

Abstract
Purpose
Gene fusions are frequently found in prostate cancer and may result in the formation of unique chimeric amino acid sequences (CASQs) that span the breakpoint of two fused gene products. This study evaluated the potential for fusion-derived CASQs to be a source of tumor neoepitopes, and determined their relationship to patterns of immune signatures in prostate cancer patients.
Experimental Design
A computational strategy was used to identify CASQs and their corresponding predicted MHC class I epitopes using RNA-Seq data from The Cancer Genome Atlas of prostate tumors. In vitro peptide-specific T cell expansion was performed to identify CASQ-reactive T cells. A multivariate analysis was used to relate patterns of in silico-predicted tumor-infiltrating immune cells with prostate tumors harboring these mutational events.
Results
Eighty-seven percent of tumors contained gene fusions with a mean of 12 per tumor. In total, 41% of fusion-positive tumors were found to encode CASQs. Within these tumors, 87% gave rise to predicted MHC class I-binding epitopes. This observation was more prominent when patients were stratified into low- and intermediate/high-risk categories. One of the identified CASQ from the recurrent TMPRSS2:ERG type VI fusion contained several high-affinity HLA-restricted epitopes. These peptides bound HLA-A*02:01 in vitro and were recognized by CD8+ T cells. Finally, the presence of fusions and CASQs were associated with expression of immune cell infiltration.
Conclusions
Mutanome analysis of gene fusion-derived CASQs can give rise to patient-specific predicted neoepitopes. Moreover, these fusions predicted patterns of immune-cell infiltration within a sub-group of prostate cancer patients.
Introduction
Recent successes using immunotherapy approaches have highlighted the potential of harnessing the immune system to treat late-stage cancers. There is an overarching view that clinical responses to immunotherapy are dependent on the presence and immune recognition of tumor-specific antigens. However, prostate cancer remains problematic given that far fewer patients demonstrate clinical responses to immunotherapy compared to other settings. For instance, the use of vaccine strategies that target over-expressed tumor associated self-antigens, such as prostatic acid phosphatase (PAP) and prostate specific antigen (PSA), have shown only moderate clinical success [1, 2]. Although encouraging, the general lack of robust anti-tumor immune responses after vaccination may be partially explained by the fact that self-antigens are often poorly immunogenic and T cells specific for such tumor-associated antigens may be tolerized. Moreover, prostate tumors generally lack an abundance of cytotoxic tumor-infiltrating lymphocytes (TILs) or have suppressed T cell response due to engagement of checkpoint inhibition pathways [3, 4].
Checkpoint blockade and adoptive transfer of tumor-specific or engineered T cells are two promising approaches under investigation. Although these strategies have shown remarkable clinical responses in other cancer settings, it is believed that tumor antigen load may, in part, explain some of the differences between responders and non-responders. For example, in highly mutated solid tumors such as melanoma and non-small cell lung carcinoma (NSCLC), the success of checkpoint blockade and adoptive cell transfer (ACT) of TILs has been largely attributed to the abundance of tumor neoantigens [5, 6]. On the other hand, these strategies have shown less success in settings with classically low tumor mutation load [7, 8], although one recent interim report found durable clinical responses to anti-PD-1 in enzalutamide-resistant prostate cancer patients [9]. Whether such responses in prostate cancer are associated with a higher frequency of mutations or the presence of a specific type of mutation remains to be determined.
At present, the vast majority of studies have focused on the identification of either recurrent or personalized somatic point mutations that generate tumor neoepitopes. However, given recent genomic efforts to define the broader tumor mutational landscape (e.g. single base substitutions, SCNA, translocations, chromothripsis, alternative splicing, epigenetic alterations, etc. [10–12]), examining other types of mutations as potential immunogenic targets warrants further investigation.
Compared to somatic point mutations, structural genomic rearrangements are considered relatively rare events in epithelial cancers. However, these genome rearrangements are the most common type of genomic abnormality in prostate cancer [13, 14]. Consequently, some of these structural genomic rearrangements are important for the pathogenesis of prostate cancer, resulting in amplification of oncogenic drivers, deletion of tumor suppressors, and fusion events that cause altered expression of tumor oncogenes [15, 16]. For instance, recurrent fusions between androgen-regulated genes and ETS family genes occur in over 50% of all prostate cancer cases [17, 18] and in many cases lead to aberrant expression of oncogenic ETS transcription factors that may contribute to prostate tumorigenesis [13, 14, 18].
Tumor mutations can generate altered protein products that are distinct from those found in normal tissues. If the resulting altered protein is immunogenic, it presents an opportunity for specific targeting of the tumor by the immune system. Indeed, personalized multi-epitope vaccines targeting non-synonymous tumor mutations have demonstrated anti-tumor efficacy in multiple pre-clinical studies [19, 20], and this concept is being further investigated in early clinical trials in numerous cancer settings [21–25]. Other reports have highlighted this proof-of-principle using adoptive transfer of expanded, mutation-specific TIL [26, 27]. However, in mutation-low tumor settings such as prostate cancer, the pool of potential tumor neoantigens arising from somatic point mutations may be limited [28–30].
Similar to non-synonymous point mutations, fusion events can generate altered protein products when the fusion occurs within a coding region. In this case, the breakpoint-spanning chimeric amino acid sequence (CASQ) of two fused genes can distinguish each gene partner from its respective wild-type counterpart. Given their specificity to the tumor, these unique CASQs have the potential to elicit tumor-specific T cell responses. For instance, a multi-peptide vaccine against the CASQ of the BCR-ABL fusion protein gave rise to measureable peptide-specific immune responses and improved disease control in chronic myelogenous leukaemia patients [31, 32]. Despite the prevalence of genomic rearrangements across prostate cancer cases, strategies to exploit the reservoir of fusion-derived CASQs as tumor neoepitopes have not been systematically explored. Here, a stringent computational approach was used to identify prospective immunogenic fusion epitopes from transcriptomics data in a cohort of prostate adenocarcinoma patients from The Cancer Genome Atlas. While a large proportion (74 out of 85) of the cases that were examined contained gene fusions, nearly half of the patients in this analysis were found to harbor CASQs, and among these, 87% of the CASQs gave rise to predicted MHC class I epitopes. These results were even more pronounced when patients were stratified into low- and intermediate/high-risk cohorts where low-risk patients contained fewer fusions and CASQs when compared to the intermediate/high-risk patients. Interestingly, there was a significant correlation between the presence of gene fusions and CASQs with distinct immune gene expression patterns in a subset of patient tumors. Thus, immunotherapy approaches in prostate cancer should consider gene fusions and their interactions with the host immune system.
Methods
Patient datasets and blood collection
RNA-Seq datasets (matched tumor and adjacent normal) from The Cancer Genome Atlas (TCGA) prostate adenocarcinoma cases were accessed from the Cancer Genomics Hub (https://cghub.ucsc.edu/; May, 2015). The patients were stratified into two groups: low-risk (PSA < 10 ng/mL Gleason Score = 6; n=35) and intermediate-high risk (PSA >10 ng/mL and Gleason Score ≥7; n=50). The available clinical information for this cohort is summarized in Table 1. A list of the TCGA identifiers for patients in this study can be found in Supplementary Data (Table S1). For in vitro studies, peripheral blood mononuclear cells (PBMC) were purified from whole blood of healthy donors using Ficoll-Paque PLUS (GE Healthcare) and cryopreserved in nitrogen vapor freezers. Healthy donors gave written informed consent to donate semi-annual 200 mL blood draws under protocols approved by the University of British Columbia-British Columbia Cancer Agency’s Research Ethics Board (REB# H07-00463). As such, this study was conducted in accordance with the Declaration of Helsinki.
Table 1.
Characteristics of the TCGA prostate adenocarcinoma patient cohort.
| Patient Characteristics | |
|---|---|
| Age at diagnosis (median) | |
| Years | 41–77 (57) |
| Gleason Score (%) | |
| 6 | 35 (41) |
| 7 | 30 (35) |
| 8 | 4 (5) |
| 9 | 16 (19) |
| Pre-operative PSA (median) | |
| 0–10 ng/mL | 35 (5.6) |
| 10–20 ng/mL | 35 (12.8) |
| >20 ng/mL | 15 (26.3) |
| pT (%) | |
| T2a | 5 (6) |
| T2b | 1 (1) |
| T2c | 34 (40) |
| T3a | 28 (33) |
| T3b | 15 (18) |
| T4 | 2 (2) |
| Regional Lymph node involvement (%) | |
| Yes | 11 (13) |
| No | 50 (59) |
| Not available | 24 (28) |
Fusion detection by defuse
Fusions were detected from RNA-Seq datasets using deFuse (Version 0.6.2) [33] based on reference genome and gene models from Ensembl GRCh37 release-75. In short, using default parameters for deFuse, candidate gene fusions were first identified by clustering spanning reads, which are defined as discordant mappings whose two mates were located within distinct genes. Spanning reads that mapped to multiple loci due to sequence homology or RNA splice variation were assigned to the most likely gene pairs based on the maximum parsimony principle. Once the set of fused genes was obtained, one-end anchored (OEA) reads whose mapped mates locate near each candidate fusion boundary were grouped together. Their unmapped mates were aligned to sequences near the fusion boundary in a dynamic programming formulation to obtain final breakpoints at nucleotide resolution. Additional confidence parameters were applied to eliminate false positive fusion calls according to recommendations for deFuse [33].
In silico translation and CASQ identification
To identify CASQs, we focused on genes that were fused with complimentary orientation and reading frame. For each predicted fusion, deFuse provides the orientation information describing how two genes are combined based on the splitting reads that support the fusion. Fusion peptides were selected as follows: First, two genes in a fusion should concatenate in a way consistent to their strands and reading frames. In other words, fusion breakpoints should always be located downstream of the first gene, and upstream of the second gene. In addition, we only considered fusions that were in-frame, and chimeric transcripts that had lost the native ATG from the 5′ gene partner were eliminated. Second, translations of chimeric peptides should pertain to the original reading frames from both parental genes. For each half of the chimeric transcript, we extract the corresponding protein sequences from the human proteome database (Ensembl GRCh37 release-75) and ensured that the translated sequence in the final chimeric peptide is identical to the peptide sequence from the same regions of the original proteins. Chimeric peptide sequences that did not share the native reading frame of their parental protein sequences were then discarded. As a result, each chimeric peptide is formed by concatenating two subsequences from known human proteins. These filtration steps ensured the prioritization of fusions encoding in-frame chimeric proteins.
MHC class I allele selection using HLAMiner
HLAMiner (Version 1.3) was used to determine the HLA haplotype of individuals at the HLA-A, -B, and -C loci from RNA-Seq [34]. The top 2 predictions at the 4-digit resolution for each locus were nominated for each given patient provided that predictions differed at the 2-digit resolution.
IEDB peptide prediction
MHC_I Binding Tool (Version 2.13) from NetMHCpan package was accessed via the Immune Epitope Database (IEDB) [35] and used to extract predicted high affinity minimal peptides from CASQs on patient autologous HLA haploptypes. Fusion-spanning 8-11mer peptides with an IEDB rank score of <2 were classified as high affinity epitopes.
Peptide library
The TMPRSS2:ERG type VI fusion was identified as a recurrent CASQ yielding predicted HLA binding epitopes and was thus selected for further in vitro validation.TMPRSS2:ERG type VI fusion peptides were synthesized and purified commercially (Genscript, NJ), reconstituted and stored in pure DMSO. We chose to evaluate overlapping decamer peptides beginning at the translational initiation site N-terminal to the fusion breakpoint plus an additional nonamer peptide, beginning at position 2, based on the predicted affinity trends for these peptides on HLA-A*02:01.
MHC stabilization assay
T2 cells were originally purchased from the American Type Culture Collection stock (CRL-1992) and obtained as a kind gift from Dr. Brad Nelson. The cells were not pathogen tested at the time they were used.T2 cells were maintained in Iscove’s Modified Dulbecco’s Medium supplemented with 20% FBS. T2 cells were plated with increasing concentrations of TMPRSS2:ERG peptides (0–80μM) for 18 hours at 26 °C. A known HLA-A*02:01 binding epitope from the melanocyte protein Melan-A/MART-1 (ELAGIGILTV) was used as the positive control. Cells were incubated for an additional 3 hours at 37 °C in the presence of brefeldin A (10μg/mL) and labeled with anti-human HLA-A2 fluorescein isothiocyanate (Clone BB7.2, BD Biosciences). The mean fluorescence index was measured using a Guava EasyCyte 8HT flow cytometer (EMD Millipore) and data were analyzed with FlowJo V10 software.
Generation of fusion-specific T cell lines
Monocyte-derived dendritic cell (DC) cultures were generated as previously described [36]. Briefly, healthy donor PBMC were plated in 6 well plates (107 cells/well) and non-adherent cells removed after 90 and 150 minutes at 37 °C. Adherent monocytes were supplemented with 800 IU/mL each granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin-4 (IL-4). On day 6, DC were matured with polyI:C (50μg/mL) for 2 days prior to harvesting for stimulation of T cells against fusion peptides. DC were pulsed with TMPRSS2:ERG fusion peptides (10μM/peptide), irradiated (32 Gy), and incubated with autologous PBMC. T cells were re-stimulated with irradiated (50 Gy) peptide-pulsed PBMC and polyI:C after 11 days and expanded with 240 IU/mL IL-2, 20 ng/mL IL-15, and 20 ng/mL IL-7 for two weeks. T cell cultures were rested for 3 days in 10 ng/mL IL-7 and screened for TMPRSS2:ERG reactivity by interferon-γ (IFNγ) ELISPOT as described [36]. Individual reactive cultures were expanded using irradiated (50 Gy) allogeneic feeder cells (PBMC) supplemented with OKT3 (30 ng/mL) and IL-2 (300 IU/mL) for 2 weeks. TMPRSS2:ERG peptide-activated T cells were enriched by fluorescence activated cell sorting on CD8+CD137+ followed by re-expansion.
Immunological correlates of CASQ
We explored correlation between total gene fusions, predicted MHC class I binding CASQ epitopes, and immunological parameters using recently published estimates that predicted immune-cell infiltrates using RNA-sequencing data [37]. For immune cell-infiltrates, we used published infiltration estimates for the patients included in this cohort generated, estimated via a single sample gene set enrichment analysis (ssGSEA) approach performed across multiple cancers [37]. These immune cell predictions were tested for associations with predicted fusions using redundancy analysis (RDA). This is a constrained extension of principal components analysis (PCA) that employs linear modelling to associate predictor variables (here the presence/absence of CASQs and total number of fusions (log +1 transformed)) and a multivariate outcome (predicted immune cell infiltrates). Model significance was assessed via permutation tests (n = 1000 permutations), all implemented in the vegan package of R v3.3.1 [38].
Results
Computational approach and strategy for RNA-Seq data analysis
An immunogenomic strategy was used to predict tumor-specific antigens contained within chimeric proteins using RNA-Seq datasets (Figure 1). This approach combined three existing computational tools: deFuse, HLAMiner, and IEDB MHC class I epitope prediction software [33–35] (www.iedb.org). First, deFuse identified a list of high-confidence genomic fusions from RNA-Seq reads using a mapping-based approach. The resulting fusion transcripts encoding predicted CASQs were translated in silico. Second, using HLAMiner, patient-specific HLA haplotypes were extracted from matched transcriptomics data. Next, IEDB was used to interrogate CASQs against patient-relevant HLAs to generate a candidate list of high-affinity MHC class I epitopes specific to the fusion-spanning regions. Stringent criteria as defined in the Methods were used at each step to uncover potential mutations with the highest probability of generating neoepitopes for in vitro validation by peptide-specific expansion of CASQ-reactive T cells from the peripheral blood of healthy donors.
Figure 1. Computational approach to identify predicted immunogenic CASQs.
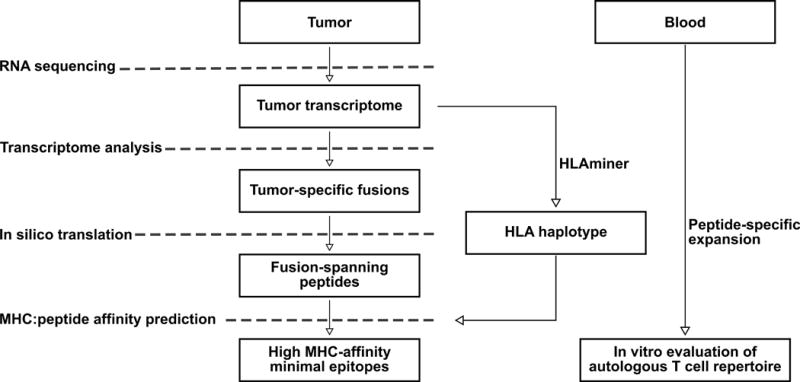
Data are generated from RNA-sequencing of tumor tissue. Transcriptomic analysis reveals tumor-specific gene fusions and in silico translation is performed to identify CASQs. Patient HLA haplotype is determined via HLAminer to generate a candidate list of MHCI:CASQ affinity prediction scores. T cells from the peripheral blood are interrogated against fusion-encoding predicted epitopes to assess existing immunoreactivity to patient-specific CASQs.
A proportion of tumor-specific gene fusions yield CASQ with predicted MHC class I epitopes
The frequency and recurrence of genomic fusion events were evaluated within a cohort of prostate adenocarcinoma cases from TCGA (n=85). Patient inclusion was based upon clinical presentation of low-risk (n=35) defined by a Gleason score of 6 and pre-operative PSA < 10 ng/mL and intermediate/high-risk (n=50), defined by a Gleason score ≥7 and pre-operative PSA ≥ 10 ng/mL (Table 1). Using the approaches described above, analysis of tumor RNA-Seq datasets revealed a unique set of fusion events from each patient (range 1–65, median 8, mean 12; Figure 2a; black bars). A complete list of gene fusions can be found in the Supplementary Data (Table S2). The prevalence of fusion events in our cohort is consistent with previously reported transcriptomic studies in similar patient groups [39]. Greater than half of the tumors (57%) in this cohort carried 10 or fewer fusions, while the top 10% of fusion-positive tumors contained greater than 25 genomic fusions. Although the mean number of putative CASQs generated by individual fusion events was low, 41% of fusion-positive tumors contained a minimum of one CASQ, with a maximum of six CASQs identified in a single case (Patient 013) (Figure 2a; grey bars). A complete list of the predicted CASQs for each patient is provided in the Supplementary Data (Table S2). When the patient cohort was analyzed based on risk, there was a significant stratification in the presence of fusions and CASQs. Of particular note, all of the tumors with no identifiable fusions belonged to the low-risk group of patients. By contrast, all 50 patient tumors in the intermediate/high-risk patients contained fusions. In the intermediate/high-risk group 44% of fusions were found to harbor at least 1 CASQ (Figure 2a; left). In contrast, only 69% of low-risk patient tumors harbored fusions, and if these, 33% harbored at least one CASQ (Figure 2a; right).
Figure 2. Gene fusions across the cohort of TCGA prostate adenocarcinoma patients.
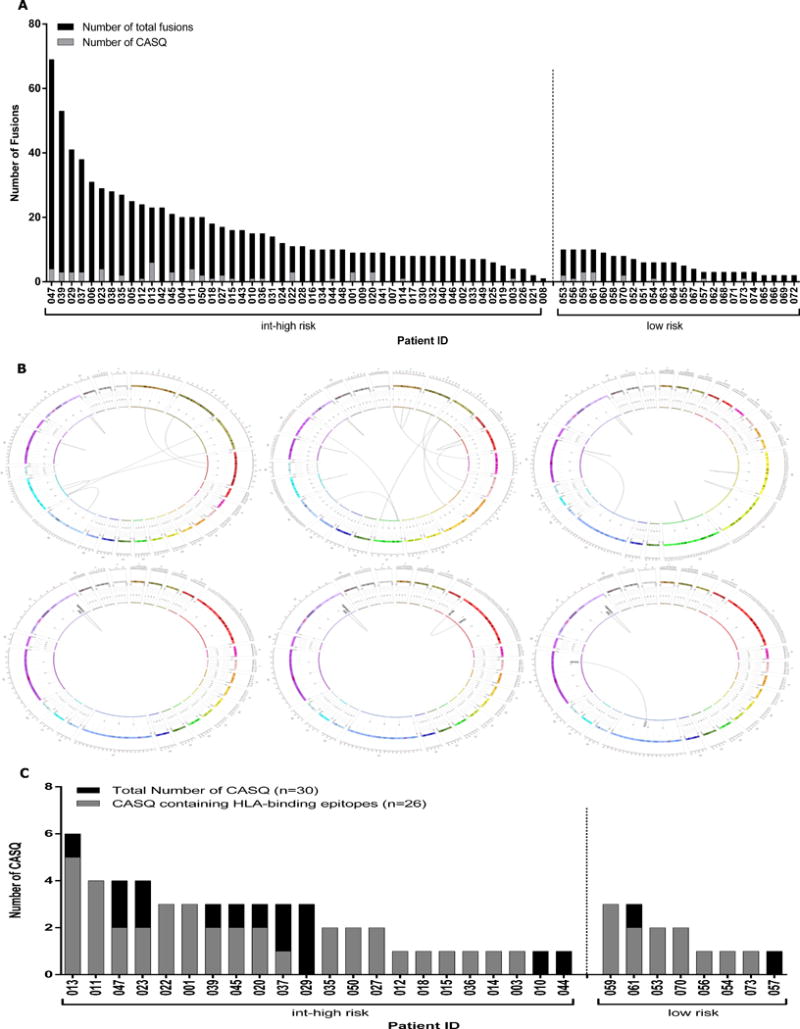
(A) The number of tumor-specific gene fusions per patient dataset by deFuse analysis using a stringent systematic filtering approach to eliminate false-positives. Fusions identified within the TCGA prostate adenocarcinoma cohort range from as many as 69 to as few as 1 (black bars). Gene fusions from 22 intermediate/high-risk (left) and 8 low-risk (right) patient tumors are predicted to encode at least one CASQ. (B) Representative Circos plots display the total number of identified fusions from 3 representative patient tumors (patients 022, 011, 020, respectively; upper panel). Circos plots display CASQs arising from those patient fusions (lower panel). (C) The total number of CASQs identified in 19 intermediate/high-risk (left) and 7 low-risk (right) patients (black bars) with a neoepitope predicted to bind to patient’s autologous HLA (grey bars). As many as 6 CASQs were identified within an individual patient tumor. The majority of CASQs encode a fusion spanning epitope predicted to bind to patients’ autologous HLA alleles. CASQs of Patients 029, 044, 010 and 057 yielded no predicted HLA-binding epitopes.
In Figure 2b, Circos plots from the analysis of three representative patients show the proportion and intra- and inter-chromosomal locations of tumor-specific fusion events leading to CASQs. Given the presence of CASQs in this cohort, patient-specific HLA haplotypes were identified using HLAMiner to predict a list of candidate MHC class I-restricted CASQ-derived epitopes. When the full patient cohort was considered, 41% (n=30) of the tumors contained a CASQ (Figure 2c; black bars). Of these, 86% (n=26) of these patients had at least one predicted high affinity MHC class I binding epitope (Figure 2c; grey bars; rank score ≤2). Regardless of patient risk, there was a high likelihood that tumors containing a CASQ had a predicted HLA-binding epitope (Figure 2c: left versus right). A complete list of each patient’s CASQ-derived epitopes is provided in Supplementary Data (Table S3).
The TMPRSS2:ERG CASQ contains patient specific HLA class I-restricted epitopes
In total, 50 gene pairs were recurrently fused throughout the cohort (Table 2). Approximately 70% of recurrent gene fusions identified within this cohort appeared to be a result of inter-chromosomal translocations based on sequence mapping across chromosomes (Supplementary Data, Table S2). Fusions between TMPRSS2 and ERG have been well annotated in prostate cancer [17]. Consistent with this finding, the most prevalent recurrent gene fusion in our cohort was between TMPRSS2 and ERG, with 13% of tumors (n=4) expressing this fusion. One of the tumors in the low-risk and 3 of the tumors in the intermediate/high-risk cohort contained the TMPRSS2 and ERG fusion. However, only 2 of the recurrent fusions encoded CASQs: CAMKK2:KDM2B and the TMPRSS2:ERG type VI fusion (exon 2:4 fusion) (Table 2), both having arisen from an intra-chromosomal fusion event. Moreover, TMPRSS2:ERG was the only recurrent CASQ that generated HLA-binding epitopes in multiple patients (n=4). While many variants of the TMPRSS2:ERG fusion have been reported, the majority of these generate ERG frameshifts or initiate transcription downstream from the fusion breakpoint, resulting in truncated ERG gene products. The TMPRSS2:ERG fusion joining exons 2 of TMPRSS2 and 4 of ERG (type VI) is the only variant in this family which encodes a CASQ that retains the native reading frame of ERG (Figure 3). Furthermore, TMPRSS2:ERG type VI is common among these fusion variants and has been reported to occur in up to 25% of cases where ERG alterations are present [40].
Table 2. Gene pairs of CASQs identified across the prostate cancer patient cohort.
CASQs arising from gene fusions were found in 22 intermediate/high-risk patients (44%) and 8 low-risk patients (33%). The maximum number of CASQs identified within a single tumor was 6. The majority of CASQs involved gene partners unique to each individual; however, two recurrent fusions were observed. CAMKK2:KDM2B generated CASQs in patients 018 and 023; TMPRSS2:ERG generated CASQs in Patients 011, 020, 022 and 070.
| Patient | Fusions | ||
|---|---|---|---|
| 001 | PMEPA1:ETV4 | TMPRSS2:MORC3 | |
| 003 | ABCD3:DPYD | ||
| 010 | MT-CYB:MT-ND3 | ||
| 011 | GPBP1:MTRR | TMPRSS2:ERG | |
| 012 | LEO1:FBN1 | ||
| 013 | KLK2:KLK3 SMYD3:TRIM58 |
MAPK9:DGKB SNAP91:BCKDHB |
SAMD5:SASH1 TMEM56-RWDD3:SNX7 |
| 014 | PRCP:RAB30 | ||
| 015 | KDM6A:ARHGAP6 | ||
| 018 | CAMKK2:KDM2B | ||
| 020 | TCF12:NPAS1 | TMPRSS2:ERG | |
| 022 | TMPRSS2:ERG | ||
| 023 | CAMKK2:KDM2B | GSK3B:ATP11B | PTPRK:ECHDC1 |
| 027 | ALDH3A2:PITPNM2 | SLC25A39:EFCAB13 | |
| 029 | SLC45A3:EPB41L4B | ||
| 035 | DLG1:CRYBG3 | UBR2:XPO5 | |
| 036 | TMPRSS2:TMEFF2 | ||
| 037 | EYA2:SYS1 | HOMER2:HDGFRP3 | PRUNE2:GNA14 |
| 039 | FAU:SRRM1 | PTBP1:UBE3C | TBC1D25:HSPA9 |
| 044 | TTC39A:MRPL37 | ||
| 045 | POLR2J:LGALS4 | VPS13B:AKAP7 | |
| 047 | KANSL3:TSGA10 SLC7A1:HRSP12 |
RPTOR:IQCH TMC6:UACA |
|
| 050 | RB1CC1:PTPN3 | ||
| 053 | HNRNPUL1:ATG10 | KLK2:KLK3 | |
| 054 | KLK2:KLK3 | ||
| 056 | KLK2:KLK3 | ||
| 057 | LIG3:PHF12 | ||
| 059 | CHD8:AP1S1 | ZMIZ1:ZCCHC24 | NXPE2:EP300 |
| 061 | PTEN:HECTD2 | KLK2:KLK3 | TOR1A:COG4 |
| 070 | DYM:KATNAL2 | TMPRSS2:ERG | |
| 073 | MOV10:ZNHIT6 |
Figure 3. The recurrent fusion TMPRSS2:ERG, present in the TCGA intermediate and high risk prostate cancer patient cohort.
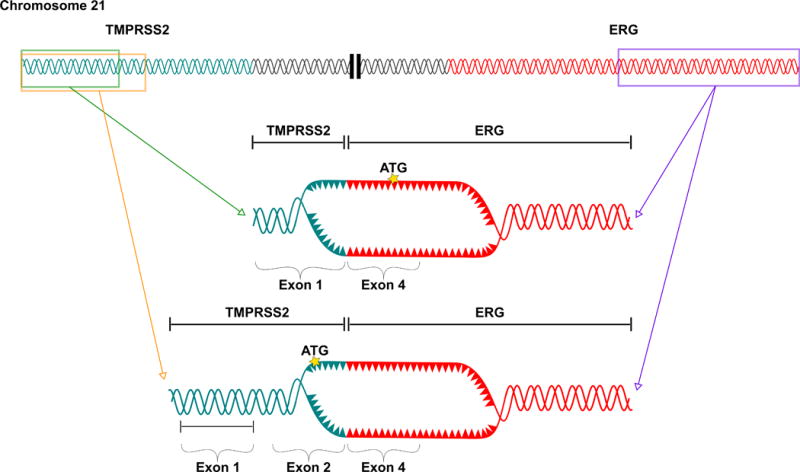
Schematic representation of two frequently recurring TMPRSS2:ERG fusions. The type VI (2:4) (bottom) fusion variant encodes an in-frame CASQ.
Of the 14 tumors that contained a TMPRSS2:ERG fusion, 23% were positive for the type VI variant (n=4; Table 2). In each of these type VI cases, in silico analysis using IEDB predicted patient-specific HLA-binding epitopes. In silico epitope predictions on common HLAs revealed fusion epitopes that were predicted to strongly bind the HLA-A*02:01 allele (rank <1; Figure 4a, b). These predictions were validated for HLA-A*02:01 binding in vitro using TAP-deficient T2 cells. Three of the TMPRSS2:ERG minimal peptides displayed no stabilization of surface MHC. In contrast, the top three ranked minimal peptides, MALNSEALSV, ALNSEALSVV, and ALNSEALSV stabilized HLA-A*02:01 above threshold in this assay (Figure 4c). One of the fusion peptides, ALNSEALSVV, bound to MHC with similar affinity to the known HLA-A*02:01-restricted epitope modified from Melan-A/MART-1 (ELAGIGILTV) [41]. The results of the binding assay for the top peptides were consistent with the in silico epitope predictions, as these three TMPRSS2:ERG fusion peptides were predicted to bind HLA-A*02:01 with the top rank scores (4.5, 0.9, and 1, respectively).
Figure 4. TMPRSS2:ERG peptides bind HLA-A*02:01.
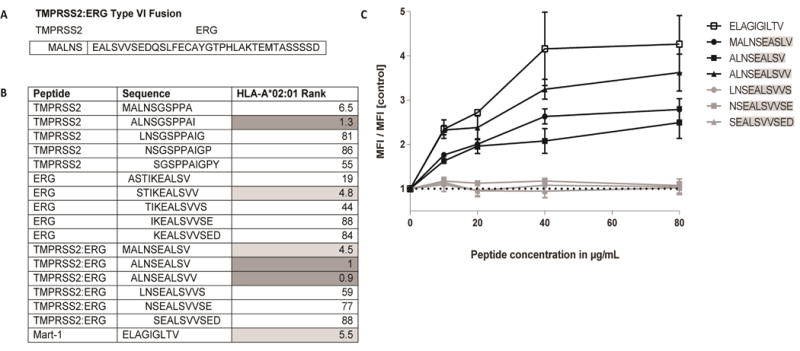
(A) The TMPRSS2:ERG CASQ. (B) Rank scores from IEDB MHC epitope predictions for each of the TMPRSS2:ERG peptides. Data includes all possible fusion-spanning 10mers, as well as the sole 9mer with a high-affinity binding score. Lower scores indicate an increased likelihood of peptide binding to HLA-A*02:01. Peptides which meet the predicted binding affinity threshold of rank ≤ 2 are highlighted in dark grey. Peptides with a predicted binding affinity of rank ≤ 6 are highlighted in light grey. (C) TMPRSS2:ERG peptides MALNSEALSV, ALNSEALSV, and ALNSEALSVV each stabilize HLA-A*02:01. T2 cells were pulsed with increasing concentrations of each peptide for 18 hours at 26°C followed by 3 hours at 37°C in the presence of 10μg/mL brefeldin A. Cells were stained with anti-HLA-A*02 FITC for 30 minutes at 4°C and analyzed for MHC stabilization by flow cytometry. The mean fluorescence index relative to unpulsed T2 cells is shown. The HLA-A*02:01-restricted peptide from MART-1, ELAGIGILTV, was used as a positive control. The dotted line indicates the MFI of T2 cells in the absence of exogenous peptide.
T cells recognize minimal peptides specific for the TMPRSS2:ERG type VI CASQ
Next, TMPRSS2:ERG minimal peptides were assessed for their ability to stimulate and expand T cells from peripheral blood of an HLA-A*02:01+ healthy donor. After two rounds of in vitro stimulation with peptide-pulsed dendritic cells, T cell cultures were monitored for antigen reactivity by IFNγ ELISPOT. Three individual peptide-reactive T cell lines were identified by this method (Figure 5a). Each T cell line recognized the 10mer peptide MALNSEALSV, as well as the 9mer, ALNSEALSV. In addition, one T cell line, 9E6, recognized the 10mer ALNSEALSVV. All three T cell lines were primarily CD8+ and up-regulated CD137 in response to peptide stimulation (Figure 5b). These responses are specific to the predicted epitope derived from the CASQ, as none of the T cell lines were cross-reactive to the corresponding native TMPRSS2 or ERG peptide sequences (Figure 5c).
Figure 5. T cells from an HLA-A*02:01+ healthy donor recognize the three TMPRSS2:ERG peptides with predicted affinity for HLA-A*02:01.
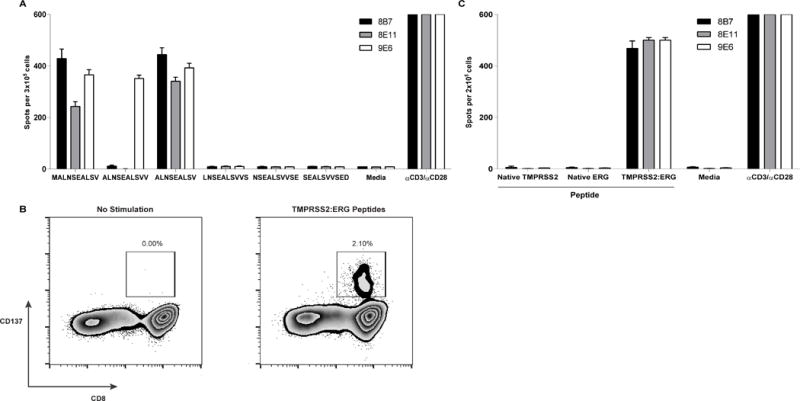
All T cells were assessed for IFNγ secretion by ELISpot after 20 hours of co-culture with pools of overlapping minimal peptides (10μM). (A) T cells secrete IFNγ in response to stimulation with TMPRSS2:ERG type VI fusion peptides. (B) TMPRSS2:ERG-specific CD8+ T cells upregulated CD137 upon stimulation by a pool of TMPRSS2:ERG peptides (MALNSEALSV, ALNSEALSVV, and ALSNSEALSV). (C) TMPRSS2:ERG-specific T cells do not cross-react with either the corresponding native TMPRSS2 or ERG peptides.
Predicted immune infiltrates are significantly associated with gene fusions
Within our cohort of low- and intermediate/high-risk patients, there was a strong stratification of risk groups according to the number of fusions (Figure 6a; Welch’s t-test on log-transformed fusions; P < 10−9). The likelihood of a tumor harboring at least one CASQ also trended towards an increase in the intermediate/high-risk patients (Fisher’s exact test; P = 0.096). Based on the CASQs and neoepitopes predicted above, one might expect that their presence or absence could impact the immune state of the tumor. This possibility was assessed with a multivariate approach to associate predicted immune cell infiltrations in these tumors with predicted fusions and CASQs using redundancy analysis. This model revealed that fusions and CASQs could predict patterns of immune cell infiltration, and together explained ~10% of the variation of the included immune markers (Figure 6b–d; RDA permutation test, P < 0.001). However, the significance of association with these immune markers was greater using fusions than CASQs in analyses with either as a single predictor (Permutation tests; P < 0.001 versus P < 0.011 respectively). When focusing on subsets of immune cells identified as associating with fusions in the RDA, there were striking negative correlations between fusion load and cytolytic signals, such as predicted NK cells and CD8+ T cells (Figure 6d; P = 6.5 × 10−9 and P = 0.039 respectively). These results were paralleled, in trends towards positive associations with markers related to anti-tumor immune suppression such as Th2 phenotypes and to a lesser extent T regulatory cells (Figure 6d; P = 0.03 and P = 0.081, respectively). While further validation is necessary in a larger case series, these data suggest that these genomic aberrations can be accompanied by profound changes in the risk stratification and predicted immunological status in a subset of prostate cancer patients.
Figure 6. Gene fusions and predicted CASQ-derived epitopes are associated with predictions of tumor infiltrating immune cells.
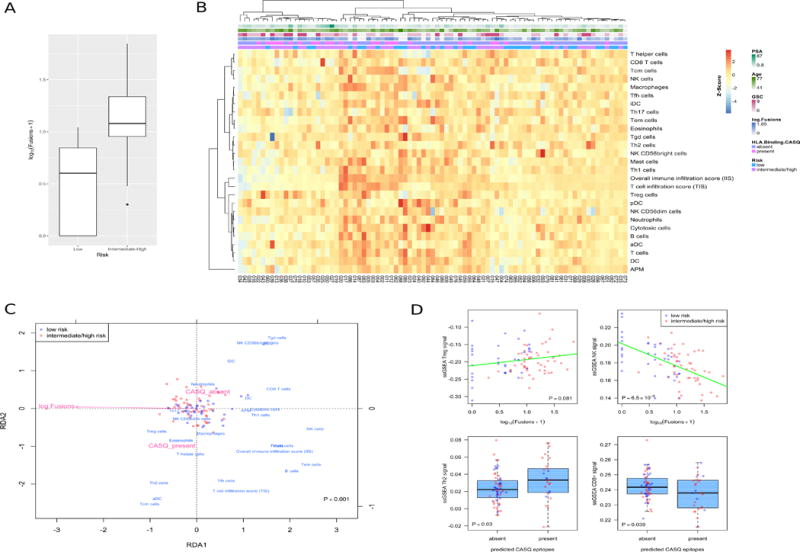
(A) Predicted number of fusions differs across risk groups (P < 10−9). (B) Heatmap of predicted immune cell infiltrate levels and immunological parameters for patients in this cohort, extracted from the ssGSEA-based predictions of Senbabaoglu et al. (2016). Annotation tracks represent the presence of predicted CASQ epitopes (CASQ), log predicted total tumor fusions, Gleason Score, patient age at diagnosis, and PSA level.
(C) Redundancy analysis associating tumor fusions and CASQ epitopes with patterns of immune cell infiltration shows a significant multivariate effect (Permutation test, P < 0.001). Triplot representing patients (red and blue circles), immune predictions (blue), and explanatory variables (light red). (D) Plots of univariate associations of predicted immune cell infiltrates with fusion and CASQ epitope predictions.
Discussion
There is increasing evidence that the presence of neoantigens contribute to the success of various modalities of immunotherapy. Several correlative studies in patients treated with anti-PD-1, -PD-L1 and -CTLA-4 suggest that tumors with a high mutation burden may be more responsive to checkpoint blockade. For example, checkpoint inhibitors have been successful in melanoma and NSCLC where the average number of non-synonymous mutations per tumor ranges from 100–230, with roughly 95% of these representing single-base substitutions [11]. On the other hand, prostate cancers harbour fewer mutation events despite the fact that a small number of durable clinical responses to anti-PD-1 have been reported [9]. However, it is not known whether these patients had a relatively higher frequency of mutations than patients who did not respond. Thus, at least for PD-1 blockade therapy the relationship between response and mutations requires further study.
Similar outcomes have been uncovered in patients who received infusions of enriched antigen-specific tumor infiltrating lymphocyte populations and went on to show dramatic clinical responses [27, 42]. However, one study in gastrointestinal cancer using similar genomic approaches described here found that out of 1,452 mutations across 10 tumors, only 18 could be recognized by CD4+ or CD8+ TIL [42]. Several other studies targeting neoantigens have focused on somatic point mutations in tumor sites where this type of genomic aberration is highly abundant. Despite the abundance of point mutations observed in such tumors, it was reported that relatively few of these give rise to authentic neoantigens [43, 44]. Given these data, it raises the question of whether other types of mutations may provide an alternative source of neoantigens that could be exploited for immune-based treatment strategies.
Gene fusions can introduce a variety of genetic alterations, including frameshifts, deletions, truncations, modified splicing patterns, differential inclusion of cryptic exons or introns, chimeric proteins, exchange or alteration of promoter regions, amongst others. In theory, the primary amino acid sequences generated from many of these events have the potential to elicit a tumor-specific immune response. Our analysis focused only on the coding region fusions that preserve the parental sequence of each gene partner across the breakpoint. While this likely results in an under-representation of the number of possible neoepitopes generated by this class of mutations in prostate tumors, this strategy is intended to streamline the identification of putative epitopes with the highest likelihood of biological relevance, allowing for a more focused selection of minimal peptides for empirical validation. By this rationale, intronic fusions were also excluded from the final analysis as these sequences would be either biologically non-functional or removed post-transcriptionally.
Based on current genome sequencing data, prostate tumors harbor an average of 3,866 somatic point mutations where the majority are unique to an individual patient. Of these, non-synonymous mutations account for only about 0.5 % of the total mutation load [45]. Furthermore, only a fraction of these would ultimately generate bona fide neoantigens, restricting the scope of potential antigens that could be targeted. Mutations arising from gene fusion events occur more frequently in prostate cancer than in other solid tumors, with reports ranging from 43 to 213 fusions per tumor [45]. Despite their prevalence, the immunogenicity of gene fusions in prostate cancer has not been explored. One potential reason may be due to the difficulties encountered during the confident identification of fusion calls from RNA-Seq data, specifically during the elimination of false-positives [33]. While multiple sequencing methods are now able to identify and resolve the breakpoint sequences of gene fusions, using RNA-Seq can be advantageous for discovering events that have a higher probability of predicting functional relevance, since only expressed and translated fusions are of interest during the identification of T cell targetable epitopes. Thus, prostate cancer is an ideal setting for evaluating the potential use of patient-specific fusion proteins as targets for T cell based immunotherapies.
Here, a computational strategy was used to evaluate the extent of CASQs generated by fusion events. This analysis identified fusions in 87% of the entire cohort. However, most of this was due to the intermediate/high-risk patients where all tumors contained fusions. By contrast, only 69% of the low-risk patients were found to harbour fusions. Similarly, 41% of all patients contained a CASQ, whereas 33% of the low- and 44% of the intermediate/high-risk patients were found to possess predicted CASQs. Within the group of patients whose tumors harbored CASQs (n=30), 86% (n=26) had at least one predicted chimeric epitope. The frequency of HLA-binding epitopes was similar between the low- and intermediate/high-risk patients. Therefore, although the proportion of fusions encoding CASQs was small, the presence of CASQs was a predictor of MHC class I binding epitopes. The majority of predicted CASQ-derived epitopes in this population were patient-specific. In fact, only two recurrent gene fusions, CAMKK2:KDM2B and TMPRSS2:ERG, yielded predicted epitopes in more than a single patient (n=2 and 4, respectively) (Figure 4a), supporting the notion that, like somatic point mutations, targeting fusion mutations may be suited as a personalized approach. Whether any of the CASQs generate aberrant cell surface protein expression is not yet known, but could represent compelling targets using chimeric antigen receptor (CAR) T cell therapy. Indeed clinical trials targeting PSMA with CAR-T cells trial are underway [46, 47].
TMPRSS2:ERG fusions were identified in 23% of tumors in our cohort and was within the range of what has been previously reported. Although several variants of this fusion have been identified, the type VI (2:4) fusion is the only known variant which preserves the native translation initiation site of TMPRSS2 while ERG remains in-frame, producing functional ERG with a 5 amino acid N-terminal extension imparted by TMPRSS2 (Figure 3) [40]. In our study, type VI fusions were present in 4 tumors (13%) and in each case led to predicted high-affinity fusion epitopes on autologous HLAs. Indeed, due to the prevalence of ERG alterations in prostate cancer, one study reported vaccine-induced immunity to ERG-derived HLA-A*02:01-restricted epitopes [48].
In this study, peptide-pulsed dendritic cells were used to expand TMPRSS2:ERG-specific T cell lines from an HLA-A*02:01+ healthy donor. However, several criteria must still be satisfied for a given mutation to produce an authentic neoepitope; for example, the parent protein must undergo endogenous antigen processing to produce the immunogenic peptides. One limitation of our study was the lack of HLA-matched tumor specimens with confirmed TMPRSS2:ERG type VI expression to test the authenticity of this fusion against TMPRSS2:ERG-specific T cell lines that were expanded in vitro.
Finally, there was a significant negative relationship between tumor fusions and the presence of cytolytic immune signatures, and the number of fusions strongly stratified patients across low and intermediate/high risk groups. In general, cytolytic immune responses such as the expression of NK cell and CD8+ T cell markers negatively correlated with tumor fusions, and, to a lesser extent, predicted CASQs. Consistent with this reduction in cytolytic indicators, there was a positive association between Th2 genes. In addition to this, there was a trend towards positive associations between T regs and the presence of fusions and CASQs. Though this exploratory analysis examined on a small subset of patients, our results suggest the possibility of functional interactions between genome rearrangements and host immune responses that may impact anti-tumor immunity. It could be speculated that the negative relationship observed between tumor fusions and cytolytic immune responses may have resulted from immune selection against these potential neoantigens during tumor evolution. A similar circumstance could also exist for these immune signatures when considering the lower frequency of both fusions and CASQ in the low- versus intermediate/high-risk patients. Indeed, such a scenario may be possible as prostate cancer risk segregates with a unique immune signature based on a specific mutation profile. However, further validation in independent cohorts is needed to confirm these findings. Nonetheless, this information could expand the potential scope of mutations, including CASQs and other gene fusions, as an alternative avenue for immune-based approaches targeting prostate cancer.
Supplementary Material
Table S1 – List of patient TCGA IDs and patient study IDs.
Table S2 – Complete list of high-confidence fusions identified in each patient.
Table S3 – Complete list of high-confidence predicted CASQ identified in each patient.
Table S4 – Complete list of CASQ-derived high-affinity HLA epitopes for each patient.
Statement of translational relevance.
The identification of T cell targets for immunotherapy remains a significant hurdle. Clinical studies have reported encouraging results targeting recurrent somatic point mutations. While their abundance is a factor that underlies some clinical responses, these mutations represent a fraction of possible neoantigens arising from the overall tumor mutational landscape. Prostate tumors exhibit a high frequency of genome rearrangements. Such an event can produce a CASQ, a unique peptide sequence spanning the junction of two fused gene products. Here, 74 of 85 tumors that were examined contained gene fusions. Of these tumors, 41% expressed CASQs and 87% of CASQs were predicted to generate HLA-restricted epitopes. In one case, T cells could recognize CASQ-derived peptides. Interestingly, gene fusions were strongly associated with distinct immune-cell signatures. These data highlight gene fusion-derived CASQs as potential sources of neoantigens, particularly in tumors with a low frequency of somatic point mutations.
Acknowledgments
The authors wish to thank Dr. Allen Zhang and Dr. Julie Nielsen for helpful feedback during the preparation of this manuscript.
Grant Support: This study was supported in whole or in part from grants from the US Department of Defence Prostate Cancer Research Program (W81XWH-15-1-0078; JJL, CC), Prostate Cancer Fight Foundation Ride for Dad (JJL), West Coast Ride for Dad (JJL), BC Cancer Foundation (JJL) and a Prostate Cancer Canada Discovery Grant (D2015-09; JJL), Indiana University Precision Health Initiative and the US NIH R01 Grant GM108348 (SCS) and the NSERC Discovery Frontiers Grant, “The Cancer Genome Collaboratory”(SCS).
Footnotes
Conflicts of Interest: The authors declare no conflict of interest
Supplementary data for this article are provided separately.
References
- 1.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 2.Kantoff PW, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiniwa Y, et al. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. 2007;13(23):6947–58. doi: 10.1158/1078-0432.CCR-07-0842. [DOI] [PubMed] [Google Scholar]
- 4.Bronte V, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. 2005;201(8):1257–68. doi: 10.1084/jem.20042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Allen EM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350(6257):207–11. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwon ED, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–12. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graff JN, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rode A, et al. Chromothripsis in cancer cells: An update. Int J Cancer. 2016;138(10):2322–33. doi: 10.1002/ijc.29888. [DOI] [PubMed] [Google Scholar]
- 11.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbieri CE, Rubin MA. Genomic rearrangements in prostate cancer. Curr Opin Urol. 2015;25(1):71–6. doi: 10.1097/MOU.0000000000000129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beltran H, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63(5):920–6. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kulda V, et al. Prognostic Significance of TMPRSS2-ERG Fusion Gene in Prostate Cancer. Anticancer Res. 2016;36(9):4787–93. doi: 10.21873/anticanres.11037. [DOI] [PubMed] [Google Scholar]
- 16.Carver BS, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet. 2009;41(5):619–24. doi: 10.1038/ng.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomlins SA, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–8. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 18.Leong M, S W, Tian J, Cho E, Raza A, Siddiqi SA, Selim A, Chen H, Zhang D. Overexpression of truncated ERG from TMPRSS2-ERG fusion and prostate cancer development. Pathol Lab Med Int. 2009;(1):13–21. [Google Scholar]
- 19.Castle JC, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 20.Yadav M, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 21.A Phase I Study With a Personalized NeoAntigen Cancer Vaccine in Melanoma. 2016 NCT01970358. [cited 2016 Dec 15]; Available from: https://clinicaltrials.gov/ct2/show/NCT01970358.
- 22.IVAC MUTANOME Phase I Clinical Trial. 2016 NCT02035956. [cited 2016 Dec 15]; Available from: https://clinicaltrials.gov/ct2/show/NCT02035956.
- 23.Safety and Immunogenicity of a Personalized Synthetic Long Peptide Breast Cancer Vaccine Strategy in Patients With Persistent Triple-Negative Breast Cancer Following Neoadjuvant Chemotherapy. 2016 NCT02427581. [cited 2016 Dec 15]; Available from: https://clinicaltrials.gov/ct2/show/NCT02427581.
- 24.Neoepitope-based Personalized Vaccine Approach in Patients With Newly Diagnosed Glioblastoma. 2016 NCT02510950. [cited 2016 Dec 15]; Available from: https://clinicaltrials.gov/ct2/show/NCT02510950.
- 25.Peptide Vaccine in Advanced Pancreatic Ductal Adenocarcinoma or Colorectal Adenocarcinoma. 2016 NCT02600949. [cited 2016 Dec 15]; Available from: https://clinicaltrials.gov/ct2/show/NCT02600949.
- 26.Robbins PF, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tran E, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown SD, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;245:743–50. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan H, Bao J, Zhou X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci Rep. 2015;5:12566. doi: 10.1038/srep12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin SD, et al. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS One. 2016;11(5):e0155189. doi: 10.1371/journal.pone.0155189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bocchia M, et al. Effect of a p210 multipeptide vaccine associated with imatinib or interferon in patients with chronic myeloid leukaemia and persistent residual disease: a multicentre observational trial. Lancet. 2005;365(9460):657–62. doi: 10.1016/S0140-6736(05)17945-8. [DOI] [PubMed] [Google Scholar]
- 32.Cathcart K, et al. A multivalent bcr-abl fusion peptide vaccination trial in patients with chronic myeloid leukemia. Blood. 2004;103(3):1037–42. doi: 10.1182/blood-2003-03-0954. [DOI] [PubMed] [Google Scholar]
- 33.McPherson A, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7(5):e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warren RL, et al. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012;4(12):95. doi: 10.1186/gm396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vita R, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015;43:D405–12. doi: 10.1093/nar/gku938. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nielsen JS, et al. Toward Personalized Lymphoma Immunotherapy: Identification of Common Driver Mutations Recognized by Patient CD8+ T Cells. Clin Cancer Res. 2016;22(9):2226–36. doi: 10.1158/1078-0432.CCR-15-2023. [DOI] [PubMed] [Google Scholar]
- 37.Senbabaoglu Y, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol. 2016;17(1):231. doi: 10.1186/s13059-016-1092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshihara K, et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015;34(37):4845–54. doi: 10.1038/onc.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, et al. Expression of variant TMPRSS2/ERG fusion messenger RNAs is associated with aggressive prostate cancer. Cancer Res. 2006;66(17):8347–51. doi: 10.1158/0008-5472.CAN-06-1966. [DOI] [PubMed] [Google Scholar]
- 41.Valmori D, et al. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J Immunol. 1998;160(4):1750–8. [PubMed] [Google Scholar]
- 42.Tran E, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350(6266):1387–90. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen CJ, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–91. doi: 10.1172/JCI82416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajasagi M, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014;1243:453–62. doi: 10.1182/blood-2014-04-567933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger MF, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470(7333):214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Junghans RP, et al. Phase I Trial of Anti-PSMA Designer CAR-T Cells in Prostate Cancer: Possible Role for Interacting Interleukin 2-T Cell Pharmacodynamics as a Determinant of Clinical Response. Prostate. 2016;76(14):1257–70. doi: 10.1002/pros.23214. [DOI] [PubMed] [Google Scholar]
- 47.Adoptive Transfer of Autologous T Cells Targeted to Prostate Specific Membrane Antigen (PSMA) for the Treatment of Castrate Metastatic Prostate Cancer (CMPC) NCT01140373. [cited 2017 July 1]; Available from: https://clinicaltrials.gov/ct2/show/NCT01140373.
- 48.Kissick HT, et al. Development of a peptide-based vaccine targeting TMPRSS2:ERG fusion-positive prostate cancer. Cancer Immunol Immunother. 2013;62(12):1831–40. doi: 10.1007/s00262-013-1482-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 – List of patient TCGA IDs and patient study IDs.
Table S2 – Complete list of high-confidence fusions identified in each patient.
Table S3 – Complete list of high-confidence predicted CASQ identified in each patient.
Table S4 – Complete list of CASQ-derived high-affinity HLA epitopes for each patient.