

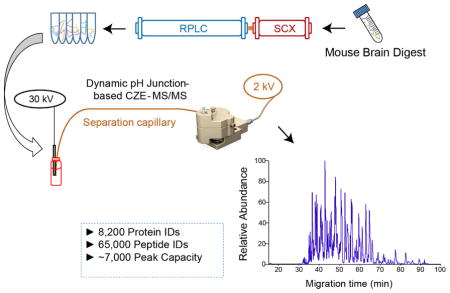
Abstract
Two-dimensional (2D) liquid chromatography (LC)-tandem mass spectrometry (MS/MS) are typically employed for deep bottom-up proteomics, and the state-of-the-art 2D-LC-MS/MS has approached over 8,000 protein identifications (IDs) from mammalian cell lines or tissues in 1–3 days of mass spectrometer time. Capillary zone electrophoresis (CZE)-MS/MS has been suggested as an alternative to LC-MS/MS for bottom-up proteomics. CZE-MS/MS and LC-MS/MS are complementary in protein/peptide ID from complex proteome digests because CZE and LC are orthogonal for peptide separation. In addition, the migration time of peptides from CZE-MS can be predicted accurately, which is invaluable for evaluating the confidence of peptide ID from the database search and even guiding the database search. However, the number of protein IDs from complex proteomes using CZE-MS/MS is still much lower than the state of the art using 2D-LC-MS/MS. In this work, for the first time, we established a strong cation exchange (SCX)-reversed phase LC (RPLC)-CZE-MS/MS platform for deep bottom-up proteomics. The platform identified around 8,200 protein groups and 65,000 unique peptides from a mouse brain proteome digest in 70 hours. The data represents the largest bottom-up proteomics dataset using CZE-MS/MS and provides a valuable resource for further improving the tool for prediction of peptide migration time in CZE. The peak capacity of the orthogonal SCX-RPLC-CZE platform was estimated to be around 7,000. SCX-RPLC-CZE-MS/MS produced comparable numbers of protein and peptide IDs with 2D-LC-MS/MS (8,200 vs. 8,900 protein groups, 65,000 vs. 70,000 unique peptides) from the mouse brain proteome digest using comparable instrument time. This is the first time that CZE-MS/MS showed its capability to approach comparable performance to the state-of-the-art 2D-LC-MS/MS for deep proteomic sequencing. SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS showed good complementarity in protein and peptide IDs and combining those two methods improved the number of protein group and unique peptide IDs by nearly 10% and over 40%, respectively, compared with 2D-LC-MS/MS alone.
Keywords: Strong cation exchange, Reversed-phase liquid chromatography, Capillary zone electrophoresis, Tandem mass spectrometry, Bottom-up proteomics
Graphical Abstract
1. Introduction
The state-of-the-art two-dimensional (2D) liquid chromatography (LC)-tandem mass spectrometry (MS/MS) has approached over 8000 protein identifications (IDs) from mammalian cell lines or tissues in 1–3 days of mass spectrometer time. [1–5] The draft human proteome containing 84% of the total annotated protein-coding genes in humans have also been generated using 2D-LC-MS/MS. [6] Over 2,000 LC-MS runs were performed for the draft human proteome, but the median protein sequence coverage was still only 28%. [6] The typical median protein sequence coverage of deep bottom-up proteomics datasets are around 25% or lower. The low sequence coverage impedes the confident identification of protein isoforms.
Alternative separation techniques that are orthogonal to LC for peptide separation will be very useful to further improve the number of peptide IDs from complex proteomes in bottom-up proteomics experiments, boosting the protein sequence coverage. Capillary zone electrophoresis (CZE)-MS/MS has been suggested as an alternative to LC-MS/MS for bottom-up proteomics. [7–18] CZE separates peptides based on their size-to-charge ratios and it is orthogonal to LC for peptide separation. CZE-MS/MS and LC-MS/MS are complementary in protein/peptide ID from complex proteome digests. [7–11] CZE tends to identify small, basic and hydrophilic peptides compared with RPLC-MS. In addition, the migration time of peptides from CZE-MS can be predicted more accurately and easily than their retention time from commonly used reversed-phase LC (RPLC)-MS.[19] The electrophoretic mobility of peptides in CZE mainly relate to their size (molecular mass) and charge, which are relatively easy to be determined accurately. The retention of peptides in RPLC can be affected by various factors, e.g., hydrophobic, hydrogen-bond, and ion-pairing interactions. Modeling those factors are very difficult. Recently, Krokhin et al. developed a simple model for CZE and approached very good correlation (R2 ~0.995) between the experimental and predicted migration time of peptides in CZE based on a large-scale peptide dataset. [19] The capability for accurate prediction of peptide migration time in CZE makes CZE-MS become a powerful tool for bottom-up proteomics because it can help us further evaluate the confidence of peptide ID from the database search and even guide the database search.
Although CZE-MS has many valuable features for bottom-up proteomics, the number of protein IDs from complex proteomes using CZE-MS/MS is still much lower than the state of the art using 2D-LC-MS/MS. Much effort has been made to improve the CZE-MS for large-scale proteomics.[7,15,20,21] Sun et al. approached 2,000 protein and 10,000 peptide IDs from a human cell line digest using single shot CZE-MS/MS with a neutrally coated separation capillary and an Orbitrap Fusion mass spectrometer.[7] Field-enhanced sample stacking was used to improve the sample loading volume to 100 nL and a 1-meter long neutrally coated separation capillary was employed to improve the peak capacity to about 300.[7] Yan et al. coupled RPLC prefractionation to CZE-MS/MS for bottom-up proteomics of Xenopus embryos, resulting in the identification of over 4,000 proteins. [20] For each CZE-MS/MS run, about 50 nL of the sample was injected for analysis. Faserl et al. coupled RPLC prefractionation to CZE-MS/MS for quantitative proteomics of yeast, leading to the identification of over 3,000 proteins. [15] A 1.5-mg yeast digest was used as the starting material and the sample loading volume for CZE-MS/MS was 40 nL. Very recently, Faserl et al. approached 6,000 protein IDs from a human cell line proteome digest by RPLC prefractionation and sequential sample injection based CZE-MS/MS with 2 mg of peptides as the starting material. [21] The sample loading volume of CZE-MS/MS was 25 nL.
In order to further improve the CZE-MS/MS for significantly deeper proteome coverage with a reasonable mass of initial protein material, we need to improve the sample loading volume of CZE-MS/MS and meantime boost the overall peak capacity of the system. The improvement in both sample loading volume and peak capacity can evidently benefit the identification of low abundant proteins. Recently, we showed that dynamic pH junction based CZE-MS/MS could approach both micro-liter scale sample loading volume and high peak capacity (up to 380) for analysis of complex peptide or protein mixtures. [22,23] In this work, we coupled online strong cation exchange (SCX)-RPLC prefractionation to the dynamic pH junction based CZE-MS/MS for deep bottom-up proteomics. The orthogonal SCX-RPLC-CZE platform approached very high peak capacity (~7,000). Because of the high peak capacity and the large sample loading volume of CZE (~0.5 μL per run), the SCX-RPLC-CZE-MS/MS system identified 8,200 protein groups and 65,000 unique peptides from a mouse brain proteome digest.
2. Experimental section
2.1 Reagents and chemicals
See supporting material I for details.
2.2 Preparation of the linear polyacrylamide-coated capillary for CZE
The inner wall of the separation capillaries for CZE was coated with linear polyacrylamide (LPA) based on references [22] and [24] in order to reduce the electroosmotic flow (EOF). The detailed protocol was described in supporting material I. After that, one end of the LPA-coated capillary was etched with hydrofluoric acid (HF) to reduce its outer diameter to ~70 μm based on the protocol in reference [14]. The LPA-coated capillary was stored at room temperature before use.
2.3 Sample preparation
See supporting material I for details.
2.4 Online SCX-RPLC fractionation of a mouse brain proteome digest
An Agilent Infinity II HPLC system with a quaternary pump was used for the experiment. A SCX trap column (Zorbax 300SCX, 4.6 mm i.d. × 12.5 mm length, 5 μm particles, Agilent Technologies) and a C18 RP column (Zorbax 300Extend-C18, 2.1 mm i.d. × 150 mm length, 3.5 μm particles, Agilent Technologies) were directly connected with a PEEK tubing and two fittings for online 2D-LC separation. 0.1% formic acid (FA) in water (mobile phase A), 0.1% FA in acetonitrile (ACN) (mobile phase B) and 890 mM ammonium acetate solution (pH=2.88) (mobile phase C) were used for separation. Mobile phase A and C were used for generation of different salt concentration for step-wise elution of peptides from the SCX column to the RPLC column. Mobile phase A and B were used for gradient separation of peptides with RPLC.
A 500-μg mouse brain proteome digest dissolved in mobile phase A were injected into a sample loop and loaded onto the SCX column by pushing the sample through the system with mobile phase A at 0.3 mL/min flow rate for 5 min. The peptides trapped on the SCX column were eluted in a step-wise fashion with different concentration of ammonium acetate for 20 min at a flow rate of 0.3 mL/min. After each salt step elution, the eluted peptides from the SCX were captured on the RPLC column, followed by RPLC gradient separation at 0.3 mL/min for 90 min: 0–20 min, 2%B; 20–22 min, 2–6%B; 22–67 min, 6–40%B; 67–72 min, 40–80% B; 72–77 min, 80%B; 77–80 min, 80-2%B; 80–90 min, 2%B. 40 fractions were collected for each RPLC run from 25 min to 71 min. From 25–31 min and from 65–71 min, we collected each fraction every 2 min; from 31 min to 65 min, we collected one fraction per min. We named the fractions based on the order of retention time from 1 to 40. Then we combined the fraction N and fraction N+20 to generate 20 fractions.
We performed two SCX-RPLC fractionation experiments. In the first experiment, we eluted the peptides from SCX with three different concentration of ammonium acetate: 150 mM, 350 mM, and 890 mM. In total, we got 60 SCX-RPLC fractions (3 salt steps× 20 fractions/salt step) from this experiment. In the second experiment, we used two salt steps for peptide elution from the SCX with salt concentration as 250 mM and 890 mM. In total, we collected 40 fractions for the second experiment. All of the collected fractions were lyophilized and stored at −80 °C for the following CZE-MS/MS experiments.
2.5 High-pH RPLC fractionation of the mouse brain proteome digest
The same Agilent Infinity II HPLC system was used for high pH RPLC fractionation. A C18 RP column (Zorbax 300Extend-C18, 2.1 mm i.d. × 150 mm length, 3.5 μm particles, Agilent Technologies) was used for separation. Mobile phase A (5 mM ammonium bicarbonate in water, pH 9) and mobile phase B (5 mM ammonium bicarbonate in 80% ACN, pH 9) were used to generate a gradient for peptide separation.
500-μg mouse brain proteome digest was injected onto the RP column for the experiment. The flow rate was 0.3 mL/min. The gradient was as follow: 0–5 min, 2% B; 5–7 min, 2–10% B; 7–67 min, 10–50% B; 67–69 min, 50–100 % B; 69–79 min, 100% B; 79–80 min, 100-2% B; 80–90 min, 2% B. In total, 60 fractions were collected from 7 min to 67 min, one fraction per min. We named the fractions based on the order of retention time from 1 to 60. Then we combined the fraction N and fraction N+30 to generate 30 fractions. Those fractions were then lyophilized and stored at −80 °C for low pH RPLC-MS/MS.
2.6 CZE-ESI-MS/MS
For CZE-ESI-MS/MS, a commercialized electro-kinetically pumped sheath flow CE-MS interface (CMP Scientific, Brooklyn NY) was employed for coupling CZE to MS. [25,26] An ECE-001 CE autosampler (CMP Scientific) was used for the automated operation of CZE. The ESI emitter was pulled from a borosilicate glass capillary (1.0 mm o.d., 0.75 mm i.d.) with a Sutter P-1000 flaming/brown micropipette puller. The orifice of the ESI emitter was 20–40 μm. The background electrolyte (BGE) was 5% (v/v) acetic acid (AA) with pH 2.4 and the sheath buffer was 0.2% (v/v) FA containing 10% (v/v) methanol. The etched end of the separation capillary was introduced into the ESI emitter, and the distance between the end of the capillary and the orifice of the ESI emitter was ~300 μm. The distance between the orifice of the emitter and the inlet of the mass spectrometer was ~2.0 mm. The voltage applied to the sample injection end of the capillary was 30 kV and ~2.2 kV was applied at the interface for ESI.
The 60 SCX-RPLC fractions from the three salt step experiment were dissolved in 4 μL of 10 mM ammonium bicarbonate (pH 8.0) for CZE-MS/MS. A 71-cm LPA-coated capillary (50-μm i.d., 360-μm o.d.) was used for CZE. Each fraction was injected into the capillary with 5-psi pressure for 63 s, corresponding to about 500-nL sample injection volume. After that, 30 kV was applied at the injection end for CZE separation for 50 min, followed by capillary flushing with BGE using 10 psi for 10 min. The 20 fractions from the second salt step (350 mM ammonium acetate) were further diluted from ~3.5 μL to 6 μL with 50 mM ammonium bicarbonate (pH 8). From those 20 fractions, we performed CZE-MS/MS analysis again using a 92-cm long LPA-coated capillary (50-μm i.d., 360-μm o.d.). For those analyses, 5 psi for 87 s was used for sample injection, corresponding to ~500-nL sample injection volume. The separation was performed with 30-kV voltage for 90 min, followed by BGE flushing with 10 psi for 15 min.
The 40 SCX-RPLC fractions from the two salt step experiment were dissolved in 4 μL of 50 mM ammonium bicarbonate (pH 8). A 94-cm long LPA-coated capillary (50-μm i.d., 360-μm o.d.) was used for CZE. The sample was injected into the capillary using 5 psi for 92 s, corresponding to about 500-nL sample injection volume. Next, 30 kV was applied at the injection end for separation for 92 min, followed by BGE flushing with 10 psi for 13 min.
A Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) was used for all of the experiments. A Top10 data-dependent acquisition (DDA) method was used. The mass resolution was set to 60,000 (at m/z 200) for both full MS scans and MS/MS scans. For full MS scans, the target value was 3E6, the maximum injection time was 50 ms and the scan range was 300 to 1500 m/z. For MS/MS scans, the target value was 1E5 and the maximum injection time was 110 ms. The ten most abundant ions in an MS spectrum with intensity higher than 1E5 were sequentially isolated in the quadrupole with isolation window as 2 m/z, followed by fragmentation in the higher energy collisional induced dissociation (HCD) cell with a normalized collision energy as 28%. Dynamic exclusion was applied and it was set to 30 s. Only ions with charge state as two or higher were considered for fragmentation.
2.7 RPLC-ESI-MS/MS
The 30 high-pH RPLC fractions were subjected to low pH RPLC-ESI-MS/MS analysis. An EASY-nLC™ 1200 system (Thermo Fisher Scientific) was used for RPLC separation. Each fraction was dissolved in 10 μL of 0.1% (v/v) FA and 2% (v/v) ACN. 3 μL of the sample was loaded onto a C18 pre-column (Acclaim PrepMapTM 100, 75-μm i.d. × 2 cm, nanoviper, 3 μm particles, 100 Å, Thermo Scientific). Then, the peptides were separated on a C18 separation column (Acclaim PrepMapTM 100, 75-μm i.d. × 50 cm, nanoviper, 2 μm particles, 100 Å, Thermo Scientific) at a flow rate of 200 nL/min. Mobile phase A (2% (v/v) ACN in water containing 0.1% (v/v) FA) and mobile phase B (80% (v/v) ACN and 0.1% (v/v) FA) was used to generate the gradient for separation. For separation, a 90-min gradient was used: 0–70 min, 8–40% B; 70–72 min, 40–100% B; 72–90 min, 100% B. The LC system required another 30 min for column equilibration between runs. Therefore, one LC-MS run required about 2 h.
The same Q-Exactive HF mass spectrometer (Thermo Fisher Scientific) was used for the RPLC-MS/MS experiments. The spray voltage was set to 1.8 kV. The other detailed parameters were the same as CZE-MS/MS described above.
2.8 Data analysis
Proteome Discoverer 2.1 software (Thermo Fisher Scientific) was used for analyses of RAW files. Sequest HT database search engine was used for database search. The mouse proteome database (UP000000589) downloaded from UniProt (http://www.uniprot.org/) was used as the database. Both the forward and reversed databases were used for database search in order to evaluate the false discovery rates (FDRs). [27,28] The enzyme was set as trypsin. The maximum number of missed cleavages was set as 2. The mass tolerance of precursor ions and fragment ions were set as 20 ppm and 0.05 Da, respectively. Oxidation (methionine) and deamination (Asparagine or Glutamine) were chosen as the dynamic modifications. The carbamidomethylation (cysteine) was set as the static modification. The peptide IDs were filtered with peptide confidence as high, corresponding to less than 1% FDR. Protein grouping was enabled, and the strict parsimony principle was applied.
The grand average of hydropathy (GARVY) value of peptides was calculated with GARVY Calculator (http://www.gravy-calculator.de/). Isoelectric points (pIs) of peptides were calculated using the “Compute pI/Mw” tool in ExPASy (http://web.expasy.org/compute_pi/). The gene ontology (GO) information of proteins was observed using the DAVID bioinformatics resources 6.8 (https://david.ncifcrf.gov/). [29,30]
3. Results and discussion
As shown in Fig. 1, proteins were extracted from mouse brains and were digested into peptides with trypsin based on the filter-aided sample preparation (FASP) method.[31] Three aliquots of mouse brain digests (500-μg peptides/aliquot) were used for prefractionation and MS/MS analysis. Two aliquots were fractionated by online SCX-RPLC. The peptides were trapped on an SCX trap column first, followed by step-wise elution from the SCX trap column to the RPLC column using three or two salt steps. The eluates were further separated by RPLC. In total, 60 fractions were collected from the three-salt-step SCX-RPLC experiment and 40 fractions were collected from the two-salt-step experiment. All of the fractions were analyzed by the CZE-MS/MS in 60 h (for the three-salt- step experiment) and 70 h (for the two-salt-step experiment). The dynamic pH junction method was used for on-line stacking of peptides to improve the sample loading capacity of CZE.[32,33] The sample loading volume for each CZE-MS/MS run was about 500 nL. The third aliquot of the mouse brain digest was fractionated by high-pH RPLC into 30 fractions, and those fractions were analyzed by low-pH RPLC-MS/MS in 60 h.
Fig. 1.

Experimental design of the work.
3.1. SCX-RPLC-CZE-MS/MS for deep bottom-up proteomics of the mouse brain
Fig. 2 presents the results from the mouse brain proteome digest using SCX-RPLC-CZE-MS/MS with three-salt-step elution (150 mM, 350 mM and 890 mM ammonium acetate, pH 2.88). The 60 SCX-RPLC fractions were analyzed by CZE-MS/MS with a 71-cm LPA-coated separation capillary in 60 h (1 h/fraction), leading to the identification of over 7,000 protein groups and 40,000 unique peptides, Fig. 2A. The LC fractions from the second salt step (350 mM) made a significantly higher contribution to the overall peptide IDs than those from other two salt steps. We made two conclusions based on this preliminary experiment. First, we should be able to boost the overall protein/peptide IDs via improving the analyses of the twenty fractions from the 350-mM salt step. Second, we need to change the concentration of the ammonium acetate for peptide elution from SCX in the following experiments to maximize the protein/peptide IDs.
Fig. 2.

Summary of the results from the mouse brain proteome digest using SCX-RPLC-CZE-MS/MS. Three salt steps were employed for step-wise elution of peptides from the SCX to the RPLC. (A) The accumulated numbers of protein group and unique peptide IDs vs. the number of fractions. A 71-cm separation capillary was used for CZE-MS/MS. (B) Comparison of the number of protein group and unique peptide IDs from the twenty LC fractions corresponding to the second salt step analyzed by the CZE-MS/MS with a 71-cm separation capillary (short) or a 92-cm separation capillary (long). (C) An electropherogram of one SCX-RPLC fraction analyzed by CZE-MS/MS with the 92-cm separation capillary. The migration time and the full width at half maximum (FWHM) of three peptides were shown in the figure.
We further analyzed the twenty LC fractions from the 350-mM salt step with CZE-MS/MS based on a much longer LPA-coated separation capillary (92 cm vs. 71 cm). The long separation capillary produced much more protein group and unique peptide IDs than the short capillary, Fig. 2B, boosting the protein group and unique peptide IDs from 6,000 to 7,100 and from about 27,000 to over 35,000, respectively. The improvement in protein and peptide IDs is most likely due to the much wider separation window from the long separation capillary (55 min vs. 30 min), leading to more tandem mass spectra. As shown in Fig. 2C, the CZE-MS system using the long separation capillary produced reasonably narrow peaks of peptides with the full width at half maximum (FWHM) ranging from 7.2 s to 36 s. The number of theoretical plates, on average, was around 240,000. The peak capacity of the CZE-MS run in Fig. 2C was estimated to be around 170 based on the FWHM of the three selected peptides. We decided to use the long separation capillary-based CZE-MS/MS for following experiments due to the much better protein/peptide IDs, although the long separation capillary required a longer time for each CZE-MS/MS run compared with the short capillary (1.75 h vs. 1 h).
Next, we tried to improve the overall protein/peptide IDs via changing the concentration of the ammonium acetate for peptide elution from SCX based on the preliminary data from the three-salt-step experiment. We fractionated another 500-μg mouse brain peptides with SCX-RPLC into 40 fractions based on two salt steps (250 mM and 890 mM ammonium acetate, pH 2.88). The SCX-RPLC fractions were analyzed by CZE-MS/MS with a 94-cm separation capillary in 70 h (1.75 h/fraction). We increased the concentration of ammonium bicarbonate (pH 8.0) in the sample buffer from 10 mM to 50 mM in order to improve the stacking performance of the dynamic pH junction method. [22,23,34] As shown in Fig. 3, the first and second salt steps made comparable contributions to the overall unique peptide IDs. In total, the platform identified nearly 8,200 protein groups and 65,000 unique peptides from the mouse brain proteome (Fig. 3), representing the largest proteomics dataset using CZE-MS/MS. CZE-MS/MS analysis of the fractions from the first salt step alone produced nearly 7,000 protein group IDs in 35 h. The data clearly suggest that CZE-MS/MS has the capability for deep sequencing of complex proteomes.
Fig. 3.
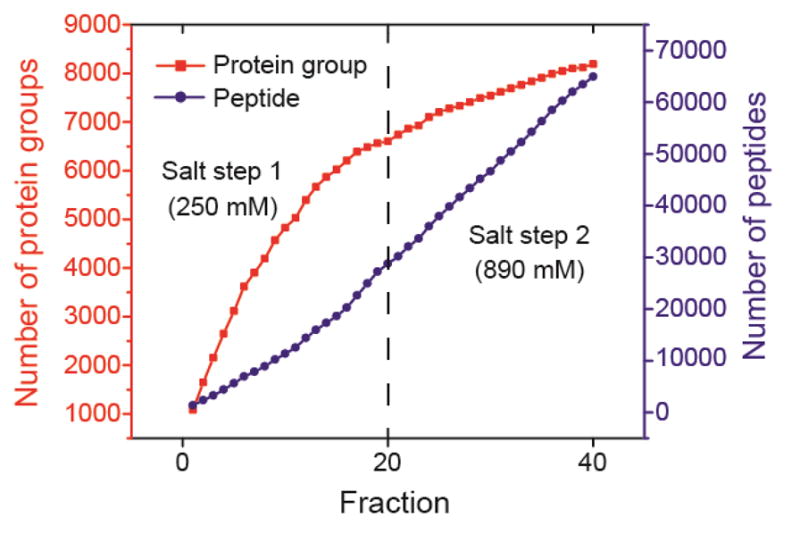
The accumulated numbers of protein group and unique peptide IDs from the mouse brain proteome digest vs. the number of SCX-RPLC fractions. Two salt steps were employed for step-wise elution of peptides from the SCX to the RPLC. CZE-MS/MS with a 94-cm separation capillary was used for analysis of the 40 SCX-RPLC fractions.
We attributed the large numbers of protein and peptide IDs from the experiment to two main reasons. First, the CZE-MS/MS system was capable of loading over 10% of the analytes in each LC fraction for analysis based on the dynamic pH junction stacking, 500 nL injection volume vs. 4 μL total sample volume. The large sample loading volume guaranteed the identification of low abundant proteins in the sample. Second, the SCX, RPLC, and CZE are orthogonal for separation of peptides based on their charge, hydrophobicity, and size-to-charge ratio. The orthogonal three-dimensional separation platform produced high peak capacity. We chose five CZE-MS runs and calculated their peak capacity based on five randomly chosen peptides with medium abundance. The peak capacity per CZE-MS run ranged from 175 to 250 based on the FWHM of those five peptides. Therefore, we estimated the overall peak capacity of the SCX-RPLC-CZE platform as at least 7,000 (175 × 40 fractions), representing the highest peak capacity of the CZE based platform until now for separation of a complex proteome digest.
The SCX-RPLC-CZE-MS/MS system combined the advantages of SCX, RPLC, and CZE-MS/MS. SCX has high sample loading capacity; RPLC can desalt the peptides and provide high-resolution separation of peptides; CZE can easily approach high separation efficiency for peptides and CZE-MS/MS can provide highly sensitive identification of peptides. [8,10,13,14] The whole platform is straightforward and no sample cleanup is required between SCX-RPLC fractionation and CZE-MS/MS. In addition, the electrophoretic mobility of peptides in CZE has been predicted accurately using a simple model based on the size (molecular mass) and charge of peptides, [19] which is invaluable for evaluating the confidence of peptide ID from the database search and even guiding the database search. The large-scale proteomic dataset from CZE-MS/MS presented in this work will be very useful for further evaluating and improving the model for prediction of electrophoretic mobility of peptides. [19]
3.2 Comparison of SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS for deep sequencing of the mouse brain proteome
Much effort has been made for comparing CZE-MS/MS and RPLC-MS/MS for bottom-up proteomics, and the results clearly showed the good complementarity of those two methods for protein/peptide ID from complex proteomes. [7–11,13,18–20,35] In general, CZE-MS/MS tended to identify small, basic and hydrophilic peptides compared with RPLC-MS/MS, most likely due to the relatively weak retention of those peptides on RPLC column. However, the highest number of protein and peptide IDs using CZE-MS/MS in those previous works was only about 4,000 and 20,000, respectively. It is still not clear whether the good complementarity between CZE- and RPLC-MS/MS in protein/peptide ID still exists or not for dramatically larger proteomic datasets.
Here we further employed 2D-LC-MS/MS (high pH RPLC-low pH RPLC) for deep sequencing of the mouse brain proteome, resulting in the identification of 8,900 protein groups and 70,000 unique peptides in 60 h of mass spectrometer time. The data represents the capability of the state-of-the-art 2D-LC-MS/MS for deep sequencing of complex proteomes. Our SCX-RPLC-CZE-MS/MS identified 8,200 protein groups and 65,000 unique peptides in 70 h using the same amount of peptides as the starting material. This is the first time that CZE-MS/MS showed its capability to approach comparable performance to the state-of-the-art 2D-LC-MS/MS for deep proteomic sequencing. The lists of identified proteins and peptides using CZE-MS/MS and LC-MS/MS are shown in supporting material II. The mass spectrometry proteomics raw files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD008432. [36,37]
We then compared the SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS on the scale of 8,000 protein groups and 65,000 peptides. The two techniques had good complementarity at both protein and peptide levels. Combination of both techniques improved the number of protein group ID to over 9700, which was nearly 10% higher than that from 2D-LC-MS/MS alone. The two techniques had even more significant complementarity at the peptide level, Fig. 4A. Combining the data from those two methods resulted in the identification of over 100,000 unique peptides, which was over 40% higher than that from 2D-LC-MS/MS alone. The median sequence coverage of the overlapped proteins between CZE-MS/MS and LC-MS/MS (~7,000 proteins) was ~22% based on the LC-MS/MS data alone, and it was boosted to ~30% by combining CZE-MS/MS and LC-MS/MS data. The data clearly indicate that combination of the SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS can significantly improve the sequence coverages of identified proteins.
Fig. 4.

Comparison of SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS in terms of the identified peptides from the mouse brain proteome digest. (A) Overlap of identified peptides. (B) Cumulative distribution of molecular weight (MW) of identified peptides. (C) Bar graph of the MW distribution of the identified peptides. (D) Correlation between migration time and MW, migration time and FWHM of identified peptides from one random CZE-MS run. The FWHM of peptides at the three different migration time were calculated based on five randomly chosen peptides. The mean and the standard deviations of the FWHM of those five peptides were shown in the figure. (E) Cumulative distribution of the isoelectric point (pI) of identified peptides. (F) Cumulative distribution of the grand average of hydropathy (GRAVY) value of the identified peptides. Negative GRAVY values indicate hydrophilic; Positive GRAVY values signify hydrophobic.
Next, we investigated the physicochemical properties of the identified peptides from the SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS. The SCX-RPLC-CZE-MS/MS tended to identify small peptides compared with 2D-LC-MS/MS, Fig. 4B and 4C. One reason is that those small peptides tend to have weak retention on RPLC column, and are most likely washed out during the sample loading step.[8] Another possible reason relates to CZE. As shown in Fig. 4D, larger peptides tend to have slower migration in the CZE separation capillary; The peptides with longer migration time tend to have wider peaks due to more significant diffusion in the capillary. Therefore, the relatively large peptides tend to have obviously wider peaks than the small peptides in CZE, leading to a more significant overlap of peptide peaks and more serious ionization suppression. As shown in Fig. 4E and 4F, the SCX-RPLC-CZE-MS/MS also tended to identify basic peptides and hydrophilic peptides. Basic peptides have more positive charges in an acidic buffer than acidic peptides, and they are more hydrophilic. Hydrophilic peptides can not be captured and separated well on the RPLC column. The different prefractionation methods used in those two platforms (SCX-RPLC vs. high pH RPLC) might also contribute to the differences in peptide IDs. In summary, the SCX-RPLC-CZE-MS/MS tended to identify small, basic and hydrophilic peptides, which agreed well with the data in the literature. [8,10,11,19,35]
We also compared the identified proteins using SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS in terms of their gene ontology (GO) information. When we performed the comparison based on all of the identified proteins, we observed that those two platforms agreed well in GO information of identified proteins, Fig. S1 in supporting material I. We further performed a biological process enrichment analysis of the genes that were uniquely identified by the SCX-RPLC-CZE-MS/MS (847 genes) or 2D-LC-MS/MS (1,476 genes). Those genes are listed in supporting material II. Surprisingly, we observed that those uniquely identified genes from those two platforms had dramatically different biological process enrichment profiles. The genes uniquely identified by the SCX-RPLC-CZE-MS/MS were enriched in potassium ion transmembrane transport, regulation of angiogenesis, bone development, covalent chromatin modification, and positive regulation of I-kappaB kinase/NF-kappaB signaling. The genes uniquely identified by 2D-LC-MS/MS were enriched in the regulation of gene expression, ribosome biogenesis, DNA methylation, nucleosome assembly, transcription, and rRNA processing. The results clearly suggest that combination of SCX-RPLC-CZE-MS/MS and 2D-LC-MS/MS not only can boost the sequence coverage of proteins but also can improve our ability for more comprehensive characterization of biological processes in cells.
4. Conclusions
In this work, for the first time, we established an SCX-RPLC-CZE-MS/MS platform for deep bottom-up proteomics, leading to the identification of 8,200 protein groups and 65,000 unique peptides from a mouse brain proteome digest. The data represents the largest bottom-up proteomics dataset using CZE-MS/MS. The orthogonal SCX-RPLC-CZE platform produced a high peak capacity as ~7,000, representing the highest peak capacity of the CZE based platform until now for separation of a complex proteome digest. The SCX-RPLC-CZE-MS/MS and the state-of-the-art 2D-LC-MS/MS showed good complementarity in protein and peptide IDs based on the comparisons performed on the scale of 8,000 proteins and 65,000 unique peptides.
We expect that the number of protein/peptide IDs from complex proteomes using the SCX-RPLC-CZE-MS/MS platform can be further significantly improved via simply increasing the number of SCX-RPLC fractions. In order to speed up the analysis of those large number of SCX-RPLC fractions, the sequential sample injection based CZE-MS/MS can be employed. [21,38,39].
Supplementary Material
Highlights.
CZE-MS/MS can identify over 8,000 protein groups from a complex proteome.
CZE-MS/MS and LC-MS/MS are comparable for deep proteomic sequencing.
Combining CZE-MS/MS and LC-MS/MS can significantly boost the proteome coverage.
The peak capacity of SCX-RPLC-CZE platform can reach ~7,000.
Acknowledgments
We thank the Prof. Chen Chen’s group at Department of Animal Science, Michigan State University for kindly providing the mouse brain samples for our research. We thank the support from the Michigan State University and the National Institute of General Medical Sciences, National Institutes of Health (NIH), through Grant R01GM125991.
Abbreviations
- ID
identification
- LPA
linear polyacrylamide
- FASP
filter-aided sample preparation
Footnotes
Conflicts of interest
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics. 2012;11:M111.014050. doi: 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding C, Jiang J, Wei J, Liu W, Zhang W, Liu M, Fu T, Lu T, Song L, Ying W, Chang C, Zhang Y, Ma J, Wei L, Malovannaya A, Jia L, Zhen B, Wang Y, He F, Qian X, Qin J. A fast workflow for identification and quantification of proteomes. Mol Cell Proteomics. 2013;12:2370–2380. doi: 10.1074/mcp.O112.025023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelstrup CD, Jersie-Christensen RR, Batth TS, Arrey TN, Kuehn A, Kellmann M, Olsen JV. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J Proteome Res. 2014;13:6187–6195. doi: 10.1021/pr500985w. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Q, Fang F, Shan Y, Sui Z, Zhao B, Liang Z, Zhang L, Zhang Y. In-Depth Proteome Coverage by Improving Efficiency for Membrane Proteome Analysis. Anal Chem. 2017;89:5179–5185. doi: 10.1021/acs.analchem.6b04232. [DOI] [PubMed] [Google Scholar]
- 6.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush MJ, Zhu G, Champion MM, Coon JJ, Dovichi NJ. Over 10,000 peptide identifications from the HeLa proteome by using single-shot capillary zone electrophoresis combined with tandem mass spectrometry. Angew Chem Int Ed. 2014;53:13931–13933. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faserl K, Sarg B, Kremser L, Lindner H. Optimization and evaluation of a sheathless capillary electrophoresis-electrospray ionization mass spectrometry platform for peptide analysis: comparison to liquid chromatography-electrospray ionization mass spectrometry. Anal Chem. 2011;83:7297–7305. doi: 10.1021/ac2010372. [DOI] [PubMed] [Google Scholar]
- 9.Ludwig KR, Sun L, Zhu G, Dovichi NJ, Hummon AB. Over 2300 phosphorylated peptide identifications with single-shot capillary zone electrophoresis-tandem mass spectrometry in a 100 min separation. Anal Chem. 2015;87:9532–9537. doi: 10.1021/acs.analchem.5b02457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Fonslow BR, Wong CC, Nakorchevsky A, Yates JR., III Improving the comprehensiveness and sensitivity of sheathless capillary electrophoresis-tandem mass spectrometry for proteomic analysis. Anal Chem. 2012;84:8505–8513. doi: 10.1021/ac301091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu G, Sun L, Yan X, Dovichi NJ. Single-shot proteomics using capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry with production of more than 1250 Escherichia coli peptide identifications in a 50 min separation. Anal Chem. 2013;85:2569–2573. doi: 10.1021/ac303750g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun L, Zhu G, Yan X, Zhang Z, Wojcik R, Champion MM, Dovichi NJ. Capillary zone electrophoresis for bottom-up analysis of complex proteomes. Proteomics. 2016;16:188–196. doi: 10.1002/pmic.201500339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lombard-Banek C, Moody SA, Nemes P. Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angew Chem Int Ed. 2016;55:2454–2458. doi: 10.1002/anie.201510411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew Chem Int Ed. 2013;52:13661–13664. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faserl K, Kremser L, Müller M, Teis D, Lindner HH. Quantitative proteomics using ultralow flow capillary electrophoresis-mass spectrometry. Anal Chem. 2015;87:4633–4640. doi: 10.1021/acs.analchem.5b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Peuchen EH, Dovichi NJ. Surface-confined aqueous reversible addition-fragmentation chain transfer (SCARAFT) polymerization method for preparation of coated capillary leads to over 10 000 peptides Identified from 25 ng HeLa digest by using capillary zone electrophoresis-tandem mass spectrometry. Anal Chem. 2017;89:6774–6780. doi: 10.1021/acs.analchem.7b01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo X, Fillmore TL, Gao Y, Tang K. Capillary electrophoresis-nanoelectrospray ionization-selected reaction monitoring mass spectrometry via a true sheathless metal-coated emitter interface for robust and high-sensitivity sample quantification. Anal Chem. 2016;88:4418–4425. doi: 10.1021/acs.analchem.5b04912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Dovichi NJ. Optimization of mass spectrometric parameters improve the identification performance of capillary zone electrophoresis for single-shot bottom-up proteomics analysis. Anal Chim Acta. 2017 doi: 10.1016/j.aca.2017.11.023. doi.org/10.1016/j.aca.2017.11.023. [DOI] [PMC free article] [PubMed]
- 19.Krokhin OV, Anderson G, Spicer V, Sun L, Dovichi NJ. Predicting electrophoretic mobility of tryptic peptides for high-throughput CZE-MS analysis. Anal Chem. 2017;89:2000–2008. doi: 10.1021/acs.analchem.6b04544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan X, Sun L, Zhu G, Cox OF, Dovichi NJ. Over 4100 protein identifications from a Xenopus laevis fertilized egg digest using reversed-phase chromatographic prefractionation followed by capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry analysis. Proteomics. 2016;16:2945–2952. doi: 10.1002/pmic.201600262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faserl K, Sarg B, Sola L, Lindner HH. Enhancing proteomic throughput in capillary electrophoresis-mass spectrometry by sequential sample injection. Proteomics. 2017;17 doi: 10.1002/pmic.201700310. [DOI] [PubMed] [Google Scholar]
- 22.Chen D, Shen X, Sun L. Capillary zone electrophoresis-mass spectrometry with microliter-scale loading capacity, 140 min separation window and high peak capacity for bottom-up proteomics. Analyst. 2017;142:2118–2127. doi: 10.1039/c7an00509a. [DOI] [PubMed] [Google Scholar]
- 23.Lubeckyj RA, McCool EN, Shen X, Kou Q, Liu X, Sun L. Single-shot top-down proteomics with capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry for identification of nearly 600 Escherichia coli proteoforms. Anal Chem. 2017;89:12059–12067. doi: 10.1021/acs.analchem.7b02532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu G, Sun L, Dovichi NJ. Thermally-initiated free radical polymerization for reproducible production of stable linear polyacrylamide coated capillaries, and their application to proteomic analysis using capillary zone electrophoresis-mass spectrometry. Talanta. 2016;146:839–843. doi: 10.1016/j.talanta.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ. Third-generation electrokinetically pumped sheath-flow nanospray interface with improved stability and sensitivity for automated capillary zone electrophoresis-mass spectrometry analysis of complex proteome digests. J Proteome Res. 2015;14:2312–2321. doi: 10.1021/acs.jproteome.5b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Simplified capillary electrophoresis nanospray sheath-flow interface for high efficiency and sensitive peptide analysis. Rapid Commun Mass Spectrom. 2010;24:2554–2560. doi: 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- 27.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 28.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 29.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 32.Aebersold R, Morrison HD. Analysis of dilute peptide samples by capillary zone electrophoresis. J Chromatogr. 1990;516:79–88. doi: 10.1016/s0021-9673(01)90206-7. [DOI] [PubMed] [Google Scholar]
- 33.Britz-McKibbin P, Chen DD. Selective focusing of catecholamines and weakly acidic compounds by capillary electrophoresis using a dynamic pH junction. Anal Chem. 2000;72:1242–1252. doi: 10.1021/ac990898e. [DOI] [PubMed] [Google Scholar]
- 34.Imami K, Monton MR, Ishihama Y, Terabe S. Simple on-line sample preconcentration technique for peptides based on dynamic pH junction in capillary electrophoresis-mass spectrometry. J Chromatogr A. 2007;1148:250–255. doi: 10.1016/j.chroma.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry as an alternative proteomics platform to ultraperformance liquid chromatography-electrospray ionization-tandem mass spectrometry for samples of intermediate complexity. Anal Chem. 2012;84:1617–1622. doi: 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, Moritz RL, Carver JJ, Wang M, Ishihama Y, Bandeira N, Hermjakob H, Vizcaíno JA. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017D;45:D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boley DA, Zhang Z, Dovichi NJ. Multisegment injections improve peptide identification rates in capillary zone electrophoresis-based bottom-up proteomics. J Chromatogr A. 2017;1523:123–126. doi: 10.1016/j.chroma.2017.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garza S, Moini M. Analysis of complex protein mixtures with improved sequence coverage using (CE-MS/MS)n. Anal Chem. 2006;78:7309–7316. doi: 10.1021/ac0612269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.