

Summary
X-linked Dystonia-Parkinsonism (XDP) is a Mendelian neurodegenerative disease that is endemic to the Philippines and associated with a founder haplotype. We integrated multiple genome and transcriptome assembly technologies to narrow the causal mutation to the TAF1 locus, which included a SINE-VNTR-Alu (SVA) retrotransposition into intron 32 of the gene. Transcriptome analyses identified decreased expression of the canonical cTAF1 transcript among XDP probands, and de novo assembly across multiple pluripotent stem cell-derived neuronal lineages discovered aberrant TAF1 transcription that involved alternative splicing and intron retention (IR) in proximity to the SVA that was anti-correlated with overall TAF1 expression. CRISPR/Cas9 excision of the SVA rescued this XDP-specific transcriptional signature and normalized TAF1 expression in probands. These data suggest an SVA-mediated aberrant transcriptional mechanism associated with XDP and may provide a roadmap for layered technologies and integrated assembly-based analyses for other unsolved Mendelian disorders.
Keywords: XDP, DYT3, SVA, retrotransposon, dystonia, Parkinson’s disease, TAF1, genome assembly, transcriptome assembly, intron retention
In Brief
A Mendelian form of parkinsonism arises from altered splicing and intron retention within a general transcription factor.
Introduction
In recent years remarkable progress has been made in Mendelian gene discovery and the potential impact of deleterious mutations in genes under strong evolutionary constraint (Samocha et al., 2014). Yet approximately half of individuals with suspected genetic disorders do not receive a diagnosis, while ~20% of Mendelian disorders have been mapped to a causal locus but the pathogenic mechanism is unknown (Chong et al., 2015; Yang et al., 2014). A few of the critical limitations that impede gene discovery in such cases include the immature functional annotation of most coding and noncoding variation, and the inability to routinely survey structural rearrangements. Another barrier is the reliance on reference-based analyses, which is an effective approach if proband and reference assemblies share gene and transcript structures, but if they differ, the methods break down. Reference-based analyses may also be insensitive to cryptic sequences that are unique to a founder haplotype. Late-onset Mendelian disorders also provide a unique interpretative challenge, as risk variants may exert subtle effects that do not impede normal development for much of the patient’s life.
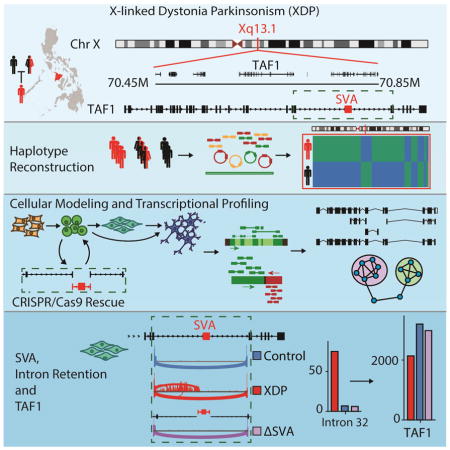
One example of such an elusive Mendelian disorder is X-linked Dystonia-Parkinsonism (XDP), an adult-onset neurodegenerative disease that has challenged conventional gene discovery for several decades. XDP is endemic to the island of Panay, Philippines, where its reported prevalence is 5.74 cases per 100,000 individuals with a mean age at onset of 39.7 years (Lee et al., 2011). The clinical phenotype combines features of dystonia and parkinsonism in a temporal progression, beginning with hyperkinetic symptoms that shift to hypokinetic movements over time (Lee et al., 2011). Conventional genetic approaches have previously mapped the XDP causal locus to the X chromosome and reported a haplotype shared by all probands that consisted of seven variants: five single nucleotide variants (SNVs), designated in the literature as Disease-specific Single-nucleotide Changes (DSC) - 1,2,3,10,12; a 48-bp deletion; and an ~2.6 kb SINE-VNTR-Alu (SVA)-type retrotransposon insertion, all of which were localized to a 449 kb region (Figure 1A) (Domingo et al., 2015; Makino et al., 2007; Nolte et al., 2003). To date, no discriminating alleles of the founder haplotype or recombination events that create partial haplotypes have been reported among XDP probands.
Figure 1. XDP associated genomic region and experimental design.

(A) Genomic segment associated with XDP on Xq13.1 with seven variants reportedly shared among probands and not observed in controls: five single nucleotide variants, annotated as Disease-specific Single-nucleotide Changes (DSCs)-1,2,3,10,12; a SINE-VNTR-Alu (SVA) retrotransposon inserted antisense to TAF1; and a 48-bp deletion. (B) Experimental workflow showing the number of XDP probands (black), carrier females (mixed), and controls (red), with the number of clones for each cell line.
Interpreting the mechanistic relevance of these previous studies in XDP has been difficult as none of the DSCs have annotated functions. Three (DSC10, 12, and the SVA) fall within introns of the TAF1 gene, while the remaining four are localized to an intergenic region 3′ to TAF1, which previous studies have proposed to include multiple unconventional exons associated with TAF1, designated as a Multiple Transcript System (MTS) (Herzfeld et al., 2007; Makino et al., 2007; Nolte et al., 2003). These observations raise the possibility that a defect in TAF1 may underlie XDP pathogenesis. TAF1 encodes TATA-Binding Protein (TBP)-Associated Factor-1 (TAF1), a subunit of the TFIID complex which mediates transcription by RNA polymerase II (RNAPII) (Thomas and Chiang, 2006). In addition to the XDP-related sequence variants, other coding variations in TAF1 have been linked to severe neurodevelopmental defects and intellectual disability (O’Rawe et al., 2015), as well as cancer (Oh et al., 2017; Zhao et al., 2013). Given the essential function of TAF1 in transcription, it is not known how sequence variation in TAF1 causes tissue-specific defects and/or specific clinical phenotypes.
Here we investigated XDP as an exemplar of an unsolved Mendelian disorder arising from a founder haplotype in an isolate population. We hypothesized that the genetic diversity of XDP has not been captured by previous approaches, and that unbiased assembly of the genome and transcriptome spanning the XDP haplotype could reveal additional sequences or aberrant transcripts unique to probands. We thus approached this problem by integrating multiple short and long-read sequencing technologies and reference-free assembly approaches in XDP cell models. Figure 1B summarizes the experimental work flow and technologies applied. Our results identified previously unknown genomic variants and assembled transcripts that were shared among XDP probands, but not observed in controls, including aberrant splicing and partial retention of intronic sequence proximal to the disease-specific SVA insertion in TAF1. This intron retention (IR) coincided with decreased exon usage in proximity to the SVA and an overall reduction in TAF1 expression, both of which were rescued by CRISPR/Cas9-based excision of the SVA. These data offer new insight into the transcript structure of TAF1 in neural cells, implicate a unique genomic mechanism for XDP, and provide a potential roadmap for integrated, reference-free genome and transcriptome assemblies in population isolates.
Results
Establishing an XDP familial cohort
We reasoned that combined genome and transcriptome analyses in a large cohort would be required to identify and interpret the XDP causal variant. To do so we evaluated 403 affected males, 23 heterozygous carrier females, 352 unaffected individuals, and 14 male haplotype carriers below the median age of onset who were asymptomatic at the time of exam, referred to as non-manifesting carriers (NMCs; Table S1). The cohort included 66 archival specimens that were previously described (Nolte et al., 2003). 78% of the probands presented initially with dystonia at a mean age at onset of 42.3 years (± 8.3), with phenotype advancement consistent with previous reports (Lee et al., 2011). All probands were positive for known XDP haplotype markers based on PCR amplification of genomic DNA. Heterozygous carrier females were positive for the haplotype but appeared neurologically normal on exam. Clinical and demographic information of subjects are summarized in Table S1.
Genome assembly and deep sequencing of the founder haplotype reveal shared sequences that narrow the causal locus
We first asked if the XDP founder haplotype includes sequences unique to the Panay population and absent from the current human reference assembly. Previous studies have reported seven variants shared by all XDP probands, with no discriminatory alleles, suggesting that the founder haplotype has never undergone recombination. We thus also probed for structural variation (SV) that might inhibit recombination of the haplotype. We initially explored these hypotheses in nine samples using four strategies (Figure 1B): (1) reference-free, de novo assembly of the XDP haplotype using Illumina paired-end 250 bp and 10X Genomics linked-read sequencing; (2) long-insert “jumping library” whole genome sequencing (liWGS) to probe for SV (Collins et al., 2017); (3) Pacific Biosciences long-read single molecule sequencing (PacBio SMRT) of bacterial artificial chromosome (BAC) clones to define the full SVA sequence; and (4) targeted capture for dense tiling and deep sequencing (CapSeq) of the XDP region. Following these exploratory analyses, we sequenced all 789 subjects using CapSeq to assess the complete allelic diversity of the region (Figure 1B).
Illumina assembly using DISCOVAR and DISCOVAR de novo (Weisenfeld et al., 2014) and 10X Genomics assembly using Supernova generated a contiguous haplotype of 410,455 bases spanning the XDP locus, including 2,106 bases not observed in the reference. PacBio sequencing of BAC clones derived from one proband covered a 200 kb segment spanning TAF1 (average read length = 10,416 bp; Figure 1B) that confirmed all Illumina results and assembled the complete SVA sequence (2,712 bp; Figure S1). The liWGS did not detect any SVs shared among probands that would suggest this region of the X chromosome may be recalcitrant to recombination.
Dense tiling and deep sequencing of the assembled segment performed well in the pilot cohort (463 kb including flanking regions, average depth = 70X, targeted bases covered = 96%) and was extended to all 789 individuals (Figure 1B, Table S1). The CapSeq and WGS assembly detected greater allelic diversity than had been recognized in XDP. We observed 1518 SNVs and 378 insertion/deletions variants (indels), including all seven known DSCs and 47 additional variants that segregated with disease status for a total of 54 variants associated with the haplotype (44 SNVs, 8 indels, the SVA, and the 48 bp deletion; Figure 2A, Table S2). DSCs identified in this study are annotated in Figure 2 as DSCn for consistency with the XDP literature and with standard human genetic nomenclature in Table S2 for integration with public reference maps. None of the missense variants linked to the neurodevelopmental TAF1 syndrome were observed in XDP patients (O’Rawe et al., 2015). We reviewed the Exome Aggregation Consortium (ExAC) data (Lek et al., 2016) for evidence of gender-specific constraint against TAF1 loss-of-function (LoF) variation, revealing TAF1 to be highly intolerant to such variation (pLI = 1.0; 50.8 expected, 2 observed). Notably, both LoF variants were observed among females but none were detected among the 33,644 males in ExAC, suggesting that complete loss of TAF1 is highly deleterious in humans.
Figure 2. Haplotypes observed among XDP probands.

(A) Allelic diversity of XDP haplotypes reconstructed from de novo assembly and CapSeq. All known DSCs (red) were detected with 47 additional variants shared among probands compared to controls for the predominant haplotype (n=373, 93% of XDP probands). Variations are shown in 5′ to 3′ orientation spanning the region. (B) Five recombinations (denoted by ®) with alleles observed for two recombinant haplotypes that narrowed the XDP causal locus. (C) Recombination between DSC1 and DSC3 in pedigree 27 produced haplotype H7, with all alleles shown. Dotted rectangle represents the narrowed XDP region shared among all haplotypes based on recombinations, with reversion to the reference allele observed at DSCn3 (See also Table S1 and Table S2). See key for all annotations.
We discovered five independent recombinations that resulted in eight distinct haplotypes among XDP probands, the first recombinations of the founder haplotype detected to date (four historical recombinations and one in pedigree 27; Figure 2, Table S1). The most common haplotype, H1 (n = 373), consisted of all 54 shared variants and most likely underwent recombination to generate the derivative haplotypes (Figure 2A). The second most frequent haplotype involved a recombination proximal to DSCn1 and reversion to the reference allele at position 70521288 (DSCn3) compared to H1 (H2, n = 16, Figure 2B), while the remaining haplotypes were less frequent (<1% of probands). Thirteen variants fully segregated with disease in all probands and were not altered by recombination (Figure 2A), defining a minimum critical region of 219.7 kb, or 203.6 kb if the DSCn3 reversion is used as the flanking point that encompasses TAF1 exclusively and likely reflects the causal locus.
Cellular modeling of XDP
To interrogate the transcript structure of this region and probe for genotypic differences in expression, we established XDP and control cell lines consisting of: (1) fibroblasts from 13 probands, 12 heterozygous female carriers, and 20 unaffected relatives; and (2) iPSCs from 5 XDP probands, 4 female carriers, and 3 unaffected relatives, with 2 clones per individual (24 total clones; Table S1). Pluripotency analysis of XDP and control iPSCs was previously reported (Ito et al., 2016) and similar characterization of iPSCs from the female carriers is depicted in Figure S3. All clones were differentiated into neural stem cells (NSCs) and induced cortical neurons (iNs) based on overexpression of neurogenin-2 (NGN2). Expression profiling of NSCs and iNs showed segregation of NSC vs. mature neuronal markers in the respective cell types (Figure 3A). Some variability in marker expression was noted across lines, but there were no consistent genotypic differences except for FOXG1, which was downregulated in XDP vs. control NSCs (Figure 3A). Neurons displayed dense processes labeled by doublecortin, MAP2, and Tuj1 (Figure 3B), and functional maturity was evaluated based on activity-dependent calcium mobilization. Neurons loaded with the calcium indicator dye, Fluo-4, exhibited robust calcium influx elicited by both KCl and the glutamate receptor agonist, kainate (Figure 3C–D), the latter of which could be blocked by the AMPA/kainate receptor antagonist CNQX, demonstrating specificity of the response.
Figure 3. Characterization of iPSC-derived NSCs and NGN2-induced cortical neurons.

(A) Heatmap of relative expression of pluripotency, neural stem cell, neuronal and glial genes in NSCs and iNs based on RNAseq. (B) Representative images from proband, carrier female, and control iNs showing processes stained with doublecortin (DCX), βIII-tubulin/Tuj, and MAP2. (C) Ca2+ mobilization in iNs visualized via Fluo-4AM. Upper and lower panels show Fluo-4AM fluorescence before and after, respectively, KCl treatment in control (left panels), carrier (middle panels) and XDP (right panels) lines. (D) Representative traces show relative change in fluorescence intensity (ΔF/F) induced by KCl (upper panels) and kainate (lower panels) in control (left), carrier (middle) and patient (right) lines. Traces represent individual cells (n = 10-15 cells).
XDP cellular models exhibit differential expression of TAF1 transcripts and partial retention of an intronic sequence proximal to the SVA
We evaluated expression changes related to the XDP haplotype and assembled the complete transcript structure of TAF1 in all cell types using: (1) strand-specific dUTP-RNAseq and Illumina sequencing (median = 39.6M paired-reads per clone); (2) targeted mRNA capture using the same 120 bp baits from the DNA CapSeq (referred to as RNA CapSeq) to tile all coding and noncoding transcripts in the region (median = 2.7M paired-end reads spanning the segment per clone, ~150-fold increase in coverage of targeted transcripts); and (3) PacBio SMRT long-reads of RNA CapSeq libraries (mean captured fragment size = 1560 bp). To assess expression changes of TAF1 features (transcripts, exons) and genome-wide differential expression in probands vs. controls, we used generalized linear mixed models (GLMMs) with individuals as a random effect to account for potential confounds of inter-iPSC clone variability. TAF1 was the only differentially expressed gene spanning the full linkage region, further supporting the likelihood that the narrowed segment encompassed the causal locus. TAF1 expression was reduced in XDP NSCs (19.9%, FDR = 1.8 x 10−6, Table S7) and fibroblasts (14.1%, FDR = 1.3 x 10−3), but not iNs. We thus focused our analyses on this locus.
De novo transcript assembly in fibroblasts and neural cells identified four TAF1 isoforms which had not been previously annotated in addition to cTAF1, the canonical transcript, and nTAF1, the neuron-specific isoform of cTAF1 that includes 6 bp derived from an alternative exon 34′ (Figure 4A, Figure S3 and Table S3). The four transcripts detected here included one isoform, annotated as ‘TAF1-32i’, that was composed of canonical exon 32 spliced to a cryptic exon in intron 32 that terminated 716 bp 5′ to the SVA (Figure 4A). We also observed a transcript 3′ to TAF1 that partially overlapped with the MTS and DSC3 (Herzfeld et al., 2007; Nolte et al., 2003) but did not splice to any TAF1 exons as previously proposed (Figure S3). Integration of Illumina assemblies with PacBio RNA CapSeq in NSCs from 3 clones confirmed each of these assembled transcripts (Figure 4A), including all junctions, and extended the transcript start site for two of them, including TAF1-32i (Figure 4A).
Figure 4. De novo assembly of TAF1 transcript structure and differential expression of splice variants.

(A) Transcript structure from de novo assembly depicts TAF1 isoforms previously annotated in Ensembl and additional splice variants detected in this study. For each transcript, boxes denote exons in black (Ensembl-annotated) or pink (this study). Brown triangle indicates genomic position of the SVA. Notation is provided for the cell type in which each transcript was detected. Extension of the transcript assembled from Illumina short reads by the PacBio data are indicated by a dashed orange line with additional exons represented by orange boxes. The genomic coordinate reflects the insertion of SVA (2627 bp). (B) Relative expression abundance of each TAF1 transcript in controls (x-axis) and relative change in TAF1 transcripts in XDP probands compared to controls (y-axis) in NSC (left) and iNs (right). Error bars reflect FDR correction of 95% confidence interval. (C) Relative expression of each exon of cTAF1 in XDP NSCs relative to controls. Black dashed line represents no change.
We next quantified expression of the assembled transcripts. cTAF1 was the predominant species in all cell types, representing 69.4%, 69.3%, and 43.1% of total TAF1 expression in fibroblasts, NSCs, and iNs, respectively (Figure 4B, S4C). nTAF1 and transcripts including exon 34′ were expressed in iNs (22.0% of total TAF1 expression and 34.1% of the expression of all exon34′ containing transcripts; Figure 4B) but was not detected in fibroblasts or NSCs (~0.49% of TAF1 in NSCs; Table S3). Moreover, cTAF1 and cTAF1-35′ were significantly downregulated in XDP fibroblasts and NSCs (cTAF1 = 19.6% decrease in XDP, FDR = 5.8 x 10−4; cTAF1-35′ = 27.7% decrease in XDP, FDR = 3.9 x 10−4; Figure 4B, S4C and Table S3), but not in iNs. This decreased expression coincided with decreased exon usage in proximity to the SVA insertion and TAF1-32i termination site, which became more pronounced in exons distal to the SVA (range = 16.68% to 28.41% decreased expression in XDP, Figure 4C, S4A, Table S4). Expression of TAF1 protein was also decreased by ~18% on average in XDP NSCs compared to controls (Figure S4, G–H), consistent with the observed mRNA expression patterns (Table S6).
The TAF1-32i transcript was rare and detected exclusively in NSCs (1.3% of overall TAF1 expression in XDP NSCs), yet distinguished probands from controls (Figure 4B and Table S3). We further scrutinized this splicing in intron 32 and found multiple rare, aberrant splice junctions and an IR pattern that was most apparent in XDP NSCs (Figure 5A). We quantified this pattern based on the: (1) proportion of aberrant splice junctions, (2) absolute expression of intron 32 and (3) relative magnitude of IR. In XDP NSCs, aberrant splicing from exon 32 to intron 32 represented on average just 5% of the normal splicing of exon 32; however, the IR results were more significant as the absolute expression of intron 32 was dramatically higher in XDP compared to controls in both fibroblasts (433%, p = 2.03 x 10−35) and NSCs (1434%, p = 1.3 x 10−10). The coverage of intron 32 relative to the overall coverage of TAF1 indicated that, at its peak, the IR pattern was equivalent to 15.8% of the average coverage of the TAF1 coding region (Figure 5A). To confirm that this IR event was an unusual expression pattern relative to null expectations, we compared transcriptome-wide IR in XDP probands and controls. We surveyed 258,852 annotated introns and observed differential retention of 80 introns (0.03%), of which the TAF1 intron 32 IR was the most statistically significant, irrespective of directionality (FDR = 3.3 x 10−6; Figure 5B, Table S5). Intron 32 expression was negatively correlated to both cTAF1 (Spearman’s ρ = −0.68, p = 1.4 x 10−3) and overall TAF1 expression in NSCs (Spearman’s ρ = −0.8, p = 2.6 x 10−5, Figure 5C), consistent with the exon usage analyses (Figure 4C). The IR was not observed in iNs or in any previous studies of neural cells from our group (Sugathan et al., 2014) (Figure 5A), and in NSCs it was not detected distal to the SVA. All results were validated by Illumina and PacBio RNA CapSeq and in a PCR-based TaqMan assay that we designed for additional confirmation (Figures 5A, S4E, 6).
Figure 5. Aberrant expression of TAF1 intron 32 and transcriptome-wide significance in XDP NSCs.

(A) Composite plot demonstrates normalized Illumina sequencing coverage of TAF1 intron 32 in control (blue) and XDP (red) samples across three cell lines. Brown triangle and the vertical brown line indicate the SVA insertion site while shadowed areas represent TAF1 coding regions. Solid horizontal lines intersecting the Y axis show the average sequencing coverage of the TAF1 coding region in control (blue) and XDP (red) samples. X axis represents the genomic coordinates of human X chromosome with the SVA inserted. (B) Transcriptome-wide levels of IR among all 258,852 annotated introns in XDP vs. control NSCs (x-axis) plotted against significance levels (y-axis, log10 transformed). Significant IR changes (FDR < 0.05) are marked in orange. (C) Expression correlations among TAF1 intron 32 expression, overall TAF1 expression and TAF1 transcripts in fibroblasts (left), NSCs (middle) and iNs (right). Colors indicate Spearman correlation coefficients. Rows and columns are clustered based on Euclidean distance.
We probed the cell type specificity of this IR by testing other neuronal cells. Because iNs derived by expression of NGN2 in iPSCs bypass the NSC stage, we differentiated XDP and control NSCs to cortical neurons without NGN2 which produced cultures similarly enriched in glutamatergic neurons (Figure S5A). Consistent with the iNs, the IR signature and aberrant splicing pattern was not detected in these neurons (Figure S4F, 6C). To test an additional lineage, we also differentiated XDP and control NSCs into GABAergic neurons and quantified the IR signature in the Taqman assay, which confirmed the highest IR expression signature in NSCs, followed by iPSCs and fibroblasts, with low but detectable levels in XDP neuronal populations, and no expression in any control cells (Figure 6, Figure S5B).
Figure 6. Excision of the SVA rescues aberrant splicing and expression in intron 32 and expression of TAF1.

(A) Sashimi plot depicting coverage and splicing in intron 32 of TAF1 in control, XDP, and SVA-excised (ΔSVA) proband NSCs. (B) Normalized RNA-Seq counts in intron 32 of TAF1 5′ to the SVA insertion (left) and TAF1 (right) in proband NSCs, corresponding ΔSVA clones, and control cells (one clone per individual). (C) Relative expression of intron 32 splice variant in fibroblasts (Fibro), iPSCs, NSCs, iNs, NSC-derived cortical neurons, and GABAergic neurons from XDP, control, and ΔSVA lines. Graphs represent mean (+SEM) from clones generated for each cell type. See methods for total numbers and biological replicates of each genotype. Unpaired two-tailed t-test (fibro) or one-way ANOVA with Tukey’s multiple comparisons test was performed on each cell type. *p<0.05, ***p<0.001, ****p<0.0001, or n.s. = not significant.
CRISPR/Cas9- mediated excision of the SVA abolishes the intron 32 retention in XDP cells
We next tested the possibility that the SVA interfered with transcription to produce these aberrant transcripts using CRISPR/Cas9-mediated gene editing to excise the SVA from three XDP iPSC lines. Four clones from these parent lines (referred to as ΔSVA) had the same precise deletion points, which removed the SVA plus a 53 nt sequence between the SVA and the flanking protospacer adjacent motif (PAM) sites (Figure S6A). These clones were differentiated to NSCs, iNs, NSC-derived neurons, and GABAergic neurons (Figure S6B–E). In NSCs, excision of the SVA rescued the intron 32 XDP signature, reduced IR to levels comparable to controls, and decreased expression of the TAF1-32i transcript so that it was no longer detectable (Figure 6). Removal of the SVA also normalized overall TAF1 expression, as levels in the edited clones were indistinguishable from that in controls (p = 0.8, Figure 6B). These data suggest that the SVA was the primary driver of the IR signature observed in XDP cells and contributed to the overall reduction in TAF1.
Transcriptome-wide XDP molecular signatures are associated with pathways related to neurodevelopment and neurodegeneration
To interrogate the transcriptional changes associated with XDP-related sequence variation, we identified differentially expressed genes (DEGs) using GLMMs as described above, functional enrichment analysis (gene ontologies and pathways), and weighted gene co-expression network analysis (WGCNA) on all samples (all gene-level results provided in Table S6).
Consistent with the magnitude of TAF1 alterations and IR expression patterns, the strongest expression changes were observed in NSCs (number of DEGs after correction for multiple testing: fibroblasts = 29; NSCs = 400; iNs = 114; NSC-derived neurons = 20). Among the lineages, eight genes were consistently altered in multiple cell types, including TAF1 (Table S6). We did not observe any enrichment for gene ontology (GO) terms at FDR <0.05, although top terms in each cell type were “GDP binding” in fibroblasts (p =1.0 x 10−3), “response to ER stress” in NSCs (p = 1.3 x 10−4), and “regulation of cell shape” in neurons (p = 4.5 x 10−4). There was statistically significant overlap between co-expression module 2 in NSCs and module 5 in iNs, and the overlapping genes within these modules were enriched for the GO terms Axon Guidance and IRE1-mediated Unfolded Protein Response, among others (Figure 7). In a highly exploratory analysis, we also noted that profiling of 400 DEGs in NSCs from the ΔSVA lines suggested an overall trend for negative correlation of the log2 fold changes in XDP/Control and ΔSVA/XDP comparisons (R2 =0.22, p = 3.7 x 10−23; Figure S7A), which was supported by the observed clustering of ΔSVA clones closer to controls and carriers than XDP probands in principal component analysis of the 400 genes (Figure S7B). Twenty DEGs achieved statistical significance in opposite directions between the comparisons, including ATF3, involved in ER stress and signaling via eukaryotic initiation factor-2 (eIF2), which was reported as a common dysfunction in dystonia (Rittiner et al., 2016).
Figure 7. Co-expression modules with strongest enrichment for DEG in NSCs and neurons are enriched for cell growth and ER stress response.

(A) Modules with significant enrichment for differentially expressed genes (DEGs) in NSCs (left) and iNs (right). The number of DEGs indicate the number of genes included in WGCNA analyses for each cell type. Color represents the significance of enrichment, and the number indicates the number of overlapping genes. Modules with the most significant enrichments for DEGs at FDR levels are outlined and the overlap between modules is represented in the Venn diagram with the corresponding enrichment p-value (center). (B) Significantly enriched gene ontology terms in 110 overlapping genes from Module 2 in NSCs and Module 5 in iNs.
Discussion
The contribution of rare noncoding variation in human disease is an area of intensive study. There are few examples of noncoding variants causally linked to Mendelian disorders, yet it is known that some dominant-acting noncoding mutations confer substantial risk (Mathelier et al., 2015). Retrotransposons are a potential source of regulatory variation, and in the human genome there are three classes which remain active: LINE, Alu, and SVA. Some have been linked to disease, including insertions that affect transcription and splicing (Kaer and Speek, 2012). Consistent with that pattern, the genome and transcriptome assembly reported here narrowed the XDP causal locus to a genomic segment including only TAF1 and discovered that an intronic SVA insertion is associated with altered splicing and expression of the host gene. These data support the notion that intronic retroelements can be associated with transcriptional interference and have significant pathogenic consequences.
The de novo transcriptome assembly with deep targeted sequencing enabled unbiased evaluation of all transcripts in the linkage region, identifying an XDP signature involving aberrant splicing and IR in proximity to the SVA. Removal of the SVA rescued this signature, suggesting it was the likely driver of these effects. Although IR events have been regarded as rare consequences of aberrant splicing (Jaillon et al., 2008; Roy and Irimia, 2008), they are in fact prevalent in mammalian transcriptomes and regulate gene expression (Braunschweig et al., 2014; Jacob and Smith, 2017; Middleton et al., 2017). This regulation may “fine tune” transcript levels, as IR-transcripts may trigger nuclear restriction, nonsense-mediated mRNA decay, and/or turnover via exosomes to prevent their translation (Ge and Porse, 2014; Jacob and Smith, 2017). As a result, IR-transcripts may undergo rapid turnover, exist at low steady-state levels, and correlate with decreased overall transcript levels, consistent with the pattern in XDP cells.
Of the 85 known diseases associated with active retroelements, seven (including XDP) are linked to SVAs, five of which are inserted in introns and induce exon skipping and/or exonization of SVA sequences (Kaer and Speek, 2012). In XDP cells, all IR in intron 32 terminated proximal to the SVA insertion site. A similar pattern was reported for an intronic SVA in CASP8 which resulted in significant IR and decreased exon expression (Stacey et al., 2016). Because intron excision occurs during transcription, its precision varies with the elongation rate of RNAPII (Fong et al., 2014; Jimeno-Gonzalez et al., 2015) which can be diminished due to (1) binding of a competing RNAPII which inhibits progression of the RNAPII transcribing the host gene; (2) changes to local chromatin; and (3) the presence of guanine-rich motifs which form quadruplex structures (Kaer and Speek, 2012; Kejnovsky and Lexa, 2014). These interactions are examples of transcriptional interference in which the RNAPII transcribing the host gene may be displaced (Hao et al., 2017; Shearwin et al., 2005). Aberrant RNA processing has also been linked to neurodegeneration due to formation of RNA foci that sequester RNA binding proteins (Gallo et al., 2005; Liu et al., 2017). In this study, the decreased exon usage downstream of exon 32 might be consistent with transcriptional interference induced by the SVA, but elucidation of the specific mechanism requires further investigation.
The TAF1 transcript reductions detected in XDP cells were relatively moderate, which may be consistent with late-onset neurodegenerative disorders such as XDP where individuals appear neurologically normal until adulthood. Larger changes in TAF1 expression may instead have severe consequences given that, in mice and C. elegans, TAF1 is expressed early in embryonic development and is required for transcription and pluripotency (Pijnappel et al., 2013; Walker et al., 2004; Wang et al., 2006). TAF1 is also under strong evolutionary constraint in males, suggesting that only moderate decreases in TAF1 expression may be tolerated yet still exert subtle effects over time. In this study, the decreased TAF1 expression and IR were detected in dividing cells but not neurons, which has multiple implications. It is possible that key pathogenic events in XDP occur primarily in neural progenitors or in glia, which we did not examine. Alternatively, the neurons differentiated here may not have recapitulated the neurons most vulnerable in XDP, either in terms of lineage and/or maturation. Further studies are warranted in other lineages, and ultimately in postmortem samples, though our studies strongly suggest that the SVA is driving the yet unknown pathogenic mechanism in this disorder. Supporting this notion, in a parallel study we have now observed that a hexameric repeat length within the SVA varies between probands, and that the age of disease onset in XDP inversely correlates with the length of this repeat (Bragg et al., 2017).
These data suggest that XDP may join a growing list of human diseases involving defective RNA splicing, IR, and transcriptional alterations driven by transposable elements. For some of these conditions, considerable progress has been made in designing strategies to correct splicing events using small molecules and antisense oligonucleotides (Faravelli et al., 2015; Shimizu-Motohashi et al., 2016). The potential to normalize this IR signature by manipulating the SVA insertion in this study, coupled with rapid advances in genome editing technologies, raises the possibility that in vivo manipulation of this sequence could eventually have clinical benefit. The observations that XDP and the previously reported TAF1 neurodevelopmental syndrome arise from different classes of perturbation within the same gene may propose a continuum of syndromic features associated with TAF1 disruption that are driven by divergent mutational mechanisms, ranging from coding mutations associated with an early onset developmental disorder to a noncoding SVA insertion with later onset neurodegeneration. These studies also illustrate the potential for layered genomic analyses to provide a roadmap for unsolved Mendelian disorders that is capable of simultaneously capturing coding and noncoding regulatory variation and interpreting their functional consequences in human disease.
STAR Methods
All critical reagents, cell lines, and software used and/or generated in this study are listed in the Key Resource Table along with corresponding vendor information and/or citations, where appropriate.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse-anti-TAF1 | This paper (Mark Timmers, University of Freiburg, Freiburg, Germany) | N/A |
| Rabbit-anti-Oct3/4 | Cell Signaling Technology | Cat# 2750S, RRID:AB_823583 |
| Rabbit-anti-Nanog | Abcam | Cat#ab21624; RRID: AB_446437 |
| Mouse-anti-SSEA4 | Millipore | Cat# MAB4304, RRID:AB_177629 |
| Mouse-anti-Tra-1-60 | Millipore | Cat# MAB4360, RRID:AB_2119183 |
| Mouse-anti-Tuj1 | Abcam | Cat# ab78078, RRID:AB_2256751 |
| Mouse-anti-MAP2 | Abcam | Cat# ab11267, RRID:AB_297885 |
| Rabbit-anti-SOX1 | Abcam | Cat# ab87775, RRID:AB_2616563 |
| Mouse-anti-NeuN | Abcam | Cat# ab104224, RRID:AB_10711040 |
| Rabbit-anti-doublecortin | Abcam | Cat# ab18723, RRID:AB_732011 |
| Rabbit-anti-GABA | Sigma | Cat# A2052, RRID:AB_477652 |
| Mouse anti-Hsp70 | Santa Cruz Biotechnology | Cat# sc-24, RRID:AB_627760 |
| Alexa Fluor®488-anti- rabbit IgG | Thermo Fisher Scientific | Cat# A-11008, RRID:AB_143165 |
| Alexa Fluor®594- anti- mouse IgG | Thermo Fisher Scientific | Cat# A-11005, RRID:AB_2534073 |
| Alexa Fluor®594-anti-rabbit IgM | Thermo Fisher Scientific | Cat# A-21044, RRID:AB_2535713 |
| HRP-conjugated anti-mouse IgG | GE Healthcare | Cat# NA931, RRID:AB_772210 |
| Bacterial and Virus Strains | ||
| Cytotune 2.0 Sendai virus Reprogramming Kit | Thermo Fisher | Cat#A16517 |
| OneShot TOP10 chemically competent E. Coli | Thermo Fisher | Cat#C404003 |
| OneShot Stbl3 chemically competent E. Coli | Thermo Fisher | Cat#C737303 |
| LV-TetO-Ngn2-PURO | Zhang et al., 2013 | N/A |
| LV-rtTA | Zhang et al., 2013 | N/A |
| Biological Samples | ||
| Human XDP male, female carrier, and control DNA | MGH Collaborative Center for XDP, this paper | N/A |
| External RNA Controls Consortium (ERCC) RNA Spike-In Mix | Thermo Fisher Scientific | Cat#4456740 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle’s Medium (DMEM) | Thermo Fisher Scientific | Cat#12634010 |
| Dulbecco’s Modified Eagle’s Medium (DMEM):F12 | Thermo Fisher Scientific | Cat#31985-070 |
| Opti-MEM Medium | Thermo Fisher Scientific | Cat#A1647801 |
| PSC Neural Induction Medium | Thermo Fisher Scientific | Cat#21103049 |
| Neurobasal Medium | Stemcell Technologies | Cat#05850 |
| mTeSR1 Medium | Stemcell Technologies | Cat#05790 |
| BrainPhys Neuronal medium | Gemini | Cat#900-20B |
| Fetal bovine serum | Thermo Fisher Scientific | Cat#10828028 |
| Knockout Serum Replacement (KOSR) | Thermo Fisher Scientific | Cat#12634010 |
| MEM-Nonessential amino acids | Thermo Fisher Scientific | Cat#11140050 |
| CultureOne | Thermo Fisher Scientific | Cat#A332020 |
| EmbryoMax 0.1% gelatin solution | Millipore/Sigma | Cat#ES-006 |
| Penicillin-Streptomycin | GE Healthcare | Cat#SV30010 |
| Accutase | Sigma | Cat#A6964-100ML |
| Y-27632 | Tocris | Cat#1254; CAS: 129830-38-2 |
| Geltrex | Thermo Fisher Scientific | Cat#12760013 |
| Glutamax supplement | Thermo Fisher Scientific | Cat#35050061 |
| N2 supplement | Thermo Fisher Scientific | Cat#17502048 |
| B27 supplement with retinol | Thermo Fisher Scientific | Cat#17504044 |
| B27 supplement w/o retinol | Thermo Fisher Scientific | Cat#12587010 |
| bFGF | Millipore | Cat#GF003; CAS: 106096-93-9 |
| BDNF | Shenandoah | Cat#100-01-100UG |
| NT-3 | Shenandoah | Cat#100-99-100UG |
| GDNF | Shenandoah | Cat#100-02-100 ug |
| rh/rActivin A | R&D Systems | Cat#338-AC |
| doxycycline | Clontech | Cat#631311; CAS: 24390-14-5 |
| Cytosine-β-D-arabinofuranoside | Sigma | Cat#C1768; CAS: 147-94-4 |
| puromycin | Clontech | Cat#631305; CAS 58-58-2 |
| Dispase II | Thermo Fisher Scientific | Cat#17105041; CAS: 42613-33-2 |
| Valproic Acid sodium salt | Millipore/Sigma | Cat#P4543; CAS: 1069-66-5 |
| Dibutyryl cAMP sodium salt | Millipore/Sigma | Cat#D0260; CAS: 16980-89-5 |
| 2-Mercaptoethanol | Millipore/Sigma | Cat#M3148; CAS: 60-24-2 |
| poly-D-lysine hydrobromide | Millipore/Sigma | Cat#P6407; 27964-99-4 CAS: |
| Laminin | Thermo Fisher Scientific | Cat#23017015; CAS: 114956-81-9 |
| RNase A | Millipore/Sigma | Cat#R4875-100MG; CAS: 9001-99-4 |
| L-Ascorbic Acid | Millipore/Sigma | Cat#A5960; CAS: 50-81-7 |
| Polybrene | EMD Millipore | Cat#TR-1003-G; CAS: 28728-55-4 |
| Ultrapure EDTA, pH 8.0 | Thermo Fisher Scientific | Cat#15575020 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000008 |
| Fluo4-AM | Thermo Fisher Scientific | Cat#F14201 |
| Kianate | Tocris | Cat#0222; CAS: 487-79-6 |
| CNQX | Tocris | Cat#0190; CAS: 115066-14-3 |
| TRIzol® | Thermo Fisher Scientific | Cat#15596026 |
| MycoZap | Lonza | Cat#LT07-818 |
| Lysis buffer AM1 | Active Motif | Cat#100566 |
| Protease inh. cocktail | Active Motif | Cat#37490 |
| Super signal West Pico substrate | Thermo Fisher Scientific | Cat#34580 |
| Critical Commercial Assays | ||
| Taqman hPSC ScoreCard™ Panel | Thermo Fisher Scientific | Cat#A15870 |
| DirectZol® RNA miniprep kit | Zymo Research | Cat#R2050 |
| High Capacity cDNA Reverse Transcription kit | Thermo Fisher Scientific | Cat#4374966 |
| PowerUp™ SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A25741 |
| Phusion® High Fidelity Polymerase | New England Biolabs | Cat#M0530L |
| BbsI restriction enzyme | New England Biolabs | Cat#R0539 |
| AccuPrime™ GC-Rich DNA Polymerase | Thermo Fisher Scientific | Cat#12337016 |
| PrimeSTAR® GXL DNA Polymerase | Takara | Cat#R050A |
| SuperScript™ III First-Strand Synthesis SuperMix | Thermo Fisher Scientific | Cat#18080400 |
| Taqman® Fast Advanced Master Mix | Thermo Fisher Scientific | Cat#4444557 |
| Custom Taqman® assay (i32) | Thermo Fisher Scientific | Cat#AJWR28J |
| GUSB Taqman® assay | Thermo Fisher Scientific | Cat#Hs009627_m1 |
| QIAquick Gel Extraction kit | Qiagen | Cat#28706 |
| TruSeq® Stranded mRNA Library kit | Illumina | Cat#RS-122-2101 |
| Library Quantification Kit | Kapa Biosystems | Cat#KK4854 |
| SureSelectXT Target Enrichment System | Agilent | Cat#G9611A |
| SureSelectXT2 Target Enrichment System | Agilent | Cat#G9621A |
| Deposited Data | ||
| dbGAP | Sequencing data from samples collected through DPRB (see Table S1) | phs001525.v1.p1 |
| Experimental Models: Cell Lines | ||
| Human embryonic kidney 293T cells | ATCC | Cat#CRL-3216 |
| CF1 mouse embryonic fibroblasts, irradiated | Thermo Fisher Scientific | Cat#A34180 |
| Human XDP male, carrier female, and control fibroblasts | MGH, this study | N/A |
| Human XDP male, carrier female, and control induced pluripotent stem cells | MGH, this study | N/A |
| Experimental Models: Organisms/Strains | ||
| Fox Chase SCID® mice | Charles River | 236 |
| Oligonucleotides | ||
| For oligonucleotide sequence information see Table S8 | This paper | N/A |
| Recombinant DNA | ||
| pGuide sgRNA expression vector | Ding et al., 2013 | Addgene plasmid 64711 |
| pCas9-GFP | gift from Kiran Musunuru | Addgene plasmid 44719 |
| pMD2.G | gift from Didier Trono | Addgene plasmid 12259 |
| pCMVR8.74 | gift from Didier Trono | Addgene plasmid 22036 |
| pTet-O-Ngn2-puro | Zhang et al., 2013 | Addgene plasmid 52047 |
| pFUW-M2rtTA | Hockemeyer et al., 2008 | Addgene plasmid 20342 |
| pBACe3.6 | BACPAC Resources, Children’s Hospital Oakland Research Institute | N/A |
| Software and Algorithms | ||
| Geneious | Biomatters | https://assets.geneious.com/documentation/geneious/release_notes.html#v10.1.3 |
| TIDE | http://tide.nki.nl | |
| DISCOVAR de novo | (Weisenfeld et al., 2014) | ftp://ftp.broadinstitute.org/pub/crd/DiscovarDeNovo/ |
| Supernova | (Weisenfeld et al., 2017) | https://support.10xgenomics.com/de-novo-assembly/software/overview/latest/welcome |
| Genome Analysis Toolkit (GATK, v3.5) | (DePristo et al., 2011) | https://software.broadinstitute.org/gatk/ |
| FastQC (v.0.11.2) | Babraham Institute, Cambridge, UK | www.bioinformatics.babraham.ac.uk |
| BWA-backtrack (v.0.7.10-r789) | (Li and Durbin, 2009) | https://github.com/lh3/bwa/blob/master/NEWS.md |
| SAMBLASTER (v.0.1.1) | (Faust and Hall, 2014) | https://github.com/GregoryFaust/samblaster |
| Sambamba (v.0.4.6) | (Tarasov et al., 2015) | http://lomereiter.github.io/sambamba/ |
| PicardTools (v1.115) | picard.sourceforge.net | https://github.com/broadinstitute/picard |
| Samtools (v1.0) | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| BamTools (v2.2.2) | (Barnett et al., 2011) | https://github.com/pezmaster31/bamtools |
| BWA-MEM (0.7.5a-r418) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| Ensembl Variant Effect Predictor (VEP, v86) | www.ensembl.org | https://useast.ensembl.org/info/docs/tools/vep/script/vep_download.html |
| SangeranalyseR | Australian National University, Lanfear Lab | https://github.com/roblanf/sangeranalyseR |
| SangerseqR | (Hill et al., 2014) | http://bioconductor.org/packages/release/bioc/html/sangerseqR.html |
| IRFinder | (Middleton et al., 2017) | https://github.com/williamritchie/IRFinder/wiki |
| 4Peaks Sequence Viewer | Nucleobytes B.V. | https://nucleobytes.com/4peaks/index.html |
| Trimmomatic (v0.36) | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR (2.5.2b) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Trinity (v2.2.0) | (Grabherr et al., 2011) | https://github.com/trinityrnaseq/trinityrnaseq/wiki |
| RSEM (v1.2.31) | (Li and Dewey, 2011) | https://deweylab.github.io/RSEM/ |
| Bowtie2 (v2.1.0) | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| HTSeq (v0.6.1) | (Anders et al., 2015) | https://htseq.readthedocs.io/en/release_0.9.1/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Fastcluster | (Mullner, 2013) | http://danifold.net/fastcluster.html |
| DynamicTreeCut | (Langfelder et al., 2008) | https://cran.r-project.org/web/packages/dynamicTreeCut/index.html |
| TopGO (v2.22.0) | (Alexa and Rahnenfuhrer, 2016) | http://bioconductor.org/packages/release/bioc/html/topGO.html |
| Weighted correlation network analysis (WGCNA | (Langfelder and Horvath, 2008) | https://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/ |
| GraphPad Prism 7 | GraphPad Software, Inc. | https://www.graphpad.com/scientific-software/prism/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Data with available consents for this study are available from dbGAP (Accession Number phs001525.v1.p1). Fibroblast lines have been deposited at the NINDS Human Cell and Data Repository (http://ninds.genetics.org; Rutgers, NJ), and iPSC clones are publicly available from WiCell (www.wicell.org). Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. M.E. Talkowski (mtalkowski@mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical Evaluation of Subjects and Sample Collection
Subjects recruited for this study included individuals who (1) had a confirmed diagnosis of XDP based on prior genetic testing; (2) exhibited clinical features consistent with XDP and reported ancestry to Panay; (3) were first-degree relatives of individuals with a confirmed or suspected diagnosis of XDP; or (4) unaffected individuals from Panay island. Participants were evaluated at Massachusetts General Hospital (Boston, MA) or in regional clinics in Panay Island affiliated with Jose Reyes Memorial Medical Center (Manila, Philippines) (Source in Table S1: DPRB-P or GCNHx). The study was approved by institutional review boards at both participating institutions, and all participants provided written informed consent. In addition, through an international XDP genomics consortium, we obtained samples from previous studies that investigated the genetic cause of XDP, including archival DNA specimens from early linkage studies provided by Dr. Ulrich Müller (University of Giessen, Giessen, Germany; Source: Archival-M) and Dr. Kirk Wilhelmsen (University of North Carolina, Chapel Hill, NC USA; Source: Archival-W), Table S1), and from more recent genotyping/sequencing efforts (Domingo et al., 2015; Source: ING-L). Collection of these samples and clinical evaluation of donor subjects were previously reported (Domingo et al., 2015; Nolte et al., 2003; Wilhelmsen et al., 1998) and performed at any of the following institutions: St. Luke’s Medical Center (Quezon City, Philippines), Metropolitan Medical Center (Manila, Philippines), and Institute of Neurogenetics (Lübeck, Germany). In addition to collecting samples from unaffected family members of XDP probands, 319 healthy control individuals (216 males, 103 females) with no history of XDP in immediate family members were included to represent an additional ethnic control group. The genotypes of all subjects were determined by evaluating haplotype markers using PCR amplification of genomic DNA (gDNA) extracted from blood, followed by Sanger sequencing of amplicons. All probands who met the inclusion criteria defined above were included in this study, as well as all haplotype-negative, unaffected control subjects. The total cohort of 792 individuals (652 males and 150 females) was stratified based on genotype and clinical disease status as follows: 352 affected XDP male probands, 403 unaffected haplotype-negative controls, 23 heterozygous XDP carrier females, and 14 XDP haplotype-positive males who were below the age of disease onset and asymptomatic at the time of exam, referred to here as nonmanifesting carriers (NMCs). The heterozygous XDP carrier females were all neurologically normal upon exam and did not exhibit any XDP-related symptoms. Table S1 provides details for all study participants, including gender/sex, pedigree relationships, age at sample collection, and available clinical data. The mean age of initial symptom manifestation was 42.31 ± 8.3 years (range = 20–67 years) among the probands for whom the age of disease onset could be determined (n = 303),. A subset of these probands (n = 263) were able to provide further clinical information about the pattern of initial symptoms, indicating that 78.7% of these individuals initially presented with dystonia while 17.1% first presented with parkinsonism (Table S1).
For the XDP and control subjects who were directly evaluated as part of this study, comprehensive neurological exams were performed by movement disorder neurologists specializing in XDP. Blood was collected for gDNA isolation from all participants. On a subset of individuals, skin biopsies for fibroblast derivation were performed as previously described (Ito et al., 2016). Tissue explants were seeded into culture dishes in growth medium [Dulbecco’s Modified Eagle Medium (DMEM) with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin] and placed under sterile coverslips to facilitate attachment. Primary fibroblasts typically migrated out from the explant over the following 2 weeks in culture. Cells were collected by trypsinization, expanded in culture, and cryopreserved pending analysis.
In vitro modeling of XDP patient cells
XDP cell models used in this study consisted of: primary skin fibroblasts, iPSCs, iPSC-derived NSCs, and iPSC-derived neurons differentiated by three different methods (NGN2-induced cortical neurons, cortical neurons generated by directed differentiation, and GABAergic neurons). For each cell type, we compared cells from affected XDP probands, heterozygous XDP carrier females, and unaffected controls. CRISPR/Cas9-based genome editing was also performed on iPSCs from affected XDP males to excise the SVA, producing four edited clones designated as ΔSVA that were compared with patient and control lines in some experiments. Table S1 indicates the individuals from whom cell lines were established, and the protocols used for propagation, iPSC reprogramming and characterization, and neural differentiation are described below (see Method Details). For iPSCs we reprogrammed fibroblasts from 5 affected XDP males, 4 XDP heterozygous carrier females, and 3 unaffected controls with 2 iPSC clones for each parent fibroblast line (24 clones total). For each experiment, the number of successfully differentiated clones used for analysis is specified in the main text.
In vivo models
For initial confirmation that iPSCs exhibited trilineage potential, a teratoma formation assay was performed in which aliquots of iPSC lines were injected into mice to evaluate the generation of tumors bearing tissue from all three germ layers. Specific pathogen-free male Fox Chase SCID mice-beige (Charles River Laboratories; Wilmington, MA USA) were used for these analyses, which were performed by the Genome Modification Facility at Harvard University under protocols approved by the Harvard Institutional Animal Care and Use Committee (IACUC). Animals were housed under standard conditions with access to food and water, and health status was monitored by approved veterinary staff. Six iPSC clones were evaluated (2 each of an affected XDP male, heterozygous XDP carrier female, and control), with each clone injected into a single mouse. Details of the assay protocol and data analysis are provided below (see Method Details). Because the teratoma assay provides only a qualitative assessment documenting the presence or absence of tissue from each germ layer within an iPSC-derived tumor, we optimized the Taqman® hPSC ScoreCard™ Panel (Thermo Fisher) which is based on qPCR of germ layer marker genes as an alternative, semi-quantitative method (see Method Details). The ScoreCard™ assay quantifies marker gene expression in embryoid bodies (EBs) derived from iPSCs in culture. The six iPSC clones evaluated in the teratoma assay were reexamined by the ScoreCard™ method, and all subsequent iPSC clones were characterized by the latter assay only.
Power Analysis
We determined power to replicate the IR results by calculating IR as a ratio of correct splicing to intron expression. Under the assumption that the log transformed IR was normally distributed, we fit a linear mixed model (LMM; described in detail in Quantification and Statistical Analysis). In the NSC experiments, where the largest IR was observed, we compared differentiated clones from 3 unaffected control subjects and 5 affected XDP probands, and observed a mean increase in the TAF1 IR ratio within intron 32 of 1434% among XDP clones relative to control cells. The power to detect this effect at FDR of 0.05 was 0.7873. Regarding the more modest changes observed in differential expression analyses, we calculated power under a negative binomial distribution assumption using our existing dataset of dispersion = 0.05838; 29 samples would be required per group to achieve 80% power to detect a fold-change of 1.5 at an adjusted FDR = 0.05.
METHOD DETAILS
Experimental design
For the genomics analyses, we first performed a pilot study to evaluate four distinct analytic methods in an initial cohort of nine samples (Figure 1B), consisting of three probands (33109, 33363, and 33808) and three members each from two families (pedigrees 22 and 27). The four methods examined in this pilot set were: (1) reference-free, de novo assembly of the XDP haplotype using PCR-free Illumina paired-end 250 bp (n=6) and 10X Genomics linked-read sequencing (n=3); (2) long-insert “jumping library” whole genome sequencing (liWGS, n=6) (3) Pacific Biosciences long-read single molecule real-time sequencing (PacBio SMRT) of bacterial artificial chromosome (BAC) clones of the XDP haplotype (n=1); and (4) targeted capture deep sequencing (CapSeq) of the XDP associated region (n=16). Based on the performance of the CapSeq assay, we expanded that sequencing to an additional batch of 101 subjects, followed by replication in a batch of 672 subjects to assess the full allelic diversity of the region (Figure 1B). To interrogate the functional impact of genomic variants identified in these analyses, we derived cell models from subjects representing ten multi-generational families which were used in both genome-wide and targeted transcriptomics experiments. These cell models consisted of: primary skin fibroblasts (13 probands, 12 heterozygous female carriers, and 20 unaffected relatives), iPSCs (5 XDP probands, 4 heterozygous XDP carrier females, 3 unaffected controls with 2 clones per individual for a total of 24 clones), iPSC-derived NSCs and neurons, and CRISPR/Cas9-based edited iPSCs in which the SVA was excised from patient cell lines (4 clones). Cell lines and donor individuals are listed in Figure 1B and Table S1, while numbers of differentiated clones used in each experiment are specified in the main text.
Genomics methods
PCR-free and linked-read deep whole-genome sequencing and assembly
DNA was extracted from blood samples taken from two affected XDP probands, two heterozygous XDP female carriers and two unaffected controls (pedigrees 22 and 27). PCR-free fragment shotgun libraries were generated using a ‘with-bead pond library’ construction protocol developed at the Broad Institute with the following modifications: 500 ng of gDNA was sheared to 500 bp fragments using a Covaris E210™ system (Covaris, Waltham, MA), followed by bead purification (Agencourt AmPure XP SPRI) and library preparation (KAPA library kit; KAPA Biosystems, Wilmington, MA), including end-repair, A-tailing, and ligation of barcoded adapters for Illumina sequencing. After final purification, libraries were eluted off the SPRI beads and quantified by qPCR using the KAPA Library Quant kit and an Agilent TapeStation. All libraries were sequenced using 250 cycle paired-end sequencing on an Illumina HiSeq 2500 (Illumina, Inc., San Diego, CA) at the Broad Institute Genomics Platform. All data were aligned to the human reference genome GRCh38 for reference-guided assembly, and reference-free assembly was performed using DISCOVAR de novo. The known XDP-specific variants were verified in the assembled region from the pedigree 22 proband.
For haplotype phasing and structural variant detection, the pedigree 22 trio was sequenced using linked-read whole-genome sequencing (10X Genomics, Pleasanton, CA). DNA was isolated from cells using an optimized salting out method. Cells were digested in lysis solution overnight at 37°C. After digestion, saturated sodiu m chloride solution was added to the reaction, mixed by inversion, and followed by centrifugation to pellet proteins. The supernatant containing the DNA was transferred to a new tube and the precipitated protein pellet was discarded. Absolute ethanol was added to the reaction and mixed by inversion, followed by centrifugation to pellet the DNA. The ethanol supernatant was discarded and the DNA pellet was re-suspended in low TE buffer (10 mM Tris-Cl; 0.5 mM EDTA; pH 9.0). The DNA was allowed to homogenize at room temperature for one hour before quantification. Linked-Read library preparation was performed using the 10x Genomics Chromium Controller Instrument and 10x Genomics Chromium Genome v2.0 by methods which are available at (https://support.10xgenomics.com/permalink/5H0Dz33gmQOea02iwQU0iK). The sequencing generated 1200 million reads per samples. De novo assembly was performed using the Supernova Linked-Read assembler (https://support.10xgenomics.com/de-novo-assembly/software/overview/welcome).
BAC generation, sequencing, and assembly
Genomic DNA from proband 33109 (pedigree 12) carrying the entire haplotype region was digested with BamHI and used to generate a BAC library (Amplicon Express; Pullman, WA) in vector pBACe3.6. Nine potentially positive clones were isolated by hybridization using an 856 bp probe located downstream of the SVA (chrX:70,674,877-70,675,733) between exons 34 and 35 of TAF1. Genotyping of the DSCs and the SVA was performed using Sanger sequencing and verified one positive clone. A second round of hybridization was carried out to obtain BAC clones containing the 5′ end of TAF1 using a 201 bp probe located upstream of the SVA (chrX:70,613,608-70,613,809) between exons 21 and 22 of TAF1. Three clones were identified but only one could be verified by PCR. BAC end sequencing using vector primers from the two verified clones suggested they spanned TAF1 from a region 40 kb upstream of the 5′UTR to 55kb downstream of the 3′ UTR. These two clones were then subjected to long-read sequencing using the PacBio SMRT technology to verify their sequence and assemble a contig. A library was made from the BACs (Amplicon Express) using the PacBio 20 Kb library preparation and sequenced on the PacBio RSII instrument (DNA Link, San Diego, CA). A single cell was used to generate 150,292 reads, at an average read length of 5,028. Reads were filtered for vector contamination and poor quality score, after which 51,210 reads with average length of 10,416 were used to assemble the region. The region was assembled through SMRT-portal (http://www.pacb.com/products-and-services/analytical-software/devnet/), using HGAP2 protocol with default parameters yet with genome size set to 200 kb. After the assembly, a 201,921bp long single contig was obtained corresponding to GRCh37 coordinates 70546230bp to 70747084bp on the X chromosome.
Long insert “Jumping Library” preparation and analysis
Custom “jumping libraries” were prepared for the pedigree 22 trio and three other probands (33109, 33363 and 33808, Table S1), using our custom “jump shear” protocol optimized to 3.5 kb inserts and sequenced on an Illumina 2000 platform with 2x51 bp reads as previously described (Redin et al., 2017; Talkowski et al., 2012). In brief, gDNA was randomly sheared on an E220evolution™ system (Covaris), and subsequently size-selected via agarose gel purification to a target fragment size of 3–5kb. These fragments were circularized with adapters containing an EcoP15I recognition site and a biotinylated thymine. The circularized DNA was EcoP15I restriction digested to capture 27 bp of fragment ends and pulled down using streptavidin beads via the biotinylated thymine. Standard Illumina Y-adapters were ligated to the ends of these fragments, and the final libraries were sequenced with 2x50bp reads on an Illumina HiSeq 2000 instrument (Illumina, San Diego, CA, USA) at The Broad Institute of MIT and Harvard. Library barcodes were de-multiplexed and filtered, and read quality was assessed with FastQC v0.11.2. Reads were trimmed and aligned with BWA-backtrack v0.7.10-r789 (Li and Durbin, 2009) to the human reference genome (GRCh37). Duplicates were marked with SAMBLASTER v0.1.1 and reads were further processed prior to SV detection using a series of tools, including sambamba v0.4.6, PicardTools v1.115, Samtools v1.0, and BamTools v2.2.2, with all algorithms and pipelines described in (Brand et al., 2015; Collins et al., 2017).
DNA Capture-Sequencing (CapSeq) assay
A capture region on the X chromosome spanning NLGN3 to CXCR3 (X:70398800-70861400) was targeted using Agilent SureSelect XT design and following the Manufacturer’s instructions (Agilent). Capture libraries were prepared from DNA extracted from each proband, carrier and unaffected subject (n = 117). Three micrograms of gDNA was sheared to approximately 175 bp fragments using the Covaris Focused-ultrasonicator (Covaris; Woburn, MA). DNA fragments were end-repaired, adenylated, ligated to adapter oligos and then amplified with 5 cycles of PCR as recommended. After quantification, 750 ng of each amplified DNA sample was hybridized overnight with the capture library. Following capture cleanup, each gDNA library was amplified with an additional 16 cycles of PCR, which also tagged each sample with an index-specific barcode. Final products were quantified using the 2200 TapeStation (Agilent) and pooled for rapid mode sequencing on the Illumina system. In the second round of capture-sequencing (n=672), the Agilent SureSelect XT2 kit and a modified bait design (based on the initial region in the first capture experiment) was used. From each sample, one microgram of genomic DNA was sheared to approximately 175-bp fragments using the Covaris Focused-ultrasonicator. DNA fragments were end-repaired, adenylated, ligated to adapter/indexing oligos and then amplified with 6 cycles of PCR. Pools of 16 samples (1500 ng total, 93.75 ng per sample) were then hybridized overnight with the capture library. Following capture cleanup, each gDNA library was amplified with 13 cycles of PCR. Final products were quantified using the 2200 TapeStation (Agilent) and 6 pools were sequenced at a time on each lane of an Illumina HiSeq2000 platform (paired-end 100 bp reads).
Read pairs were aligned to GRCh37 with BWA-MEM 0.7.5a-r418 (Li and Durbin, 2009). Picard Tools and samtools were used to sort and index the alignments and mark duplicates. The CapSeq data resulted in 2.7M reads per sample (read counts range 0.5M - 12.8M per sample) (Table S2). The sequencing data covered 96% of bases of the targeted region with a median coverage of 70X. The Genome Analysis Toolkit (GATK) Haplotype caller v3.5 was used for base quality recalibration, indel realignment, single nucleotide variant (SNV) and indel calling, and genotyping as per published best practice protocols. Because the region of interest is on the X chromosome, the haploidy option was used in GATK (1 for males, 2 for females). Scalpel (Fang et al., 2016) was also used to call indels and overlapping calls from two tools were used. GATK haplotype caller detects or reports variants based on alignments, in our case BWA-mem, while Scalpel performs localized micro-assembly of specific regions of interest with the goal of detecting mutations with high accuracy and increased power. SNVs and indels were annotated using Ensembl Variant Effect Predictor (VEP) tool. Five sample with low coverage, eight misclassified samples and four duplicate samples were excluded from the further analysis and haplotype reconstruction. The females X-chromosomes were phased based on the genotypes in male members of the family whenever possible.
Annotation of XDP haplotypes
The annotated H1 haplotype included all 54 shared variants and was the most frequent among XDP probands. All six additional haplotypes were variants of the H1 haplotype that were generated following recombinations (Figure 2A). The second most frequent haplotype among probands was annotated H2, and involved a recombination between nucleotide positions 70439816 and 70482458 on the X chromosome, as well as a reversion of a deletion (DSCn3) to the reference allele at base position 70521288 compared to the canonical H1 haplotype. The annotated H3 haplotype involved an apparent historical recombination between DSCr5 and DSCr6 occurring 5′ to GJB1. All remaining haplotypes involved at least one historical recombination and one or more altered alleles. There was an observed recombination in one of our pedigrees that was annotated as H7, and this recombination defined the distal site of the core shared region among probands as it occurred in a carrier mother between DSC1 and DSC3 (between 70733510 and 70749635, H7, Table S1, pedigree 27, 34427; Figure 2C). Haplotypes for all XDP probands and NMCs are listed in Table S1. Allele frequencies in probands, controls and carriers along with predicted functional consequence of all variations observed are provided in Table S2.
Validation of novel DSCs by Sanger sequencing
Validation experiments were performed to estimate specificity for a subset of DSCs detected in this study. These analyses were performed for 17 variants with sub-threshold sequencing depth across all samples from CapSeq, with all variants and data provided in Table S2. Primers for both PCR and Sanger sequencing were designed to +/− 500 bp flanking regions of each variant using Primer3 (v. 4.0). If flanking regions were dominated by low-complexity elements (e.g., LINE, SINE, AT-rich, simple repeats), a nested PCR strategy was employed by designing an outer set of PCR primers, flanking +/− 5 kb of variant, in conjunction with inner PCR/sequencing primers flanking +/− 500 bp of variant. PCR was performed using either Phusion® High-Fidelity Polymerase or PrimeSTAR® GXL DNA Polymerase, per manufacturer’s recommendations; annealing temperatures were empirically determined using gradient PCRs (from 60–72°C) and the final annealing temperatures are indicated in Table S8). Bands of appropriate size were gel extracted using QIAquick Gel Extraction Kit according to manufacturer’s instructions. Purified PCR products were Sanger sequenced, and results were analyzed using sangeranalyseR and sangerseqR packages in R (Hill et al., 2014), as well as by visual inspection of the electropherograms, to confirm polymorphic and/or heterozygous sites. Validation rates for detected variants was 100%. Primer sequences are documented in Table S8.
Cell model development and differentiation
Fibroblast cell culture, iPSC reprogramming, and characterization
XDP and control fibroblasts were cultured as previously described (Ito et al., 2016). Briefly, cells were propagated in growth medium (DMEM with 20% FBS and 1% penicillin/streptomycin), and passaged every 4–5 days by trypsinization. For iPSC reprogramming, fibroblasts were plated at a density of 2x104/cm2 and after 24 hrs transduced with Sendai viruses encoding Oct4, Klf4, Sox2 and c-Myc at multiplicities of infection = 3. Cells were fed every other day until day 7, at which point they were replated onto 0.1% gelatin-coated 10 cm plates containing mouse embryonic fibroblasts (MEFs) and switched to hESC medium (7 μL/liter β-mercaptoethanol, 20% Knockout Serum Replacement [KOSR], 2% L-glutamine, 1% Non-essential amino acids [NEAA] and 10 ng/mL basic fibroblast growth factor [bFGF] in DMEM/F12). Colonies were picked by manual dissection, transferred to fresh MEFs, and expanded using mechanical and enzymatic passaging.
Pluripotency of iPSC clones was confirmed based on RT-qPCR and immunostaining for standard markers (Table S8). For RT-qPCR, RNA was isolated from iPSCs using Zymo DirectZol® RNA miniprep kit, reverse transcribed to cDNA using High Capacity cDNA Reverse Transcription kit, and amplified with primers against NANOG, OCT4, hTERT, REX1, SOX2 and DNMT3B using PowerUp™ SYBR Green Master Mix, all as recommended. For immunofluorescence, iPSCs were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 minutes at room temperature, washed 3 times in PBS/0.05% Tween 20, permeabilized in PBS/0.1% Triton X-100 for 15 minutes, and then blocked in 4% donkey serum/PBS for 1 hour at room temperature. Cultures were then incubated overnight in primary antibodies against Oct3/4 (1:200 in blocking buffer), Nanog (1:50), SSEA4 (1:200), or Tra-1-60 (1:200). The next day cells were washed 3 times in PBS/0.05% Tween 20), incubated for 1 hr in Alexa Fluor®-conjugated secondary antibodies (1:1000 in PBS), washed again in PBS/0.05% Tween 20, and then counterstained with DAPI to visualize nuclei. Images were acquired on a Nikon Eclipse TE2000-U microscope with 20x magnification.
To analyze trilineage potential of iPSC clones, iPSCs were allowed to form embryoid bodies bearing cells of all three germ layers. Cells were incubated in Accutase (1:3 dilution in PBS) for 3 min at 37°C, washed in PBS, and then switched to EB medium (DMEM + 10% KOSR + 1% Pen/Strep) and scraped with trituration to generate small clumps. Clumps were allowed to settle for 5–10 minutes, after which medium was aspirated and cells were gently resuspended in EB medium with the ROCK inhibitor, Y-27632 (4 μM). Cells were seeded onto ultra-low attachment plates to promote EB formation. After 24 hrs, cell suspensions were collected into 15-ml tubes, EBs were allowed to settle, and medium was exchanged to remove ROCK inhibitor before replating. The process was repeated every other day until day 7, at which point they were collected and seeded onto gelatin-coated plates in DMEM + 10% FBS + 1% penicillin/streptomycin. Media was exchanged every 3 days until day 14 after initial plating. EBs were then collected, with RNA isolation and cDNA generation performed as described above. Expression of germ layer markers was quantified using Taqman® hPSC ScoreCard™ Panel as recommended. In parallel, some iPSC clones were also evaluated by teratoma formation in mice. For these lines, approximately 1 x 106 cells in Matrigel/PBS were implanted transcutaneously at multiple sites in Fox Chase SCID® mice by the Harvard Genome Modification Facility. Mice were euthanized after 6–8 weeks, and tumors were collected, paraffin embedded, sectioned, and stained with hematoxylin/eosin to identify cells of the three germ layers based on morphology.
Lentiviral vector generation
For packaging of VSVG-pseudotyped lentiviral vectors encoding NGN2 or rtTA, HEK-293T cells were plated onto 10-cm dishes the day before transfection at a density of 2.5x106 cells/dish. 24 hrs later cells were co-transfected using calcium phosphate with 3.5 μg of envelope vector pMD2.G, 6.5 μg of packaging vector pCMVR8.74, and 10 μg of transfer vector pTet-O-Ngn2-puro or pM2rtTA. Transfections were done in Opti-MEM medium with complete exchange to mTeSR1 media after 24 hrs. Supernatants containing un-concentrated lentiviral particles were collected at 48 and 72 hrs, filtered through a 0.45 μm syringe filter, and stored at −80°C.
Neural differentiation and characterization
For conversion to NSCs, iPSC clones on Geltrex were cultured in PSC Neural Induction Medium for 7 days, with media exchanges every other day. After seven days, cells were collected via Accutase and seeded onto fresh Geltrex-coated plates in Neural Expansion Medium (1:1 PSC Neural Induction Medium: DMEM:F12) with Y-27632 (5 μM). Medium was exchanged 24 hrs later to remove the ROCK inhibitor. Cells were propagated in culture for up to four passages, as NSC-like morphology typically improved over this period and any residual pluripotent-like cell colonies initially present would be depleted. Neurons were generated using a previously reported protocol (Zhang et al., 2013) with few modifications. Briefly, iPSCs were plated at clonal density on Geltrex in mTesR1 containing Y-27632 (10 μM). 24 hrs later, cultures were infected with NGN2- and rtTA lentiviral vectors by placing cells in undiluted inoculum + polybrene (8 μg/ml) for 4 hrs, followed by a medium exchange, and then a second round of infection the next day. 24 hrs after the second infection, cells were switched to DMEM:F12 medium + N2 supplement, NEAA, human brain-derived neurotrophic factor (BDNF; 10 ng/ml), human neurotrophin-3 (NT-3; 10 ng/ml) and doxycycline (2 μg/ml). The next day cells were treated with puromycin (1 μg/ml). After selection for 48 hrs, the neural population was collected via Accutase and seeded onto poly-D-lysine/laminin-coated plasticware in Neurobasal/Glutamax medium supplemented with B27, BDNF, NT-3, doxycycline, and Y-27632 (5 μM). The next day cells received a 50% medium exchange including additional selection with cytosine-β-D-arabinofuranoside (Ara-C) to deplete any non-neural cells resistant to puromycin. Ara-C was removed after 48 hrs, and cells continued to receive 50% media exchanges every other day until DIV 14.
Alternatively, mature neurons were generated from NSCs using methods described previously (Yan et al., 2013), with few modifications. Briefly, NSCs at passage 3 were dissociated with Accutase™ and plated onto poly-D-lysine/laminin coated plates at a density of 5x104 cells/cm2 in PSC Neural Expansion Medium supplemented with 5 μM ROCK inhibitor Y27632. The next day (DIV 0), cells were given a medium exchange with B27/Neurobasal/Glutamax supplemented with nonessential amino acids, BDNF (20 ng/mL), GDNF (20 ng/mL), and L-ascorbic acid (200 μM, Sigma-Aldrich). Every other day subsequently, 50% of the media was exchanged. On DIV 7, cells were treated with 2uM Ara-C to eliminate the remaining dividing cells, and cells were harvested at DIV 17.
GABA-ergic neurons were generated based on a previously reported protocol (Arber et al., 2015). Briefly, NSCs were plated into a Geltrex-coated 6-well plate at a density of 2x105 cells/cm2 in NSC expansion media with media replaced the following day. After 48 hrs. Media was changed and replaced with 4 mL of retinol-free N2B27 media supplemented with Activin A (25 ng/mL). The following day (Day1), cells were passaged en bloc 1:1 using dispase onto poly-D-lysine (10 μg/mL) and laminin (15 ng/mL) coated 6-well plates in 2 mL/well N2B27 media supplemented with Activin A (25 ng/mL) and Y-27632 (5 μM). Next day (Day 2), media was changed to remove Y-27632 by adding 5 mL N2B27 media supplemented with Activin A (25 ng/mL). This media change was repeated every other day for a week, after which cells were single cell passaged using Accutase, and resuspended in 2 mL/well N2B27 supplemented with Activin A (25 ng/mL) and Y-27632 (5 μM). Cells were then replated on poly-D-lysine (10 μg/mL) and laminin (15 ng/mL) coated plates at a density of 3x105 cells/cm2. The following day the same amount of BrainPhys™/B27 (with retinol)/CultureOne™ supplemented with Activin A (25 ng/mL), BDNF (20 ng/mL), GDNF (20 ng/mL), NT-3 (20 ng/mL), valproic acid (VPA; 2 mM), ascorbic acid (400 μM) and dbcAMP (400 mM) was added to each well and incubated for 3 days. After that, 50% of media was changed every 3rd day until Day34 with BrainPhys™/B27 (with retinol)/CultureOne™ supplemented with Activin A (50 ng/mL; until DIV 20) BDNF (20 ng/mL), GDNF (20 ng/mL), NT-3 (20 ng/mL), valproic acid (VPA; 2 mM), ascorbic acid (400 μM; until DIV 17) and dbcAMP (400 mM).
NSC, NSC-derived and NGN2-induced neuronal identity was confirmed by RNAseq. NSC-derived and NGN2-induced neurons (iNs) were labeled with antibodies against doublecortin, SOX1, Tuj1, MAP2, and GABA. Images were captured on a Nikon Eclipse TE2000-U epifluorescence microscope using an Andor-Zyla sCMOS camera and on a Zeiss LSM 510 confocal laser scanning microscope. To further assess functional maturity of iNs, cells seeded during differentiation into 35-mm dishes (6.6x104 cells/cm2) were loaded at DIV14 with the Ca2+ indicator dye, Fluo4-AM, at room temperature for 40 min, rinsed 3 times in PBS, then transferred to a Na2+-based extracellular solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 D-Glucose, 10 Hepes; pH 7.4. Cells were imaged using a Nikon Eclipse Ti microscope, Andor Zyla CMOS camera with a PE4000 Cool-LED light source. Exposure times were 40–60 ms and images were taken every 1 s. KCl (40 mM) or kainate (10 mM) were added for 10 s after 1 min baseline imaging recording. Individual cells were selected with Nikon software and resulting Ca2+ responses were calculated and graphed in Matlab as relative change in fluorescence intensity (ΔF/F).
CRISPR/Cas9 nuclease-mediated genome editing
For cloning and characterization of sgRNAs targeting sequences 5′ and 3′ of the SVA,, 20-nucleotide oligo sequences preceding S.pyogenes Cas9 (SpCas9) NGG PAM sites were designed immediately 5′ and 3′ of the SVA using Geneious DNA analysis software. BbsI overhang-containing oligos were synthesized for ligation into a BbsI-linearized pGuide sgRNA expression vector under control of the U6 promoter. After ligation, bacterial transformation and miniprep plasmid DNA isolation, sgRNA vectors targeting the SVA were verified by Sanger sequencing. Sequence-verified sgRNA plasmids (250 ng) and human codon-optimized pCas9-GFP (750 ng), containing SpCas9 nuclease fused to GFP under control of a CAG promoter, were transfected into HEK293T cells via Lipofectamine 3000 as recommended. Cells were maintained in culture for 72 hrs, and gDNA was isolated and subjected to PCR with primers amplifying a segment spanning the targeted region. PCR amplicons were purified and Sanger sequenced to confirm the presence of double-stranded DNA breaks (DSBs). Cleavage efficiency based on Sanger sequencing traces was estimated using TIDE (Tracking Indels by Decomposition).
From that analysis, two guides targeting sites flanking the SVA insertion site were used to excise the retrotransposon from three XDP iPSC clones, 33363-D, 33109-2B and -2G. Cells grown to 70–80% confluence on Geltrex were collected via Accutase, triturated to single cells in Opti-MEM media, and transfected in suspension (1x106 cells) with 1.5 μg pCas9-GFP, 0.75 μg of each SVA sgRNA-encoding pGuide, 5 μL P3000 reagent, and 3.75 μL Lipofectamine 3000. After 15 minutes, fresh mTeSR1 with ROCK inhibitor Y-27632 (10 μM) was added to each transfection reaction, and cells were plated on Geltrex. A complete medium exchange was performed the next day to remove ROCK inhibitor, and cells were maintained for an additional 48 hrs. GFP-positive cells were then collected as single cells via Accutase in 250 - 750 μL DPBS with Y-27632 (10 μM), filtered through a 35-μm mesh cell strainer, and sorted on a BD FACSAria™ Fusion SORP Cell Sorter using a 100 μm nozzle at a pressure of 20 psi. Sorted cells were collected and plated at clonal density of 2.5 – 3.0 x 104 cells per 10-cm dish in mTeSR1 with Y-27632 (10 μM) and MycoZap™ (1:250 dilution). A complete medium exchange was performed the next day to remove ROCK inhibitor and every day thereafter until single cell-derived colonies reached 2–4 mm in size. Single colonies were manually picked into individual wells of a 96-well plate, propagated until approximately 90% confluent, then collected using Accutase and divided into two 96-well duplicate plates: one to which freezing media (80% FBS/20% DMSO) was added 1:1 to each well and stored at −80°C (clone recovery plate); the other kept in culture with cells propagated until reaching approximately 90% confluency again. Cells were then lysed overnight in 50 μL lysis buffer (10 mM Tris-HCl, pH 7.5–8.0; 10 mM disodium EDTA; 10 mM NaCl; 0.5% [w/v] sarcosyl) at 56°C. The next day gDNA was precipitated with ice cold ethanol (95% v/v) for 2 hr at −20°C, pelleted, washed with ethanol (70% v/v), and resuspended in 30 μL ddH2O + 0.1 mg/mL RNase A. Samples were screened by PCR with primers amplifying a segment spanning the SVA insertion site, and successful excision of the SVA was detected based on the size of the amplicon (~0.6 kb vs. 3.0 kb with or without the SVA, respectively) determined by electrophoresis. Positive amplicons lacking the SVA were subsequently confirmed by Sanger sequencing. Successfully edited clones were then recovered from the sister plate and propagated as described above, confirmed to retain normal karyotype and expression of pluripotency markers, and differentiated to NSCs, iNs, and NSC-derived, glutamatergic and GABAergic neurons.
Transcriptomics
Strand-specific dUTP RNAseq library preparation
RNASeq libraries (n=112) were prepared using TruSeq® Stranded mRNA Library Kit (Illumina) and prepared per manufacturer’s instructions. In brief, RNA sample quality (based on RNA Integrity Number, RIN) and quantity was determined on an Agilent 2200 TapeStation and between 500-100 ng of total RNA was used to prepare libraries. 1 uL of diluted (1:100) External RNA Controls Consortium (ERCC) RNA Spike-In Mix (Thermo Fisher) was added to each sample alternating between mix 1 and mix 2 for each well in batch. PolyA bead capture was used to enrich for mRNA, followed by stranded reverse transcription and chemical shearing to make appropriate stranded cDNA inserts for library. Libraries were finished by adding both sample specific barcodes and adapters for Illumina sequencing followed by between 10–15 rounds of PCR amplification. Final concentration and size distribution of libraries were evaluated by 2200 TapeStation and/or qPCR, using Library Quantification Kit (KK4854, Kapa Biosystems), and multiplexed by pooling equimolar amounts of each library prior to sequencing. RNASeq libraries were sequenced to 40–100 million reads per library with 85–97% covered bases included in annotated mRNA.
RNA Capture-Sequencing (CapSeq) library preparation
RNA CapSeq libraries were prepared using a combination of TruSeq® Stranded mRNA Library Kit (Illumina) and SureSelectXT kit with a SureSelectXT custom capture library targeting the region of 400 kb on the X chromosome from OGT to CXCR3. cDNA was made from RNA using the TruSeq® Stranded mRNA Library Kit (Illumina). 500-100 ng of total RNA was used to prepare libraries that were first PolyA-bead captured to enrich for mRNA, followed by stranded reverse transcription and chemical shearing to generate appropriate stranded ~175 bp length cDNA inserts. These cDNA inserts were end-repaired, adenylated, ligated to adapter oligos and then amplified with 5 cycles of PCR according to manufacturer’s instructions. After quantification, 750ng of each amplified DNA sample was hybridized overnight with the Capture Library. Following capture cleanup, each gDNA library was amplified with additional 16 cycles of PCR, which also tagged each sample with an index-specific barcode. Final products were quantified using the TapeStation 2200 and pooled for rapid mode sequencing on the Illumina. RNA CapSeq libraries were sequenced at the Broad Institute Genomic Services as 101bp paired-end reads on an Illumina HiSeq2000 platform.
Single molecule, real-time (SMRT) sequencing of captured RNA molecules using IsoSeq
One microgram of total RNA from three NCS cell lines (33113, 32517 and 33363) was used for PacBio Iso-Seq library preparation. Briefly, the polyA+ RNA were purified with oligo-dT magnetic beads (Dynal, ThermoFisher) and was reverse transcribed (20 uL reaction with 100U Maxima RNaseH- RT, ThermoFisher) with a modified oligo-dT SMARTer (Clontech) oligonucleotide containing a 16bp barcode between the Clontech primer IIA sequence and the oligo-dT. A template switch oligo (TSO) was included in the reaction at: 5′–AAGCAGTGGTATCAACGCAGAGTACNNNGG+G was modified with a 3′ LNA-G as in (Picelli et al., 2013). Reverse transcription (2 hrs, 45°C) was heat killed (5 min, 85°C) and the cDNA was purified by a 1X AMPure PB purification. PCR amplification of the cDNA library with Takara LA Taq and enrichment were performed according to PacBio-IDT protocol (http://www.pacb.com/wp-content/uploads/Unsupported-Protocol-Full-length-cDNA-Target-Sequence-Capture-IDT-xGen-Lockdown-Probes.pdf) with the following modifications: No size selection was employed and the oligo capture bait library used in CapSeq was used for IsoSeq enrichment. Final enriched cDNA samples were PCR amplified on-bead with Kapa HiFi Master mix and the Clontech primer IIA (5′-5′-AAGCAGTGGTATCAACGCAGAGTAC-3′). The PCR output was purified with a 0.6X AMPure PB bead purification and converted to SMRTbell sequencing templates using the PacBio template prep kit and protocol. Libraries were quantified using the Qubit High Sensitivity kit (Thermo Fisher Scientific) and approximate sizes measured using a High Sensitivity DNA Bioanalyzer kit (Agilent Genomics). Libraries were prepared for sequencing using the P6-C4 chemistry and MagBead loading standard methods at 20–25 pM concentration on plate (Pacific Biosciences). Data acquisition proceeded with 6 hour movies on the PacBio RSII instrument at the University of Washington PacBio Sequencing Services facility. Data from the RSII runs were imported into SMRT Link version 5.0.1 (Pacific Biosciences) and converted to BAM files. Data were subjected to CCS2 Circular Consensus Sequencing analysis using default parameters (minimum 3 full passes and predicted accuracy of 0.9) except maximum sub-read length was extended from 7 kb to 10 kb.
Analyses for RNA CapSeq and total RNAseq
Read pairs of RNA CapSeq and RNAseq were trimmed using trimmomatic v0.36 for Illumina Truseq adapters and primers. The trimmed read pairs were then aligned to human genome (GRCh37, Ensembl release 75) with SVA inserted at position X:70660363 by STAR 2.5.2b (Dobin et al., 2013) allowing 5% mismatches with a unique mapping and with following parameters “--outFilterMultimapNmax 1 --outFilterMismatchNoverLmax 0.05 --alignEndsType EndToEnd”.
The preprocessed consensus reads from PacBio Iso-seq reads (CCS2 reads) were trimmed using cutadapter 1.14. The first round of trimming was applied with “-g AAGCAGTGGTATCAACGCAGAG -a CTCTGCGTTGATACCACTGCTT -e 0.05” option to remove PCR primer at 5′ and 3′ end respectively, allowing 5% mismatch. The second round of trimming involved hard clipping 16 bp from both ends to remove potential barcodes and retain remaining reads no less than 25bp using “-u 16 -u -16 -m 25”. The trimmed Iso-seq reads were then aligned by STARlong algorithm of STAR 2.5.2b with “--runMode alignReads --runThreadN 8 --outSAMattributes NH HI NM MD --outFilterMultimapScoreRange 20 --outFilterScoreMinOverLread 0 --outFilterMatchNminOverLread 0.95 --outFilterMismatchNmax 1000 --winAnchorMultimapNmax 200 --scoreGapNoncan -20 --scoreGapGCAG -4 --scoreGapATAC -8 --scoreDelOpen -1 --scoreDelBase -1 --scoreInsOpen -1 --scoreInsBase -1 --alignEndsType Local --seedSearchStartLmax 50 --seedPerReadNmax 100000 --seedPerWindowNmax 1000 --alignTranscriptsPerReadNmax 100000 --alignTranscriptsPerWindowNmax 10000 ” options.
De novo TAF1 transcript assembly
For de novo assembly, alignments of each sample (both total RNAseq and RNA CapSeq) within the capture region were de-multiplexed, duplicate reads were removed, and reads pairs from the same cell types were then merged. Transcripts were assembled by bowtie-0.12.8 and Trinity v2.2.0 on each cell type (fibroblasts, NSCs, and iNs) with the following parameters: default settings “--SS_lib_type RF”, “--genome_guided_bam” and “--genome_guided_max_intron 100000” (Grabherr et al., 2011). After the preliminary assembly, transcripts from the three cell types were compared and merged to a non-redundant list. These non-redundant de novo transcripts were then compared individually against human genome GRCh37.75 using BLAT to resolve internal splicing structures. For each transcript, we required the whole set of splice junctions to be supported by more than half of the RNAseq samples of any category combining cell types and genotype (controls, carriers and probands), or the de novo transcript was disregarded.
Nomenclature of de novo transcripts
All de novo transcripts were annotated by their structures relative to the canonical TAF1 transcript, cTAF1. There were three de novo transcripts that differed from the canonical cTAF1 based on usage of exons 34′ (6 bp) and/or 35′ (102 bp). Compared to cTAF1, nTAF1 (cTAF1-34′) was distinguished by the extension of exon 34, cTAF1-35′ contained an extended exon 35′, and cTAF1-34′-35′ incorporated both exon 34′ and exon 35′ (Figure S3). In addition to these transcripts, there were two truncated transcripts that involved IR, which we annotated as TAF1-28i and TAF1-32i. TAF1-28i involved an expressed sequence previously annotated as part of canonical intron 28, while TAF1-32i involved a transcribed sequence in canonical intron 32 that terminated 716 bp proximal to the insertion site of the XDP-specific SVA. All the de novo transcripts and their structure were listed in Table S3.
Confirmation of de novo TAF1 transcripts
Ensembl reported transcripts were used to confirm de novo transcripts from Illumina and PacBio reads. The Ensembl TAF1-202 differs from the de novo transcript cTAF1-35′ by loss of 63 bp at the 5′ end of exon 5. Similarly, de novo TAF1-008 (cTAF1) differs from Ensembl TAF1-009 by the trimmed exon 5. This trimmed exon 5 (exon 5′) was confirmed in PacBio reads, although the full transcript structure could not be fully reconciled as the reads did not extend beyond exon 20. Ensembl transcripts, TAF1-005, TAF1-006, TAF1-019, TAF1-020 and TAF1-022, are transcripts with retained introns 6, 14, 18, 17 and 37, respectively, and all but TAF1-022 were recovered by PacBio sequencing. Similarly, TAF1-018 may be a longer transcript than annotated based on PacBio reads.
Annotation of de novo transcripts with MTS and other reported transcripts
All previously described MTS transcripts (Nolte et al., 2003), except Var.4 and Var.2d, are recorded in Ensembl as TAF1-011, TAF1-012, TAF1-013, TAF1-016, TAF1-021 and TAF1-023. All MTS transcripts reportedly shared the two exonic regions located 67.7 kb downstream of the 3′ end of exon 37 of TAF1, with the exception of Var.4, which is also composed of these two exonic regions but with a 633 bp extension at the 5′ end (Figure S3). A subsequent study found MTS transcripts could be further extended to exon 26 of TAF1 from the originally reported exon 30 (one such transcript is recorded as Ensembl TAF1-010) (Herzfeld et al., 2007).
The only de novo transcript detected downstream of TAF1 was Var-trimmed (based on nomenclature used by Nolte et al., 2003), had two exons overlapping with previously proposed MTS transcripts. This transcript was not connected to TAF1 exon 37 in the de novo assembly.
Western blot analysis
Cells were collected by scraping and centrifugation, then washed 3X in cold phosphate-buffered saline (PBS) prior to lysis in Lysis Buffer AM1 (Active motif) supplemented with protease inhibitor cocktail (Active motif). Equivalent amounts of total protein from each lysate were prepared in Novex Bolt™ lithium dodecyl sulfate (LDS) sample buffer, resolved by electrophoresis on Novex Bolt™ bis-acrylamide (4–12%) gels and transferred to polyvinylidene fluoride (PVDF) membranes (all from Thermo Scientific). Membranes were blocked with 5% BSA in TBS-T (150 mM NaCl, 50 mM Tris pH 7.9, 0.5% TWEEN) and incubated overnight in primary antibodies diluted in 2.5% BSA in TBS-T. Blots were washed 3X in TBS-T and incubated in HRP-conjugated secondary antibodies, with labeled proteins visualized via chemiluminescence using SuperSignal West Pico Substrate™ (Thermo Scientific).
Antibodies and dilutions used for western blotting were: mouse anti-TAF1 (#134; 1:2000; generated by M. Timmers); mouse anti-Hsp70 (sc-24; 1:50,000; Santa Cruz); HRP-conjugated anti-mouse IgG (NA931; 1:6,000; GE Life Sciences). The human TAF1 134 mAb was raised in mice against the cTAF1 peptide: MTPGPYTPQPPDLYDTNT. It displays a weak cross reactivity with the nTAF1-specific peptide: MTPGPYTPQAKPPDLYDTNT.
Quantification and statistical analyses
Quantification and filtering of expressed features
Gene counts were generated using HTSeq (v.0.6.1) with “-p yes -s reverse” for genes and exons. Relative normalization factors among samples were calculated by applying DESeq2 function estimateSizeFactor() on the count matrix of each gene across all samples.
The abundance of assembled transcripts was measured in each total RNAseq sample using kallisto 0.43.0 with “--rf-stranded” (Bray et al., 2016). The absolute expression of each TAF1 isoform in a sample was calculated as the product of abundance of each transcript and overall normalized TAF1 expression in that sample.
Quantification of transcriptome-wide intron retention
The IRFinder from Middleton et al. was applied to estimate genome-wide intron retention in each sample (Middleton et al., 2017). The package can be found on GitHub (https://github.com/williamritchie/IRFinder/wiki). In general, the intronic expression levels were represented by the median depth of each intronic region without considering the pre-calculated low mappability regions and any intronic region that overlaps with expression features such as microRNAs and snoRNAs. The intron retention level of each intron was then estimated as a ratio of the intronic expression to the number of reads that connect the flanking exon junctions.
Quantitative RT-PCR for intron 32 retention
RNA isolated from XDP, control, and ΔSVA samples was reverse transcribed into cDNA using Superscript III First Strand Synthesis SuperMix (Thermo Scientific) with oligo(dT). A custom Taqman® primer/probe was generated to detect the presence of the exon 32/intron 32 splice site, such that the forward and reverse primers bound to exon 32 and intron 32, respectively, and the probe spanned the splice junction. The reference gene GUSB was detected using an inventoried Taqman® gene expression assay (Thermo Scientific). Samples were run on a StepOne Plus™ Real-Time PCR System (Applied Biosystems) as recommended, using Taqman Fast Advanced Master Mix (Thermo Scientific). Each reaction assayed 50 ng RNA in 20 μL total volume for an initial holding step of 95°C (20 sec) followed by 40 cycles of 95°C (1 sec) and 60°C (20 sec). Raw Ct values were normaliz ed to the geometric mean of the reference gene. Any Ct values 35 or greater were considered undetectable. RT-PCR expression data were graphed using GraphPad Prism® 7 software (GraphPad Software). Statistical analysis was based on the following numbers from each cell type. Fibroblasts: n=10 for XDP and n=6 for controls. iPSCs: n=27 for XDP, n=17 for controls, and n=9 for dSVA. NSCs: n=26 for XDP, n=18 for controls, and n=7 for dSVA. NGN2-induced cortical neurons: n=8 for XDP, n=6 for controls, and n=4 for dSVA. NSC-derived cortical neurons: n=7 for XDP, n=6 for controls, and n=1 for dSVA. GABA-ergic neurons: n=7 for XDP, n=6 for controls, and n=4 for dSVA.
Linear mixed modeling of expressed features and differential feature analysis
Generalized linear mixed models (GLMMs) were used to model the count-based features such as gene/transcript/exon as follows: For each tissue, genes with normalized median counts < 5 in all genotypes were filtered out. Mixed models were fit as:
where
Kij - raw counts observed in gene/transcript/exon i in sample j
sj- sequencing depth adjustment parameter (size factor) for sample j
cij - counts for gene/transcript/exon i in sample j, normalized by the sequencing depth
Fixed effects:
Bj- vector, indicating the batch of sample j
Gj - vector, indicating the genotype of sample j
Random effects:
Ij - random effect of individual of sample j, Ij ~ N(0, σI)
Cj - random effect of clone of sample j, Cj ~ N(0, σC)
Parameters β0, βB and βG and variances σI and σC were estimated using R package 1me4.
As intron retention is calculated as the ratio of a continuous feature (trimmed mean of intronic depth) and a count-based feature (counts of splicing at two flanking junctions) and the aim is to compare the change of this ratio between XDP and control, a linear mixed model (LMM) equation was applied here:
where
Kij - raw counts observed at intronic region or splice junction of intron i in sample j
sj - sequencing depth adjustment parameter (size factor) for sample j
cij - counts for gene/transcript/exon i in sample j, normalized by the sequencing depth
Fixed effects:
Bj - vector, indicating the batch of sample j
Gj - vector, indicating the genotype of sample j
S - vector, indicating the whether it’s counts for intron or counts for splice junction
Random effects:
Ij - random effect of individual of sample j, Ij ~ N(0,σI)
Cj - random effect of clone of sample j, Cj ~ N(0,σC)
Parameters β0, βG and βI and variances σI and σC were estimated using R package 1me4.
The models were fit by cell type, and in each experiment ‘n’ refers to the number of cellular clones in a given group. Control samples were treated as baseline (interception) in all models. Before model fitting, features with low counts were filtered. The filter is described as at least half of the samples in any of control, carrier or XDP genotype should be no less than a specific threshold. The thresholds for gene, transcript exon and IR were 5 counts, 1000 TPM (1% of overall gene expression), 1 count and 1% IR ratio respectively.
After model fitting, differential feature analysis was carried out by applying Wald test on corresponding parameters in the model. A Wald test was applied on Genotype XDP parameter for differential gene/transcript/exon expression. To compare differential IR events, the Wald test was applied on the contrast of the two parameters representing interaction terms.
Correlations, co-expression networks, gene ontologies and pathways analysis
Pearson correlation of the transcripts was calculated based on log2 counts adjusted for batch effects in fibroblasts, NSCs and iNs, and, carrier genotype effects in Fibroblasts and iNs. Enrichment for gene ontologies was tested using R package topGO (version 2.22.0), using only curated gene ontology assignments (excluding evidence with codes ND, IEA, NR) with algorithm “weight01.” Co-expression network analysis was performed using R package WGCNA (Langfelder and Horvath, 2008) for each cell type separately using unsigned network type. Only genes counts > 10 in at least half of the samples within a cell type were considered. Normalized counts were adjusted for Carrier genotype and Batch using parameters estimated from the mixed models described above, and the log transformation was used as log2(counts_adjusted +1) Soft power was selected such that the scale-free topology fit (R2) > 0.8. Merge of the modules with similar eigen genes was performed. Module membership for each gene was reevaluated based on the module membership p-value; if p-value > 0.01, the gene was marked as unassigned (Module 0). Correlation of modules to TAF1 transcripts/exons/intron32 expression was calculated using Spearman’s correlation.
Data and Software Availability
DNA and RNA sequencing data generated for this study is available in the dbGAP repository with accession phs001525.v1.p1.
Supplementary Material
Multiple sequence alignment of the XDP-specific SVA (XDP_SVA_insert) described by (Makino et al., 2007) and the SVA assembled from the BAC clone (BAC_SVA) in this study using PacBio SMRT sequencing of the XDP BAC clone. Shown is the region different between the two SVA sequences.
All samples analyzed by various NGS technologies, including probands, non-manifesting carriers (NMCs), female carriers, and controls, as well as information on cell lines and the specific sequencing details. Pedigree information for probands with available clinical data specifying age at onset, initial symptom, and initial region affected are listed under the heading “Clinical Data”. The technologies applied on each sample along with raw read counts obtained per sample in various technologies are documented under “Genomics Data” heading. Haplotypes are reported for probands and NMCs. Data for samples labelled “DPRB-P” in the Source column are deposited in dbGaP. Samples were collected through the MGH Collaborative Center for XDP (Boston, MA), from the following projects/organizations/institutions: DPRB – Dystonia Partners Research Bank; GCNHx – Overarching study on the Natural History of XDP, being conducted at MGH; Archival – archival samples from Drs. Müller (Giessen, Germany) and Wilhelmsen (Chapel Hill, NC USA); ING-L – Institute of Neurogenetics, Lübeck, Germany.
List of variants discovered in CapSeq and WGS in 792 samples, their public database identifiers, chromosomal positions, as well as frequencies in probands, controls, and carrier groups. The table also documents the predicted consequences of the variation on the resultant transcripts and proteins, along with global minor allele frequency (MAF) and the MAFs in the major world population. The XDP-associated variants are included with XDP-associated names (bold), status in previous studies, and PCR-validated status.
Transcript structures and summaries from generalized linear mixed model including expression levels in controls, log-transformed fold changes in XDP and test-related statistics cited in the main text.
Summaries from a generalized linear mixed model including expression levels in controls, log-transformed fold changes in XDP probands, and test-related statistics cited in the main text.
Summaries from a generalized linear mixed model including IR levels in controls, log-transformed fold changes in XDP and test-related statistics cited in the main text.
Expression values, fold changes and associated test statistics for all genes. Genes that were filtered due to low expression in a particular cell types have all the values set to NA.
(A) Immunocytochemistry for stemness marker expression Oct4, Nanog, SSEA-4 (all in green) and Tra-1-60 (red). All iPSC colonies were counterstained with the nuclear stain DAPI (blue). Scale bar: 100 μm. (B) Expression of pluripotency-related genes OCT4, NANOG, SOX2, REX1, DNMT3B, and hTERT in each XDP-carrier iPSC line compared with its parental fibroblast line as determined by qRT-PCR. Heatmap is based on ΔCt values. Red, high expression; blue, low expression. (C) Representative XDP-carrier teratomas formed in mice after iPSC implantation. Scale bar: 50 μm. (D) Tri-lineage differentiation potential of all XDP-carrier iPSC clones as determined by Taqman hPSC Scorecard™ assay of RNA isolated from iPSC-derived embryoid bodies. Heatmap depicts expression of 94 marker genes related to pluripotency, primitive mesendoderm, definitive ectoderm, mesoderm and endoderm. Values represent mean fold changes (fc) for each gene relative to reference pluripotent stem cell lines as calculated by hPSC Scorecard™ analysis software. Red, high expression; blue, low expression. (E) Karyotype analysis revealed no chromosomal abnormalities in any of the XDP-carrier iPSC clones used for this study.
All de novo assembled transcripts were compared to Ensembl annotation and transcripts revealed by previous studies. Within each transcript, boxes denote exons either in black (Ensembl-annotated exon) or purple (novel). The colored circles (blue, fibroblast; purple, NSC; and green, iN) indicate cell type in which each de novo assembled transcript was expressed. The longer transcripts supported by PacBio are extended by dashed orange lines and additional exons are represented by orange boxes. The genomic coordinates reflect SVA insertion (2627 bp) at 70660362.
(A) Relative change in exon expression between XDP and control samples in fibroblasts (red), iNs (blue) and NSC-derived neurons (orange). Cross bars indicate exons that were not expressed. (B) Relative change in expression of the transcript Var-trimmed in XDP versus its expression level in control iNs. (C) and (D) Expression of transcripts in XDP cells relative to expression in corresponding control fibroblasts (C) and NSC-derived neurons (D). Black dashed line indicates no change, and error bars reflect FDR-corrected 95% confidence interval (A - D). (E) Average PacBio RNA CapSeq coverage (normalized) from exon 32 to the SVA insertion site (the brown triangle and the vertical brown line) for one control (Pedigree 12 in blue) and two XDP samples (Pedigree 17 in solid red line and Pedigree 22 in dashed red line) in NSCs. The X axis represents genomic coordinates of human X chromosome with the SVA inserted. (F) The curved lines demonstrate Illumina sequencing coverage from exon 32 to the SVA insertion site (the brown triangle and the vertical brown line), in control (blue) and XDP (red) samples in NSC-derived neurons. The shadowed area represents the TAF1 coding seqeunce. The solid horizontal lines intersecting with Y axis show the average sequencing coverage of TAF1 coding region in control (blue) and XDP (red) samples. The X axis represents genomic coordinates of human X chromosome with the SVA inserted. (G) Western blot for TAF1 protein expression in XDP (red) and control (blue) NSCs. (H) Normalized TAF1 protein expression relative to HSP70 in XDP and control NSCs.
(A) Heatmap of relative expression of pluripotency, neural stem cell, mature neuronal and glial genes in NSCs, NSC-derived and iNs based on RNAseq reads. (B) Representative images from XDP, ΔSVA, and unaffected control of NSC-derived cortical neurons showing comparable morphologies with dense networks of processes immunopositive for βIII-tubulin/Tuj1 and MAP2, as well as SOX1 positive progenitor cells (upper panels in B). Differentiation of NSCs using Activin A generated MAP2- and GABA-immunopositive neurons (lower panel in B). Scale bar = 50μm (Related to Figure 3 and Figure 6).
(A) Schematic of TAF1 with the targeted region in intron 32 is indicated. Annotated sequence contains sgRNAs (underlined) targeting loci 5′ and 3′ to the SVA insertion site (blue, arrowhead) and PAM sites (red). Below the schematic is the Sanger sequence of XDP iPSC clones with the SVA excised. (B) Expression of pluripotency-related genes OCT4, NANOG, SOX2, DNMT3B, and hTERT in each ΔSVA-XDP iPSC line compared with its unedited parent clone and parental fibroblast line as determined by qRT-PCR. Heatmap is based on ΔCt values. Red, high expression; blue, low expression. (C) SVA-edited clones 3C2, 3A12, 1A4 and 2F2 differentiated into NSC and NGN2-induced neurons showed expected expression of NSC vs. neuron markers, based on RNAseq. (D) Immunocytochemistry for mature neuronal markers DCX (green, left panel), Tuj1 (red, middle panel) and MAP2 (red, right panel) showing expression of these markers in NGN2-induced neurons generated from ΔSVA clones. Scale bar is 50 μm. (E) Karyotype analysis by g-banding revealed no apparent chromosomal abnormalities in the SVA-excised XDP iPSC lines.
(A) Scatter plot of log2 fold changes in XDP vs controls (X axis) and ΔSVA vs parental XDP (Y axis) and corresponding linear regression line and 95% confidence intervals for the slope of the regression line. Negative correlation suggests the reciprocal changes in gene expression in XDP lines upon SVA excision. (B) First (X axis) and second (Y axis) principal components of 400 DEGs in NSC, ΔSVA samples cluster closer to the controls and carriers.
Table S7. Results of differential expression analysis of ΔSVA vs corresponding XDP lines in NSCs and iNs (Related to Figure 6).
The Oligonucleotide sequences used in STAR method section for validation of novel DSCs by Sanger sequencing, CRISPR/Cas9 nuclease-mediated genome editing and cell model development and differentiation. Tm: Annealing temperature (in °C); ET: Extension time (seconds).
Highlights.
Genome assembly identifies novel recombinations and narrows the causal XDP locus to TAF1
An XDP-specific SVA insertion induces intron retention and down-regulation of TAF1
CRISPR/Cas9 excision of SVA rescues aberrant splicing and cTAF1 expression in XDP
Expression profiling implicates neurodevelopment and dystonia pathways in XDP
Acknowledgments
Flow cytometry services were supported by grants 1S10OD012027-01A1, 1S10OD016372-01, 1S10RR020936-01, and 1S10RR023440-01A1 at the MGH Department of Pathology Flow and Image Cytometry Research Core. We thank Daniel MacArthur for review of mutation patterns in ExAC and Nikka Keivanfar for DNA extractions for the 10X genomics library preparation. Funding for this study was provided by the MGH Collaborative Center for X-Linked Dystonia-Parkinsonism (D.C.B., M.E.T., N.S.) and by National Institutes of Health grants R01NS102423 (M.E.T. and D.C.B.), 5P01NS087997 (D.C.B., N.S., L.J.O., X.O.B.), and UM1HG008900 (M.E.T.). Dr. Eichler is an investigator of the Howard Hughes Medical Institute. Dr. Talkowski is the Desmond and Ann Heathwood MGH Research Scholar.
Footnotes
Declaration of Interests
J.U. is employed by Pacific Biosciences, Inc. D.M.C., S.R.W., S.G., N.W., and D.J. are employed by 10X Genomics. E.E.E. is on the scientific advisory board of DNAnexus, Inc. The authors declare no other competing interests.
Author contributions
Conceptualization, M.E.T., D.C.B., X.O.B., L.J.O.; Methodology, T.A., W.T.H, R.Y., D.S., D.G., A.S., B.C., J.U., H.B., C.A., A.R., N.I., N.W., B.J.W., D.J., D.C.B., M.E.T.; Formal Analysis; T.A., R.Y., D.G., R.L.C., A.D., T.M.B., S.R.W., S.G., A.S., B.C., H.B., C.H., C.A., A.R., J.G., G.L., M.E.T.; Investigation, T.A., W.T.H., R.Y., D.S., D.G., C.A.V., R.L.C., A.D., E.B.P., A.S., B.C., H.B., C.H., C.A., J.G., G.L., J.D. L.C., Y.S., B.J.W., D.H.; Resources, C.G., M.D., P.A., T.M.B., C.K., R.D.G.J., U.M., K.C.W., N.S., R.L.R., D.C., E.E.E., H.T.M.T., N.S.; Writing, T.A., W.T.H., R.Y., D.G., D.C.B., M.E.T.; Supervision, M.E.T., D.C.B., L.J.O. X.O.B.; Funding Acquisition, D.C.B., M.E.T., N.S., L.J.O, X.O.B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber C, Precious SV, Cambray S, Risner-Janiczek JR, Kelly C, Noakes Z, Fjodorova M, Heuer A, Ungless MA, Rodriguez TA, et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–1386. doi: 10.1242/dev.117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, Dhakal J, Ton ML, Cheng A, Russo CT, et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1712526114. [DOI] [PMC free article] [PubMed]
- Brand H, Collins RL, Hanscom C, Rosenfeld JA, Pillalamarri V, Stone MR, Kelley F, Mason T, Margolin L, Eggert S, et al. Paired-Duplication Signatures Mark Cryptic Inversions and Other Complex Structural Variation. Am J Hum Genet. 2015;97:170–176. doi: 10.1016/j.ajhg.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014;24:1774–1786. doi: 10.1101/gr.177790.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, McMillin MJ, Wiszniewski W, Gambin T, et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet. 2015;97:199–215. doi: 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Brand H, Redin CE, Hanscom C, Antolik C, Stone MR, Glessner JT, Mason T, Pregno G, Dorrani N, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol. 2017;18:36. doi: 10.1186/s13059-017-1158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo A, Westenberger A, Lee LV, Brænne I, Liu T, Vater I, Rosales R, Jamora RD, Pasco PM, Cutiongco-Dela Paz EM, et al. New insights into the genetics of X-linked dystonia-parkinsonism (XDP, DYT3) Eur J Hum Genet. 2015;23:1334–1340. doi: 10.1038/ejhg.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Bergmann EA, Arora K, Vacic V, Zody MC, Iossifov I, O’Rawe JA, Wu Y, Jimenez Barron LT, Rosenbaum J, et al. Indel variant analysis of short-read sequencing data with Scalpel. Nat Protoc. 2016;11:2529–2548. doi: 10.1038/nprot.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faravelli I, Nizzardo M, Comi GP, Corti S. Spinal muscular atrophy--recent therapeutic advances for an old challenge. Nat Rev Neurol. 2015;11:351–359. doi: 10.1038/nrneurol.2015.77. [DOI] [PubMed] [Google Scholar]
- Faust GG, Hall IM. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics. 2014;30:2503–2505. doi: 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014;28:2663–2676. doi: 10.1101/gad.252106.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo JM, Jin P, Thornton CA, Lin H, Robertson J, D’Souza I, Schlaepfer WW. The role of RNA and RNA processing in neurodegeneration. J Neurosci. 2005;25:10372–10375. doi: 10.1523/JNEUROSCI.3453-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Porse BT. The functional consequences of intron retention: alternative splicing coupled to NMD as a regulator of gene expression. Bioessays. 2014;36:236–243. doi: 10.1002/bies.201300156. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Palmer AC, Dodd IB, Shearwin KE. Directing traffic on DNA-How transcription factors relieve or induce transcriptional interference. Transcription. 2017;8:120–125. doi: 10.1080/21541264.2017.1285851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzfeld T, Nolte D, Muller U. Structural and functional analysis of the human TAF1/DYT3 multiple transcript system. Mamm Genome. 2007;18:787–795. doi: 10.1007/s00335-007-9063-z. [DOI] [PubMed] [Google Scholar]
- Hill JT, Demarest BL, Bisgrove BW, Su YC, Smith M, Yost HJ. Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev Dyn. 2014;243:1632–1636. doi: 10.1002/dvdy.24183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, Hendriks WT, Dhakal J, Vaine CA, Liu C, Shin D, Shin K, Wakabayashi-Ito N, Dy M, Multhaupt-Buell T, et al. Decreased N-TAF1 expression in X-linked dystonia-parkinsonism patient-specific neural stem cells. Dis Model Mech. 2016;9:451–462. doi: 10.1242/dmm.022590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob AG, Smith CWJ. Intron retention as a component of regulated gene expression programs. Hum Genet. 2017;136:1043–1057. doi: 10.1007/s00439-017-1791-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Bouhouche K, Gout JF, Aury JM, Noel B, Saudemont B, Nowacki M, Serrano V, Porcel BM, Segurens B, et al. Translational control of intron splicing in eukaryotes. Nature. 2008;451:359–362. doi: 10.1038/nature06495. [DOI] [PubMed] [Google Scholar]
- Jimeno-Gonzalez S, Payan-Bravo L, Munoz-Cabello AM, Guijo M, Gutierrez G, Prado F, Reyes JC. Defective histone supply causes changes in RNA polymerase II elongation rate and cotranscriptional pre-mRNA splicing. Proc Natl Acad Sci U S A. 2015;112:14840–14845. doi: 10.1073/pnas.1506760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaer K, Speek M. Intronic retroelements: Not just “speed bumps” for RNA polymerase II. Mob Genet Elements. 2012;2:154–157. doi: 10.4161/mge.20774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kejnovsky E, Lexa M. Quadruplex-forming DNA sequences spread by retrotransposons may serve as genome regulators. Mob Genet Elements. 2014;4:e28084. doi: 10.4161/mge.28084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics. 2008;24:719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LV, Rivera C, Teleg RA, Dantes MB, Pasco PM, Jamora RD, Arancillo J, Villareal-Jordan RF, Rosales RL, Demaisip C, et al. The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”) Int J Neurosci. 2011;121(Suppl 1):3–11. doi: 10.3109/00207454.2010.526728. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu EY, Cali CP, Lee EB. RNA metabolism in neurodegenerative disease. Dis Model Mech. 2017;10:509–518. doi: 10.1242/dmm.028613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, Matsumoto S, Tabuena MD, Maranon E, Dantes M, et al. Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am J Hum Genet. 2007;80:393–406. doi: 10.1086/512129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Shi W, Wasserman WW. Identification of altered cis-regulatory elements in human disease. Trends Genet. 2015;31:67–76. doi: 10.1016/j.tig.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ, Bomane A, Cosson B, Eyras E, Rasko JE, et al. IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol. 2017;18:51. doi: 10.1186/s13059-017-1184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullner D. fastcluster: Fast Hierarchical, Agglomerative Clustering Routines for R and Python. Journal of Statistical Software. 2013;53:18. [Google Scholar]
- Nolte D, Niemann S, Müller U. Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proc Natl Acad Sci U S A. 2003;100:10347–10352. doi: 10.1073/pnas.1831949100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rawe JA, Wu Y, Dorfel MJ, Rope AF, Au PY, Parboosingh JS, Moon S, Kousi M, Kosma K, Smith CS, et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am J Hum Genet. 2015;97:922–932. doi: 10.1016/j.ajhg.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh HR, An CH, Yoo NJ, Lee SH. Frameshift Mutations in the Mononucleotide Repeats of TAF1 and TAF1L Genes in Gastric and Colorectal Cancers with Regional Heterogeneity. Pathol Oncol Res. 2017;23:125–130. doi: 10.1007/s12253-016-0107-0. [DOI] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Pijnappel WW, Esch D, Baltissen MP, Wu G, Mischerikow N, Bergsma AJ, van der Wal E, Han DW, Bruch H, Moritz S, et al. A central role for TFIID in the pluripotent transcription circuitry. Nature. 2013;495:516–519. doi: 10.1038/nature11970. [DOI] [PubMed] [Google Scholar]
- Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2017;49:36–45. doi: 10.1038/ng.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittiner JE, Caffall ZF, Hernandez-Martinez R, Sanderson SM, Pearson JL, Tsukayama KK, Liu AY, Xiao C, Tracy S, Shipman MK, et al. Functional Genomic Analyses of Mendelian and Sporadic Disease Identify Impaired eIF2alpha Signaling as a Generalizable Mechanism for Dystonia. Neuron. 2016;92:1238–1251. doi: 10.1016/j.neuron.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW, Irimia M. Intron mis-splicing: no alternative? Genome Biol. 2008;9:208. doi: 10.1186/gb-2008-9-2-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnstrom K, Mallick S, Kirby A, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearwin KE, Callen BP, Egan JB. Transcriptional interference--a crash course. Trends Genet. 2005;21:339–345. doi: 10.1016/j.tig.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu-Motohashi Y, Miyatake S, Komaki H, Takeda S, Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am J Transl Res. 2016;8:2471–2489. [PMC free article] [PubMed] [Google Scholar]
- Stacey SN, Kehr B, Gudmundsson J, Zink F, Jonasdottir A, Gudjonsson SA, Sigurdsson A, Halldorsson BV, Agnarsson BA, Benediktsdottir KR, et al. Insertion of an SVA-E retrotransposon into the CASP8 gene is associated with protection against prostate cancer. Hum Mol Genet. 2016;25:1008–1018. doi: 10.1093/hmg/ddv622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci U S A. 2014;111:E4468–4477. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- Walker AK, Shi Y, Blackwell TK. An extensive requirement for transcription factor IID-specific TAF-1 in Caenorhabditis elegans embryonic transcription. J Biol Chem. 2004;279:15339–15347. doi: 10.1074/jbc.M310731200. [DOI] [PubMed] [Google Scholar]
- Wang K, Sun F, Sheng HZ. Regulated expression of TAF1 in 1-cell mouse embryos. Zygote. 2006;14:209–215. doi: 10.1017/S0967199406003704. [DOI] [PubMed] [Google Scholar]
- Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. Direct determination of diploid genome sequences. Genome Res. 2017;27:757–767. doi: 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenfeld NI, Yin S, Sharpe T, Lau B, Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et al. Comprehensive variation discovery in single human genomes. Nat Genet. 2014;46:1350–1355. doi: 10.1038/ng.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Shin S, Jha BS, Liu Q, Sheng J, Li F, Zhan M, Davis J, Bharti K, Zeng X, et al. Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl Med. 2013;2:862–870. doi: 10.5966/sctm.2013-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, Ward P, Braxton A, Wang M, Buhay C, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–1879. doi: 10.1001/jama.2014.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Choi M, Overton JD, Bellone S, Roque DM, Cocco E, Guzzo F, English DP, Varughese J, Gasparrini S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc Natl Acad Sci U S A. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multiple sequence alignment of the XDP-specific SVA (XDP_SVA_insert) described by (Makino et al., 2007) and the SVA assembled from the BAC clone (BAC_SVA) in this study using PacBio SMRT sequencing of the XDP BAC clone. Shown is the region different between the two SVA sequences.
All samples analyzed by various NGS technologies, including probands, non-manifesting carriers (NMCs), female carriers, and controls, as well as information on cell lines and the specific sequencing details. Pedigree information for probands with available clinical data specifying age at onset, initial symptom, and initial region affected are listed under the heading “Clinical Data”. The technologies applied on each sample along with raw read counts obtained per sample in various technologies are documented under “Genomics Data” heading. Haplotypes are reported for probands and NMCs. Data for samples labelled “DPRB-P” in the Source column are deposited in dbGaP. Samples were collected through the MGH Collaborative Center for XDP (Boston, MA), from the following projects/organizations/institutions: DPRB – Dystonia Partners Research Bank; GCNHx – Overarching study on the Natural History of XDP, being conducted at MGH; Archival – archival samples from Drs. Müller (Giessen, Germany) and Wilhelmsen (Chapel Hill, NC USA); ING-L – Institute of Neurogenetics, Lübeck, Germany.
List of variants discovered in CapSeq and WGS in 792 samples, their public database identifiers, chromosomal positions, as well as frequencies in probands, controls, and carrier groups. The table also documents the predicted consequences of the variation on the resultant transcripts and proteins, along with global minor allele frequency (MAF) and the MAFs in the major world population. The XDP-associated variants are included with XDP-associated names (bold), status in previous studies, and PCR-validated status.
Transcript structures and summaries from generalized linear mixed model including expression levels in controls, log-transformed fold changes in XDP and test-related statistics cited in the main text.
Summaries from a generalized linear mixed model including expression levels in controls, log-transformed fold changes in XDP probands, and test-related statistics cited in the main text.
Summaries from a generalized linear mixed model including IR levels in controls, log-transformed fold changes in XDP and test-related statistics cited in the main text.
Expression values, fold changes and associated test statistics for all genes. Genes that were filtered due to low expression in a particular cell types have all the values set to NA.
(A) Immunocytochemistry for stemness marker expression Oct4, Nanog, SSEA-4 (all in green) and Tra-1-60 (red). All iPSC colonies were counterstained with the nuclear stain DAPI (blue). Scale bar: 100 μm. (B) Expression of pluripotency-related genes OCT4, NANOG, SOX2, REX1, DNMT3B, and hTERT in each XDP-carrier iPSC line compared with its parental fibroblast line as determined by qRT-PCR. Heatmap is based on ΔCt values. Red, high expression; blue, low expression. (C) Representative XDP-carrier teratomas formed in mice after iPSC implantation. Scale bar: 50 μm. (D) Tri-lineage differentiation potential of all XDP-carrier iPSC clones as determined by Taqman hPSC Scorecard™ assay of RNA isolated from iPSC-derived embryoid bodies. Heatmap depicts expression of 94 marker genes related to pluripotency, primitive mesendoderm, definitive ectoderm, mesoderm and endoderm. Values represent mean fold changes (fc) for each gene relative to reference pluripotent stem cell lines as calculated by hPSC Scorecard™ analysis software. Red, high expression; blue, low expression. (E) Karyotype analysis revealed no chromosomal abnormalities in any of the XDP-carrier iPSC clones used for this study.
All de novo assembled transcripts were compared to Ensembl annotation and transcripts revealed by previous studies. Within each transcript, boxes denote exons either in black (Ensembl-annotated exon) or purple (novel). The colored circles (blue, fibroblast; purple, NSC; and green, iN) indicate cell type in which each de novo assembled transcript was expressed. The longer transcripts supported by PacBio are extended by dashed orange lines and additional exons are represented by orange boxes. The genomic coordinates reflect SVA insertion (2627 bp) at 70660362.
(A) Relative change in exon expression between XDP and control samples in fibroblasts (red), iNs (blue) and NSC-derived neurons (orange). Cross bars indicate exons that were not expressed. (B) Relative change in expression of the transcript Var-trimmed in XDP versus its expression level in control iNs. (C) and (D) Expression of transcripts in XDP cells relative to expression in corresponding control fibroblasts (C) and NSC-derived neurons (D). Black dashed line indicates no change, and error bars reflect FDR-corrected 95% confidence interval (A - D). (E) Average PacBio RNA CapSeq coverage (normalized) from exon 32 to the SVA insertion site (the brown triangle and the vertical brown line) for one control (Pedigree 12 in blue) and two XDP samples (Pedigree 17 in solid red line and Pedigree 22 in dashed red line) in NSCs. The X axis represents genomic coordinates of human X chromosome with the SVA inserted. (F) The curved lines demonstrate Illumina sequencing coverage from exon 32 to the SVA insertion site (the brown triangle and the vertical brown line), in control (blue) and XDP (red) samples in NSC-derived neurons. The shadowed area represents the TAF1 coding seqeunce. The solid horizontal lines intersecting with Y axis show the average sequencing coverage of TAF1 coding region in control (blue) and XDP (red) samples. The X axis represents genomic coordinates of human X chromosome with the SVA inserted. (G) Western blot for TAF1 protein expression in XDP (red) and control (blue) NSCs. (H) Normalized TAF1 protein expression relative to HSP70 in XDP and control NSCs.
(A) Heatmap of relative expression of pluripotency, neural stem cell, mature neuronal and glial genes in NSCs, NSC-derived and iNs based on RNAseq reads. (B) Representative images from XDP, ΔSVA, and unaffected control of NSC-derived cortical neurons showing comparable morphologies with dense networks of processes immunopositive for βIII-tubulin/Tuj1 and MAP2, as well as SOX1 positive progenitor cells (upper panels in B). Differentiation of NSCs using Activin A generated MAP2- and GABA-immunopositive neurons (lower panel in B). Scale bar = 50μm (Related to Figure 3 and Figure 6).
(A) Schematic of TAF1 with the targeted region in intron 32 is indicated. Annotated sequence contains sgRNAs (underlined) targeting loci 5′ and 3′ to the SVA insertion site (blue, arrowhead) and PAM sites (red). Below the schematic is the Sanger sequence of XDP iPSC clones with the SVA excised. (B) Expression of pluripotency-related genes OCT4, NANOG, SOX2, DNMT3B, and hTERT in each ΔSVA-XDP iPSC line compared with its unedited parent clone and parental fibroblast line as determined by qRT-PCR. Heatmap is based on ΔCt values. Red, high expression; blue, low expression. (C) SVA-edited clones 3C2, 3A12, 1A4 and 2F2 differentiated into NSC and NGN2-induced neurons showed expected expression of NSC vs. neuron markers, based on RNAseq. (D) Immunocytochemistry for mature neuronal markers DCX (green, left panel), Tuj1 (red, middle panel) and MAP2 (red, right panel) showing expression of these markers in NGN2-induced neurons generated from ΔSVA clones. Scale bar is 50 μm. (E) Karyotype analysis by g-banding revealed no apparent chromosomal abnormalities in the SVA-excised XDP iPSC lines.
(A) Scatter plot of log2 fold changes in XDP vs controls (X axis) and ΔSVA vs parental XDP (Y axis) and corresponding linear regression line and 95% confidence intervals for the slope of the regression line. Negative correlation suggests the reciprocal changes in gene expression in XDP lines upon SVA excision. (B) First (X axis) and second (Y axis) principal components of 400 DEGs in NSC, ΔSVA samples cluster closer to the controls and carriers.
Table S7. Results of differential expression analysis of ΔSVA vs corresponding XDP lines in NSCs and iNs (Related to Figure 6).
The Oligonucleotide sequences used in STAR method section for validation of novel DSCs by Sanger sequencing, CRISPR/Cas9 nuclease-mediated genome editing and cell model development and differentiation. Tm: Annealing temperature (in °C); ET: Extension time (seconds).