

Abstract
To investigate whether mutation profiling and microsatellite instability (MSI) status were associated with clinicopathological features and the prognosis in metastatic colorectal cancer (mCRC), mutations in RAS (including KRAS, NRAS, and HRAS) and BRAF were determined by Sanger sequencing. Tumor mismatch repair proteins and MSI status were examined using immunohistochemistry and polymerase chain reaction, respectively. The clinical value of these abnormalities was statistically analyzed, and prognostic value of different treatment regimens was also evaluated. Among 461 mCRC patients, mutations in RAS, BRAF, and MSI-high (MSI-H) status were observed in 45.3% (209/461), 5.6% (26/461), and 6.5% (30/461) of cases, respectively. Brain metastasis and high carcinoembryonic antigen level were highly correlated with KRAS mutation (P = 0.011 and P < 0.001), and tumors from females or located in the right colon tended to harbor BRAF mutation (P = 0.039 and P = 0.001). RAS/BRAF mutations may predict brain and/or lung metastases. Although neither clinical nor prognostic importance of MSI status was identified in our study, KRAS and BRAF mutations were demonstrated to be independent prognostic factors for overall survival and progression-free survival. Besides, in wild-type group, patients treated with chemotherapy plus targeted therapy exhibited the most favorable prognosis. Therefore, RAS/BRAF mutations may serve as indicators for prognosis and treatment options in mCRC.
1. Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy and the fourth most frequent cause of cancer-associated mortality worldwide [1]. Previous evidence has indicated that liver and lung metastases were quite common in metastatic CRC (mCRC), accounting for approximately 20–30% of all patients when initially diagnosed [2]. As the understanding of molecular mechanisms underlying tumorigenesis and progression of CRC develops, genetic analyses and targeted therapy have already become popular alternatives, representing a significant landmark towards individually tailored treatment.
It is usually admitted that epidermal growth factor receptor (EGFR) is an imperative molecular target in mCRC [3]. In general, the monoclonal antibody against EGFR, cetuximab or panitumumab, is capable of competitively blocking EGFR from binding to its ligand, thus suppressing efficiently downstream RAS/Raf/MAPK pathway activity and improved outcomes [4]. However, mutations of RAS (including KRAS, NRAS, and HRAS) and BRAF genes may bring about constitutive activation of the pathway, independent of EGFR inhibition, which is associated with resistance to anti-EGFR therapy [5]. Therefore, the screening of a full gene mutation profiling contributes to select suitable candidates for appropriate therapeutic regimens and regular surveillance.
Microsatellite instability (MSI), a genetic change resulted from mismatch repair (MMR) deficiencies during DNA replication, involves with the pathogenesis of CRC [6]. MSI-high (MSI-H) is known to occur in about 10% of sporadic CRCs and 3% hereditary CRCs [7]. Recently, Le et al. [8] reported a high response rate of mCRC with MSI-H to programmed death-1 (PD-1) inhibitor therapy, indicating that MSI status could be a useful checkpoint for immune therapy.
Multiple researches have documented that KRAS mutations were common in a diverse range of human neoplasms, such as lung adenocarcinoma [9], pancreatic cancer [10], and thyroid cancer [11]. Especially in CRC, the rate of KRAS mutations is nearly 40%, although NRAS or HRAS mutations only for less than 3% or 1% [12–14]. Due to high homology and close correlation with KRAS, NRAS and HRAS behave as typical oncogenes [12]. Increasing evidence revealed that CRC patients with NRAS mutations had relatively favorable prognosis compared with those with KRAS or BRAF mutations [15]. However, the clinical importance of HRAS mutation remained unclear in CRC because of its rarity [14]. Additionally, as a downstream member of KRAS, BRAF encodes a serine/threonine protein kinase which plays an important role in cell division and secretion [16]. Cancers with BRAF mutation are closely related to tumor location and lower survival, especially for those together with MSI-low (MSI-L) or microsatellite stable (MSS) [17]. Nevertheless, information available about the abnormalities of these oncogenes and the MSI status in mCRC have not been convincingly elucidated.
Here, we comprehensively characterized RAS/BRAF mutations and MSI status as well as evaluated the prognostic value of different treatment regimens in mCRC patients, which can provide an optimal insight between gene abnormalities and patient survival in Chinese population.
2. Materials and Methods
2.1. Patients and Clinical Data
The observational model was developed in 461 clinicopathologically confirmed mCRC patients at Guangdong General Hospital (Guangzhou, China) between March 2011 and December 2014. All participants received genetic testing as a part of integrated care. Information on clinicopathological and therapeutic data were obtained from medical archive; tumor classification and grading were based on the World Health Organization criteria. Overall survival (OS) and progression-free survival (PFS) were defined from enrollment start time until death/censoring and tumor progression/censoring, respectively. An outpatient follow-up was conducted every 3 months in accordance with Response Evaluation Criteria in Solid Tumors (RECIST 1.1) in the first 2 years after clinical treatments, followed by every 6 months, until the study endpoint or death. Informed consent was obtained from all individual participants included in the study, and authorization was acquired from the Ethics Committee of Guangdong General Hospital.
2.2. Tissue Sampling and Mutation Assessment
Comprehensive genomic profiling was analyzed on 461 formalin-fixed paraffin-embedded (FFPE) primary CRCs retrieved from surgical/endoscopic biopsies and 247 metastases from surgical/percutaneous needle biopsies. Genomic DNA was isolated from each FFPE specimen with QIAamp DNA FFPE Tissue Kit Qiagen (Hilden, Germany) based on the manufacturer's recommendations. Besides that, cancer cell-rich regions were identified in advance by application of hematoxylin-eosin (H&E) staining to ascertain all cases tested enrichment of ≥70% malignant cells. Extracted DNA concentration was determined in a ND-1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). Mutations in the KRAS (exons 2, 3, and 4), NRAS (exons 2, 3, and 4), HRAS (exon 2), and BRAF (exon 15) of each tumor specimen were examined. AmpliSeq Designer v.1.2.6 software (Life Technologies) was used to design primer pairs for these gene amplifications [18]. DNA amplification was performed by using GoTaq® Hot Start Polymerase (Promega, Madison, WI) and 0.2 lM of each primer with the GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA) under the cycling conditions as described previously [19]. Amplicons were finally Sanger sequenced bidirectionally on an ABI 3730XL genetic analyzer (Invitrogen Life Technologies, Carlsbad, CA, USA), and detailed procedures were the same as reported earlier [20].
2.3. MMR Proteins Determination
Immunohistochemistry (IHC) analysis of the four most frequent MMR proteins (i.e., MLH1, MSH2, MSH6, and PMS2) was conducted on FFPE tumor specimens following standard IHC protocols [21]. Representative tumor areas were carefully selected and marked before paraffin blocks were longitudinally sliced to 4 μm thick sections. Immunostaining was carried out with mouse monoclonal antibodies MLH1 (liquid, 1 : 150 dilution; BD, New Jersey, USA), MSH2 (lyophilized, 1 : 100 dilution; BD, New Jersey, USA), MSH6 (liquid, 1 : 150 dilution; BD, New Jersey, USA), and PMS2 (liquid, 1 : 150 dilution; BD, New Jersey, USA). Normal protein expression presented nuclear staining of tumor cells, while negative result showed no nuclear staining in tumor cells with concurrent positive controls within surrounding cells. Tumors were classified as MMR deficiency (MMR-D) when any MMR protein expression was negative and MMR intact (MMR-I) when all MMR proteins were positively expressed. The results were judged by two independent pathologists.
2.4. Analysis of Microsatellite Instability (MSI) Status
Extracted DNA samples from primary CRCs and paired metastases were also used for MSI analysis. Briefly, MSI status was examined with the panel of five microsatellite markers (BAT25, BAT26, NR21, NR24, and NR27) by fluorescence-based PCR. Primer pairs for amplification were designed using the software package mentioned above. DNA was amplified in a 20 μL volume with GoTaq Hot Start Polymerase (Promega, Madison, WI), starting with an initial 5-minute denaturation step at 95°C, then 35 cycles at 95°C for 30 seconds, annealing at 60°C for 30 seconds, and extension at 72°C for 30 seconds and finally an extension at 72°C for 10 minutes. The PCR products were analyzed on a Genetic Analyzer (Applied Biosystems 3500, ABI), and allelic sizes were determined with the GeneMapper Software (Applied Biosystems). Patients were defined as MSI-L if a single marker presented instability, MSI-H if two or more of the five studied markers showed instability, and MSS if no marker showed instability.
2.5. Statistical Analysis
The data analysis was performed by SPSS version 19.0 (SPSS Inc., Chicago, IL, USA). The correlation between gene status and clinicopathological variables was compared with Pearson's Chi-square (χ 2) test. Logistic regression was done to identify potential predictors for brain/lung metastases, and the area under the receiver operating characteristic (ROC) was used to estimate the predictive value of the clinical factors. Survival curves were plotted by Kaplan-Meier method with a log-rank test. Univariate and multivariate proportional Cox models were employed to assess independent prognostic factors. The statistically significant difference was set at 0.05.
3. Results
3.1. Frequency of Gene Mutations in Primary Lesions and Corresponding Metastases
Among 461 primary CRCs, 231 (50.1%) were RAS/BRAF wild-type. KRAS, NRAS, and HRAS mutations were observed in 43.6% (201/461), 2.8% (13/461), and 0.2% (1/461) of cases, respectively. Besides, as another indispensible incidence of EGFR pathway, BRAF mutations were present in 5.6% (26/461) cases. Notably, gene mutations in primary lesions were highly coincident with those in matched metastases except two patients, whose KRAS mutations occurred in primary tumors rather than metastases. The most frequently noted mutation occurred in exon 2 (codons 12 and 13) of KRAS (37.1%, 171/461). Detailed distribution of mutation subtypes was summed up in Table 1.
Table 1.
Mutation subtype frequency distribution of RAS and BRAF genes.
| Genes | Codon | Mutation | Cases (% of 461) |
|---|---|---|---|
| Total cases with RAS mutation | 230 (49.9%) | ||
| Total cases with KRAS mutation | 201 (43.6%) | ||
| KRAS | 12 | p.G12D | 74 (16.1%) |
| 12 | p.G12V | 35 (7.6%) | |
| 12 | p.G12C | 15 (3.3%) | |
| 12 | p.G12A | 6 (1.3%) | |
| 12 | p.G12R | 9 (1.9%) | |
| 12 | p.G12S | 1 (0.2%) | |
| 13 | p.G13D | 31 (6.7%) | |
| 59 | p.A59T | 4 (0.9%) | |
| 61 | p.Q61H | 2 (0.4%) | |
| 146 | p.A146T | 16 (3.5%) | |
| 146 | p.A146V | 3 (0.6%) | |
| Others | Others | 5 (1.1%) | |
| Total cases with NRAS mutation | 13 (2.8%) | ||
| NRAS | 12 | p.G12D | 4 (0.9%) |
| 12 | p.G12S | 2 (0.4%) | |
| 18 | p.A18T | 1 (0.2%) | |
| 61 | p.Q61L | 4 (0.9%) | |
| 61 | p.Q61R | 2 (0.4%) | |
| Total cases with HRAS mutation | 1 (0.2%) | ||
| HRAS | 12 | p.G12D | 1 (0.2%) |
| Total cases with BRAF mutation | 26 (5.6%) | ||
| BRAF | 600 | V600E | 26 (5.6%) |
Particularly, mapping correlations between different gene mutations demonstrated that 6 patients carried both KRAS and NRAS mutations, and in another 5 patients, KRAS and BRAF mutations concomitantly existed. However, no cooccurring mutations of NRAS with BRAF were observed in our study, nor did HRAS and other genes (Figure 1).
Figure 1.
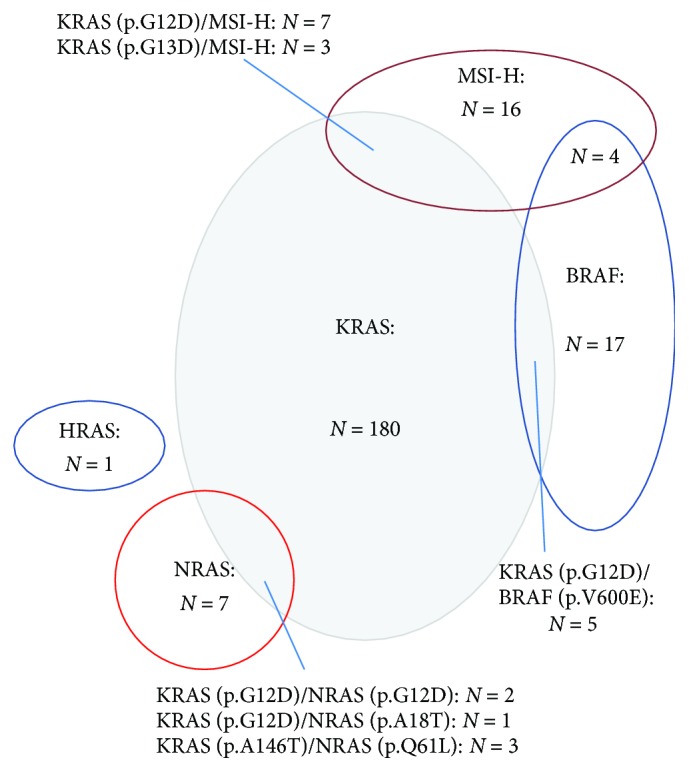
Set diagram illustrates the associations among KRAS, NRAS, HRAS, and BRAF mutations and MSI-H status. Mutations in KRAS and NRAS are not mutually exclusive, and neither are KRAS and BRAF. MSI-H status cooccurred with KRAS or BRAF mutations. MSI-H: microsatellite instability-high.
3.2. Frequency of Loss of MMR Protein Expression and MSI Status Detection
Among the entire study population, 32 cases (6.9%) were MMR-D phenotype, while 429 cases (93.1%) were MMR-I phenotype in primary CRCs. In the MMR-D cases, MLH1 expression loss was the most common (46.9%, 15/32) (Figures 2(a)–2(d)). Moreover, the specimens were also tested by PCR, the gold standard for confirming MSI status. Results showed that 30 primary tumors (6.5%) were with MSI-H, 45 (9.8%) were with MSI-L, and 386 (83.7%) were with MSS (Figures 2(e) and 2(f)). Similarly, there was a high concordance of MMR protein expression (98.8%, 244/247) and MSI status (98.4%, 243/247) between primary lesions and corresponding metastases. Specifically, three cases carrying MMR-I primary lesions exhibited the MMR-D phenotype in metastases. Of the four discordant cases with MSS primary tumors, three carried MSI-L metastases and one carried MSI-H metastases. Besides, MSI-H and KRAS/BRAF mutations can coexist according to our data (Figure 1).
Figure 2.

MMR protein determination and MSI status analysis. Immunohistochemical staining pattern of MSI-L colorectal carcinoma samples with isolated loss of MLH1 (a) and intact staining of MSH2 (b), MSH6 (c), and PMS2 (d); examples of fluorescence-based PCR of mononucleotide repeats and typical profiles of a MSS tumor (e) and a MSI-H case (f). MMR: mismatch repair; MSI: microsatellite instability; MSI-L: MSI-low; MSI-H: MSI-high; MSS: microsatellite stability; PCR: polymerase chain reaction.
3.3. Clinical Significance of RAS/BRAF Mutations and the MSI Status in mCRC Patients
All analyses were carried out in terms of sequencing outcomes in primary lesions. KRAS mutations were closely correlated with brain metastasis (P = 0.011) and high carcinoembryonic antigen (CEA) level (P < 0.001), and BRAF revealed a higher mutation rate in female patients (P = 0.039) and the right colon (P = 0.001). As for NRAS mutations, no significant relevance with the characteristics was observed. HRAS mutation was too rare to further explore. Moreover, no remarkable association between MSI status and gene mutations was identified in our study (P > 0.05) (Table 2).
Table 2.
Correlation between mutation profile and clinicopathological features in 461 patients with metastatic colorectal cancer.
| Clinicopathological features | n | KRAS | Status | BRAF | Status | NRAS | Status | All wild-type | Any mutation | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild-type (n = 260, %) | Mutation (n = 201, %) | P | Wild-type (n = 435, %) | Mutation (n = 26, %) | P | Wild-type (n = 448, %) | Mutation (n = 13, %) | P | (n = 231, %) | (n = 230, %) | P | ||
| Gender | |||||||||||||
| Male | 250 | 143 (57.2) | 107 (42.8) | 0.706 | 241 (96.4) | 9 (3.6) | 0.039 | 245 (98.0) | 5 (2.0) | 0.247 | 133 (53.2) | 117 (46.8) | 0.148 |
| Female | 211 | 117 (55.5) | 94 (44.5) | 194 (91.9) | 17 (8.1) | 203 (96.2) | 8 (3.8) | 98 (46.4) | 113 (53.6) | ||||
| Age, years | |||||||||||||
| <65 | 241 | 140 (58.1) | 101 (41.9) | 0.443 | 231 (95.9) | 10 (4.1) | 0.147 | 237 (98.3) | 4 (1.7) | 0.115 | 131 (54.4) | 110 (45.6) | 0.056 |
| ≥65 | 220 | 120 (54.5) | 100 (45.5) | 204 (92.7) | 16 (7.3) | 211 (95.9) | 9 (4.1) | 100 (45.5) | 120 (54.5) | ||||
| Tumor location | |||||||||||||
| Left colon | 209 | 123 (58.9) | 86 (41.1) | 0.509 | 193 (92.3) | 16 (7.7) | 0.001 | 205 (98.1) | 4 (1.9) | 0.097 | 105 (50.2) | 104 (49.8) | 0.565 |
| Right colon | 55 | 32 (58.2) | 23 (41.8) | 48 (87.3) | 7 (12.7) | 51 (92.7) | 4 (7.3) | 24 (43.6) | 31 (56.4) | ||||
| Rectum | 197 | 105 (53.3) | 92 (46.7) | 194 (98.5) | 3 (1.5) | 192 (97.5) | 5 (2.5) | 102 (51.8) | 95 (48.2) | ||||
| Primary tumor size | |||||||||||||
| <5 cm | 381 | 216 (56.7) | 165 (43.3) | 0.781 | 357 (93.7) | 24 (6.3) | 0.181 | 368 (96.6) | 13 (3.4) | 0.094 | 189 (49.6) | 192 (50.4) | 0.638 |
| ≥5 cm | 80 | 44 (55.0) | 36 (45.0) | 78 (97.5) | 2 (2.5) | 80 (100.0) | 0 (0.0) | 42 (52.5) | 38 (47.5) | ||||
| Differentiation | |||||||||||||
| Well/moderate | 289 | 166 (57.4) | 123 (42.6) | 0.559 | 269 (93.1) | 20 (6.9) | 0.122 | 278 (96.2) | 11 (3.8) | 0.097 | 142 (49.1) | 147 (50.9) | 0.588 |
| Poor | 172 | 94 (54.7) | 78 (45.3) | 166 (96.5) | 6 (3.5) | 170 (98.8) | 2 (1.2) | 89 (51.7) | 83 (48.3) | ||||
| Histological type | |||||||||||||
| Papillary/tubular adenocarcinoma | 380 | 211 (55.5) | 169 (44.5) | 0.413 | 358 (94.2) | 22 (5.8) | 0.763 | 369 (97.1) | 11 (2.9) | 0.834 | 186 (48.9) | 194 (51.1) | 0.280 |
| Mucinous/signet ring cell | 81 | 49 (60.5) | 32 (39.5) | 77 (95.1) | 4 (4.9) | 79 (97.5) | 2 (2.5) | 45 (55.6) | 36 (44.4) | ||||
| Depth of invasion | |||||||||||||
| T1 | 2 | 0 (0.0) | 2 (100.0) | 0.266 | 2 (100.0) | 0 (0.0) | 0.557 | 2 (100.0) | 0 (0.0) | 0.53 | 0 (0.0) | 2 (100.0) | 0.300 |
| T2 | 30 | 14 (46.7) | 10 (53.3) | 28 (93.3) | 2 (6.7) | 30 (100.0) | 0 (0.0) | 12 (40.0) | 18 (60.0) | ||||
| T3 | 347 | 158 (57.6) | 126 (42.4) | 325 (93.7) | 22 (6.3) | 335 (96.5) | 12 (3.5) | 175 (50.4) | 172 (49.6) | ||||
| T4 | 82 | 38 (56.1) | 26 (43.9) | 80 (97.6) | 2 (2.3) | 81 (98.8) | 1 (1.2) | 44 (53.7) | 38 (46.3) | ||||
| Nodal stage | |||||||||||||
| N0 | 32 | 15 (46.9) | 17 (53.1) | 0.143 | 32 (100.0) | 0 (0.0) | 0.319 | 32 (100.0) | 0 (0.0) | 0.680 | 15 (46.9) | 17 (53.1) | 0.224 |
| N1 | 285 | 171 (60.0) | 114 (40.0) | 265 (93.0) | 20 (7.0) | 276 (96.8) | 9 (3.2) | 150 (46.9) | 135 (53.1) | ||||
| N2a | 95 | 52 (54.7) | 43 (45.3) | 91 (95.8) | 4 (4.2) | 93 (97.9) | 2 (2.1) | 48 (50.5) | 47 (49.5) | ||||
| N2b | 49 | 22 (44.9) | 27 (55.1) | 47 (95.9) | 2 (4.1) | 47 (95.9) | 2 (4.1) | 18 (36.7) | 31 (63.3) | ||||
| Metastatic site | |||||||||||||
| Brain | 19 | 4 (21.1) | 15 (78.9) | 0.011 | 17 (89.5) | 2 (10.5) | 0.172 | 19 (100.0) | 0 (0.0) | 0317 | 3 (15.8) | 16 (84.2) | 0.002 |
| Lung | 121 | 66 (54.5) | 55 (45.5) | 112 (92.6) | 9 (7.4) | 115 (95.0) | 6 (5.0) | 53 (43.8) | 68 (56.2) | ||||
| Liver | 233 | 136 (58.4) | 97 (41.6) | 219 (94.0) | 14 (6.0) | 227 (97.4) | 6 (2.6) | 122 (52.4) | 111 (47.6) | ||||
| Others | 88 | 54 (61.4) | 34 (38.6) | 87 (98.9) | 1 (1.1) | 87 (98.9) | 1 (1.1) | 53 (60.2) | 35 (39.8) | ||||
| COX-2 expression | |||||||||||||
| Negative | 128 | 63 (49.2) | 65 (50.8) | 0.054 | 123 (96.1) | 5 (3.9) | 0.317 | 124 (96.9) | 4 (3.1) | 0.806 | 58 (45.3) | 70 (54.7) | 0.202 |
| Positive | 333 | 197 (59.2) | 136 (40.8) | 312 (93.7) | 21 (6.3) | 324 (97.3) | 9 (2.7) | 173 (52.0) | 160 (48.0) | ||||
| C-MET expression | |||||||||||||
| Negative/weak | 178 | 109 (61.2) | 69 (38.8) | 0.097 | 171 (96.1) | 7 (3.9) | 0.208 | 171 (96.1) | 7 (3.9) | 0.252 | 99 (55.6) | 79 (44.4) | 0.061 |
| Moderate/strong | 283 | 151 (53.4) | 132 (46.6) | 264 (93.3) | 19 (6.7) | 277 (97.9) | 6 (2.1) | 132 (46.6) | 151 (53.4) | ||||
| Initial CEA (ng/mL) | |||||||||||||
| <20 | 70 | 54 (77.1) | 16 (22.9) | <0.001 | 68 (97.1) | 2 (2.9) | 0.273 | 70 (100.0) | 0 (0.0) | 0.122 | 52 (74.3) | 18 (25.7) | <0.001 |
| ≥20 | 391 | 206 (52.7) | 185 (47.3) | 367 (93.9) | 24 (6.1) | 378 (96.7) | 13 (3.3) | 179 (45.8) | 212 (54.2) | ||||
| MSI | |||||||||||||
| MSI-H | 30 | 20 (66.7) | 10 (33.3) | 0.241 | 26 (86.7) | 4 (13.3) | 0.059 | 30 (100.0) | 0 (0.0) | 0.335 | 16 (53.3) | 14 (46.7) | 0.715 |
| MSI-L/MSS | 431 | 240 (55.7) | 191 (44.3) | 409 (94.9) | 22 (5.1) | 418 (97.0) | 13 (3.0) | 215 (49.9) | 216 (50.1) | ||||
COX-2 = cyclooxygenase-2; CEA = carcinoembryonic antigen; C-MET = mesenchymal-epithelial transition factor; MSI = microsatellite instability; MSI-H = MSI-high; MSI-L = MSI-low; MSS = microsatellite stability.
3.4. Predictors of Brain and/or Lung Metastases according to the Clinical Factors
Unconditional logistic regression revealed that RAS/BRAF mutations and moderate/strong C-MET expression were both significantly correlated with the occurrence of brain and/or lung metastases [odds ratio (OR): 4.027, P < 0.001 and OR: 3.901, P < 0.001, respectively (Table 3)].
Table 3.
Logistic regression analysis of the factors associated with brain and/or lung metastases in metastatic colorectal cancer patients.
| Characteristics | OR | 95% CI | P value |
|---|---|---|---|
| C-MET expression: negative/weak versus moderate/strong | 3.901 | 2.496–6.098 | <0.001 |
| RAS/BRAF genes: all wild-type versus any mutation | 4.027 | 2.551–6.358 | <0.001 |
| Constant | 0.111 |
P < 0.05 is statistically significant. CI: confidence interval; OR: odds ratio; C-MET: mesenchymal-epithelial transition factor.
With ROC curve analysis, the sensitivity and specificity of RAS/BRAF mutations alone, C-MET expression alone, or their combination for predicting brain and/or lung metastases within mCRC patients were evaluated. The findings which indicated a combination of RAS/BRAF mutations and C-MET expression [area under curve (AUC): 0.711, 95% CI: 0.659–0.763, P < 0.001] exhibited a better predictive value compared with single RAS/BRAF mutations (AUC: 0.638, 95% CI: 0.584–0.693, P < 0.001) or C-MET expression (AUC: 0.634, 95% CI: 0.578–0.690, P < 0.001) (Figure 3).
Figure 3.
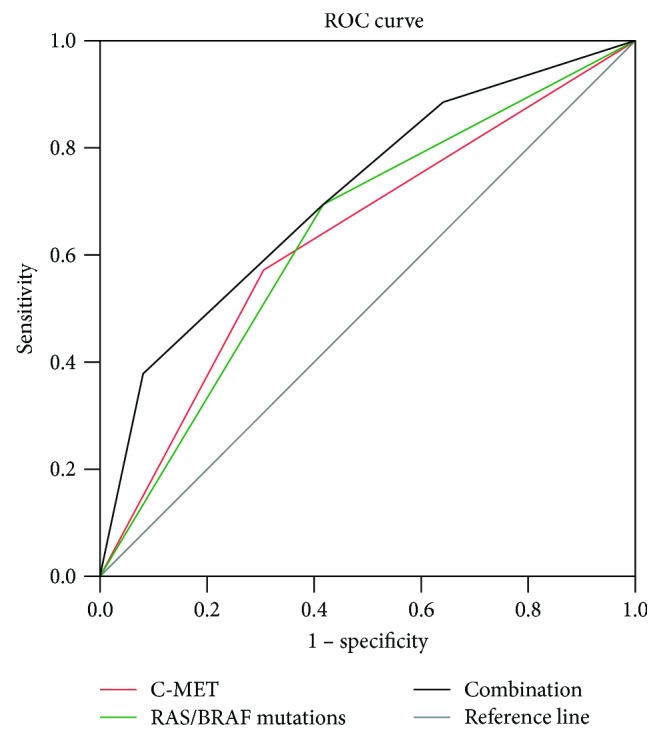
ROC curves for the predictive value of RAS/BRAF mutations and C-MET expression for brain and/or lung metastasis. ROC: receiver operating characteristic curve.
3.5. Survival Analysis
By the cutoff day on October 1, 2017, 257 (56.2%) of the enrolled patients had demised during the follow-up period. The median follow-up period was 24.3 months (range, 0.6–62 months), while 24 (5.2%) patients lost to follow-up. The potential influence of gene mutations and MSI status on survival was assessed with the Kaplan-Meier method. It was concluded that OS and PFS for patients with RAS/BRAF mutations were significantly shortened than those of cases with all wild-type. Particularly, cases exhibiting BRAF mutations had the worst prognosis (median OS and PFS: 12.8 months and 8.6 months), instead the any-other-RAS-mutated group had longer median OS and PFS (25.9 months and 21.6 months) than the other two mutational groups (Figures 4(a) and 4(b)). However, patients with different MSI status did not significantly differ in OS and PFS (χ 2 = 1.165, P = 0.280 and χ 2 = 2.717, P = 0.099; Figures 4(c) and 4(d)).
Figure 4.

Kaplan-Meier survival curves of metastatic colorectal carcinoma patients. (a) OS and (b) PFS of patients with different gene mutations; (c) OS and (d) PFS (MSI-L/MSS versus MSI-H) of entire study population. OS: overall survival; PFS: progression-free survival; MSI: microsatellite instability; MSI-L: MSI-low; MSI-H: MSI-high; MSS: microsatellite stability.
Furthermore, clinical value of various prognostic factors was estimated using Cox proportional hazards model. As confirmed by multivariate analyses, KRAS or BRAF mutation emerged as an independent risk factor for OS [hazard ratio (HR): 1.826, 95% confidence interval (CI): 1.361–2.450, P < 0.001 and HR: 4.798, CI: 2.989–7.700, P < 0.001; Table 4] and PFS (HR: 2.082, CI: 1.545–2.805, P < 0.001 and HR: 3.864, CI: 2.375–6.287, P < 0.001). In brief, our findings revealed that RAS/BRAF mutations played an essential role in patients' survival.
Table 4.
Univariate and multivariate analyses of OS and PFS for 461 metastatic colorectal cancer patients.
| Parameter | Variable | OS univariate | Analysis | OS multivariate | Analysis | PFS univariate | Analysis | PFS multivariate | Analysis |
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | HR (95% CI) | P value | ||
| Gender | Male versus female | 0.998 (0.773–1.287) | 0.985 | 1.026 (0.795–1.325) | 0.841 | ||||
| Age, years | <65 versus ≥65 | 1.310 (1.013–1.694) | 0.039 | 1.330 (0.998–1.772) | 0.052 | 1.067 (0.827–1.376) | 0.619 | ||
| Tumor location | Left/right colon versus rectum | 1.013 (0.884–1.162) | 0.849 | 1.089 (0.950–1.249) | 0.220 | ||||
| Primary tumor size, cm | <5 versus ≥5 | 1.077 (0.783–1.480) | 0.650 | 1.311 (0.955–1.800) | 0.094 | ||||
| Differentiation | Well/moderate versus poor | 1.031 (0.790–1.345) | 0.823 | 1.322 (1.016–1.721) | 0.038 | 0.395 (0.145–1.075) | 0.069 | ||
| Histological type | Papillary/tubular adenocarcinoma versus mucinous/signet ring cell | 1.142 (0.829–1.575) | 0.416 | 1.280 (0.929–1.764) | 0.131 | ||||
| Depth of invasion | T1 + T2 versus T3 + T4 | 0.952 (0.728–1.244) | 0.717 | 1.184 (0.912–1.536) | 0.205 | ||||
| Nodal stage | N0 + N1 versus N2a + N2b | 1.162 (0.980–1.378) | 0.084 | 1.245 (1.053–1.474) | 0.011 | 1.119 (0.920–1.361) | 0.260 | ||
| Metastatic site | Brain + lung versus liver + others | 1.540 (1.174–2.020) | 0.002 | 1.536 (1.130–2.088) | 0.006 | 1.728 (1.321–2.260) | <0.001 | 1.481 (1.094–2.006) | 0.011 |
| COX-2 expression | Negative versus positive | 1.046 (0.779–1.406) | 0.765 | 0.945 (0.704–1.270) | 0.709 | ||||
| Initial CEA (ng/mL) | <20 versus ≥20 | 3.103 (1.913–5.034) | <0.001 | 2.257 (1.366–3.730) | 0.001 | 2.659 (1.641–4.310) | <0.001 | 1.838 (1.113–3.036) | 0.017 |
| MSI | MSI-H versus MSI-L/MSS | 0.782 (0.493–1.238) | 0.294 | 0.688 (0.435–1.089) | 0.110 | ||||
| C-MET expression | Negative/weak versus moderate/strong | 1.690 (1.275–2.240) | <0.001 | 1.429 (1.052–1.940) | 0.022 | 1.495 (1.129–1.979) | 0.005 | 1.351 (0.993–1.839) | 0.055 |
| KRAS mutation | Yes versus no | 2.112 (1.620–2.754) | <0.001 | 1.826 (1.361–2.450) | <0.001 | 2.050 (1.574–2.671) | <0.001 | 2.082 (1.545–2.805) | <0.001 |
| BRAF mutation | Yes versus no | 8.615 (5.537–9.045) | <0.001 | 4.798 (2.989–7.700) | <0.001 | 4.458 (2.935–6.771) | <0.001 | 3.864 (2.375–6.287) | <0.001 |
| NRAS mutation | Yes versus no | 0.620 (0.230–1.668) | 0.344 | 0.462 (0.472–1.242) | 0.126 | ||||
| Anti-EGFR therapy | Yes versus no | 0.599 (0.401–0.894) | 0.012 | 0.742 (0.463–1.189) | 0.215 | 0.694 (0.465–1.036) | 0.074 | ||
| Bevacizumab therapy | Yes versus no | 0.713 (0.529–0.963) | 0.027 | 0.663 (0.469–0.937) | 0.020 | 0.758 (0.562–1.022) | 0.069 | 0.682 (0.484–0.961) | 0.029 |
| Surgery | Yes versus no | 0.702 (0.532–0.927) | 0.013 | 0.758 (0.531–1.082) | 0.127 | 0.712 (0.539–0.941) | 0.017 | 0.745 (0.525–1.057) | 0.099 |
OS = overall survival; PFS = progression-free survival; HR = hazard ratio; CI = confidence interval; COX-2 = cyclooxygenase-2; C-MET = mesenchymal-epithelial transition factor; CEA = carcinoembryonic antigen; MSI = microsatellite instability; MSI-H = MSI-high; MSI-L = MSI-low; MSS = microsatellite stability.
3.6. Prognostic Value of Different Treatment Regimens and Efficacy of Anti-EGFR Therapies
Of 461 mCRC patients, 452 (98.0%) received oxaliplatin-based or irinotecan-based chemotherapy, including 159 cases treated with chemotherapy alone, 118 combined with surgery, and 169 combined with targeted therapies (Table 5). Further analyses revealed in RAS/BRAF mutant group, different treatment regimens showed no significant difference on OS and PFS (χ 2 = 4.621, P = 0.099 and χ 2 = 2.882, P = 0.237; Figures 5(a) and 5(b)). In contrast, among wild-type patients, chemotherapy plus targeted therapies exhibited more favorable prognosis than the other treatment options (Figures 6(c)–6(f)), although there was no significant difference on survival (OS: χ 2 = 0.007, P = 0.933; PFS: χ 2 = 0.001, P = 0.988; Figures 6(a) and 6(b)) between chemotherapy alone and chemotherapy plus surgery groups. Moreover, bevacizumab therapy has been confirmed to be an independent prognostic factor for improved outcomes (Table 4).
Table 5.
Treatment details of metastatic colorectal cancer patients.
| Treatment methods | n (% of 461) | n (any mutation) | n (all wild-type) |
|---|---|---|---|
| Chemotherapy alone | 159 (34.5%) | 82 | 77 |
| 1 line | 21 (4.6%) | 15 | 6 |
| 2 lines | 79 (17.1%) | 52 | 27 |
| ≥3 lines | 59 (12.8%) | 15 | 44 |
| Chemotherapy combined with surgery | 118 (25.7%) | 73 | 45 |
| Primary lesion resection | 63 (13.7%) | 36 | 27 |
| Metastasectomy | 22 (4.8%) | 16 | 6 |
| Both | 33 (7.2%) | 21 | 12 |
| Chemotherapy combined with radiotherapy | 4 (0.8%) | 2 | 2 |
| Chemotherapy combined with targeted therapy | 169 (36.6%) | 65 | 104 |
| Bevacizumab therapy | 113 (24.5%) | 61 | 52 |
| Anti-EGFR therapy | 52 (11.3%) | 4 | 48 |
| Both | 4 (0.8%) | 0 | 4 |
| Chemotherapy combined with surgery and targeted therapy (primary lesion resection with anti-EGFR therapy) | 2 (0.4%) | 0 | 2 |
| Chemotherapy for the entire population | 452 (98.0%) | 222 | 230 |
| 1 line | 63 (13.7%) | 48 | 15 |
| 2 lines | 210 (45.5%) | 119 | 91 |
| ≥3 lines | 179 (38.8%) | 55 | 124 |
Figure 5.

Kaplan-Meier survival curves of mutant group stratified according to treatment regimens. (a) OS and (b) PFS of patients treated with different regimens. OS: overall survival; PFS: progression-free survival.
Figure 6.

Kaplan-Meier survival curves of wild-type group stratified according to treatment regimens. (a) OS and (b) PFS chemotherapy alone versus chemotherapy combined with surgery, (c) OS and (d) PFS chemotherapy alone versus chemotherapy combined with targeted therapy, and (e) OS and (f) PFS chemotherapy combined with surgery versus chemotherapy combined with targeted therapy. OS: overall survival; PFS: progression-free survival.
Among wild-type participants, 48 were treated by chemotherapy plus anti-EGFR agents. Data showed that the disease control rate (DCR) was 72.9% (35/48), with no patient for complete response (CR), 11 patients for partial response (PR), and 24 cases for stable disease (SD) for the first response evaluation at 3 months. In addition, 4 subjects with gene mutations also received cetuximab treatment (1 with BRAF mutation and 3 with KRAS exon 4 mutation), but DCR was 0.0% (0/4). Thus, the DCR and the response rate (including CR and PR) of wild-type patients were relatively better than those of cases with RAS/BRAF mutations (72.9% versus 0.0% and 22.9% versus 0.0%), although no statistical significance was attained.
4. Discussion
As a pathologically and clinically heterogeneous malignancy, CRC presented high aggressiveness and an accompanying worse prognosis on account of its aggressive nature. Despite the complexity of carcinogenesis, the discovery of extensive molecular markers for CRC has attracted special interests. As a result, gene detection has been attached to important connections with CRC evaluation and targeted therapy. However, the predictive and prognostic value of RAS/BRAF mutations and the MSI status in human mCRC has not previously been comprehensively elucidated.
Based on our data, the prevalence of gene mutations or the MSI-H status was in line with previous publications [12, 13, 22–24]. Meanwhile, there was a high concordance between primary CRCs and corresponding metastases, demonstrating that RAS/BRAF abnormalities emerged early in CRC tumorigenesis [25], and tumor cells kept their MSI status during development [6]. Different from intratumoral heterogeneity of KRAS mutations and rare NRAS or HRAS mutation, BRAF mutation showed relative intratumoral homogeneity [26, 27]. In addition, the present study also demonstrated that mutations in RAS/BRAF were not mutually exclusive, although the finding conflicted with several studies from other populations [28, 29]. One likely explanation may be the disparity of included cases and sample sources (Chinese versus European population). Regarding the MSI status, Fujiyoshi et al. [6] proposed that MSI-H status and RAS/BRAF mutations could coexisted. Similarly, our results corroborated the fact. Given increasing data on mutation profiling was accumulated, associations among RAS/BRAF genes will be further expounded.
Moreover, we characterized RAS/BRAF mutations and MSI status, and results revealed that RAS or BRAF mutation possessed clinical significance in promoting the development and metastasis of mCRC. In brief, KRAS mutations may be important indicators to identify subsets with increased CEA level and brain metastases. The viewpoints were partially different from literatures published, in which KRAS mutations were related to older age, differentiation degree, and later clinical stage [22, 29, 30]. The variability in various researches probably attributed to geographical distribution and ethnicities. Until now, the significance of NRAS or HRAS mutations remained controversial due to their rarity. A recent CRC study [12] proposed that NRAS mutations were found to be tilted to the right colon and MSI-L cancers. Nevertheless, no clinical relevance of NRAS mutations was observed in our research; HRAS mutation was too rare to further explore. Recently, Zhang et al. [26] reported that BRAF mutations were observed more frequently in the right colon and female patients, which supported the conclusions of our study. Particularly, no significant association was found between the MSI status and RAS/BRAF mutations, albeit a recent report [6] showed that MSI-H linked with BRAF mutations. This bias might be caused by the limited data and the different detection techniques.
The initiation and progression of CRC are a multistep process accompanied by inactivation of tumor suppressors and accumulation of gene mutations, especially somatic changes in RAS/BRAF, which are driver mutations and represent the principle aspect of gene abnormalities in CRC [31]. Another focus of our research was searching for the predictive value of RAS/BRAF mutations and MSI status. Numerous experimental model systems have confirmed that RAS/BRAF abnormalities contributed to cell invasion and apoptosis suppression during metastatic cascade, which may bring about organ involvement and tumor progression [4, 32]. In one previous study [33], KRAS exon 2-mutated CRC patients exhibited an obvious propensity for lung metastases. Similar results have also been described by Morris et al. [34], in which cases with RAS/BRAF mutations harbored the trend towards lung metastases. Here, our data for the first time revealed that RAS/BRAF mutations were significant predictors for higher risk of brain metastases, followed by lung metastases, suggesting its value in distinguishing CRC with highly aggressive behavior from low metastatic ability. Thus, the emergence of RAS/BRAF mutations provided powerful insight into the complexity of tumor foci genotype and gained useful clues for treatment option.
Unfortunately, when it came to the MSI status, neither predictive nor prognostic relevance was observed in mCRC. This phenomenon was concordant with studies issued [24]. But for stage II or III cases, MSI-H contributed to the favorable prognosis [7]. Because of too few MSI-H cases restrained the discovery of potential clinical and prognostic value of MSI status, more focusing on the issue was desired.
Mutation in KRAS was regarded as an adverse predictors for disease-specific survival more early in 1990 [35]. Not until the last ten years, prognostic ability of RAS/BRAF aberrations in CRC has spurred much more attention. In agreement with previous series [15, 34], our data also revealed that patients with gene mutations, especially BRAF mutation, suffered inferior prognosis compared with wild-type counterparts. Interestingly, cases carrying NRAS mutations showed relatively better survival than those with other RAS mutations. Besides that, as the National Comprehensive Cancer Network (NCCN) recommends targeted therapies for mCRC patients, our analysis suggested that chemotherapy combined with targeted therapy could remarkably improve the prognosis of wild-type patients. Importantly, bevacizumab had been considered as an independent prognostic factor according to our data, which accorded with some meta-analyses and randomized controlled trials [36, 37]. Meantime, RAS/BRAF mutations were emphasized to be predictive biomarkers of resistance to therapies against EGFR, and only wild-type CRC patients may gain survival benefit from cetuximab and panitumumab.
Owing to the retrospective nature, there have been inevitably selection bias in our outcomes. Firstly, some participants and their medical record documentation may be lost to follow-up, especially for those who were not hospitalized after first-line chemotherapy. Secondly, the patients were heterogeneous and selected according to availability of molecular detection, which limited the data analyses. Therefore, more prospective studies are required to confirm our conclusions.
5. Conclusions
Altogether, RAS/BRAF mutations may serve as significant predictors of malignant behavior. Accordingly, radiological diagnosis combined with gene detection may help to evaluate the prognosis of novel CRC cases and devised optimal individualized medicine in the future.
Acknowledgments
This work was supported by a grant from the Program of Health and Family Planning Commission Foundation of Guangzhou City, Guangdong Province, China (Grant no. A2017418), and a grant from the Program of Science and Technology Commission Foundation of Guangzhou City, Guangdong Province, China (Grant no. 20140705).
Conflicts of Interest
The authors declare that they have no conflict of interest.
Supplementary Materials
Table 1: primer sequences and fragment length of RAS, BRAF, and EGFR gene amplification.
References
- 1.Siegel R., Desantis C., Jemal A. Colorectal cancer statistics, 2014. CA: A Cancer Journal for Clinicians. 2014;64(2):104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Kawai M., Komiyama H., Hosoya M., et al. Impact of chromosome 17q deletion in the primary lesion of colorectal cancer on liver metastasis. Oncology Letters. 2016;12(6):4773–4778. doi: 10.3892/ol.2016.5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scaltriti M., Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clinical Cancer Research. 2006;12(18):5268–5272. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 4.McCubrey J. A., Steelman L. S., Abrams S. L., et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Advances in Enzyme Regulation. 2006;46(1):249–279. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Vakiani E., Solit D. B. KRAS and BRAF: drug targets and predictive biomarkers. The Journal of Pathology. 2015;223(2):219–229. doi: 10.1002/path.2796. [DOI] [PubMed] [Google Scholar]
- 6.Fujiyoshi K., Yamamoto G., Takahashi A., et al. High concordance rate of KRAS/BRAF mutations and MSI-H between primary colorectal cancer and corresponding metastases. Oncology Reports. 2017;37(2):785–792. doi: 10.3892/or.2016.5323. [DOI] [PubMed] [Google Scholar]
- 7.Yan W. Y., Hu J., Xie L., et al. Prediction of biological behavior and prognosis of colorectal cancer patients by tumor MSI/MMR in the Chinese population. OncoTargets and Therapy. 2016;Volume 9:7415–7424. doi: 10.2147/OTT.S117089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le D. T., Uram J. N., Wang H., et al. PD-1 blockade in tumors with mismatch-repair deficiency. The New England Journal of Medicine. 2015;372(26):2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mascaux C., Iannino N., Martin B., et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. British Journal of Cancer. 2005;92(1):131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ginestà M. M., Mora J., Mayor R., et al. Genetic and epigenetic markers in the evaluation of pancreatic masses. Journal of Clinical Pathology. 2013;66(3):192–197. doi: 10.1136/jclinpath-2012-201123. [DOI] [PubMed] [Google Scholar]
- 11.Chai L., Li J., Lv Z. An integrated analysis of cancer genes in thyroid cancer. Oncology Reports. 2016;35(2):962–970. doi: 10.3892/or.2015.4466. [DOI] [PubMed] [Google Scholar]
- 12.Ogura T., Kakuta M., Yatsuoka T., et al. Clinicopathological characteristics and prognostic impact of colorectal cancers with NRAS mutations. Oncology Reports. 2014;32(1):50–56. doi: 10.3892/or.2014.3165. [DOI] [PubMed] [Google Scholar]
- 13.Yokota T., Ura T., Shibata N., et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. British Journal of Cancer. 2011;104(5):856–862. doi: 10.1038/bjc.2011.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Downward J. Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 15.Foltran L., De Maglio G., Pella N., et al. Prognostic role of KRAS, NRAS, BRAF and PIK3CA mutations in advanced colorectal cancer. Future Oncology. 2015;11(4):629–640. doi: 10.2217/fon.14.279. [DOI] [PubMed] [Google Scholar]
- 16.Amaral T., Sinnberg T., Meier F., et al. MAPK pathway in melanoma part II-secondary and adaptive resistance mechanisms to BRAF inhibition. European Journal of Cancer. 2017;73:93–101. doi: 10.1016/j.ejca.2016.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Kadowaki S., Kakuta M., Takahashi S., et al. Prognostic value of KRAS and BRAF mutations in curatively resected colorectal cancer. World Journal of Gastroenterology. 2015;21(4):1275–1283. doi: 10.3748/wjg.v21.i4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ion Ampliseq Designer. [ https://www.ampliseq.com/browse.action]
- 19.Xie C. H., Xie F., Wu P., et al. The mutation rates of EGFR in non-small cell lung cancer and KRAS in colorectal cancer of Chinese patients as detected by pyrosequencing using a novel dispensation order. Journal of Experimental & Clinical Cancer Research. 2015;34(1):p. 63. doi: 10.1186/s13046-015-0179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J., Wu H., Wang L., et al. Validation of targeted next-generation sequencing for RAS mutation detection in FFPE colorectal cancer tissues: comparison with Sanger sequencing and ARMS-Scorpion real-time PCR. BMJ Open. 2016;6(1, article e009532) doi: 10.1136/bmjopen-2015-009532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeinalian M., Emami M. H., Naimi A., Salehi R., Hashemzadeh-Chaleshtori M. Immunohistochemical analysis of mismatch repair proteins in Iranian colorectal cancer patients at risk for lynch syndrome. Iranian Journal of Cancer Prevention. 2015;8(1):11–17. [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z. Z., Wang F., Zhang Z. C., et al. Mutation profiling in chinese patients with metastatic colorectal cancer and its correlation with clinicopathological features and anti-EGFR treatment response. Oncotarget. 2016;7(19):28356–28368. doi: 10.18632/oncotarget.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Russo A. L., Borger D. R., Szymonifka J., et al. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer. 2014;120(10):1482–1490. doi: 10.1002/cncr.28599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein J., Tran B., Ensor J., et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H) Annals of Oncology. 2014;25(5):1032–1038. doi: 10.1093/annonc/mdu100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fearon E. R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J., Zheng J., Yang Y., et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancer patients: analysis of 1,110 cases. Scientific Reports. 2015;5(1, article 18678) doi: 10.1038/srep18678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Roock W., Claes B., Bernasconi D., et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 28.Hawkes E., Cunningham D. Relationship between colorectal cancer biomarkers and response to epidermal growth factor receptor monoclonal antibodies. Journal of Clinical Oncology. 2010;28(28):e529–e531. doi: 10.1200/JCO.2010.29.5626. [DOI] [PubMed] [Google Scholar]
- 29.Kawazoe A., Shitara K., Fukuoka S., et al. A retrospective observational study of clinicopathological features of KRAS, NRAS, BRAF and PIK3CA mutations in Japanese patients with metastatic colorectal cancer. BMC Cancer. 2015;15(1):p. 258. doi: 10.1186/s12885-015-1276-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen Y., Wang J., Han X., et al. Effectors of epidermal growth factor receptor pathway: the genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS One. 2013;8(12, article e81628) doi: 10.1371/journal.pone.0081628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Roock W., De Vriendt V., Normanno N., Ciardiello F., Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. The Lancet Oncology. 2011;12(6):594–603. doi: 10.1016/S1470-2045(10)70209-6. [DOI] [PubMed] [Google Scholar]
- 32.Mendelsohn J., Baselga J. Epidermal growth factor receptor targeting in cancer. Seminars in Oncology. 2006;33(4):369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 33.Kim M. J., Lee H. S., Kim J. H., et al. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer. 2012;12(1):p. 347. doi: 10.1186/1471-2407-12-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris V. K., Lucas F. A., Overman M. J., et al. Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Annals of Oncology. 2014;25(10):2008–2014. doi: 10.1093/annonc/mdu252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slebos R. J., Kibbelaar R. E., Dalesio O., et al. K-Ras oncogene activation as a prognostic marker in adenocarcinoma of the lung. New England Journal of Medicine. 1990;323(9):561–565. doi: 10.1056/NEJM199008303230902. [DOI] [PubMed] [Google Scholar]
- 36.Hurwitz H. I., Tebbutt N. C., Kabbinavar F., et al. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. The Oncologist. 2013;18(9):1004–1012. doi: 10.1634/theoncologist.2013-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sasaki Y., Akasu T., Saito N., et al. Prognostic and predictive value of extended RAS mutation and mismatch repair status in stage III colorectal cancer. Cancer Science. 2016;107(7):1006–1012. doi: 10.1111/cas.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table 1: primer sequences and fragment length of RAS, BRAF, and EGFR gene amplification.