

SUMMARY
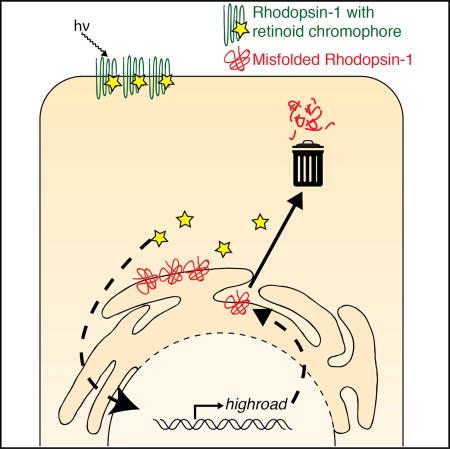
Rhodopsins require retinoid chromophores for their function. In vertebrates, retinoids also serve as signaling molecules, but whether these molecules similarly regulate gene expression in Drosophila remains unclear. Here, we report the identification of a retinoid-inducible gene in Drosophila, highroad, which is required for photoreceptors to clear foldingdefective mutant Rhodopsin-1 proteins. Specifically, knockdown or genetic deletion of highroad blocks the degradation of folding-defective Rhodopsin-1 mutant, ninaEG69D. Moreover, loss of highroad accelerates the age-related retinal degeneration phenotype of ninaEG69D mutants. Elevated highroad transcript levels are detected in ninaEG69D flies, and interestingly, deprivation of retinoids in the fly diet blocks this effect. Consistently, mutations in the retinoid transporter, santa maria, impairs the induction of highroad in ninaEG69D flies. In cultured S2 cells, highroad expression is induced by retinoic acid treatment. These results indicate that cellular quality-control mechanisms against misfolded Rhodopsin-1 involve regulation of gene expression by retinoids.
In Brief
Folding-defective mutant rhodopsins undergo degradation in photoreceptors, but the underlying mechanism was unclear. Huang et al. identify highroad as a factor required for mutant Drosophila Rhodopsin-1 degradation. Loss of highroad accelerates retinal degeneration caused by mutant Rhodopsin-1, and highroad expression is dependent on retinoids.
INTRODUCTION
As in other metazoans, Drosophila has several rhodopsin genes, including ninaE, that encode Rhodopsin-1 (Rh1) (O’Tousa et al., 1985; Zuker et al., 1985). Once synthesized, Rh1 becomes conjugated to the 11-cis-3-hydroxyretinal chromophore to detect light in the outer photoreceptors of the eye (Ahmad et al., 2006).
Certain types of mutations in human rhodopsin underlie autosomal dominant retinitis pigmentosa (ADRP), a disorder of agerelated retinal degeneration (Dryja et al., 1990; Sung et al., 1991). This disease has been modeled in Drosophila through similar mutations in ninaE, including the G69D and P37H alleles, which trigger age-related retinal degeneration (Colley et al., 1995; Galy et al., 2005; Kurada and O’Tousa, 1995). The encoded mutant proteins fail to fold properly in the endoplasmic reticulum (ER) and therefore impose stress in this organelle and activate the unfolded protein response (UPR) (Ryoo et al., 2007).
At the same time, healthy cells are equipped with quality-control mechanisms that act against such misfolded proteins. In the ER, a network of proteins is involved in the detection, retro-translocation, and ubiquitination of misfolded peptides for proteasomal degradation in the cytoplasm, a process referred to as ER-associated degradation (ERAD) (Brodsky, 2012; Ruggiano et al., 2014). We had previously shown that overexpression of the central ubiquitin ligase involved in ERAD, hrd1, strongly delayed retinal degeneration in the Drosophila ninaEG69D mutant (Kang and Ryoo, 2009). In addition to ERAD, recent studies indicate that mutant and wild-type rhodopsins are partly degraded in the lysosome (Chiang et al., 2012; Chinchore et al., 2009; Wang et al., 2014).
Without the retinal chromophore and its precursors, rhodopsins cannot function properly and fail to undergo proper maturation (Harris et al., 1977; Ozaki et al., 1993; Gu et al., 2004; Wang and Montell, 2005; Wang et al., 2007). In vertebrates, retinoids also have a second role as transcriptional regulators whose effects are mediated by the nuclear hormone receptor proteins (Mangelsdorf and Evans, 1995). Although previous studies reported that Drosophila that are deprived of the retinoid precursor vitamin A in the diet have altered levels of opsin and fatty-acid-binding glycoprotein transcripts (Picking et al., 1996; Shim et al., 1997), the biological role and the mechanism of retinoid-mediated gene expression control in Drosophila remain unclear.
In this study, we report the identification of highroad (hiro), a gene that is required for mutant Rh1 degradation in Drosophila that also affects the course of age-related retinal degeneration. Furthermore, our data indicate that hiro transcript levels increase in ninaEG69D mutant flies, and that this is dependent on retinoid availability in vivo. These observations suggest that the degradation of mutant Rh1 is associated with retinoid-mediated gene expression control in Drosophila.
RESULTS AND DISCUSSION
Adult Eye Morphology-Based RNAi Screen for Genetic Interactors of ninaEG69D
We previously established a facile genetic assay system to assess cellular stress caused by ninaEG69D overexpression through the eye-specific GMR promoter (henceforth referred to as GMR-Rh1G69D). In these flies, Rh1G69D is overexpressed in the early stages of eye development, resulting in adults with malformed eyes (Kang and Ryoo, 2009; Kang et al., 2012; Figure S1). This phenotype can be attributed to increased misfolded proteins in the ER, since co-overexpression of the ERAD-mediating gene hrd1 almost completely suppresses the external eye phenotype (Kang and Ryoo, 2009; Figure S1).
To identify other factors involved in misfolded Rh1 quality control, we screened for RNAi lines that impaired the protective effects of hrd1 overexpression against GMR-Rh1G69D (Figure S1A; see also Experimental Procedures). A total of 80 RNAi lines were tested, many of which targeted Drosophila homologs of mammalian genes with known roles in ERAD, or those that are found in protein complexes with human HRD1 and its associated proteins (Christianson et al., 2011). We also included RNAi lines that targeted annotated membrane proteases and carboxypeptidases in Drosophila (the full list of RNAi lines is in Table S1).
RNAi knockdown of hrd1 in the developing eye did not impair eye development when expressed alone, but aggravated the eyes of flies co-expressing Rh1G69D and hrd1 (Figure S1B). A number of other lines gave rise to phenotypes similar to hrd1 knockdown. These included not only the lines that targeted Drosophila homologs of known ERAD genes, but also genes with no previous associations with ERAD, including CG32441, asrij, and CG3344 (Figure S1B).
highroad Is a Gene Required to Reduce Mutant Rh1 Levels in Photoreceptors
As a secondary assay for validation, we turned to the classical ninaEG69D allele with a mutation in the endogenous ninaE locus that dominantly reduces total Rh1 levels in newly enclosed adult flies (Colley et al., 1995; Kurada and O’Tousa, 1995). Candidate RNAi lines from the primary screen were expressed in the photoreceptors of ninaEG69D/+ flies, and we found that one particular RNAi line (VDRC 110402) almost fully restored Rh1 levels in the ninaEG69D/+ background to wild-type levels (Figures 1A and 1B). This line targets a previously uncharacterized carboxypeptidase, CG3344, homologous to a mammalian protein known as retinoid-inducible serine carboxypeptidase or serine carboxypeptidase 1 (SCPEP1) (Chen et al., 2001). Neither CG3344 nor its mammalian homolog has known roles in ERAD. Based on the loss-of-function phenotype, we henceforth refer to CG3344 as high rhodopsin-accelerated degeneration or highroad (hiro). Knockdown of known ERAD components did not restore Rh1 levels significantly in this system, which is consistent with what we reported previously (Kang and Ryoo, 2009). Although hiro was identified through a genetic interaction screen with hrd1, the precise relationship between the two genes remains unclear.
Figure 1. hiro Is Required for Photoreceptors to Reduce Mutant Rh1 Levels.

(A) Shown are western blots of adult head extracts with the indicated antibodies. In the ninaE wild-type (lane 1) or in the ninaEG69D/+ genetic background (lanes 2–7), the indicated genes were knocked down with Rh1-Gal4 driver.
(B) Quantification of the band intensities shown in (A).
(C) A schematic diagram of the hiro locus. The bar above indicates sequences deleted in Df(3L)5. Below is a schematic diagram of the hiroF1 allele.
(D and E) Validation of the hiro RNAi phenotype through classical alleles. Anti-Rh1 (top) and anti-β-tubulin (bottom) western blot from adult fly head extracts of the indicated genotypes.
(D) The effect of Df(3L)5 deletion on wild-type (lanes 1–3) or ninaEG69D/+ flies (lanes 4–6).
(E) hiroF1−/− increases Rh1 levels in ninaEG69D/+ heads (lane 3 and 6), but introduction of the BAC rescue transgene CH321-70D3 that contains the hiro sequence reverses this effect (lane 7).
(F) Anti-Rh1 (lanes 1-3) and anti-Hsv (lanes 4, 5) western blots on 21-day-old Rh1p > ninaEP37H fly head extracts in the indicated genotypes.
Error bars represent SEM.
Knockdown of other Drosophila carboxypeptidases, such as CG4572 or CG32821, did not result in the recovery of Rh1 levels in ninaEG69D/+ flies (Figure S2A). We also knocked down genes that mediate autophagy or late endosome trafficking in the ninaEG69D/+ photoreceptors, but overall Rh1 levels did not recover under those conditions (Figure S2B).
To further validate the RNAi result from the screen, we used two hiro mutant alleles: (1) Df(3L)5 deficiency, in which hiro and its neighboring gene, earthbound1, are deleted (Benchabane et al., 2011); and (2) an allele generated by CRISPR-Cas9-mediated deletion of the hiro locus (Figure 1C; see also Experimental Procedures), which we refer to henceforth as hiroF1. hiroF1−/− or hiroF1/Df(3L)5 flies were viable, did not exhibit any obvious external morphology defects, and did not affect total Rh1 levels in the ninaE wild-type background (Figures 1D, lanes 1 and 2, and 1E, lane 1). We also introduced the UPR reporter XBP1- EGFP (Coelho et al., 2013) into the background of adult flies that contain Df(3L)5−/− mosaic clones in the eye. We found no evidence of excessive ER stress and abnormal UPR activation in these photoreceptors (Figure S3).
Loss of hiro in the ninaEG69D/+ background, however, had clear effects on Rh1 levels. Specifically, loss of hiro in the Df(3L)5−/−, Df(3L)/hiroF1, or hiroF1−/− backgrounds resulted in the recovery of Rh1 levels in ninaEG69D/+ eyes to a degree that was comparable to that of ninaE wild-type flies (Figures 1D, lane 6, and 1E, lanes 3 and 6). This effect of hiroF1 loss of function on Rh1 was reversed by the introduction of a transgenic bacterial artificial clone (BAC), CH321-70D3, containing the hiro locus DNA (Figure 1E, lane 7). Together, these results validate that hiro is genetically required to reduce Rh1 levels in ninaEG69D/+ mutants.
hiro Affects the Levels of Other Rh1 Mutant Alleles
In humans, the most widespread rhodopsin allele associated with ADRP is the P23H mutation, which encodes a misfolding-prone rhodopsin that undergoes degradation in the ER and the lysosome (Chiang et al., 2012; Dryja et al., 1990; Liu et al., 1996). We examined a previously generated herpes simplex virus (HSV)-tagged ninaE transgenic line with the equivalent Drosophila mutation, P37H (Galy et al., 2005). Unlike the G69D mutants, we found that Rh1 levels were not noticeably lower in newly enclosed P37H flies (data not shown), but decreased significantly by 21 days post-enclosure (Figure 1F, lane 2). Similar to results with the G69D mutants, Rh1 levels in P37H mutants were almost restored to wild-type levels in the Df(3L)5−/− background (Figure 1F, lane 3). Consistently, western blotting with anti-HSV to specifically detect P37H mutant protein (rather than total Rh1) showed higher P37H-HSV in the Df(3L)5−/− background (Figure 1F, lanes 4, 5). These results show that the effect of hiro loss on Rh1 is not specific to the ninaEG69D allele but is applicable to other disease-relevant ninaE mutant alleles.
hiro Does Not Affect the Levels of Another ER-Stress-Causing Protein, alpha-1 antitrypsinNHK
alpha-1 antitrypsin (a1at) encodes a secreted human protein, and the NHK mutant allele underlies alpha-1 antitrypsin deficiency due to its propensity of undergoing rapid degradation through ERAD. We had previously generated a uas-a1atNHK transgenic fly line that activates the UPR and also undergoes ERAD when expressed through a Gal4 driver in Drosophila tissues (Kang and Ryoo, 2009). We were able to detect this protein from adult head extracts when driven with the GMR-Gal4 driver, but its levels did not increase when hiro was knocked down through RNAi (Figure S2C). These results indicate that hiro does not affect all misfolding-prone proteins in the ER.
hiro Expression Pattern
To visualize the localization of Hiro protein, we inserted a GFP trangene into the hiro coding sequence within its genomic locus using minos-mediated integration cassette (MiMIC)-based protein trap technology (Nagarkar-Jaiswal et al., 2015; see Experimental Procedures). This resulted in a hiro-GFP protein trap hybrid gene with GFP in frame with hiro that we refer to as hiroPT (Figure S4A). The N-terminal signal peptide of hiro remained intact after GFP insertion. The fluorescence from the resulting GFP was readily detectable in a confined area of the larval intestine (Figures S4B and S4C). The mammalian homolog of hiro is reportedly a lysosomal protein, but the GFP signal did not overlap with LysoTracker (Figure S4D). The signal peptide within hiro indicates that the protein must be synthesized in the ER before reaching its ultimate subcellular site, and consistently, there was partial overlap of the GFP signal with the ER marker anti-Calnexin (Figure S4E). However, there were also foci with intense GFP signals that did not overlap with the ER, and we interpret that Hiro is ultimately trafficked out of the ER to this final destination.
hiro Mutants Increase Rh1 Protein Levels in the Photoreceptor Rhabdomeres and along the Secretory Pathway Organelles
To determine the sub-cellular localization of Rh1 protein in hiro mutants, we labeled adult ommatidia with anti-Rh1 antibody and performed initial analysis through confocal microscopy. Whereas ninaEG69D/+ eyes had most anti-Rh1 signals coming from the rhabdomeres in trapezoidal arrangements, ninaEG69D, Df(3L)5/Df(3L)5 eyes showed additional anti-Rh1 signals in the cell body. We performed co-localization experiments with various subcellular organelle markers, including that of the ER and the lysosome (Figures 2A–2C), but none of those markers fully overlapped with the cytoplasmic anti-Rh1 signals. Some of the anti-Rh1 signals in the cytoplasm appeared to be juxtaposed or associated with sub-fractions of the ER (Figure 2B), but the resolution provided by fluorescent microscopy was insufficient to draw conclusions.
Figure 2. Subcellular Localization of Rh1 in the hiro Mutant Background.

(A–C) Confocal microscopy imaging of adult Drosophila ommatidia labeled with anti-Rh1 (green) together with other subcellular organelle markers (red).
(A) ninaEG69D/+ ommatidia.
(B and C) The ninaEG69D, Df(3L)5/Df(3L)5 ommatidia, where Rh1 is not only detected in rhabdomeres, but also in the cell body.
(B’ and B”) A double-labeled image with the ER marker calenxin (B” is the calnexin-only channel).
(C and C”) Images with the lysosome marker ATP6V1B1.
(D–H) Immunogold EM with anti-Rh1 antibody. Signals appear as black dots and are indicated with arrows.
(D) ninaEG69D/+ adult fly ommatidium with seven photoreceptors labeled as R1–R7. Note that anti-Rh1 signal is not detected in the R7 rhabdomere.
(E) A ninaEG69D/+ photoreceptor.
(F–H) In ninaEG69D, Df(3L)5/Df(3L)5 photoreceptors, Rh1 is detected in the rhabomeres (B and D) and in additional sites of the cell body. These include the ER (G) and other organelles (F). Neither the lysosome nor the mitochondria show obvious anti-Rh1 labeling (H). Subcellular structures are abbreviated as follows: R, rhabodomere; M, mitochondria; ER, endoplasmic reticulum; L, lysosome
To obtain higher resolution images of anti-Rh1 labels, we performed immunogold electron microscopy (EM) with rabbit anti-Rh1 antibody. The anti-Rh1 signal was specific based on the fact that rhabodomeres of photoreceptors R1–R6 showed immuno-labeling, whereas the signal was lacking in R7 photoreceptors that do not normally express Rh1 (Scavarda et al., 1983; Figure 2D). In ninaEG69D/+ flies, most anti-Rh1 signals came from the rhabdomeres (Figures 2D and 2E). In the Df(3L)5−/− background, ninaEG69D/+ ommatidia still had most anti-Rh1 signals emanating from the rhabodomeres, but there were additional subcellular sites where anti-Rh1 signals were detected (Figures 2F–2H). One of those was the ER (Figure 2G), but other subcellular sites also showed anti-Rh1 signals (Figure 2F). We did not detect any anti-Rh1 signals from the lysosome or the mitochondria (Figure 2H). Because Rh1 is ultimately trafficked to rhabdomeres in this genotype, we interpret that Rh1 levels increase in multiple subcellular organelles along the secretory pathway.
ninaEG69D/+ Flies Show Accelerated Retinal Degeneration in the Absence of hiro
Because our data indicated that hiro mutants fail to properly regulate Rh1 levels, we decided to examine if this affected the age-related retinal degeneration of ninaEG69D/+ flies. The integrity of the retinal photoreceptors can be determined by dissecting and labeling fly eyes with phalloidin, which marks rhabdomeres of photoreceptors (Figure 3A). In wild-type flies, phalloidin labeling showed typical trapezoidal patterns of seven photoreceptors in regular arrays (Figure 3A). In ninaEG69D/+ flies up to 20 days old, this pattern was maintained in most dissected flies (Figures 3A). On the other hand, ninaEG69D, hiroF1/hiroF1 fly eyes lost the trapezoidal rhabdomere arrangement by day 20, which is indicative of retinal degeneration (Figure 3A, lower rightmost panel). We independently validated this using the Rh1-GFP reporter that allows the examination of photoreceptor integrity in live flies over a time course of 30 days. A majority of ninaEG69D/+ flies (n = 49) showed an organized trapezoidal pattern of Rh1-GFP for up to 20 days before showing signs of degeneration (Figure 3B). In the hiroF1 mutant background, ninaEG69D/+ flies showed earlier signs of retinal degeneration (n = 31, p < 0.0001, Chi-square = 178.1, when compared with ninaEG69D/+), with more than 75% of flies showing disorganized Rh1-GFP patterns at 20 days old (Figure 3B). The control hiro−/− flies in the ninaE wild-type background showed no signs of retinal degeneration at these time points (n = 54). Although ninaE is only expressed in six photoreceptors (R1–R6) (Scavarda et al., 1983), all photoreceptors degenerated under these conditions, which is consistent with previous studies reporting that even R7 and R8 photoreceptor degeneration occurs in these flies due to indirect effects (Colley et al., 1995; Kurada and O’Tousa, 1995; Leonard et al., 1992).We also performed the converse experiment of hiro overexpression using the Rh1-Gal4/ uas-hiro system (n = 50 in each genotype group). Although hiro was expressed as detected by the V5 epitope associated with the transgene, such conditions were not sufficient to delay the course of age-related retinal degeneration (Figure 3C). Taken together, these results indicate that hiro is necessary but not sufficient to protect ninaEG69D/+ photoreceptors from age-related retinal degeneration.
Figure 3. Loss of hiro Accelerates the Course of Age-Related Retinal Degeneration in ninaEG69D/+ Flies.

(A) Representative images of dissected ommatidia labeled with phalloidin that labels rhabdomeres of photoreceptors (red). The genotypes are indicated on top of each panel. Intact ommatidia show a trapezoidal arrangement of seven photoreceptor rhabdomeres (one example marked with asterisks on the upper left panel). Twenty-day-old ninaEG69D, hiroF1−/− eyes (lower rightmost panel) lack the trapezoidal pattern of phalloidin labeling. The asterisks in that panel indicate the expected positions of phalloidin-positive rhabdomeres in an ommatidium.
(B and C) Retinal degeneration was assessed in live flies containing Rh1-GFP to visualize photoreceptors, quantified, and exhibited as a graph.
(B) The effect of hiro loss of function on retinal degeneration. The genotype and the numbers (n) are indicated.
(C) The effect of hiro overexpression. UAS-hiro-V5 was expressed in ninaEG69D/+ photoreceptors using Rh1-Gal4. (Left) Western blot showing V5 epitope detection in the adult fly head extracts (top gel) and hiro-V5’s effect on Rh1 levels (middle gel). Anti-β-tubulin was used as a loading control. (Right) Comparison of the course of age-related retinal degeneration in control ninaEG69D/+ flies with those that express hiro-V5 in an otherwise identical genetic background.
hiro Is Inducible by Retinoids
The mammalian homolog of hiro is a gene whose expression is induced by retinoic acids (Chen et al., 2001). To test whether Drosophila hiro similarly responds to retinoic acids, we challenged Drosophila S2 cells with commercially available all-trans retinoic acid. We found that 20 min of retinoic acid treatment resulted in increased hiro transcript levels as evidenced by semiquantitative RT-PCR as well as qPCR analyses (Figures 4A and 4B).
Figure 4. hiro Transcript Levels Are Regulated by Retinoids.

(A) Semiquantitative RT-PCR of hiro (top) and the control RP49 (bottom) from cultured Drosophila S2 cells with or without retinoic acid treatment.
(B) qPCR of hiro from S2 cells treated with DMSO (left) as control or with retinoic acids (right).
(C) qPCR of hiro from fly head extracts of indicated genotypes. Flies were either raised in regular cornmeal medium or in vitamin-A-deficient food.
(D) qPCR analysis of hiro induction in wild-type (WT) and in santa maria1 mutants that impair retinoid transport to the nervous system (right).
Error bars represent SEM.
qPCR analysis of hiro from adult Drosophila heads also detected higher signals from ninaEG69D/+ samples, as compared with the wild-type controls (Figure 4C, lanes 1 and 2). To determine if such hiro induction in fly heads is due to retinoid-induced gene expression, we repeated the semiquantitative RT-PCR analysis under conditions that deprived retinoids in the fly visual system. Because metazoans require dietary vitamin A to produce retinoids and related metabolites, one way to achieve retinoid deprivation is to rear flies on vitamin-A-deficient food (Blomhoff et al., 1990; Harris et al., 1977; Ozaki et al., 1993). We found that such conditions blocked the increase in hiro transcripts in ninaEG69D/+ heads (Figure 4C, lane 3). To independently validate the role of retinoids in the regulation of hiro transcript levels, we used the santa maria mutant that has impaired vitamin A/b-carotene transport to the photoreceptors (Wang et al., 2007). Similar to the results obtained with flies reared under vitamin-A-deficient food, loss of santa maria impaired hiro induction in the ninaEG69D/+ fly heads (Figure 4D). These results support an unexpected idea that retinoids regulate gene expression in Drosophila, and one such regulated gene, hiro, is involved in the clearance of mutant Rh1.
It is noteworthy that most studies on Drosophila retinoids have centered around their roles as rhodopsin chromophores (Gu et al., 2004; Harris et al., 1977; Kiefer et al., 2002; Ozaki et al., 1993; Wang and Montell, 2005; Wang et al., 2007). Interesting recent studies indicate that retinoids have additional roles in mediating an X-ray irradiation response (Halme et al., 2010), but whether those effects are due to retinoic-acid-mediated gene expression changes remain unknown. As normally folded Rh1 is bound to an 11-cis hydroxylretinal (Ahmad et al., 2006), our results raise the possibility that these retinoids are released to the cytoplasm of photoreceptors with Rh1 folding mutations, thereby serving as a second messenger to instruct the expression of quality-control genes that clear misfolded Rh1. Such a speculative idea awaits more in-depth examination in future studies, along with a search for the transcription factors that directly bind retinoids for gene expression regulation in Drosophila.
EXPERIMENTAL PROCEDURES
Fly Genetics
The following flies used in this study were reported previously: GMR-Rh1G69D and uas-alpha one antitrypsinNHK (Kang and Ryoo., 2009), ninaEG69D (Colley et al., 1995), Rh1-GFP (Pichaud and Desplan, 2001), Rh1-Gal4 (Mollereau et al., 2000), santa maria1 (Wang et al., 2007), Df(3L)5 (Benchabane et al., 2011), Rh1p > ninaEP37H (Galy et al., 2005), and uas-dicer2 (Dietzl et al., 2007). The P[acman]-derived BAC transgenic line CH321-70D3 (sequence information for this clone is available from www.flybase.org) was purchased from GenetiVision.
For the RNAi screen, inverted repeat UAS (UAS-RNAi) lines from the Vienna Stock Center were crossed to the female virgins of the following genotype: GMR-Gal4; ey-Gal4, GMR-Rh1G69D, uas-hrd1/CyO; uas-dicer2. As a negative control, we also crossed the UAS-RNAi lines to GMR-Gal4; ey-Gal4/CyO; uasdicer2 flies. To validate the hits with the ninaEG69D endogenous allele, UASRNAi lines were crossed to the virgin females of the genotype: Rh1-Gal4; uas-dicer2; ninaEG69D/TM6B. Non-TM6B progeny were collected to generate head extracts for anti-Rh1 western blot analysis.
hiroPT flies were generated using recombinase-mediated cassette exchange (RMCE) following a published protocol (Nagarkar-Jaiswal et al., 2015; further details in Supplemental Experimental Procedures).
To generate hiro deletion mutants, we followed the homology-directed repair CRISPR-Cas9 protocol described in www.flycrispr.molbio.wisc.edu (further details in Supplemental Experimental Procedures). The deletion of hiro was further confirmed through genomic PCR. This hiroF1 allele was recombined with ninaEG69D to generate hiroF1, ninaEG69D/TM6B, which was used in subsequent crosses to make hiro homozygous mutants with ninaEG69D mutation in the background.
Immunofluorescence and Western Blots
Standard protocols were followed for western blots and whole-mount immuno-labeling. The following primary antibodies were used in this study: mouse monoclonal 4C5 anti-Rh1 (Developmental Studies Hybridoma Bank, used at 1:500 for whole mount and 1:5,000 for western blots), rabbit anti- Rh1 polyclonal antibody (generated in Charles Zuker’s laboratory [Hsiao et al., 2013], used at 1:100), anti-β-tubulin antibody (Covance catalog no. MMS-410P), rabbit anti-ATP6V1Ba antibody (Abgent catalog no. AP11538C, used at 1:20), mouse anti-Calnexin 99 (Riedel et al., 2016; used at 1:100), guinea pig anti-Hsc3 (Ryoo et al., 2007; used at 1:25). Rhodamine-conjugated Phalloidin (Molecular Probes catalog no. R415) was used to detect rhabdomeres in whole-mount stainings.
Lysosomes were stained using LysoTracker Red DND-99 (Thermo Fisher Scientific) according to a published protocol with minor modifications (Yacobi-Sharon et al., 2013).
EM
Immunogold EM of Drosophila ommatidia was done following a previously described protocol (Colley et al., 1991). Rabbit polyclonal anti-Rh1 was used as the primary antibody, and 18 nm Colloidal gold anti-rabbit antibody was used as the secondary.
RT-PCR
qRT-PCR was performed using the Power SYBR green master mix kit (Thermo Fisher Scientific). The primer sequence can be found in the Supplemental Experimental Procedures.
Retinal Degeneration Assay
The retinal degeneration assays in Figures 3B and 3C were done in the cn, bw−/− background to eliminate eye pigments. To generate the retinal degeneration graphs in Figures 3B and 3C, green fluorescent pseudopupils appearing from Rh1-GFP were analyzed in live flies.
Statistics
We quantified anti-Rh1 band intensities in Figure 1 by measuring the average pixel intensities of western blot bands using ImageJ software and normalizing them to anti-α-tubulin bands. The data are representative of the average of three independent experiments and p values were calculated through a paired Student’s t test. For retinal degeneration analysis, we applied the Log-rank (Mantel-Cox) test. The graphs of Figures 3B and 3C were made by following the GraphPad Prism program using the Kaplan-Meier method and calculating the 95% confidence interval for “fractional survival.” All error bars represent SEM. The n value in Figure 3 represents the number of adult flies analyzed. In other figures, n represents the number of times the experiment was repeated.
Supplementary Material
Highlights.
highroad is required for the degradation of folding-defective Rhodopsin-1
highroad loss accelerates retinal degeneration in ninaEG69D mutant flies
highroad mRNA is induced by retinoic acids in cultured Drosophila cells
highroad mRNA expression is induced in ninaEG69D flies
Acknowledgments
We thank J. Wildonger, M.J. Kang, and W.H. Yoon for technical advice and A. Galy, C. Montell, and C. Desplan (VDRC and Bloomington Stock Centers) for fly strains and antibodies. Immuno-EM images were collected at the Microscopy Core at New York University Langone Medical Center. This work was supported by NIH grant R01 EY020866 to H.D.R. B.B. was supported by NIH grants T32 HD007520 and T32GM007308. P.M.D. was supported by FCT (Portugal) grants LISBOA-01-0145-FEDER-007660, FCT-ANR/NEU-NMC/0006/2013, PTDC/NEU-NMC/2459/2014, and IF/ 00697/2014.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.032.
AUTHOR CONTRIBUTIONS
All authors were involved in the design of the experiments. H.-W.H., B.B., J.C., and P.M.D. conducted the experiments. H.D.R. wrote the manuscript based on input from all authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ahmad ST, Joyce MV, Boggess B, O’Tousa JE. The role of Drosophila ninaG oxidoreductase in visual pigment chromophore biogenesis. J. Biol. Chem. 2006;281:9205–9209. doi: 10.1074/jbc.M510293200. [DOI] [PubMed] [Google Scholar]
- Benchabane H, Xin N, Tian A, Hafler BP, Nguyen K, Ahmed A, Ahmed Y. Jerky/Earthbound facilitates cell-specific Wnt/Wingless signalling by modulating β-catenin-TCF activity. EMBO J. 2011;30:1444–1458. doi: 10.1038/emboj.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomhoff R, Green MH, Berg T, Norum KR. Transport and storage of vitamin A. Science. 1990;250:399–404. doi: 10.1126/science.2218545. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. Cleaning up: ER-associated degradation to the rescue. Cell. 2012;151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Streb JW, Maltby KM, Kitchen CM, Miano JM. Cloning of a novel retinoid-inducible serine carboxypeptidase from vascular smooth muscle cells. J. Biol. Chem. 2001;276:34175–34181. doi: 10.1074/jbc.M104162200. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Messah C, Lin JH. IRE1 directs proteasomal and lysosomal degradation of misfolded rhodopsin. Mol. Biol. Cell. 2012;23:758–770. doi: 10.1091/mbc.E11-08-0663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchore Y, Mitra A, Dolph PJ. Accumulation of rhodopsin in late endosomes triggers photoreceptor cell degeneration. PLoS Genet. 2009;5:e1000377. doi: 10.1371/journal.pgen.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2011;14:93–105. doi: 10.1038/ncb2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho DS, Cairrão F, Zeng X, Pires E, Coelho AV, Ron D, Ryoo HD, Domingos PM. Xbp1-independent Ire1 signaling is required for photoreceptor differentiation and rhabdomere morphogenesis in Drosophila. Cell Rep. 2013;5:791–801. doi: 10.1016/j.celrep.2013.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley NJ, Baker EK, Stamnes MA, Zuker CS. The cyclophilin homolog ninaA is required in the secretory pathway. Cell. 1991;67:255–263. doi: 10.1016/0092-8674(91)90177-z. [DOI] [PubMed] [Google Scholar]
- Colley NJ, Cassill JA, Baker EK, Zuker CS. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc. Natl. Acad. Sci. USA. 1995;92:3070–3074. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gaser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Galy A, Roux MJ, Sahel JA, Léveillard T, Giangrande A. Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum. Mol. Genet. 2005;14:2547–2557. doi: 10.1093/hmg/ddi258. [DOI] [PubMed] [Google Scholar]
- Gu G, Yang J, Mitchell KA, O’Tousa JE. Drosophila ninaB and ninaD act outside of retina to produce rhodopsin chromophore. J. Biol. Chem. 2004;279:18608–18613. doi: 10.1074/jbc.M400323200. [DOI] [PubMed] [Google Scholar]
- Halme A, Cheng M, Hariharan IK. Retinoids regulate a developmental checkpoint for tissue regeneration in Drosophila. Curr. Biol. 2010;20:458–463. doi: 10.1016/j.cub.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WA, Ready DF, Lipson ED, Hudspeth AJ, Stark WS. Vitamin A deprivation and Drosophila photopigments. Nature. 1977;266:648–650. doi: 10.1038/266648a0. [DOI] [PubMed] [Google Scholar]
- Hsiao HY, Jukam D, Johnston R, Desplan C. The neuronal transcription factor erect wing regulates specification and maintenance of Drosophila R8 photoreceptor subtypes. Dev. Biol. 2013;381:482–490. doi: 10.1016/j.ydbio.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M-J, Ryoo HD. Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proc. Natl. Acad. Sci. USA. 2009;106:17043–17048. doi: 10.1073/pnas.0905566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Chung J, Ryoo HD. CDK5 and MEKK1 mediate proapoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nat. Cell Biol. 2012;14:409–415. doi: 10.1038/ncb2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer C, Sumser E, Wernet MF, Von Lintig J. A class B scavenger receptor mediates the cellular uptake of carotenoids in Drosophila. Proc. Natl. Acad. Sci. USA. 2002;99:10581–10586. doi: 10.1073/pnas.162182899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurada P, O’Tousa JE. Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron. 1995;14:571–579. doi: 10.1016/0896-6273(95)90313-5. [DOI] [PubMed] [Google Scholar]
- Leonard DS, Bowman VD, Ready DF, Pak WL. Degeneration of photoreceptors in rhodopsin mutants of Drosophila. J. Neurobiol. 1992;23:605–626. doi: 10.1002/neu.480230602. [DOI] [PubMed] [Google Scholar]
- Liu X, Garriga P, Khorana HG. Structure and function in rhodopsin: correct folding and misfolding in two point mutants in the intradiscal domain of rhodopsin identified in retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 1996;93:4554–4559. doi: 10.1073/pnas.93.10.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mollereau B, Wernet MF, Beaufils P, Killian D, Pichaud F, Kühnlein R, Desplan C. A green fluorescent protein enhancer trap screen in Drosophila photoreceptor cells. Mech. Dev. 2000;93:151–160. doi: 10.1016/s0925-4773(00)00287-2. [DOI] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S, DeLuca SZ, Lee PT, Lin WW, Pan H, Zuo Z, Lv J, Spradling AC, Bellen HJ. A genetic toolkit for tagging intronic MiMIC containing genes. eLife. 2015;4:e08469. doi: 10.7554/eLife.08469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Tousa JE, Baehr W, Martin RL, Hirsh J, Pak WL, Applebury ML. The Drosophila ninaE gene encodes an opsin. Cell. 1985;40:839–850. doi: 10.1016/0092-8674(85)90343-5. [DOI] [PubMed] [Google Scholar]
- Ozaki K, Nagatani H, Ozaki M, Tokunaga F. Maturation of major Drosophila rhodopsin, ninaE, requires chromophore 3-hydroxyretinal. Neuron. 1993;10:1113–1119. doi: 10.1016/0896-6273(93)90059-z. [DOI] [PubMed] [Google Scholar]
- Pichaud F, Desplan C. A new visualization approach for identifying mutations that affect differentiation and organization of the Drosophila ommatidia. Development. 2001;128:815–826. doi: 10.1242/dev.128.6.815. [DOI] [PubMed] [Google Scholar]
- Picking WL, Chen DM, Lee RD, Vogt ME, Polizzi JL, Marietta RG, Stark WS. Control of Drosophila opsin gene expression by carotenoids and retinoic acid: northern and western analyses. Exp. Eye Res. 1996;63:493–500. doi: 10.1006/exer.1996.0139. [DOI] [PubMed] [Google Scholar]
- Riedel F, Gillingham AK, Rosa-Ferreira C, Galindo A, Munro S. An antibody toolkit for the study of membrane traffic in Drosophila melanogaster. Biol. Open. 2016;5:987–992. doi: 10.1242/bio.018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo HD, Domingos PM, Kang MJ, Steller H. Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J. 2007;26:242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scavarda NJ, O’tousa J, Pak WL. Drosophila locus with gene-dosage effects on rhodopsin. Proc. Natl. Acad. Sci. USA. 1983;80:4441–4445. doi: 10.1073/pnas.80.14.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim K, Picking WL, Kutty RK, Thomas CF, Wiggert BN, Stark WS. Control of Drosophila retinoid and fatty acid binding glycoprotein expression by retinoids and retinoic acid: northern, western and immunocytochemical analyses. Exp. Eye Res. 1997;65:717–727. doi: 10.1006/exer.1997.0383. [DOI] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Hennessey JC, Maumenee IH, Jacobson SG, Heckenlively JR, Nowakowski R, Fishman G, Gouras P, Nathans J. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 1991;88:6481–6485. doi: 10.1073/pnas.88.15.6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Montell C. Rhodopsin formation in Drosophila is dependent on the PINTA retinoid-binding protein. J. Neurosci. 2005;25:5187–5194. doi: 10.1523/JNEUROSCI.0995-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Jiao Y, Montell C. Dissection of the pathway required for generation of vitamin A and for Drosophila phototransduction. J. Cell Biol. 2007;177:305–316. doi: 10.1083/jcb.200610081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Tan KL, Agosto MA, Xiong B, Yamamoto S, Sandoval H, Jaiswal M, Bayat V, Zhang K, Charng WL, et al. The retromer complex is required for rhodopsin recycling and its loss leads to photoreceptor degeneration. PLoS Biol. 2014;12:e1001847. doi: 10.1371/journal.pbio.1001847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacobi-Sharon K, Namdar Y, Arama E. Alternative germ cell death pathway in Drosophila involves HtrA2/Omi, lysosomes, and a caspase-9 counterpart. Dev. Cell. 2013;25:29–42. doi: 10.1016/j.devcel.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Zuker CS, Cowman AF, Rubin GM. Isolation and structure of a rhodopsin gene from D. melanogaster. Cell. 1985;40:851–858. doi: 10.1016/0092-8674(85)90344-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.