

Summary
Proteolysis Targeting Chimera (PROTAC) technology has emerged over the last two decades as a powerful tool for targeted degradation of endogenous proteins. Herein we describe the development of PROTACs for receptor tyrosine kinases, a protein family yet to be targeted for induced protein degradation. The use of VHL-recruiting PROTACs against this protein family reveals several advantages of degradation over inhibition alone: direct comparisons of fully-functional, target degrading PROTACs with target-inhibiting variants that contain an inactivated E3 ligase-recruiting ligand show that degradation leads to more potent inhibition of cell proliferation, a more durable and sustained downstream signalling response, and thus addresses the kinome re-wiring challenge seen with many RTK inhibitors. Combined, these findings demonstrate the ability to target receptor tyrosine kinases for degradation using the PROTAC technology and outline the advantages of this degradation-based approach.
Keywords: PROTAC, Targeted Degradation, Receptor Tyrosine Kinases, EGFR, HER2, c-Met, VHL
eTOC Blurb
Burslem, Smith et al. describe the development of PROTACs capable of degrading transmembrane receptor tyrosine kinases and further highlight the advantages of degradation over inhibition in terms of potency, duration of effect, and combating compensatory signalling.
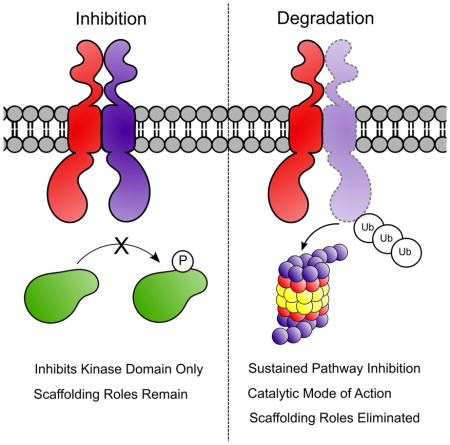
Introduction
The discovery of small molecule receptor tyrosine kinase (RTK) inhibitors greatly enabled the study of these key proteins in normal and oncogenic signalling (Takeuchi and Ito, 2011). Eukaryotic cell proliferation is driven by RTK activation following binding of cognate growth factors (Lemmon and Schlessinger, 2010), and many forms of cancer are driven by the hyperactivation of specific RTKs due either to overexpression of the protein to super-physiological levels, or to mutations that confer growth factor-independent activation (Gschwind et al., 2004). In order to obtain RTK inhibition over an extended time, exposure to small molecule kinase inhibitors at sustained and saturating concentrations is required (Pratz et al., 2009). We postulated that small molecule-mediated degradation of the protein itself rather than inhibition of the kinase domain could provide advantages, such as reduced drug exposure time required to suppress signalling and the ability to target kinase-independent functions. Studies have shown that cancerous cells can co-opt other existing RTK signalling pathways in order to permit the inhibited RTK to continue to exist as a node, thereby restoring downstream oncogenic signalling (Engelman et al., 2007; Graves et al., 2013; Pillay et al., 2009; Thelemann et al., 2005; Xu and Huang, 2010). Degradation of the RTK, as opposed to inhibition of the kinase activity, is a strategy with the potential to yield a more complete and lasting inactivation of downstream signalling and circumvent the problem of “kinome re-wiring”, whereby inhibition of receptor signalling leads to compensatory feedback activation via alternate kinases (Graves et al., 2013; Kurimchak et al., 2016). Indeed, RTK elimination would prevent the inactive kinase from persisting as a scaffolding node for oncogenic signalling. We postulate that this more sustained loss of function can be accomplished using Proteolysis Targeting Chimeras (PROTACs)), a technology for post-translational protein degradation that our lab has developed and refined for nearly two decades (Bondeson et al., 2015; Lai et al., 2016; Lu et al., 2015; Sakamoto et al., 2001; Schneekloth et al., 2008). By chemically tethering ligands for two different proteins – an E3 ubiquitin ligase and a protein of interest – a new pharmacological entity is created that facilitates the ubiquitination and proteasomal degradation of the protein of interest. Its net effect on target protein levels is similar to that achievable using RNAi technology; however, the small-molecule approach of PROTACs does not have the inherent liabilities of proposed nucleic-based modalities. Indeed, PROTACs are comparable to RTK inhibitors, in that both are amenable to adjustable dosing and can offer temporal control to achieve the desired level of signal inactivation, nor does it require any genetic manipulations/modification of cells in order to work.
To date, our group and others have successfully developed PROTACs that can degrade enzymes, steroid receptors, transcription factors, membrane-tethered scaffolding proteins and several kinases (Bondeson et al., 2015; Buckley et al., 2015; Gadd et al., 2017; Hines et al., 2013; Lai et al., 2016; Lebraud et al., 2016; Lu et al., 2015; Raina et al., 2016; Sakamoto et al., 2001; Schiedel et al., 2017; Schneekloth et al., 2008; Zengerle et al., 2015; Zhou et al., 2017). An outstanding question, however, has been whether the PROTAC methodology is able to induce degradation of transmembrane-spanning proteins, given their restricted cellular localization and the questionable accessibility of membrane-bound receptors for ubiquitination by the cytosolic machinery. The hydrophobic tagging approach, also developed in our laboratory (Neklesa and Crews, 2012; Neklesa et al., 2011), has been previously used to degrade an inactive RTK (Xie et al., 2014), however PROTACs could provide key advantages such as improved physicochemical properties, reduced toxicity, facile modular design, and a defined mechanism of degradation. As some of the most compelling anti-cancer targets are RTKs, the demonstration of their susceptibility to PROTAC-mediated degradation has remained an important question. Given their well-defined role in human cancers and the broad understanding of their regulation and downstream signalling pathways, we focused our efforts on EGFR, HER2 and c-Met as potential PROTAC targets of interest. Herein, we show effective PROTAC-mediated degradation of these RTKs, including a number of relevant oncogenic mutant isoforms. Our results strongly suggest that not only are RTKs viable substrates for post-translational degradation, but also that the signalling inactivation and growth inhibition achieved by PROTACs is more potent, more sustained, and less susceptible to kinome re-wiring than that achieved via RTK inhibition.
Results and Discussion
Epidermal Growth Factor Receptor (EGFR), also known as ErbB1/HER1, is a proto-oncogene that has been implicated in a range of cancers including glioblastoma multiforme, head and neck, and non-small cell lung cancer (Yewale et al., 2013). Overexpression and/or activating mutations of EGFR are associated with a poor prognosis, therefore significant effort has focused on targeting EGFR with both small molecule and antibody-based therapies (Dassonville et al., 2007). Small molecule kinase inhibitors competitively bind to the kinase domain, thereby preventing signalling, while antibodies are capable of preferentially binding the cognate ligand recognition site, thus preventing kinase activation. Furthermore, degradation of EGFR by FDA-approved antibodies has been implicated in their clinical success, suggesting that degradation may be advantageous (Perez-Torres et al., 2006). With this rationale in mind, we sought to create a small molecule capable of inducing EGFR degradation by developing an EGFR-targeting PROTAC.
By conjugating an EGFR binding element -- initially the kinase inhibitor, lapatinib (Tykerb) (Konecny et al., 2006) -- to a ligand developed in our laboratory (Buckley et al., 2012a; Buckley et al., 2012b) that binds to the E3 ligase, VHL, we were able to synthesize a molecule (Compound 1) capable of penetrating the cell membrane and inducing EGFR degradation at low nanomolar concentrations (Figure 1A and Figure S1). Crucially, inversion of the hydroxyproline stereochemistry on the VHL-recruiting moiety of the PROTAC abrogates its ability to degrade EGFR (Figure 1B) by abolishing its ability to bind VHL. This diastereomeric version (Compound 2) of the PROTAC provides an excellent control compound with nearly identical physicochemical properties (see Figure S1) but capable of only inhibiting EGFR (Figure S2C); an ideal tool with which to directly assess the advantages of EGFR degradation over kinase inhibition without variations in probe solubility, membrane permeability, or chemical stability obscuring a head-to-head comparison (as would be the case if comparisons were drawn directly with lapatinib itself).
Figure 1. Small molecule induced degradation of EGFR and mutants.

A–F – Immunoblots of cells expressing different EGFR variants treated with increasing doses of the indicated compound for 24 hours. A – OVCAR8 cells treated with lapatinib-based PROTAC 1. B – OVCAR8 cells treated with compound 2, an inactive diastereomer of lapatinib-based PROTAC 1. C – HeLa cells expressing FLAG-tagged exon 20 ins (ASV duplication) EGFR treated with lapatinib-based PROTAC 1. D – HCC827 cells expressing exon 19 del EGFR treated with gefitinib-based PROTAC 2. E – H3255 cells expressing L858R EGFR treated with gefitinib-based PROTAC 3. F – H1975 cells expressing double mutant (L858R/T790M) EGFR treated with afatinib-based PROTAC 4. G – Summary table of DC50 (the concentration at which half-maximal degradation is achieved) and Dmax (the maximum percentage of degradation) values. See also Figure S1 and S2.
Having demonstrated that recruitment of VHL to EGFR via a lapatinib-based PROTAC is capable of efficiently inducing degradation of a receptor tyrosine kinase, we sought to expand on this finding by employing different EGFR-binding elements to degrade different clinically relevant forms of EGFR. As shown in Figure 1C, the lapatinib-based PROTAC, Compound 1, is also capable of degrading an exon-20 insertion mutant form of EGFR (ASV duplication) (Yasuda et al., 2013). Switching to a warhead based on mutant-EGFR selective gefitinib (Compound 3) (Iressa) (Anido et al., 2003) enabled the degradation of both exon-19 deletion EGFR as well as the mutant isoform containing the L858R activating point mutation (Figure 1D/E), while sparing the WT EGFR (see Figure S2). Finally employing the second-generation inhibitor afatinib (Gilotrif) (Solca et al., 2012) yielded a PROTAC (Compound 4) capable of degrading the gefitinib-resistant double mutant (L858R/T790M) EGFR (Figure 1F). We have previously shown that choice of warhead is crucial for successful target degradation (Lai et al., 2016); here we demonstrate that careful selection of the recruiting element can also allow degradation of proteins in different mutational states. For some of the aforementioned PROTACs, we observed a “hook effect” on substrate degradation, which has been previously reported and results from the formation of unproductive dimers (rather than productive ‘trimers’) at higher concentrations (Douglass et al., 2013). We postulate that lack of this ‘hook effect’ in other PROTACs arises from additive target:E3 ligase protein-protein interactions that they induce.
Since lapatinib is also a potent binder to other HER family RTKs, we sought to explore the potential for HER2 degradation by lapatinib-based PROTACs. Similar to EGFR, HER2 overexpression is an oncogenic driver of many forms of cancer including ovarian, breast, and gastric cancers (Iqbal and Iqbal, 2014). Immunoblotting analysis revealed that Compound 1, which utilizes a diethylene glycol linker to tether lapatinib to the VHL recruiting element, could concurrently degrade both EGFR and HER2 (Figure 2A). However, extension of the linker by an additional ethylene glycol unit to create Compound 5 enabled the selective degradation of EGFR while sparing HER2. This observation suggests that affinity for both target protein and E3 ligase is not sufficient for the development of a successful PROTAC (see accompanying paper Bondeson, Smith et al.), and that a more complex dynamic process may be responsible.
Figure 2. Selective PROTAC-mediated degradation of HER2 and implications for kinome re-wiring.

A – Employing different linker lengths imparts PROTAC selectivity for EGFR over HER2. OVCAR8 cells were treated with PROTAC 1 or 5 for 24 hours before being lysed and probed for EGFR, HER2 and tubulin (as a loading control). B – Cell proliferation assay in SKBr3 cells after 72 hours of treatment with the indicated compound concentrations. C - Treatment of SKBr3 cells with sub-lethal concentrations (500 nM) of PROTAC 1 or diastereomer 2 over 48 hours shows a gradual increase in downstream signalling consistent with kinome re-wiring, previously observed in SKBr3 cells, with diastereomer but not with PROTAC. D – Immunoblotting analysis of c-Met phosphorylation after 48 hours with 500 nM PROTAC 1 or diastereomer 2. See also Figure S2.
The advantages of PROTAC-mediated degradation over kinase inhibition become apparent when the effect on cell proliferation is compared (Figure 2B). SKBr3 cells, a HER2-driven breast cancer cell line, are more responsive to PROTAC 1 than the cognate diastereomeric control 2 that has equivalent cell permeability and kinase inhibitory properties but is incapable of inducing HER2 degradation (Figure S2 B/C). PROTAC 1 treatment induces a greater response in terms of anti-proliferative efficacy than does diastereomer 2, and at a greater potency (PROTAC 1 IC50 = 102 nM, diastereomer 2 IC50 = 171 nM).
Comparison of PROTAC 1 with diastereomer 2 also reveals additional advantages of degradation over inhibition following an extended treatment period. Inhibition of EGFR/HER2 in SKBr3 cells has previously been shown to rapidly induce kinome “re-wiring”, whereby alternative, uninhibited kinases are recruited as heterodimerization partners leading to the phosphorylation of the same downstream effectors to restore oncogenic signalling via RTK crosstalk (Stuhlmiller et al.). To investigate this phenomenon, we treated SKBr3 cells with equal, saturating concentrations (Figure S2 B/C) of either PROTAC 1 or diastereomer 2, which provided a direct and head-to-head comparison of degradation versus inhibition given their similar physicochemical properties. Interestingly, degradation appears to have a protective effect against kinome re-wiring, particularly in the case of ERK1/2 phosphorylation and specific HER3 and AKT phosphorylation sites. Cells treated with the inhibitor diastereomer 2 led to transient suppression of activation of ERK1/2, phosphorylation of Akt (Thr308), c-Met and HER3, the latter being a major node for preventing apoptosis and promoting survival (Figure 2C) (Engelman et al., 2007). However, within 24 to 48 hours, this suppression was reversed despite continued presence of the inhibitor. Quite strikingly, treatment with an equivalent concentration of PROTAC 1 itself yielded sustained suppression of downstream signalling, suggesting that removal of the target RTKs discourages kinome re-wiring and permits longer sustained growth suppression. Inhibition transiently prevents downstream signalling but degradation may also impact the scaffolding roles exhibited by RTKs, particularly in instances of kinome re-wiring by receptor cross-talk (Jo et al., 2000). For example, heterodimerization of EGFR with c-Met and signalling via the c-Met kinase domain has been implicated in resistance to some inhibitors/antibodies (Engelman et al., 2007). Analysis of the c-Met phosphorylation status after 48 hours of PROTAC 1 or diastereomer 2 treatment revealed a substantial increase in signalling via c-Met kinase domain in diastereomer-treated cells compared to the PROTAC treated cells, presumably by trans-activation (Figure 2D). These results demonstrate the advantages gained from RTK degradation compared to kinase inhibition with regards to prevention of downstream signalling and delayed onset of kinome rewiring.
Since PROTACs proved successful in degrading both EGFR and HER2, we sought to exploit this technology against a different RTK family. C-Met is the receptor for Hepatocyte Growth Factor (HGF), which is also known as the “scatter factor” for its ability to promote tumor metastasis (Birchmeier et al., 2003; Gherardi et al., 2012; Tsao et al., 2001). Upon binding of HGF, c-Met dimerizes and trans-phosphorylates on tyrosine residues within its kinase domain (Y1234 and Y1235) as well as on its unique c-terminal multifunctional docking domain (Y1313, Y1349, Y1356 and Y1365) (Organ and Tsao, 2011). The docking domain contains recognition sites for diverse cellular effectors such as Src, Gab1, Crk, Grb2, SHC and PI-3 kinase, which potently activate downstream mitogenic pathways. Inhibitors, such as foretinib, which competitively displace ATP from the c-Met kinase domain block HGF-stimulated activation of ERK and Akt, the primary downstream effectors of c-Met signalling. Despite this, however, small molecule c-Met inhibitors have performed disappointingly in clinical trials, (Scagliotti et al., 2015; Sequist et al., 2011) suggesting the possibility of a kinase-independent function driving oncogenesis and highlighting the potential advantage of c-Met degradation over inhibition.
Employing the c-Met inhibitor foretinib (Zillhardt et al., 2011) as a recruiting element, we developed a PROTAC, Compound 7, capable of recruiting VHL to, and thereby inducing degradation of c-Met in a dose- and time-dependent fashion. MDA-MB-231 cells treated with increasing concentrations of foretinib-based PROTAC 7 (Figure 3A) or its cognate diastereomer 8 (Figure 3B), in the presence or absence of a HGF pulse, demonstrated that treatment with approximately 10-fold higher concentration of diastereomer is required to completely inhibit agonist-driven AKT phosphorylation. We observed a similar level of inhibition of Akt phosphorylation in GTL16 cells, a c-Met-overexpressing cell line, when grown and treated in full serum with PROTAC 7 and diastereomer 8 in (Figure S3A/B). The reduced potency differential between PROTAC 7 and diastereomer 8 inhibition of Akt phosphorylation likely results from the steady-state versus agonist-challenged activation of the signalling cascade between the two cell lines (GTL16 and MDA-MB-231, respectively). The foretinib-based PROTAC 7 is also greater than two-fold more potent than its corresponding diastereomer 8 at inhibiting the proliferation of GTL16 cells (Figure 3C), again highlighting the advantages of developing probes capable of protein degradation, especially in oncogene-addicted contexts.
Figure 3. PROTAC mediated degradation of c-Met.

A–B – MDA-MB-231 cells treated for 24 hours with increasing concentrations of foretinib-based PROTAC 7 (A) or diastereomer 8 (B). C – Cell proliferation assay in GTL16 cells (PROTAC 7 IC50 = 66.7 nM, diastereomer 8 IC50 = 156 nM) D – Time course of c-Met degradation by foretinib-based PROTAC (500 nM) 7. E – PROTAC effects are longer lasting in cell culture. Cells were treated for 24 hours with 500 nM PROTAC 7 or diastereomer 8 before re-plating on new plastic, in fresh media for 24 or 48 hours. Excess VHL (25 μM) ligand was added to the indicated wells. See also Figure S3 and S6.
Additionally, clearance of WT c-Met from the cell is relatively rapid, requiring only 6 hours of treatment to significantly reduce protein levels in MDA-MB-231 cells (Figure 3D), providing a temporal advantage over RNA-mediated knockdown techniques as well as abrogating the requirement for transfection reagents or exogenous selection pressure that may interfere with other normal cellular activities.
We have previously demonstrated the catalytic nature of PROTACs, (Bondeson et al., 2015) and as such we sought to explore the duration of their effect upon wash-out. Cells were treated for 24 hours with DMSO control, foretinib-based PROTAC 7 or its corresponding diastereomer 8 before being dissociated from the culture dishes, rinsed with PBS to wash-out extracellular treatment compound, and re-plated into fresh media and on new culture dishes for additional 24 or 48 hour periods followed by lysis. PROTAC-treated cells exhibited a prolonged reduction in c-Met levels out to 48 hours post wash-out (Figure 3E and Supplemental Figure 3). Crucially, in cells treated with PROTAC 7, c-Met levels could be rescued by treatment with 50-fold excess free VHL ligand (Figure S3E) following the wash-out. The free VHL ligand prevents E3 ligase recruitment to the RTK by PROTAC 7, indicating that the sustained knockdown in PROTAC treated cells is mediated post-translationally by disrupting PROTAC 7:Met:VHL complexes remaining in the cell (catalytic mode of action) rather than a response at the translational level, as has been observed with other small molecules (Field et al., 2017). Moreover, when MDA-MB-231 cells are co-treated with PROTAC 7 and increasing doses of free VHL ligand, the ability of the PROTAC to degrade c-Met is hindered, further demonstrating the necessity and specificity for VHL-recruitment (Figure S3F). Additionally, foretinib-based PROTAC 7 is also capable of preventing inhibitor-induced compensatory signalling in a similar way as the aforementioned lapatinib-based PROTACs. GTL16 cells display profound kinome re-wiring after 48 hours, as evidenced by ERK1/2 phosphorylation in diastereomer 8 treated cells, but not in PROTAC 7 treated cells (Figure S3C).
Having proven that RTKs could be degraded via the PROTAC technology we sought to elucidate the mechanism of degradation. Initially, we performed quantitative real time PCR over time to demonstrate that the observed decrease in c-Met protein levels in response to PROTAC treatment occurs at the post-transcriptional level (Figure S4A). Additionally, co-treatment with pharmacological agents which inhibit either the proteasome (epoxomicin) (Bo et al., 2005) or the ubiquitination cascade (NEDD8 activating enzyme E1 inhibitor, MLN-4924) (Soucy et al., 2009) were able to restore protein levels to untreated levels, demonstrating not only that ubiquitination is crucial for c-Met degradation but also that it progresses via the proteasome (Figure S4B/C). Furthermore, RTKs are known to rely on the HSP90 chaperone for proper folding as well as being cycled between the plasma membrane and early endosomes in a HSP90-dependent fashion (Mahalingam et al., 2009). As such we sought to explore the effect of the HSP90 inhibitor 17-AAG (17-N-allylamino-17-demethoxygeldanamycin) on PROTAC-mediated degradation. Co-treatment with PROTAC 7 and 17-AAG has an additive effect on c-Met protein degradation in the MDA-MB-231 cell line (Figure S4D), suggesting that HSP90 may be intercepting c-Met in a separate compartment from PROTAC 7, thereby enhancing the degradation of total c-Met within the cell. Previous work has shown c-Met to be a client protein of HSP90 and that geldanamycin and 17-AAG could promote its ubiquitination and proteasome-dependent degradation (Koga et al., 2007; Miyajima et al., 2013; Wang et al., 2009).
Next, we sought to determine whether PROTAC-targeted RTKs were removed directly from the cell surface or were intercepted at some point in the secretory pathway to the membrane. Employing a cell surface biotinylation degradation assay, we demonstrated that PROTAC 7 induces the degradation of the c-Met mature form (145 kDa) from the cell surface, suggesting that VHL recruitment is capable of – either directly or indirectly – inducing RTK internalization (Figure 4A/B). We confirmed PROTAC 7-mediated RTK internalization by confocal immunofluorescence microscopy: untreated cells exhibit cell surface c-Met immunofluorescence but treatment with PROTAC 7 (Figure 4C) or HGF (Supplemental Figure 5) shows internalization and localization to a perinuclear compartment as previously described (Kermorgant et al., 2003). This perinuclear compartment also stains positive for the early endosome antigen 1 (EEA1) and appears to be distinct from the Golgi apparatus, which is c-Met positive in untreated cells (Figure S5C). These large EEA1 positive vesicle-like structures have previously been reported in MDA-MB-231 cells (Pellinen et al., 2006). This provides, to the best of our knowledge, the first evidence of small molecule-induced internalization of an endogenous RTK and further suggests sorting into endosomes prior to degradation via the proteasome (Figure S4B). Interestingly, preliminary siRNA experiments suggest that this process is clathrin-independent (Figure S5E).
Figure 4. PROTAC mediated internalization.

A/B – Cell surface proteins were labelled with a cell membrane impermeant biotin reagent prior to treatment with 500 nM foretinib-based PROTAC 7 (A) or 100 ng/ml HGF (B) for the indicated times and lysed. Biotinylated proteins were enriched by streptavidin pulldown and immunoblotted for c-Met. Biotinylated proteins represent the cell surface fraction. Corresponding whole cell lysates are also shown C - Representative confocal microscopy images of c-Met (green) internalization in response to PROTAC 7 (500 nM) treatment for the indicated times (DAPI nuclear stain - blue). See also Figure S5.
We sought to further make evident the advantages of degradation over inhibition in the Hs746T gastric cancer cell line, which expresses an exon 14 splice variant of c-Met (Asaoka et al., 2010). Exon 14 skipping results in the expression of c-Met lacking the juxtamembrane domain recruitment site (Y1003) for Cbl, the endogenous E3 ligase that promotes HGF-dependent internalization and subsequent degradation of c-Met (Abella et al., 2005; Peschard et al., 2001). This clinically-relevant mutation results in prolonged downstream signalling since the naturally occurring “off-switch” for HGF-induced signalling is no longer present. The lack of this regulatory domain also increases the intrinsic stability of c-Met protein in the absence of any other degradation signal (Drilon, 2016). Cycloheximide chase experiments (Figure 5A) revealed that WT c-Met has a basal half-life of 4.4 hours, while the basal half-life of the exon 14 mutant c-Met is >8 hours. HGF treatment results in rapid degradation of WT c-Met (Figure 5A/C) but not exon 14-deleted c-Met (Figure 5A/B/D); this lack of internalization/degradation results in sustained downstream signalling (Figure 5C/D) enhancing the cell proliferation and tumorigenesis of exon 14 mutant c-Met expressing cells (Kong-Beltran et al., 2006). Interestingly, PROTAC 7 treatment can induce the degradation of exon 14-deleted c-Met (Figure 5A/E) despite that fact that it is not degraded by the major natural mechanism (i.e. HGF). The degradation half-life of PROTAC 7 treated exon 14-deleted c-Met is only marginally longer than that of PROTAC 7 treated WT c-Met (4.2 hr vs. 2.5 hr, respectively), in contrast to the wide differential in their respective HGF-induced, native degradation rates (>8 hr vs. 1.66 hr, respectively) (Figure 5B). The fact that PROTAC 7 is able to degrade exon 14-deleted c-Met suggests that this process in Cbl-independent, as well as clathrin-independent.
Figure 5. Exon 14-deleted c-Met has increased stability and resistance to HGF-mediated degradation that can be combated by foretinib-based PROTAC 7.

A – Quantitation of WT c-Met or exon 14-deleted c-Met degradation upon treatment with HGF, PROTAC 7 or DMSO control in the presence of cycloheximide (CHX). B – Table of calculated half-lives. C – Representative CHX time course of WT c-Met degradation and signalling in MDA-MB-231 cells treated with HGF. D – Representative CHX time course of exon 14 deleted c-Met degradation and signalling in Hs746T cells treated with HGF. E - Representative CHX time course of exon 14-deleted c-Met degradation in Hs746T cells treated with PROTAC 7. F and G – Both MDA-MB-231 and Hs746T cells were treated with either DMSO or PROTAC for 18 hours before the addition of HGF and lysis at the indicated time points following stimulation. H – Immunoprecipitation of c-Met from PROTAC 7 (1μM) or DMSO treated Hs746T cells followed by immunoblotting for Ubiquitin. I – Tandem Ubiquitin Binding Entity 1 (TUBE1) pulldown from PROTAC 7 (1μM) or DMSO treated Hs746T cells followed by immunoblotting for c-Met See also Figure S6.
This provided another instance whereby target degradation by PROTAC might prove advantageous over inhibition alone, in that inhibition can temporarily block signalling at the level of kinase activity, but only degradation can provide a lasting “off-switch” for the receptor itself as demonstrated in Figure 5F/G. Pre-treatment of Hs746T cells, which express an exon 14 mutant c-Met, with PROTAC 7 reduces HGF-induced activation of Akt. There remains a brief elevation of phospho-Akt in these cells at 0.5 hr following HGF-treatment; more importantly, however, is that the sustained signalling observed in DMSO treated cells (up to 6 hr) is not observed in the PROTAC 7 treated cells (Figure 5G). This restoration of a wild-type phenotype to a mutant protein via a PROTAC is intriguing and the approach could prove potentially advantageous in cancer patients bearing exon 14 splice variants of c-Met (Wislez et al., 2016). As a result of this apparent restoration of a wild-type phenotype to a mutant protein via treatment with PROTAC, we sought to investigate the PROTAC 7-induced ubiquitination state of c-Met through immunoprecipitation experiments in the exon 14 mutant cells. After 4 hours of treatment with PROTAC 7, immunoprecipitated c-Met is ubiquitinated to a greater extent than vehicle control samples (Figure 5H). Furthermore, Hs746T lysate was subjected to Tandem Ubiquitin Binding Entity 1 (TUBE1) immunoprecipitation in an effort to enrich for polyubiquitinated substrates within the cell. PROTAC 7 treated Hs746T cells display marked TUBE1 enrichment of c-Met when compared to vehicle control treated cells (Figure 5I) (Hjerpe et al., 2009). These experiments provide evidence that PROTAC 7 induces polyubiquitination of c-Met, even in an exon 14 skipped context lacking the natural phosphodegron.
While the general applicability of PROTAC-mediated degradation to RTKs may be inferred beyond the specific examples described in this study, we are continuing to investigate other instances of this broad superfamily of proteins as well as the larger considerations of harnessing the PROTAC approach to the entire proteome (see accompanying paper Bondeson, Smith et al). Additionally, the advantages of inducing degradation over inhibition of target proteins gleaned from this “case study” provide a strong foundation for future PROTAC-based paradigms.
Significance
We demonstrate for the first time that PROTACs are capable of inducing the degradation of active receptor tyrosine kinases and provide examples of successful degradation of three separate RTKs – EGFR, HER2, and c-Met, including multiple mutants of EGFR and c-Met. Subsequently we show that degradation may provide advantages over inhibition in several key ways. In most cases compounds capable of degradation inhibit downstream signalling and cell proliferation at lower concentrations than similar compounds that only inhibit. Furthermore, degradation provides a more sustained reduction in signalling as evidenced by the reduction in kinome re-wiring observed previously with EGFR, HER2 and c-Met inhibitors, as well as the sustained duration of response even after washout. We also demonstrate that PROTACs are capable of disposing of proteins that are mutated to avoid their natural “off-switch”. This work significantly expands upon the potential protein targets of PROTACs to include transmembrane proteins and establishes that recruitment of VHL to RTKs is capable of efficiently removing this class of protein targets from the membrane in a similar fashion to their response to growth factor. We demonstrate through control experiments using the inactive diastereomeric compounds with identical physicochemical properties that degradation is leveraged over inhibition alone, highlighting the potential advantages of this pharmacologic modality.
STAR Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGFR | SantaCruz | 1005 |
| FLAG | Sigma | F1804 |
| Tubulin | Sigma | T9026 |
| HER2 | Cell Signaling | 2165S |
| p-EGFR (Y1068) | Abcam | ab40815 |
| p-HER2 (Y1221/1222) | Cell Signaling | 2243S |
| p-AKT (T308) | Cell Signaling | 2965S |
| p-ERK 1/2 (T202/204) | Cell Signaling | 4370 |
| HER3 | Cell Signaling | 12708 |
| pHER3 (Y1197) | Cell Signaling | 4561 |
| pHER3 (Y1289) | Cell Signaling | 4791 |
| pGSK-3b (S9) | Cell Signaling | 9331 |
| c-Met | Cell Signaling | 8198 |
| c-Met | Cell Signaling | 3127 |
| p-Met (Y1245/1235) | Cell Signaling | 3126 |
| p-AKT (S473) | Cell Signaling | 4060 |
| Ubiquitin (P4D1) | Cell Signaling | 3936 |
| p230 Trans Golgi | BD Biosciences | 611281 |
| EEA1 | BD Biosciences | 610456 |
| Clathrin Heavy Chain | SantaCruz | sc-12734 |
| Alexa Fluor-546 conjugated anti-mouse | ThermoFisher | A-21143 |
| Alexa Fluor-488 conjugated anti-rabbit | ThermoFisher | A-11008 |
| HRP linked Mouse IgG | GE Life Sciences | NA931 |
| HRP Linked Rabbit IgG | GE Life Sciences | NA934 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | C104450 |
| MLN4924 | Sigma | 5.05477 |
| PR-619 | LifeSensors | SI9619 |
| EZ-link Sulfo-NHS-SS-Biotin | Thermo | 21331 |
| Pierce NeutrAvidin Agarose beads | Thermo | 29200 |
| Agarose-TUBE 1 | LifeSensors | UM401 |
| Protein A-Sepharose 4B, Fast Flow beads | Sigma | P9424 |
| Recombinant Human HGF Protein | R&D Systems | 294-HG-250 |
| Critical Commercial Assays | ||
| CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) | Promega | G5421 |
| Experimental Models: Cell Lines | ||
| OVCAR8 | Joyce Liu, Dana Farber | |
| MDA-MB-231 | ATCC | HTB-26 |
| HeLa | ATCC | CCL-2 |
| HCC827 | ATCC | CRL-2868 |
| H3255 | Katerina Politi, Yale | |
| H1975 | ATCC | CRL-5908 |
| SKBr3 | ATCC | HTB-30 |
| GTL16 | F. Maina, Developmental Biology Institute of Marseille-Luminy | |
| Hs746T | ATCC | HTB-135 |
| Oligonucleotides | ||
| Clathrin Heavy Chain siRNA | SantaCruz | sc-35067 |
| Software and Algorithms | ||
| Image Lab 6.0 | Biorad | N/A |
| Graphpad | Prism | N/A |
Contact for Reagent and Resource Sharing
Further information and request for resources and reagents should be directed and will be fulfilled by Craig Crews (craig.crews@yale.edu).
Experimental Model and Subject Details
MDA-MB-231, SKBr-3, HCC-827, and H1975 cells were obtained from the American Type Culture Collection (ATCC). OVCAR8 cells were a gift from Joyce Liu (Dana Farber). H3255 cells were a gift from Katerina Politi (Yale University). All of the aforementioned cell lines were cultured in RPMI-1640 (1X) medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin and grown in a humidified incubator at 37°C, 5% CO2. GTL-16 cells were a gift from F. Maina (Developmental Biology Institute of Marseille-Luminy) and similarly grown in RPMI-1640 medium containing 10% FBS and 1% penicillin-streptomycin. To generate an Exon 20-insertion EGFR stable cell line, HeLa cells (ATCC) were transduced with a lentiviral mammalian expression vector pD2119-EFs-3xFLAG-EGFR-Exon20ins (purchased from DNA 2.0) and selected with 2 ug/ml puromycin in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS. This vector contains a 767 ASV duplication of exon 20.
Method Details
Immunoblotting
Cells were treated with the indicated concentrations of PROTAC or corresponding inhibiting diastereomer for the specified time and then harvested in lysis buffer (25 mM Tris·HCl pH 7.5 with 1% NP-40 and 0.25% deoxycholate, supplemented with 10 mM sodium pyrophosphate, 20 mM β-glycerophosphate, 10 mM sodium fluoride, 1 mM sodium orthovanadate, 0.1 mM phenylarsine oxide, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 30 μg/ml bestatin, 0.3 trypsin inhibitor units/ml aprotinin and 1 mM PMSF). Following centrifugation at 16,000 × g for 10 min at 4°C to pellet insoluble materials, the protein concentrations of the supernatants were quantitated by BCA assay (Thermo Fisher Scientific). Protein samples were resolved by 8% SDS-PAGE, electrophoretically transferred to nitrocellulose and probed with the antibodies listed above. Immunoblots were developed using enhanced chemiluminescence and visualized using a Bio-Rad Chemi-Doc MP Imaging System and quantitated with Image Lab v.5.2.1 software (Bio-Rad Laboratories).
Cell Proliferation assays
Following PROTAC or diastereomer treatment of cells as indicated, culture medium was supplemented with 330 μg/ml MTS (Promega Corp., Madison, WI) and 25 μM phenazine methosulfate (Sigma, St. Louis, MO) and incubated at 37°C. Mitochondrial reduction of MTS to the formazan derivative was monitored by measuring the medium’s absorbance at 490 nm using a Wallac Victor2 platereader (Perkin-Elmer Life Sciences, Waltham, MA). Data analysis and statistics performed using Prism v7.0 software (GraphPad Software).
Cell surface biotinylation degradation assay
A protocol was adapted from Joffre et. al to measure the removal of c-Met from the cell surface of MDA-MB-231 cells (Joffre et al., 2011). Cells were plated in full serum, allowed to adhere, and switched to serum-free RPMI-1640 for 16 hr. After this time, cells were placed on ice and rinsed with ice-cold 1X PBS-CM (0.1 mM CaCl2, 1 mM MgCl2) twice and incubated with PBS-CM for 5 min at 4°C. PBS-CM was aspirated, at which point cells were labelled with a cell membrane impermeant reagent, EZ-link Sulfo-NHS-SS-biotin at 0.5 mg/ml for 30 min at 4°C with gentle rocking. This step enabled covalent labelling of all cell surface proteins. All of the following were carried out at 4°C to prevent trafficking of said proteins. Cells were subsequently rinsed with ice-cold PBS-CM twice and excess biotin was quenched with Tris-glycine buffer (100 mM Tris pH 8.0, 150 mM NaCl, 0.1 mM CaCl2, 1 mM MgCl2 10 mM glycine, 1% BSA) for 15 min at 4°C with gentle rocking. Cells were then rinsed with ice-cold PBS-CM three times before being chased with warm serum-free RPMI-1640 medium containing either HGF (100 ng/ml) or PROTAC (500 nM) and placed in a humidified incubator at 37°C for the indicated amount of time, at which point the cells were lysed with lysis buffer (50 mM Tris, pH 7.5, 100 mM NaCl, 10% glycerol, 1% NP-40, 1 mM EDTA) supplemented with 1X protease inhibitors (Roche). Lysates were spun down at 14,000 × g at 4°C for 10 min and protein content was measured by BCA assay. Protein lysate was normalized and aliquoted onto pre-equilibrated NeutrAvidin agarose beads for 2 hrs at 4°C, with gentle rotation. Beads were washed three times with wash buffer (100 mM Tris, pH 7.5, 300 mM NaCl, and 1% Triton X-100) and resuspended in 2X elution buffer (62.5 mM Tris, pH 6.8, 3% SDS, 10% glycerol, 0.02% bromophenol blue, 160 mM DTT). Protein was eluted off of the beads by heating at 95°C for 5 min and the supernatant was run on an SDS-PAGE gel and evaluated for the presence of cell surface c-Met protein. Whole-cell lysate refers to the lysate loaded onto NeutrAvidin beads, thereby representing the total c-Met protein.
Cycloheximide chase assay
MDA-MB-231 cells were plated at 3×105 cells per well in a 6-well dish, allowed to adhere, and switched to serum-free RPMI-1640 for 16 hr. Cells were then pre-treated with cycloheximide (Sigma) at 100 ug/ml for 1 hr prior to addition of either HGF (100 ng/ml), PROTAC (500 nM), or vehicle. At the indicated time points, cells were immediately placed on ice, rinsed with PBS, lysed, and boiled.
Chemical Syntheses
Details of Chemical Syntheses can be found in Methods S1.
Immunofluorescence Microscopy
MDA-MB-231 cells were plated at a density of 1×105 cells/ml onto 12 mm round coverslips, cultured overnight, switched to serum free media for >12 hours and then treated with 500 nM PROTAC 7 or 100 ng/ml HGF for the indicated times before washing with PBS. Cells were fixed with 4% formaldehyde for 20 minutes at room temperature, washed with ice-cold PBS, permeabilized and blocked with 0.25% Triton X-100/1% BSA in PBS for 30 minutes. Fixed cells were incubated with c-Met Antibody (1:3000 dilution, Cell Signalling #8198) for 1 hour, washed three times with PBS for 5 minutes, incubated with Alexa Fluor-488 conjugated anti-rabbit antibody (1:1000 dilution, ThermoFisher A-11008) for 1 hour washed three times with PBS for 5 minutes and mounted in vectashield containing DAPI. Imaged on Zeiss Axio Observer Z1 inverted microscope.
siRNA Experiments
The siRNA (4μL of 10 μM stock solution, 40 pMol) was diluted with Opti-MEM media (150 μL) then added to a solution of Lipofectamine RNAiMAX (9 μL in 150 μL in Opti-MEM) and incubated for 10 minutes before being added to MDA-MB-231 cells at ~ 80% confluency. The following day, the transfected cells were plated out and used for experiments as described above.
Immunoprecipitation Experiments
Hs746T cells (2.5 × 106) were seeded into 10 cm dishes, allowed to adhere, switched to serum-free DMEM media for 16hr. After this time, cells were pre-treated with 2 uM epoxomicin for 30 minutes at 37°C. After this pre-treatment, 10 cm plates were treated with either 1 uM Compound 7 or vehicle for 4 hours at 37 °C, after which they were placed on ice, rinsed twice with ice-cold 1X PBS and lysed with 500 uL modified 1X RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing 5 mM 1,10-phenanthroline monohydrate, 10 mM N-ethylmaleimide, 20 uM PR-619, and 1X cOmplete protease inhibitor cocktail (Roche). Lysates were spun down at 14,000 × g at 4°C for 10 min and protein content was measured by BCA assay. Protein lysate was normalized and 500 ug of lysate was aliquoted onto naked Protein A-Sepharose 4B beads (Sigma), and pre-cleared for 1 hr at 4°C with gentle rotation. After this 1 hr incubation, samples were spun down at 3,000 × g at 4°C for 2 min and the normalized, pre-cleared lysate were subsequently loaded onto Protein A-Sepharose 4B beads coupled with 5 ug of Met (CST, #8198) antibody. MET was immunoprecipitated from Hs746T lysates for 2 hr at 4°C with gentle rotation, after which samples were spun down at 3,000 × g at 4°C for 2 min, flow-thru was collected to assess pulldown efficiency (see Fig. S6D), and the beads were washed once with ice-cold lysis buffer and thrice with ice-cold 1X TBS-T (137 mM NaCl, 2.7 mM KCl, 19 mM Tris-HCl pH 7.5, 0.02% Tween-20). The beads were resuspended in 1X LDS sample buffer containing 5% BME. Immunoprecipitated protein was eluted off of the beads by heating at 95°C for 5 min and the supernatant was run on an SDS-PAGE gel and evaluated for the presence of immunoprecipitated total Met (CST, #3127), as well as ubiquitinated Met (CST, #3936). Whole-cell lysate refers to the normalized, input lysate loaded onto Protein A-Sepharose beads.
TUBE1 Immunoprecipitation Experiments
TUBE1 immunoprecipitations were carried out exactly as described in the previous section (Immunoprecipitation Experiments), except for the fact that 1 mg of Hs746T lysate was used and loaded onto 20 uL TUBE1 agarose (LifeSensors) resin per sample.
Supplementary Material
Highlights.
PROTACs are capable of degrading transmembrane receptor tyrosine kinases (RTKs)
PROTACs prove superior to corresponding diastereomeric inhibitors
Degradation abrogates scaffolding roles of RTKs thus suppressing kinome re-wiring
Acknowledgments
We acknowledge members of the Crews laboratory for useful discussions. G.M.B. is a supported by an Leukemia and Lymphoma Society Career Development Award. J.H. is supported by an R50 Research Specialist Award from the NCI. D.B. is supported by an NCI Predoctoral to Postdoctoral Fellow Transition Award. A.C.L acknowledges support from the NIH (MSTP NIH/NIGMS T32GM007205). C.M.C. gratefully acknowledges support from the Leukemia and Lymphoma Society and the NIH (R35CA197589). C.M.C. is founder, consultant and shareholder in Arvinas, LLC, which supports research in his lab. H.D., Y.Q., J.W. and A.P.C. are employees of and shareholders in Arvinas LLC.
Footnotes
Author Contributions
Investigation, G.M.B., B.E.S., A.C.L. J.H., D.M. and D.B.; Resources and Reagents: S.J.F., M.T., A.P.C., H.D., Y.Q and J.W.; Writing - Original Draft, G.M.B, B.E.S. and J.H.; Writing - Review and Editing, all authors; Supervision, C.M.C; Funding Acquisition, C.M.C.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbé S, Park M. Met/Hepatocyte Growth Factor Receptor Ubiquitination Suppresses Transformation and Is Required for Hrs Phosphorylation. Mol Cell Biol. 2005;25:9632–9645. doi: 10.1128/MCB.25.21.9632-9645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anido J, Matar P, Albanell J, Guzmán M, Rojo F, Arribas J, Averbuch S, Baselga J. ZD1839, a Specific Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor, Induces the Formation of Inactive EGFR/HER2 and EGFR/HER3 Heterodimers and Prevents Heregulin Signaling in HER2-overexpressing Breast Cancer Cells. Clinical Cancer Research. 2003;9:1274–1283. [PubMed] [Google Scholar]
- Asaoka Y, Tada M, Ikenoue T, Seto M, Imai M, Miyabayashi K, Yamamoto K, Yamamoto S, Kudo Y, Mohri D, et al. Gastric cancer cell line Hs746T harbors a splice site mutation of c-Met causing juxtamembrane domain deletion. Biochem Biophys Res Commun. 2010;394:1042–1046. doi: 10.1016/j.bbrc.2010.03.120. [DOI] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Bo KK, Fonseca FN, Crews CM. Development and characterization of proteasome inhibitors. Methods Enzymol. 2005;399:585–609. doi: 10.1016/S0076-6879(05)99039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM. Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α. Angew Chem Int Ed. 2012a;51:11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Raina K, Darricarrere N, Hines J, Gustafson JL, Smith IE, Miah AH, Harling JD, Crews CM. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem Biol. 2015;10:1831–1837. doi: 10.1021/acschembio.5b00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1α Interaction. J Am Chem Soc. 2012b;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: Monoclonal antibodies versus tyrosine kinase inhibitors. Critical Reviews in Oncology/Hematology. 2007;62:53–61. doi: 10.1016/j.critrevonc.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Douglass EF, Miller CJ, Sparer G, Shapiro H, Spiegel DA. A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J Am Chem Soc. 2013;135:6092–6099. doi: 10.1021/ja311795d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A. <em>MET</em> Exon 14 Alterations in Lung Cancer: Exon Skipping Extends Half-Life. Clinical Cancer Research. 2016;22:2832–2834. doi: 10.1158/1078-0432.CCR-16-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. <em>MET</em> Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- Field SD, Arkin J, Li J, Jones LH. Selective Downregulation of JAK2 and JAK3 by an ATP-Competitive pan-JAK Inhibitor. ACS Chem Biol. 2017;12:1183–1187. doi: 10.1021/acschembio.7b00116. [DOI] [PubMed] [Google Scholar]
- Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Graves Lee M, Duncan James S, Whittle Martin C, Johnson Gary L. The dynamic nature of the kinome. Biochemical Journal. 2013;450:1–8. doi: 10.1042/BJ20121456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Hines J, Gough JD, Corson TW, Crews CM. Posttranslational protein knockdown coupled to receptor tyrosine kinase activation with phosphoPROTACs. Proc Natl Acad Sci USA. 2013;110:8942–8947. doi: 10.1073/pnas.1217206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO reports. 2009;10:1250–1258. doi: 10.1038/embor.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal N, Iqbal N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Molecular Biology International. 2014;2014:9. doi: 10.1155/2014/852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between Epidermal Growth Factor Receptor and c-Met Signal Pathways in Transformed Cells. J Biol Chem. 2000;275:8806–8811. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- Joffre C, Barrow R, Menard L, Calleja V, Hart IR, Kermorgant S. A direct role for Met endocytosis in tumorigenesis. Nat Cell Biol. 2011;13:827–837. doi: 10.1038/ncb2257. [DOI] [PubMed] [Google Scholar]
- Kermorgant S, Zicha D, Parker PJ. Protein Kinase C Controls Microtubule-based Traffic but Not Proteasomal Degradation of c-Met. J Biol Chem. 2003;278:28921–28929. doi: 10.1074/jbc.M302116200. [DOI] [PubMed] [Google Scholar]
- Koga F, Tsutsumi S, Neckers LM. Low Dose Geldanamycin Inhibits Hepatocyte Growth Factor- and Hypoxia-Stimulated Invasion of Cancer Cells. Cell Cycle. 2007;6:1393–1402. doi: 10.4161/cc.6.11.4296. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Pegram MD, Venkatesan N, Finn R, Yang G, Rahmeh M, Untch M, Rusnak DW, Spehar G, Mullin RJ, et al. Activity of the Dual Kinase Inhibitor Lapatinib (GW572016) against HER-2-Overexpressing and Trastuzumab-Treated Breast Cancer Cells. Cancer Research. 2006;66:1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, Holcomb T, Pujara K, Stinson J, Fu L, et al. Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Research. 2006;66:283–289. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- Kurimchak Alison M, Shelton C, Duncan Kelly E, Johnson Katherine J, Brown J, O’Brien S, Gabbasov R, Fink Lauren S, Li Y, Lounsbury N, et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Reports. 2016;16:1273–1286. doi: 10.1016/j.celrep.2016.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, Crews CM. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebraud H, Wright DJ, Johnson CN, Heightman TD. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Central Science. 2016 doi: 10.1021/acscentsci.6b00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, Giles FJ. Targeting HSP90 for cancer therapy. Br J Cancer. 2009;100:1523–1529. doi: 10.1038/sj.bjc.6605066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima N, Tsutsumi S, Sourbier C, Beebe K, Mollapour M, Rivas C, Yoshida S, Trepel JB, Huang Y, Tatokoro M, et al. The HSP90 Inhibitor Ganetespib Synergizes with the MET Kinase Inhibitor Crizotinib in both Crizotinib-Sensitive and -Resistant MET-Driven Tumor Models. Cancer Research. 2013;73:7022–7033. doi: 10.1158/0008-5472.CAN-13-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neklesa TK, Crews CM. Chemical biology: Greasy tags for protein removal. Nature. 2012;487:308–309. doi: 10.1038/487308a. [DOI] [PubMed] [Google Scholar]
- Neklesa TK, Tae HS, Schneekloth AR, Stulberg MJ, Corson TW, Sundberg TB, Raina K, Holley SA, Crews CM. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat Chem Biol. 2011;7:538–543. doi: 10.1038/nchembio.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organ SL, Tsao MS. An overview of the c-MET signaling pathway. Therapeutic Advances in Medical Oncology. 2011;3:S7–S19. doi: 10.1177/1758834011422556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellinen T, Arjonen A, Vuoriluoto K, Kallio K, Fransen JAM, Ivaska J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of β1-integrins. The Journal of Cell Biology. 2006;173:767–780. doi: 10.1083/jcb.200509019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Torres M, Guix M, Gonzalez A, Arteaga CL. Epidermal Growth Factor Receptor (EGFR) Antibody Down-regulates Mutant Receptors and Inhibits Tumors Expressing EGFR Mutations. J Biol Chem. 2006;281:40183–40192. doi: 10.1074/jbc.M607958200. [DOI] [PubMed] [Google Scholar]
- Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, Langdon WY, Park M. Mutation of the c-Cbl TKB Domain Binding Site on the Met Receptor Tyrosine Kinase Converts It into a Transforming Protein. Molecular Cell. 2001;8:995–1004. doi: 10.1016/s1097-2765(01)00378-1. [DOI] [PubMed] [Google Scholar]
- Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG. The Plasticity of Oncogene Addiction: Implications for Targeted Therapies Directed to Receptor Tyrosine Kinases. Neoplasia. 2009;11:448–IN442. doi: 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratz KW, Cortes J, Roboz GJ, Rao N, Arowojolu O, Stine A, Shiotsu Y, Shudo A, Akinaga S, Small D, et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938–3946. doi: 10.1182/blood-2008-09-177030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scagliotti G, Pawel Jv, Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non–Small-Cell Lung Cancer. Journal of Clinical Oncology. 2015;33:2667–2674. doi: 10.1200/JCO.2014.60.7317. [DOI] [PubMed] [Google Scholar]
- Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Oláh J, Ovádi J, Sippl W, Jung M. Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals) J Med Chem. 2017 doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008;18:5904–5908. doi: 10.1016/j.bmcl.2008.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Pawel Jv, Garmey EG, Akerley WL, Brugger W, Ferrari D, Chen Y, Costa DB, Gerber DE, Orlov S, et al. Randomized Phase II Study of Erlotinib Plus Tivantinib Versus Erlotinib Plus Placebo in Previously Treated Non–Small-Cell Lung Cancer. Journal of Clinical Oncology. 2011;29:3307–3315. doi: 10.1200/JCO.2010.34.0570. [DOI] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- Stuhlmiller Timothy J, Miller Samantha M, Zawistowski Jon S, Nakamura K, Beltran Adriana S, Duncan James S, Angus Steven P, Collins Kyla AL, Granger Deborah A, Reuther Rachel A, et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Reports. 11:390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Ito F. Receptor Tyrosine Kinases and Targeted Cancer Therapeutics. Biol Pharm Bull. 2011;34:1774–1780. doi: 10.1248/bpb.34.1774. [DOI] [PubMed] [Google Scholar]
- Thelemann A, Petti F, Griffin G, Iwata K, Hunt T, Settinari T, Fenyo D, Gibson N, Haley JD. Phosphotyrosine Signaling Networks in Epidermal Growth Factor Receptor Overexpressing Squamous Carcinoma Cells. Molecular & Cellular Proteomics. 2005;4:356–376. doi: 10.1074/mcp.M400118-MCP200. [DOI] [PubMed] [Google Scholar]
- Tsao MS, Yang Y, Marcus A, Liu N, Mou L. Hepatocyte growth factor is predominantly expressed by the carcinoma cells in non–small-cell lung cancer. Human Pathology. 2001;32:57–65. doi: 10.1053/hupa.2001.21133. [DOI] [PubMed] [Google Scholar]
- Wang S, Pashtan I, Tsutsumi S, Xu W, Neckers L. Cancer cells harboring MET gene amplification activate alternative signaling pathways to escape MET inhibition but remain sensitive to Hsp90 inhibitors. Cell Cycle. 2009;8:2050–2056. doi: 10.4161/cc.8.13.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wislez M, Domblides C, Cortot A, Lemoine A. Mutations at the splice sites of exon 14 of MET gene: a new target for sarcomatoid carcinomas? Annals of Translational Medicine. 2016;4 doi: 10.21037/atm.2016.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Lim SM, Westover KD, Dodge ME, Ercan D, Ficarro SB, Udayakumar D, Gurbani D, Tae HS, Riddle SM, et al. Pharmacological targeting of the pseudokinase Her3. Nat Chem Biol. 2014;10:1006–1012. doi: 10.1038/nchembio.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AM, Huang PH. Receptor Tyrosine Kinase Coactivation Networks in Cancer. Cancer Research. 2010;70:3857–3860. doi: 10.1158/0008-5472.CAN-10-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda H, Park E, Yun CH, Sng NJ, Lucena-Araujo AR, Yeo WL, Huberman MS, Cohen DW, Nakayama S, Ishioka K, et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Science Translational Medicine. 2013;5:216ra177–216ra177. doi: 10.1126/scitranslmed.3007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials. 2013;34:8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]
- Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang C-Y, Zhao Y, et al. Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression. J Med Chem. 2017 doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed]
- Zillhardt M, Park SM, Romero IL, Sawada K, Montag A, Krausz T, Yamada SD, Peter ME, Lengyel E. Foretinib (GSK1363089), an Orally Available Multikinase Inhibitor of c-Met and VEGFR-2, Blocks Proliferation, Induces Anoikis, and Impairs Ovarian Cancer Metastasis. Clinical Cancer Research. 2011;17:4042–4051. doi: 10.1158/1078-0432.CCR-10-3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.