
Column A (genomicPosition) lists the strand-specific genomic position of the mismatch. Column B (mutationType) lists the type of mismatch, where X>Y indicates the reference (X) and read (Y) nucleotide of the mismatch. Column C (editType) indicates whether the mismatch is ADAR1- or ADAR1p150- associated. Columns D (StringtieGenes) and E (StringtieAssociatedEnsembleGeneIDs) indicate which Stringtie-determined gene was annotated at the genomic position, and which Ensembl genes were associated with the Stringtie determined gene, respectively. Column G (EnsemblAssociatedGeneIDs) indicates which Ensembl genes are present at the genomic position. The gene names from the Ensembl gene IDs in columns E and G are given in columns F (StringtieAssociatedEnsembleGenenames) and H (EnsemblAssociatedGenenames), respectively. Column I (RepeatMaskerElements) indicates any repeat sequence that was present at the genomic position. Columns J-AA list for each sample the number and fraction of total reads that are mismatched; e.g. if 13 As, 10 Gs and 2 Cs are found at a position in a specific sample, the A>G edit frequency is 10/25(0.4).
For each Stringtie-annotated gene (column A, StringtieGene), the number of ADAR1- and ADAR1p150- associated read:reference mismatch sites are listed in columns D (ADAR1- associated mismatch count) and E (ADAR1p150-associated mismatch count), respectively. Ensembl gene IDs that were associated by Stringtie to the Stringtie annotated gene are listed in column B (StringtieAssociatedEnsembleGeneIDs). Gene names from the Ensembl gene IDs in column B are listed in column C (StringtieAssociatedEnsembleGenenames).
All read:reference that map to the ORF of a transcript in the Ensembl annotations are listed here. Column A (Mismatch in highest expressed transcript isoform) indicates if the mismatch is in a transcript isoform that was most highly expressed in comparison to the other transcript isoforms of the same gene. The chromosome, position, and strand of the mismatch are listed in columns B (Chromosome), C (position), and D (strand), respectively. The Ensembl transcript ID and gene name are listed in columns F (transcript IDs) and E (gene), respectively. Column G (Alu sequence) indicates whether an Alu sequence was present at the mismatch position. Column H (mismatch association) indicates whether the mismatch was ADAR1- or ADAR1p150- associated.
For different comparisons as described in column A (comparison), differentially enriched (p-value<0.05 and FDR<0.1) gene sets are listed. Column B (gene set name) lists the name of the gene set. Column C (gene set size) lists the number of genes in the set. Column D (Expression up/down) indicates whether the set gene expression was up or down regulated in the comparison. The p-value and FDR of the test for up/down regulation, as determined using CAMERA, are listed in columns E (p-value) and F (FDR), respectively.