

Abstract
Background
Turner syndrome (TS) patients with hypoplastic left heart syndrome (HLHS) have poor single ventricle palliation outcomes; therefore, consideration of other potential management strategies is important. Little is known about heart transplantation (HTx) in this group, as standard HTx databases do not allow for identification of TS. This study describes experiences and outcomes of HTx in TS using a unique linkage between the Scientific Registry of Transplant Recipients (SRTR) and the Pediatric Health Information System (PHIS) databases.
Methods
All pediatric HTx recipients (2002–2016) with TS were identified in the database using ICD-9 code 758.6 (gonadal dysgenesis) in conjunction with female sex. Patient characteristics and outcomes were described.
Results
Fourteen patients with TS were identified who underwent 16 HTx procedures at 8 centers. For initial HTx, HLHS was the most common indication (10/14) with a median age of 10 months (IQR 3–73 months). Median transplant-free survival following initial HTx was 4.1 years (IQR: 16 days–10.5 years), with all deaths occurring in the first year post-HTx. For patients that survived past 1 year (8/14), follow-up ranged from 4.1 to 10.9 years (median 8.0 years) with no deaths observed.
Conclusions
Our cohort demonstrates that while there is a clear risk for early mortality, there is the potential for favorable outcomes following HTx in patients with TS. Therefore, TS should not be viewed as an absolute contraindication to HTx, but careful assessment of candidate risk is needed. Primary palliation with HTx for HLHS and TS may be a reasonable consideration given the poor outcomes of single ventricle palliation in this group. Further research is needed to fully delineate the outcomes and characteristics of this unique population.
Keywords: Heart transplant, Pediatric, Turner Syndrome, Outcomes, Hypoplastic Left Heart Syndrome
Introduction
Turner syndrome (TS) is a genetic syndrome characterized by monosomy X (45,XO) in phenotypic females, affecting approximately 1 in 2,100–2,500 live female births [1,2]. Common features include growth failure, pubertal delay, edema of the hands and feet, nuchal folds, low hairline, low-set ears, characteristic facies, high arched palate, nail hypoplasia, cubitus valgus, and a spectrum of congenital heart defects [3]. In fact, congenital heart disease is present in 23–50% of individuals with TS and most commonly involves left-sided heart lesions, including hypoplastic left heart syndrome (HLHS), which is estimated to occur in 10% of live born females with TS [4].
HLHS in patients with TS is associated with significant morbidity and mortality. Reiss et al. reported outcomes in 10 such patients, with only 2 surviving to second stage palliation [5]. A more recent single center study demonstrated significant mortality in the first year of life in patients with HLHS and TS (75%), while an analysis from the Pediatric Cardiac Care Consortium similarly reported an overall mortality of 90.4% in this group [6,7]. Given the poor outcomes of this patient population, determining the optimal management strategy is challenging.
There is limited experience with solid organ transplantation in patients with TS reported in the literature. Four prior cases of liver transplantation in TS have been reported, all in adults [8–11]. Of the three patients with reported outcomes, 2 were alive at last follow-up while one patient died 3 months following transplantation from respiratory failure and fungal urosepsis [9–11]. To our knowledge, there are no published reports of heart transplantation (HTx) in patients with TS. Current HTx databases do not readily allow for identification of patients with TS. We sought to describe the experience and outcomes of HTx in children with TS, with a particular focus on infants with TS and HLHS, using a unique linkage between the Scientific Registry of Transplant Recipients (SRTR) and the Pediatric Health Information System (PHIS) databases.
Methods
We utilized a unique linked dataset of the SRTR and PHIS databases that contains data from nearly 3000 pediatric HTx recipients transplanted within the U.S. between 2002 and 2016. The methods for linkage and description of these data have been previously reported [12]. The SRTR data system includes data on all donor, wait-listed candidates, and transplant recipients in the U.S., submitted by the members of the Organ Procurement and Transplantation Network (OPTN), and has been described elsewhere [13]. The Health Resources and Services Administration, U.S. Department of Health and Human Services, provides oversight to the activities of the OPTN and SRTR contractors. The SRTR collects and maintains data regarding organ transplantation in the U.S. and includes data from every organ transplant and waitlist addition since October 1987. The PHIS database is an administrative dataset that collects clinical and resource utilization data from hospital encounters at 49 large children’s hospitals. This includes encounter-level data from inpatient hospitalizations, observation, ambulatory surgery, and emergency department visits. PHIS provides a rich dataset for assessing patient diagnoses, procedures, and resource utilization through collection of ICD diagnosis and procedure codes, as well as hospital charge data [12].
The linked dataset was queried for all female patients with ICD-9 code of 758.6 (gonadal dysgenesis) documented at any encounter to identify patients with TS who have undergone HTx. One patient was excluded who had ICD-9 code 279.11 (DiGeorge syndrome) documented on multiple encounters and a single occurrence of ICD-9 code 758.6, that was thought to represent a coding error. Demographics of TS patients were assessed and reported. Patient characteristics assessed at the time of HTx included age, ethnicity, diagnosis, listing status at the time of HTx, history of prior surgery, and the need for mechanical ventilation or mechanical circulatory support (ventricular assist device or extracorporeal membrane oxygenation) at the time of HTx. Post-HTx data analyzed included freedom from death or re-transplantation and freedom from various ctube drainage for more than 2 weeks after HTx, and the need for dialysis, cardiac re-operation, and other surgical procedures. All analyses were performed using STATA (version 14; College Station, TX; StataCorp LP).
Results
A total of 14 unique female HTx recipients with TS were identified. Among the included patients, there were 16 total HTx procedures as 2 patients required re-transplantation (at 4.1 and 10.5 years post initial HTx). Patient demographics are shown in Table 1. Age at HTx ranged from 0 to 17 years; however, the majority (57%) of patients were <1 year of age at the time of initial HTx. Most were Caucasian (11/14 patients, 79%), OPTN status 1A at HTx (14/16 HTx, 88%), and had an underlying cardiac diagnosis of HLHS (10/14 patients, 71%). All but 3 patients were known to have had cardiac surgery prior to their first HTx. Four patients were mechanically ventilated and two additional patients were supported with a ventricular assist device at the time of transplant. No patients were supported with extracorporeal membrane oxygenation (ECMO) at the time of HTx. Reporting of further pre-operative characteristics including measures of sensitization were limited by missing data in this small cohort.
Table 1.
Patient demographics, pre-operative characteristics, and follow-up status in pediatric Turner syndrome patients undergoing heart transplantation; Hypoplastic left heart syndrome (HLHS), total anomalous pulmonary venous return (TAPVR), pulmonary artery (PA), endocardial fibroelastosis (EFE)
| Patient # |
Age at transplant |
Race | Transplant indication |
Prior Surgery |
Mechanical ventilation |
Ventricular assist device |
Listingstatus | Follow- up statusb |
Follow- up time |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0m a10y 6m |
Caucasian | HLHS Cardiac allograft vasculopathy |
- Y |
N N |
N N |
1A 1A |
R A |
10y 179d 3y 364d |
| 2 | 1m | Hispanic | HLHS | N | N | N | 1A | D | 110d |
| 3 | 2m | Caucasian | HLHS, TAPVR, PA anomaly | Y | Y | N | 1A | A | 4y 350d |
| 4 | 3m | Caucasian | HLHS | Y | N | N | 1A | A | 10y 323d |
| 5 | 7m | Hispanic | HLHS, PAPVR, EFE | Y | N | N | 1A | A | 7y 351d |
| 6 | 10m | Caucasian | HLHS | Y | Y | N | 1A | A | 5y 333d |
| 7 | 10m | Caucasian | HLHS, coronary anomaly | Y | Y | N | 1A | A | 365d |
| 8 | 10m a4y 11m |
Caucasian | HLHS Cardiac allograft vasculopathy |
Y Y |
Y N |
N N |
1A 1B |
R D |
4y 40d 20d |
| 9 | 3y | Caucasian | Other single ventricle | Y | N | N | 1A | D | 16d |
| 10 | 5y 1m | African American | HLHS | Y | N | N | 2 | D | 11d |
| 11 | 6y 1m | Caucasian | HLHS | Y | N | N | 1A | D | 12d |
| 12 | 8y 7m | Caucasian | Idiopathic dilated cardiomyopathy | - | N | N | 1A | D | 27d |
| 13 | 10y 9m | Caucasian | Congenital heart defect with prior surgery | Y | N | Y | 1A | D | 14d |
| 14 | 17y 2m | Caucasian | Idiopathic dilated cardiomyopathy | Y | N | Y | 1A | A | 10y 4d |
Re-transplantation in same patient,
Alive (A), died (D), re-transplanted (R)
Post-transplant outcomes are shown in Figure 1. Median length of follow-up after primary HTx was 2.6 years (IQR 18 days-8.0 years), and median transplant-free survival was 4.1 years (IQR 16 days-10.5 years). For patients that survived past 1 year (8/14), follow-up ranged from 4.1 to 10.9 years (median 8.0 years), during which no deaths were observed; however 2 patients underwent re-transplant at 4.1 and 10.5 years. Following initial HTx, 6 patients died, all in the first year following HTx, with 5 occurring within the first month. Three of these patients had HLHS and TS. Two deaths were from primary graft failure at 11 and 14 days post-HTx, 2 occurred following cardiac arrest (1 with ventricular failure at 12 days post-HTx and 1 with anoxic brain injury 16 days post-HTx), and 2 were from multi-organ failure (1 in the setting of bacterial infection) at 27 and 110 days post-HTx. Regarding the two patients that underwent re-transplantation at 4.1 and 10.5 years after primary HTx, 1 died 20 days post-HTx from multi-organ failure in the setting of sepsis and 1 was alive at last follow-up of 3.9 years.
Fig. 1.
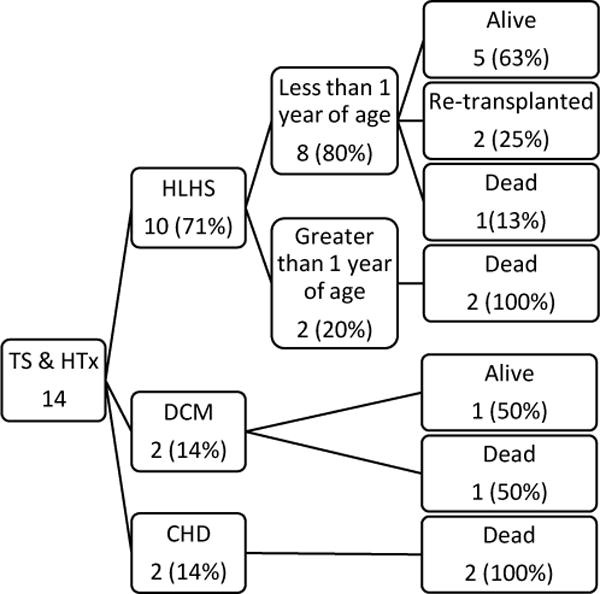
Heart transplant outcomes in pediatric Turner syndrome patients; Turner syndrome (TS), Heart transplantation (HTx), Hypoplastic left heart syndrome (HLHS), Idiopathic dilated cardiomyopathy (DCM), other congenital heart disease (CHD)
Five of the 6 deaths following initial HTx occurred in patients that were >1 year of age. Only 1 death occurred amongst the 8 patients <1 year of age. All of the patients <1 year of age carried a diagnosis of HLHS. Figure 2 is a visual representation of the freedom from death or re-transplantation in HLHS patients <1 year of age with and without TS. Given the small number of patients in the TS cohort, the study was underpowered to detect a clinically significant difference between groups, and therefore, a statistical comparison was not performed. Amongst the 6 patients with TS >1 year of age at HTx, HLHS was the indication for two patients. Both patients >1 year of age transplanted for HLHS died within the first month post-HTx due to primary graft failure and cardiac arrest with ventricular failure, respectively.
Fig. 2.
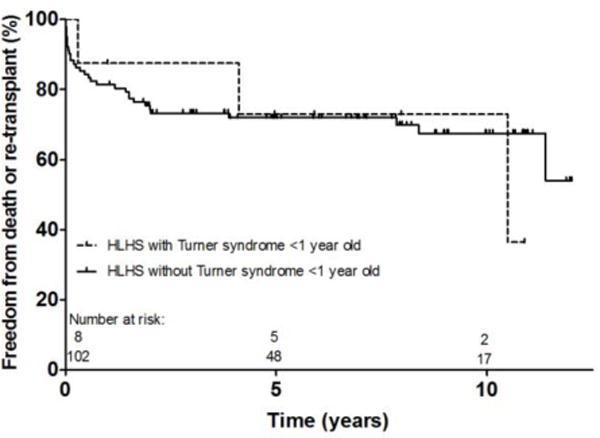
Freedom from death or re-transplant following heart transplant for patients less than 1 year old with hypoplastic left heart syndrome; Hypoplastic left heart syndrome (HLHS)
Post-HTx complications prior to hospital discharge are summarized in Table 2. Drug treated infection was the most common complication, occurring in 50% of patients. Five patients required post-HTx ECMO. Median days of post-HTx mechanical ventilation was 13 (IQR 3–18 days). Median days of post-HTx intensive care was 14 (IQR 10–24 days).
Table 2.
Post-operative complications and characteristics following heart transplantation in pediatric Turner syndrome patients; Hypoplastic left heart syndrome (HLHS), Extracorporeal membrane oxygenation (ECMO)
| All Patients | Patients with HLHS <1 year old | |
|---|---|---|
| Drug treated rejection | 2 (12.5) | 0 (0) |
| Drug treated infection | 8 (50) | 5 (63) |
| Stroke | 0a (0) | 0 (0) |
| Dialysis | 4 (25) | 1 (12.5) |
| Cardiac re-operation | 2b (12.5) | 1 (12.5) |
| Other surgical procedures | 2b (12.5) | 1 (12.5) |
| Pacemaker | 0 (0) | 0 (0) |
| Chest tube drainage >2 weeks | 1 (6.25) | 1 (12.5) |
| ECMO | 5b (31.25) | 1a (12.5) |
| Intensive care days [median (range)] | 14 (2–66) | 17 (5–66) |
| Ventilator days [median (range)] | 13 (0–103) | 14 (1–103) |
1 patient’s status unknown,
2 patient’s status unknown; Presented as number of patients (%), unless otherwise stated
Discussion
To our knowledge, this represents the first report of HTx in patients with TS to date. Overall outcomes in our cohort were mixed, with a clear risk of early mortality. All deaths occurred within 110 days of HTx, and 6 of the 7 deaths within the first the 27 days. This is similar to overall pediatric HTx outcomes with the greatest risk of mortality in the first year following HTx [14]. While overall median survival in the TS cohort appears suboptimal, it is better than that reported for TS undergoing single ventricle palliation. Given the small number of patients included in our analysis, it remains unclear if HTx outcomes are different compared to non-TS patients with congenital heart disease. However, our analysis demonstrates the potential for favorable outcomes, and therefore TS should not be considered an absolute contraindication to HTx.
It is difficult to identify characteristics that may place a TS patient at increased risk for early death following HTx. Interestingly, early mortality was skewed toward older patients in our cohort, with all but one early death occurring in patients >1 year of age. Certainly, this finding may not be clinically significant given the small sample size. However, one must wonder if there are inherent differences in the older sub-population, such as having more co-morbidities or worse functional status. Unfortunately, the size of our cohort and the data available limit our ability to identify risk factors for early mortality.
Given the association of TS with left-sided obstructive cardiac lesions, it is not surprising that HLHS was the most common indication for HTx in this cohort. HLHS with TS is associated with significant morbidity and mortality. Specifically, Reiss et al. reported that in a cohort of 10 infants with HLHS and TS who underwent first stage palliation, only two survived and underwent second stage palliation [5]. In a cohort of 4 patients with HLHS and TS, Cramer et al. described 1 death following initial palliation, 2 deaths following second stage palliation, and 1 surviving patient with significant medical issues who was in palliative care at the time of the report [6]. Two additional analyses, one utilizing the Texas Birth Defects Registry and the other using the Pediatric Cardiac Care Consortium, demonstrated 90% mortality in HLHS with TS (9/10 and 19/21 patients, respectively) [7,15]. The fact that many pediatric cardiologists recommend hospice care for infants with HLHS and a chromosomal abnormality further demonstrates the lack of favorable management strategies in this population [16]. Clearly, the current management approach to HLHS with TS is suboptimal and suggests the need for alternate management strategies, such as HTx, that may result in improved long term survival.
The significant early mortality in our cohort, with 6 deaths occurring following initial HTx, may cause some hesitation when considering HTx in TS patients. However, the outcomes demonstrated in our analysis compare favorably to the current outcomes of single ventricle palliation in this group [5–7,15]. HTx may offer better long term outcomes for patients with HLHS and TS, but further research is needed to define risk factors for early mortality to optimize patient outcomes. Additionally, careful consideration of candidate risk is needed prior to pursuing HTx given the limited availability of donor organs.
Although they remain unexplored at this time, the inherent co-morbidities related to TS coupled with standard post-HTx care raise the question for potential unique management considerations. TS portends an increased risk of diabetes mellitus that may be heightened by post-HTx immunosuppression including calcineurin inhibitors and corticosteroids [17–20]. Despite decreasing use of corticosteroids post-HTx in the recent era, corticosteroid use post-HTx may increase the risk of osteoporosis in TS [21,22]. Approximately 30–40% of TS patients have structural renal abnormalities [3]. Although TS patients do not typically have renal dysfunction, the resulting increased risk of renal infections with these structural abnormalities coupled with known nephrotoxicity of post-HTx immunosuppressive regimens may place these patients at increased risk of renal dysfunction relative to other pediatric HTx recipients [23]. Both TS and HTx are independently associated with increased risk of autoimmune diseases [17,24–29]. Clearly, the overlapping risk profile of TS and HTx patients raises the question of unique challenges post-HTx.
Limitations
Our study has inherent limitations. While the linkage between SRTR and PHIS provides a platform to assess HTx outcomes in this population, it does not contain data from all pediatric HTx centers and therefore may miss some patients with TS who have undergone HTx. In addition to this, the SRTR-PHIS linkage does not include patients who were listed but not transplanted and therefore waitlist outcomes for this group cannot be assessed. Also, there may be a selection bias in patients with TS listed for HTx, with only the healthiest patients progressing to HTx. However, the dataset is multicenter and reporting for all patients cared for at these centers is mandatory, limiting the chance of a post-HTx selection bias as could be seen in single center case series. While ICD coding allowed identification of patients with TS, there may be inaccuracies in coding and limited options to verify these data. The limited patient numbers prohibited performance of a robust multivariable analysis, as well as further comparison of HTx outcomes to patients without TS.
Conclusions
There is limited experience with HTx in patients with TS. Although there is a risk of early mortality, our cohort demonstrates the potential for favorable outcomes. Given the poor outcomes associated with single ventricle palliation in this group, it is reasonable to consider HTx in the discussion of management options for TS patients, although careful consideration of candidate risk is needed to optimize outcomes with this limited resource. Further research is needed to determine the optimal management of this complex group of patients and to define risk factors for early mortality following HTx.
Acknowledgments
This project was supported through internal funding from the Katherine Dodd Faculty Scholar Program at Vanderbilt University (JG). Research reported in this publication was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number K23HL123938 (Bethesda, MD) (JS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Compliance with Ethical Standards:
Disclosures: The data reported here have been supplied by the Minneapolis Medical Research Foundation (MMRF) as the contractor for the Scientific Registry of Transplant Recipients (SRTR). The interpretation and reporting of these data are the responsibility of the author(s) and in no way should be seen as an official policy of or interpretation by the SRTR or the U.S. Government.
Ethical Approval: As this is a retrospective analysis of a linked database, this article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent: For this type of study, formal consent is not required.
Conflict of Interest: The authors declare that they have no conflicts of interest related to this report.
References
- 1.Nielsen J, Wohlert M. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Birth Defects Orig Artic Ser. 1990;26(4):209–223. [PubMed] [Google Scholar]
- 2.Nielsen J, Sillesen I. Incidence of chromosome aberrations among 11148 newborn children. Humangenetik. 1975;30(1):1–12. doi: 10.1007/BF00273626. [DOI] [PubMed] [Google Scholar]
- 3.Bondy CA, Turner Syndrome Study G Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92(1):10–25. doi: 10.1210/jc.2006-1374. [DOI] [PubMed] [Google Scholar]
- 4.Bondy CA. Congenital cardiovascular disease in Turner syndrome. Congenit Heart Dis. 2008;3(1):2–15. doi: 10.1111/j.1747-0803.2007.00163.x. [DOI] [PubMed] [Google Scholar]
- 5.Reis PM, Punch MR, Bove EL, van de Ven CJ. Outcome of infants with hypoplastic left heart and Turner syndromes. Obstet Gynecol. 1999;93(4):532–535. doi: 10.1016/s0029-7844(98)00462-1. [DOI] [PubMed] [Google Scholar]
- 6.Cramer JW, Bartz PJ, Simpson PM, Zangwill SD. The spectrum of congenital heart disease and outcomes after surgical repair among children with Turner syndrome: a single-center review. Pediatr Cardiol. 2014;35(2):253–260. doi: 10.1007/s00246-013-0766-5. [DOI] [PubMed] [Google Scholar]
- 7.Madriago E, Nguyen T, McFerson M, Larson EV, Airhart N, Moller JH, Silberbach M. Frequency and outcomes of cardiac operations and catheter interventions in Turner syndrome. Am J Cardiol. 2012;110(4):580–585. doi: 10.1016/j.amjcard.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 8.Roulot D, Degott C, Chazouilleres O, Oberti F, Cales P, Carbonell N, Benferhat S, Bresson-Hadni S, Valla D. Vascular involvement of the liver in Turner’s syndrome. Hepatology. 2004;39(1):239–247. doi: 10.1002/hep.20026. [DOI] [PubMed] [Google Scholar]
- 9.Aucejo F, Ibrahim Z, Hashimoto K, Quintini C, Kelly D, Vogt D, Eghtesad B, Fung J, Miller C, Tuthill R. Cruveilhier-Baumgarten disease in a patient with Turner syndrome: case report of a rare indication for liver transplantation. Liver Transpl. 2008;14(3):299–302. doi: 10.1002/lt.21374. [DOI] [PubMed] [Google Scholar]
- 10.Chintanaboina J, Shah PR, Riley TR., 3rd An unusual occurrence of hepatic granulomas and secondary sitosterolemia in turner syndrome. Case Rep Med. 2015;2015:186718. doi: 10.1155/2015/186718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawabata S, Sakamoto S, Honda M, Hayashida S, Yamamoto H, Mikami Y, Inomata Y. Liver transplantation for a patient with Turner syndrome presenting severe portal hypertension: a case report and literature review. Surg Case Rep. 2016;2(1):68. doi: 10.1186/s40792-016-0194-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Godown J, Thurm C, Dodd D, Soslow J, Feingold B, Smith A, Mettler B, Thompson B, Hall M. A Unique Linkage of Administrative and Clinical Registry Databases to Expand Analytic Possibilities in Pediatric Heart Transplantation Research. American Heart Journal. 2017;194:9–15. doi: 10.1016/j.ahj.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leppke S, Leighton T, Zaun D, Chen SC, Skeans M, Israni AK, Snyder JJ, Kasiske BL. Scientific Registry of Transplant Recipients: collecting, analyzing, and reporting data on transplantation in the United States. Transplant Rev (Orlando) 2013;27(2):50–56. doi: 10.1016/j.trre.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Rossano JW, Dipchand AI, Edwards LB, Goldfarb S, Kucheryavaya AY, Levvey RnBJ, Lund LH, Meiser B, Yusen RD, Stehlik J, International Society for H. Lung T. The Registry of the International Society for Heart and Lung Transplantation: Nineteenth Pediatric Heart Transplantation Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant. 2016;35(10):1185–1195. doi: 10.1016/j.healun.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 15.Lara DA, Ethen MK, Canfield MA, Nembhard WN, Morris SA. A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit Heart Dis. 2017;12(1):105–112. doi: 10.1111/chd.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yates AR, Hoffman TM, Boettner B, Feltes TF, Cua CL. Initial counseling prior to palliation for hypoplastic left heart syndrome. Congenit Heart Dis. 2011;6(4):347–358. doi: 10.1111/j.1747-0803.2011.00525.x. [DOI] [PubMed] [Google Scholar]
- 17.Gravholt CH, Juul S, Naeraa RW, Hansen J. Morbidity in Turner syndrome. J Clin Epidemiol. 1998;51(2):147–158. doi: 10.1016/s0895-4356(97)00237-0. [DOI] [PubMed] [Google Scholar]
- 18.Hathout EH, Chinnock RE, Johnston JK, Fitts JA, Razzouk AJ, Mace JW, Bailey LL. Pediatric post-transplant diabetes: data from a large cohort of pediatric heart-transplant recipients. Am J Transplant. 2003;3(8):994–998. doi: 10.1034/j.1600-6143.2003.00186.x. [DOI] [PubMed] [Google Scholar]
- 19.Salgin B, Amin R, Yuen K, Williams RM, Murgatroyd P, Dunger DB. Insulin resistance is an intrinsic defect independent of fat mass in women with Turner’s syndrome. Horm Res. 2006;65(2):69–75. doi: 10.1159/000090907. [DOI] [PubMed] [Google Scholar]
- 20.Ye X, Kuo HT, Sampaio MS, Jiang Y, Reddy P, Bunnapradist S. Risk factors for development of new-onset diabetes mellitus in adult heart transplant recipients. Transplantation. 2010;89(12):1526–1532. doi: 10.1097/TP.0b013e3181dd6bd9. [DOI] [PubMed] [Google Scholar]
- 21.Freriks K, Timmermans J, Beerendonk CC, Verhaak CM, Netea-Maier RT, Otten BJ, Braat DD, Smeets DF, Kunst DH, Hermus AR, Timmers HJ. Standardized multidisciplinary evaluation yields significant previously undiagnosed morbidity in adult women with Turner syndrome. J Clin Endocrinol Metab. 2011;96(9):E1517–1526. doi: 10.1210/jc.2011-0346. [DOI] [PubMed] [Google Scholar]
- 22.Rossano JW, Cherikh WS, Chambers DC, Goldfarb S, Khush K, Kucheryavaya AY, Levvey BJ, Lund LH, Meiser B, Yusen RD, Stehlik J, International Society for H. Lung T. The Registry of the International Society for Heart and Lung Transplantation: Twentieth Pediatric Heart Transplantation Report-2017; Focus Theme: Allograft Ischemic Time. J Heart Lung Transplant. 2017 doi: 10.1016/j.healun.2017.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Feingold B, Zheng J, Law YM, Morrow WR, Hoffman TM, Schechtman KB, Dipchand AI, Canter CE, Pediatric Heart Transplant Study I Risk factors for late renal dysfunction after pediatric heart transplantation: a multi-institutional study. Pediatr Transplant. 2011;15(7):699–705. doi: 10.1111/j.1399-3046.2011.01564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonamico M, Pasquino AM, Mariani P, Danesi HM, Culasso F, Mazzanti L, Petri A, Bona G, Italian Society Of Pediatric Gastroenterology H, Italian Study Group for Turner S Prevalence and clinical picture of celiac disease in Turner syndrome. J Clin Endocrinol Metab. 2002;87(12):5495–5498. doi: 10.1210/jc.2002-020855. [DOI] [PubMed] [Google Scholar]
- 25.El-Mansoury M, Bryman I, Berntorp K, Hanson C, Wilhelmsen L, Landin-Wilhelmsen K. Hypothyroidism is common in turner syndrome: results of a five-year follow-up. J Clin Endocrinol Metab. 2005;90(4):2131–2135. doi: 10.1210/jc.2004-1262. [DOI] [PubMed] [Google Scholar]
- 26.Kulikowska A, Boslaugh SE, Huddleston CB, Gandhi SK, Gumbiner C, Canter CE. Infectious, malignant, and autoimmune complications in pediatric heart transplant recipients. J Pediatr. 2008;152(5):671–677. doi: 10.1016/j.jpeds.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 27.Livadas S, Xekouki P, Fouka F, Kanaka-Gantenbein C, Kaloumenou I, Mavrou A, Constantinidou N, Dacou-Voutetakis C. Prevalence of thyroid dysfunction in Turner’s syndrome: a long-term follow-up study and brief literature review. Thyroid. 2005;15(9):1061–1066. doi: 10.1089/thy.2005.15.1061. [DOI] [PubMed] [Google Scholar]
- 28.Price WH. A high incidence of chronic inflammatory bowel disease in patients with Turner’s syndrome. J Med Genet. 1979;16(4):263–266. doi: 10.1136/jmg.16.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouledoux JH, Albers EL, Lu Z, Saville BR, Moore DJ, Dodd DA. Clinical predictors of autoimmune and severe atopic disease in pediatric heart transplant recipients. Pediatr Transplant. 2014;18(2):197–203. doi: 10.1111/petr.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]