

Abstract
Uridine, a pyrimidine nucleoside present at high levels in the plasma of rodents and humans, is critical for RNA synthesis, glycogen deposition, and many other essential cellular processes. It also contributes to systemic metabolism, but the underlying mechanisms remain unclear. We found that plasma uridine levels are regulated by fasting and refeeding in mice, rats, and humans. Fasting increases plasma uridine levels, and this increase relies largely on adipocytes. In contrast, refeeding reduces plasma uridine levels through biliary clearance. Elevation of plasma uridine is required for the drop in body temperature that occurs during fasting. Further, feeding-induced clearance of plasma uridine improves glucose metabolism. We also present findings that implicate leptin signaling in uridine homeostasis and consequent metabolic control and thermoregulation. Our results indicate that plasma uridine governs energy homeostasis and thermoregulation in a mechanism involving adipocyte-dependent uridine biosynthesis and leptin signaling.
Graphical abstract
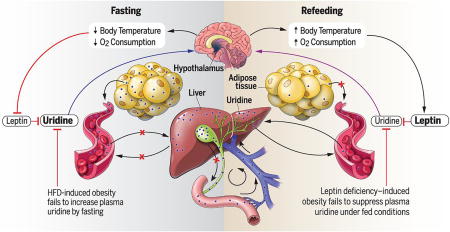
A regulatory model of energy homeostasis during fasting/refeeding. The liver is the predominant biosynthetic organ and contributor to plasma uridine in the fed state, whereas the adipocyte dominates uridine biosynthetic activity in the fasted state. Biliary excretion is the primary mechanism for plasma uridine clearance. Because nutrient intake triggers bile release, plasma uridine levels are elevated during fasting and drop rapidly in the postprandial state. The fasting-associated increase of plasma uridine elicits a hypothalamic response culminating in body temperature lowering, whereas bile-mediated uridine release promotes a decline of plasma uridine and enhances insulin sensitivity.
Uridine is a uracil nucleoside that plays a critical role as a building block for RNA and DNA biosynthesis (1, 2). It also serves to generate pyrimidine-lipid and pyrimidine-sugar conjugates required for glycogen deposition, protein and lipid glycosylation, extracellular matrix biosynthesis, and detoxification of xenobiotics (1, 2). These anabolic reactions are critical for normal cellular function and survival. In addition, uridine catabolism by uridine phosphorylase may regulate events important to systemic metabolism, such as body temperature and circadian rhythm (3). Despite its important physiologic and pharmacological roles, uridine has received much less research emphasis than adenosine (3).
Plasma uridine concentrations are tightly regulated in both humans and rodents, but the mechanisms underlying homeostasis of circulating uridine are unknown (2). The liver is considered the major organ controlling plasma uridine levels, mediating de novo biosynthesis within hepatocytes and uridine clearance via Kupffer cells (4, 5). The majority of plasma uridine is degraded by the liver after uptake through the portal vein. However, the processes by which the liver regulates uridine degradation, and the biological effects of this massive clearance process, have not been studied.
A constant supply of circulating uridine is required for a number of biological functions (2, 6), and disruption of plasma uridine homeostasis through uridine supplementation has profound effects on systemic metabolism (7). Short-term supplementation of uridine in food improves insulin sensitivity in mice, but long-term administration causes fatty liver and promotes development of pre-diabetes. Thus, control of plasma uridine is tightly coupled to energy homeostasis.
Previous work suggests that uridine affects both body temperature and feeding behavior. For example, injection of high doses of uridine decreases body temperature in rodents (8). Co-administration of uridine and benzylacyclouridine, a compound that inhibits uridine degradation, partially prevents this temperature drop (8, 9). In addition, elevated plasma uridine can increase brain (hypothalamic) levels of uridine diphosphate (UDP), which then promotes food intake via P2Y2-dependent activation of AgRP (Agouti-related protein) neurons (10). Here, we investigated mechanisms by which feeding behavior regulates body temperature and plasma uridine levels.
Fasting and refeeding regulate plasma uridine concentrations
Because fasting leads to a number of physiological adaptations, we set out to determine whether this process is associated with dynamic regulation of plasma uridine. In C57BL/6 mice, plasma uridine levels doubled upon 24 hours of fasting (4.96 ± 0.76 µM versus 8.89 ± 1.02 µM, P < 0.0001) and recovered to basal levels 4 hours after refeeding (3.86 ± 0.54 µM) (Fig. 1A). Rats manifested a similar elevation in plasma uridine after an overnight fast (5.61 ± 0.71 µM versus 9.74 ± 2.24 µM, P < 0.001), but the levels did not return to basal levels even 24 hours after refeeding (Fig. 1B). In healthy human subjects (table S1), a single meal following an overnight fast reduced plasma uridine levels by 40.3 ± 10.3% within 4 hours (4.63 ± 0.75 µM after overnight fast versus 1.84 ± 0.44 µM 4 hours after meal, P < 0.001) (Fig. 1C). There was no postprandial change in plasma uric acid concentrations in the human subjects (Fig. 1D), indicating that the change in plasma uridine levels that results from fasting and refeeding is unlikely to be due to generalized nucleotide catabolism.
Fig. 1. Plasma uridine dynamics during fasting and refeeding.

(A) Plasma uridine levels in male C57BL/6 mice in a fasting/refeeding study (n = 7). (B) Plasma uridine levels in male Sprague-Dawley rats in a fasting/refeeding study (n = 7). (C and D) Plasma uridine and uric acid levels in healthy women after subjects were fasted for ~12 hours overnight, and at regular intervals after they consumed a breakfast meal at 7 a.m. (47). Plasma uridine and uric acid levels after overnight fasting were considered as basal for each subject for statistical analysis (n = 6). Data were analyzed with paired t test. ***P < 0.001, ****P < 0.0001; ns, not significant. Error bars denote SEM.
Plasma uridine links thermoregulation to fasting and refeeding
In rodents, fasting and refeeding triggers body temperature changes that allow the animal to adapt to the altered nutrient status. Previous reports show that administration of high-dose uridine (3500 mg/kg) resulted in severe hypothermia of 6° to 10°C in mice (8). To test whether uridine plays a role in thermoregulation, we injected mice intraperitoneally (i.p.) with uridine at a reduced dose (1000 mg/kg) to avoid severe hypothermia. We found that this treatment rapidly lowered body temperature [37.9° ± 0.9°C, 34.9° ± 0.7°C, and 34.0° ± 1.6°C at 0, 15, and 30 min after uridine injection, P < 0.01 compared to phosphate-buffered saline (PBS) injection]. This drop in temperature was then partially recovered within 60 min (36.0° ± 0.9°C, P < 0.01 compared to PBS injection) (Fig. 2A).
Fig. 2. Plasma uridine dynamics correlates with body temperature fluctuation.

(A) Body temperature was monitored in male C57BL/6 mice after intraperitoneal injection of PBS or uridine (1 g/kg) (n = 6 per group). (B) Body temperature was monitored in a fasting/refeeding study using male wild-type (WT), ob/ob, and 6 weeks HFD-fed animals (n = 6 per group). (C) Male WT and ob/ob mice fed on chow or HFD (10 weeks) were monitored for plasma uridine levels during a fasting/refeeding study (n = 6 per group). (D) PALA prevented the drop of body temperature by fasting in ob/ob mice (n = 7 per group). Statistical analysis was performed for each condition using time 0 or the fed state of that group as base line if not specified. Data in (A) to (C) were analyzed with paired t test, and data in (D) were analyzed with two-tailed Student t test. *P < 0.05, **P < 0.01. Error bars denote SEM.
Obesity, a pandemic now seen throughout the world (11), displays characteristic abnormalities in thermoregulation (12). To test this in a preclinical model and to explore the potential role of leptin, we studied two animal models: leptin-deficient ob/ob mice and wild-type mice on a high-fat diet (HFD). We found that when wild-type mice fed a chow diet were fasted for 24 hours, their body temperature dropped 1.8° ± 0.5°C (n = 6, P < 0.05). However, when chow-fed leptin-deficient ob/ob mice were fasted for 24 hours, the effect was more striking: Their body temperature dropped by 7.3° ± 2.7°C (n = 6, P < 0.01) (Fig. 2B). These findings confirm that leptin-deficient mice have defects in maintaining normal body temperature (13); however, it is unclear whether this is the direct result of leptin deficiency or a general phenomenon due to obesity.
To address this, we compared the body temperature response to fasting and refeeding in HFD-fed wild-type mice and HFD-fed ob/ob mice. In HFD-fed wild-type mice, the body temperature drop triggered by 24 hours of fasting (1.2° ± 0.6°C, n = 6, P < 0.01) was similar to that elicited by fasting in chow-fed wild-type mice (1.8° ± 0.5°C, n = 6, P < 0.05) (Fig. 2B). By contrast, when ob/ob mice were fed HFD for 6 weeks, their body temperature drop triggered by 24 hours of fasting was reduced to 2.3° ± 1.3°C (n = 6, P < 0.01) (Fig. 2B). Thus, although both the HFD and ob/ob mice are obese and are widely used for studies of insulin resistance, the fasting response in HFD-fed wild-type animals differs from the fasting response in ob/ob mice. Together, these data lend credence to a model in which leptin signaling participates in fasting-induced temperature declines in a manner blunted by HFD exposure.
To test whether uridine participates in thermoregulation during fasting and refeeding, we analyzed plasma uridine levels in HFD-fed ob/ob and wild-type mice. We detected higher levels of plasma uridine in ob/ob mice than in wild-type mice; conversely, obese HFD-fed wild-type mice harbored plasma uridine levels comparable to those in lean chow-fed wild-type mice (Fig. 2C). Fasting further increased plasma uridine levels in ob/ob mice in the first 4 hours after food removal, but fasting had no effect on plasma uridine levels in HFD-fed wild-type mice (Fig. 2C). Upon refeeding, ob/ob mice exhibited a rapid decline in plasma uridine levels, whereas no significant changes were observed in obese HFD-fed wild-type mice (Fig. 2C). These data reveal a strong correlation between plasma uridine levels and thermoregulation and further highlight the distinct effects of HFD and leptin-deficiency on body temperature that is mediated through plasma uridine.
Uridine-triggered temperature declines in rodents rely on the activity of uridine phosphorylase (8), the enzyme responsible for initiation of uridine catabolism. We hypothesized that increases in plasma uridine levels during fasting mediate the temperature drop by increasing uridine availability for degradation. To test this, we injected ob/ob mice with N-(phosphonacetyl)-l-aspartate (PALA). This compound is an inhibitor of aspartate transcarbamylase (14), which is the rate-limiting enzyme for uridine biosynthesis (15) and is part of the trifunctional protein Cad (carbamoyl phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase). In this context, PALA prevented both the drop in body temperature in ob/ob mice (Fig. 2D) and the elevation of plasma uridine after 24-hour fasting. These findings support a model in which plasma uridine levels govern core body temperature.
Uridine links thermoregulation with leptin
Temperature exchange with the environment depends on the difference in temperature between the subject and surrounding environment and relies on three mechanisms: conduction, convection, and radiation. To examine whether the acute temperature drop triggered by uridine is mediated by temperature exchange with the environment, we housed mice in a near-thermoneutral environment (29°C), 7°C above the ambient room temperature used for the studies depicted in Fig. 2A. Wild-type mice injected with PBS showed a slight increase in body temperature after they were moved to 29°C (37.5° ± 0.24°C, 38.3° ± 0.2°C, and 38.1° ± 0.2° C at 0, 15, and 30 min, respectively; n = 6) (Fig. 3A). When mice were injected with uridine and subsequently transferred to the 29°C incubator, the temperature drop seen at ambient room temperature (Fig. 2A) was no longer observed; rather, the mice displayed a body temperature only slightly lower than that observed with the PBS injection under the same conditions (37.4° ± 0.3°C versus 38.3° ± 0.2°C at 15 min, 36.9° ± 0.4°C versus 38.1° ± 0.2°C at 30 min; n = 6, P < 0.05) (Fig. 3A). From these experiments, we conclude that the thermoregulation effects of uridine rely on temperature exchange with the ambient environment.
Fig. 3. Thermoregulation effects of uridine.

(A) Male C57BL/6 mice were injected intraperitoneally (i.p.) with PBS or uridine (1 g/kg) at room temperature (22° to 25°C), then transferred to a chamber at 29°C, and monitored for their body temperature (n = 6 per group). (B to D) Injection of uridine (i.p., 1 g/kg) reduced the rates of O2 consumption and CO2 production, but increased RER of male C57BL/6 mice in an indirect calorimetry study (n = 5 per group). The arrows indicate the changes due to cage opening. Values of VO2 and VCO2 were plotted relative to the baseline measurements. (E) Body temperature was monitored in male WT and ob/ob mice after uridine i.p. injection (1 g/kg) at the indicated time point. The HFD group consisted of male WT mice fed with HFD for 15 weeks (n = 5 per group). (F) Plasma leptin levels in WT mice (HFD for 15 weeks) were measured before and after uridine i.p. injection (1 g/kg) (n = 6 per group). (G) Plasma leptin levels in WT and ob+/− mice were measured before and 15 min after uridine i.p. injection (1 g/kg) (n = 6 per group). Statistical analysis was performed for different treatments or genotypes at indicated time points if not specified. Data were analyzed with two-tailed Student t test. *P < 0.05, **P < 0.01. Error bars denote SEM.
To determine whether uridine reduces heat production through effects on metabolism, we housed mice in metabolic cage units and measured O2 consumption, CO2 production, and respiratory exchange ratio (RER). After two baseline recordings, we injected the mice i.p. with PBS or uridine and then performed a third recording 5 min later. Because the cages had to be opened for injections, there was a sudden transient change in all parameters. Upon equilibration, the rates of O2 consumption and CO2 production rapidly recovered to normal levels in the vehicle group, indicating that metabolic rates were not affected. In contrast, uridine-injected mice displayed a reduced rate of recovery of O2 consumption and CO2 production, reflecting a suppression of the metabolic rate (Fig. 3, B and C). A reduced energy demand is another way to reduce heat production under stress conditions such as hypoxia (16). To determine whether a modulation of energy demand contributes to uridine-related thermoregulation, we measured the RER, which was increased within 1 hour after uridine injection (Fig. 3D). This increase suggests that uridine administration leads to an increase in carbohydrate consumption instead of lipid use (17). Indeed, a correlation has been established between elevated RER and lower energy demand (18). Combined, these data indicate that the uridine-induced drop in body temperature is caused by metabolic rate suppression due to lowered energetic demand.
One of the major sites of leptin action is the hypothalamus (19), which is also the master regulator of core body temperature (20). To examine whether the uridine-mediated drop in body temperature involves leptin, we injected uridine intraperitoneally into ob/ob and into HFD-fed wild-type mice. In both cases, uridine caused a rapid drop in body temperature (2.9° ± 0.3°C and 4.2° ± 0.9°C, n = 3, P < 0.01, respectively, 30 min after uridine injection). However, ob/ob mice manifested a significant delay in the recovery of normal body temperature (Fig. 3E). These results suggest that the uridine-induced drop in body temperature is leptin-independent, but that leptin is required to recover body temperature after hypothermia. Consistent with this hypothesis, we observed a robust increase by nearly a factor of 2 in plasma leptin levels within 30 min after uridine injection (Fig. 3F).
To determine whether the change in circulating leptin derives from increased secretion or reduced clearance, we studied heterozygous ob/+ mice, which are known to harbor reduced plasma levels of leptin (21). If uridine-stimulated leptin elevation is due to impaired clearance, the response in ob/+ mice would be expected to be similar to that occurring in wild-type mice. However, we found that increases in plasma leptin were diminished in the ob/+ mice after uridine injection (Fig. 3G), which suggests that the effect of uridine on circulating leptin is likely due to increased secretion.
Plasma uridine is cleared by bile
The accumulation of plasma uridine upon fasting, coupled with its rapid postprandial drop, led us to examine whether bile might be involved in plasma uridine clearance, because bile excretion is increased by food intake. Indeed, we found that bile extracted from the gallbladder of wild-type mice contained uridine levels that were higher than levels present in plasma under fasting conditions by a factor of ≤8 (Fig. 4A). Moreover, bile uridine levels trended toward a decrease in fasted mice, but within 4 hours of refeeding, bile uridine concentrations increased 27% (n = 6, P < 0.05) and 100% (n = 10, P < 0.01) in male and female wild-type mice, respectively (Fig. 4B); these findings suggest that mice under refed conditions have a much higher rate of uridine flux from blood to the gallbladder than fasted mice. And consistent with changes in plasma uridine observed in HFD and ob/ob mice (Fig. 2C), bile from HFD mice harbored lower uridine content under fasted conditions (Fig. 4C). Conversely, bile isolated from ob/ob mice had higher uridine levels that declined when they were exposed to a HFD (Fig. 4D).
Fig. 4. Biliary release of uridine.

(A) Uridine concentrations in plasma and bile from 24 hour–fasted male C57BL/6 mice (n = 6). (B) Uridine concentration in bile from male and female C57BL/6 mice (n = 6 to 10 for each time point). (C) Biliary uridine levels in 24 hour–fasted male C57BL/6 mice fed with chow or HFD for 15 weeks (n = 6 per group). (D) Biliary uridine levels in 24 hour–fasted male WT and ob/ob mice fed with chow or HFD for 15 weeks (n = 6 per group). (E) Male C57BL/6 mice were administered with [5-3H]uridine by tail vein injection or oral gavage (gav). Plasma from tail tip and bile from gallbladder were harvested at indicated time points (n = 4 per time point). (F) Male C57BL/6 mice were administered with [3H]uridine by oral gavage. Plasma from tail tip and portal vein and bile from gallbladder were harvested 2 min after gavage (n = 6). Data were analyzed with two-tailed Student t test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars denote SEM.
To study the dynamics of plasma uridine under fed conditions, we injected wild-type mice with a trace amount of [3H]uridine (25 µCi) via the tail vein. Rapid turnover (half-life ~3 min) of plasma uridine has been reported in rats and dogs (22, 23), and we observed similar values in mice. Notably, we found a concomitant accumulation of [3H]uridine in bile (Fig. 4E), with levels in bile as high as those in plasma within 15 min of tail vein injection (Fig. 4E). In contrast, when [3H]uridine was gavaged orally, no significant radioactivity was detected in plasma, whereas rapid accumulation of [3H]uridine was detected within 2 min in bile (Fig. 4E).
Together, these data suggest that orally supplied uridine is efficiently channeled through the liver to the gallbladder with a negligible amount of transit through the peripheral circulation. To test this further, we collected blood from the portal vein 2 min after [3H]uridine gavage. Here, we observed a significant burst of radioactivity within the portal vein, comparable to levels detected in the gallbladder, within 2 min (Fig. 4F). In contrast, radioactivity detected in plasma was less than 50% of that in bile, confirming that orally supplied uridine is efficiently channeled to the liver through the portal vein.
Biliary uridine clearance improves glucose tolerance
To begin to explore whether uridine plays a role in metabolism beyond thermoregulation, we examined its effect on glucose homeostasis. First, we found that oral co-administration of uridine and glucose to wild-type mice on a HFD led to significant improvements in glucose tolerance (Fig. 5A); this improvement, however, was not observed in ob/ob mice (Fig. 5B). We next tested whether parenteral administration of uridine mimics the effects of oral uridine. We injected uridine i.p. (at the same dose that induced a temperature drop) into HFD-fed wild-type mice and into ob/ob mice 15 min prior to an oral glucose tolerance test (OGTT). The HFD-fed wild-type mice also manifested a significant improvement in glucose tolerance after uridine injection (Fig. 5C). In contrast, and consistent with an active role of leptin in this process, ob/ob mice manifested deterioration in their glucose tolerance after uridine injection (Fig. 5D). Together, these results suggest that leptin is a key mediator of uridine’s effect on glucose metabolism.
Fig. 5. Uridine effects on glucose metabolism.

(A and B) Plasma glucose levels from male WT mice (HFD for 25 weeks) or ob/ob mice were measured in oral glucose tolerance tests with glucose or glucose-uridine solution (n = 6 per group). (C and D) Plasma glucose levels from male WT mice (HFD for 25 weeks) or ob/ob mice were measured in oral glucose tolerance tests with PBS or uridine intraperitoneal injection 15 min before glucose gavage (n = 6 per group). (E) Plasma glucose levels from male C57BL/6 mice (18 months old) were measured in oral glucose tolerance tests with glucose or glucose-uridine solution (n = 6 per group). (F) Plasma glucose levels from male C57BL/6 mice (18 months old) were measured in oral glucose tolerance tests with PBS or uridine intraperitoneal injection 15 min before glucose gavage separately (n = 6 per group). (G and H) Plasma glucose levels and body weight were measured in male C57BL/6 mice (HFD for 30 days) before and after i.p. injection with PALA or vehicle (Veh) (n = 5 per group). Statistical analysis was performed for different treatments at indicated time points if not specified. Data were analyzed with two-tailed Student t test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Error bars denote SEM.
Aging is often associated with development of insulin resistance and obesity. In a study of older wild-type mice (18 months of age), we found that uridine administration markedly improved glucose tolerance (Fig. 5, E and F), which suggests that both aging-associated and HFD-associated insulin resistance can be attenuated by uridine supplementation. To explore this further, we used PALA to inhibit uridine biosynthesis (14). Prior toxicological evaluation of this compound in C57BL/6 mice revealed that a 50% lethal dose corresponds to 1587 mg/kg (24). Given this report, we studied the effect of a much lower single dose of 62.5 mg/kg. No gross abnormalities were observed after drug administration in chow-fed mice. However, PALA led to an acute elevation of plasma glucose in HFD-fed mice (Fig. 5G). Upon more prolonged monitoring of these mice, after receiving just this single dose of PALA, the mice displayed a significant loss of body weight and died within 2 weeks of treatment (Fig. 5H)—an observation not seen in the chow-fed animals. We interpret these observations with a uridine biosynthesis inhibitor to suggest that endogenous uridine synthesis is critical for both blood glucose control and survival of mice when exposed to HFD.
Enteral uridine delivery does not reduce body temperature
Our results indicate that enteral provision of uridine does not culminate in plasma increases in uridine (Fig. 4E) but improves glucose tolerance similar to that elicited by i.p. administration (Fig. 5C). To explore the underlying mechanisms, we first evaluated core body temperature. Here, we observed that uridine gavaged orally in HFD-fed mice elicited marginal changes in body temperature and led to temporary elevation of body temperature in ob/ob mice (Fig. 6A). This is in sharp contrast to the effects of i.p. administration of the same dose of uridine (Fig. 3E). Thus, uridine administration through an oral route did not provoke hypothermia. Further, we noted that glucose gavage did not lead to a decrease in body temperature in either HFD-fed wild-type or ob/ob mice, irrespective of co-administration of uridine (Fig. 6, B and C). This suggests that uridine-induced improvements in glucose tolerance are not mediated by thermoregulation.
Fig. 6. Thermoregulation effects of enteral administration of uridine.

(A) Body temperature of male WT (chow or HFD for 10 weeks) and age-matched ob/ob mice was monitored before and after oral administration of uridine (1 g/kg, n = 6 per group). (B and C) Body temperature of male WT (HFD for 10 weeks) or age-matched ob/ob mice was monitored before and after oral administration of glucose or glucose-uridine solution (n = 6 per group). Data were analyzed by two-way ANOVA and no significant difference was detected between each group. Error bars denote SEM.
Adipocytes are a key regulator for plasma uridine
Our findings indicate that biliary clearance is critical for the declines in plasma uridine elicited by refeeding. We proposed that fasting-induced elevations in plasma uridine could derive from reduced biliary clearance during the fasting state. Further, the abnormal dynamics of plasma uridine observed in ob/ob and HFD-fed mice suggested that adipose tissue might participate in the elevations of plasma uridine triggered by fasting. To test this, we evaluated the impact of acute loss of adipocytes on plasma uridine levels. First, we used our previously characterized FAT-ATTAC mice, a model in which adipocytes in adult animals can be selectively eliminated by induced apoptosis (25). Three days after loss of viable adipocytes, FAT-ATTAC mice under fed conditions displayed a modest but statistically significant drop in plasma uridine levels relative to wild-type mice (8.16 ± 1.07 µM in wild type, n = 6 versus 6.34 ± 1.11 µM in FAT-ATTAC; n = 6, P = 0.016) (Fig. 7A). Upon fasting, the FAT-ATTAC mice manifested no increase in uridine levels relative to control mice (Fig. 7A). In contrast, refeeding led to similar reductions in plasma uridine concentrations in both groups. Beyond a small reduction in fat mass (0.15 ± 0.05 g in wild type, −0.39 ± 0.13 g in FAT-ATTAC), no significant changes in body weight were detected between the two groups either before or 3 days after induction of adipocyte apoptosis (fig. S1).
Fig. 7. Adipocytes are critical for plasma uridine supply.

(A) Plasma uridine levels in male WT and FAT-ATTAC mice in a fasting/refeeding study (n = 6 per group). (B) Relative plasma uridine levels in male WT and Agpat2 KO mice in a fasting/refeeding study (n = 4 per group). (C) qPCR quantification of genes involved in pyrimidine biosynthesis in liver from male C57BL/6 mice (n = 5 per group). (D) qPCR quantification of Cad in liver, epididymal fat (eWAT), subcutaneous fat (sWAT), and brown fat (BAT) from male C57BL/6 mice (n = 5 per group). (E) Uridine contents in subcutaneous adipose tissue biopsies from metabolically healthy subjects were reduced 5 hours after breakfast (n = 6). (F to I) Male C57BL/6 mice were treated with streptozotocin (STZ) or vehicle (CTL) and monitored for plasma glucose levels and body weight up to 7 weeks. The uridine concentrations in plasma, bile, and tissues were measured from mice fasted for 24 hours (n = 5 or 6 per group). Statistical analysis was performed for each group or treatment using the fed state or CTL treatment of that group as baseline if not specified. Data were analyzed with two-tailed Student t test. *P < 0.05, **P < 0.01, ***P < 0.001. Error bars denote SEM.
To test this further, we studied mice selectively silenced for Agpat2 (1-acylglycerol-3-phosphate-O-acyltransferase 2), a mutant line that lacks functional adipose tissue and serves as a model of congenital lipodystrophy (26). In these animals, fasting did not elicit an increase in plasma uridine levels (Fig. 7B). These data lend additional credence to a model in which adipose tissue is required for fasting-induced increases in plasma uridine.
The liver is widely held to be the major organ maintaining plasma uridine supply (4). However, even though fasting triggered increases in plasma uridine, we found that the genes responsible for de novo uridine synthesis were all downregulated by fasting in the liver (Fig. 7C). In contrast, Cad, which encodes the rate-limiting enzyme for uridine biosynthesis, was highly expressed in three different adipose tissue depots when examined under both fed and fasted conditions (Fig. 7D). Furthermore, the genes encoding the other two enzymes in the uridine biosynthetic pathway, Dhodh and Umps, manifested expression patterns similar to Cad (fig. S2). This suggests that adipose tissue is indeed an important site of uridine synthesis contributing to fasting-induced rises in plasma uridine. Consistent with this, biopsy specimens of subcutaneous fat from subjects depicted in Fig. 1C uniformly harbored reduced uridine content in the postprandial state (Fig. 7E).
To explore further the role of adipose tissue in plasma uridine homeostasis, we studied a streptozotocin (STZ)–induced model of type 1 diabetes. As expected, STZ administration caused hyperglycemia (Fig. 7F) and a rapid loss of fat mass (fig. S3). After the initial loss of body mass, mice subsequently reached a plateau of body weight that was maintained for the subsequent 7 weeks (Fig. 7G). Under these conditions, both plasma and bile uridine levels were reduced to half the levels observed in the control vehicle-treated group (Fig. 7H). Similarly, uridine concentrations in both brown adipose tissue (BAT) and the heart were reduced (Fig. 7I). In contrast, uridine levels in livers isolated from STZ-treated mice were comparable to those in the control group (Fig. 7I), which suggests that uridine biosynthesis in the liver remains functional even in the absence of circulating insulin. However, the seemingly unaltered production of uridine in the liver is insufficient to maintain normal levels of plasma uridine under fasting conditions.
Although our observations support the idea that adipose tissue plays a critical role in plasma uridine homeostasis, the inducible and constitutive loss of adipose tissue as well as lowering of systemic insulin production through the loss of β cells are associated with a multitude of metabolic changes, so the effects on uridine could be indirect. To test more specifically whether adipose tissue supplies uridine through pyrimidine biosynthesis, we generated mice selectively silenced for Cad in adipocytes. Upon 24 hours of fasting, the Cad knockout mice manifested no increase in uridine levels relative to the control group (fig. S4), which suggests that uridine biosynthesis in adipocytes is necessary for a fasting-induced increase in plasma uridine. However, more experiments are warranted to examine confounding effects from compensatory alteration in liver uridine biosynthesis and/or degradation in response to reduced adipocyte Cad expression.
Discussion
Uridine and thermoregulation
Temperature homeostasis is a tightly regulated process in mammals. The human body retains a set temperature of ~37°C, with complications arising when the temperature increases by little more than 3°C (27). During fasting, both rodents and humans manifest a reduction in body temperature, a process regulated by the sympathetic nervous system (20, 28). Our findings indicate that plasma uridine is a metabolite critically involved in this process.
The dynamic regulation of plasma uridine is unusual for a metabolite, particularly one with the range of vital biological functions associated with uridine. Characterizing the dynamics of plasma uridine as part of the systemic response to fasting and refeeding has led us to propose a model that integrates feeding, carbohydrate metabolism, and thermoregulation through a novel adipo-biliary-uridine axis. In studies with leptin-deficient ob/ob mice, coupled with measurements of circulating leptin in response to uridine administration, we also uncovered a role for leptin signaling in the governance of uridine homeostasis and fasting-induced declines in body temperature.
Plasma-bile-gut uridine homeostasis
Our results unveil pivotal roles for uridine biosynthesis and transport to and from bile in the metabolic control of body temperature. Bile is produced by the liver and released into the small intestine to enhance the digestion and absorption of fat (29). Recently, components of bile have been shown to mediate effects on energy metabolism and blood glucose regulation, and even to shape the composition of the gut flora (30). Our findings indicate that uridine released from bile can increase glucose assimilation. Although the detailed mechanism is unclear, intestinal nutrient absorption is a process tightly linked to the function of biliary uridine. Because of the limited capacity for de novo synthesis of nucleotides (31, 32), the salvage pathway for utilization of exogenous nucleosides is of particular importance in epithelial cells in the small intestine.
How, then, is uridine transported across these systems? Nucleosides are transported into cells by plasma membrane carriers that belong to two gene families, CNT and ENT (33–36). Whereas CNT1- and CNT2-related proteins are responsible for the concentrative Na-dependent high-affinity transport of pyrimidine (33–36), less is known about the transport of uridine. Our findings point to excretion of uridine into bile canaliculi by hepatocytes, as well as transport of plasma uridine into the gallbladder, suggesting the presence of nucleoside carriers on both canalicular and sinusoidal membranes of hepatocytes. Interestingly, bile acids can increase CNT2-related activity in the liver through the recruitment of CNT2 from intracellular stores to the plasma membrane (37); this implies that uridine may be taken up by hepatocytes during the fed state and subsequently shunted to the gallbladder.
Adipocyte-dependent uridine biosynthesis
A role for adipocytes in plasma uridine homeostasis has not been appreciated to date. Our observations reveal that fasting-induced elevation and maintenance of elevated plasma uridine levels rely critically on adipocytes. The uridine levels in fat biopsies from HIV-infected patients that suffer from HIV-associated partial lipodystrophy are significantly elevated (38), indicating that excessive uridine production might lead to the loss of adipose tissue. Our studies have drawn a consistent correlation between increased plasma uridine and increased biosynthesis in adipocytes; however, regardless of the consistency and strength of the correlation, this does not establish causality.
Detailed mechanisms underlying the leptin-mediated plasma uridine clearance also remain unclear. However, a connection between leptin transcription and the hexosamine biosynthetic pathway end product UDP-N-acetylglucosamine (UDP-GlcNAc) has been reported (39), suggesting the involvement of a signaling pathway from uridine to UDP-GlcNAc. Given the complexity of plasma uridine homeostasis and the pivotal role of uridine in systemic metabolism reported here, inhibition of uridine supplied from one tissue is expected to alter the supply, salvage, and clearance of uridine in other tissues. Indeed, our findings point to a critical role of adipocytes in plasma uridine homeostasis. Additional studies combining tissue-specific loss and gain of function of enzymes responsible for pyrimidine de novo synthesis are warranted to characterize the contribution of adipocytes to the plasma uridine supply.
Future directions
Our description of an adipo-biliary-uridine axis raises interesting questions for future investigation. What are the effects of feeding-induced reductions in uridine levels in organs that rely heavily on uridine uptake from plasma, such as the heart? Given the established links among feeding, metabolism, and circadian rhythmicity (40, 41), does uridine play a central role in these processes? Whether bariatric surgery, the only obesity treatment that achieves substantial and permanent weight loss (42), is associated with alterations in plasma uridine homeostasis due to altered recycling of uridine from gut to blood also merits investigation.
Recently, diabetes has been linked to impairments in temperature control during exposure to thermal stress (43). Our studies reveal a direct link between temperature regulation and metabolism, indicating that a uridine-centered model of energy homeostasis may pave the way for future studies on uridine homeostasis and diabetes as well as other metabolic diseases.
Materials and methods
Animals and experimental protocol
Adult C57BL/6 inbred mice (WT) and leptin deficient mice (ob/ob) were housed in individually ventilated cages with constant temperature on a standard 12:12 light cycle, and were fed standard rodent chow diet (2916, Teklad) with free access to water. Male C57BL/6 mice that were used for high fat diet study were switched to high fat diet (D12492, Research Diet) after their body weight reached 20 g (8 to 10 weeks old). Age-matched ob/ob mice were used, with both male and female data pooled in the analysis. Male Sprague-Dawley rats were obtained from the Charles River Laboratory and fed with rat diet (Teklad). All procedures were approved by The Institutional Animal Care and the Use Committee of UT Southwestern Medical Center.
Study subjects and human samples
This study was approved by the Institutional Review Board of Washington University School of Medicine in St. Louis, MO, and written informed consent was obtained from all subjects before their participation. Subjects were admitted to the Clinical Research Unit in the evening before the study, and consumed a standard dinner (12 kcal per kg fat-free mass) at 1800 hours. At 0700 hours the next morning, after subjects fasted for 12 hours overnight, they consumed a liquid mixed meal, provided in 5 equal aliquots every 5 min for 20 min. The meal contained one-third of each subject’s estimated total daily energy requirement and was comprised of 55% of total energy as carbohydrates, 15% as protein, and 30% as fat. Arterial blood samples were obtained 10 min before and immediately before, and at 20, 40, 60, 120, 180, 240, and 300 min after initiating meal consumption. Human subject information is shown in table S1.
Fasting/refeeding studies in rodents
Male WT mice (10 to 35 weeks old) and male rats (10 weeks old) were used for fasting/refeeding studies at ambient temperature (22° to 25°C). Age-matched ob/ob mice were used which include both males and females. Male Agpat KO (8 to 10 weeks old) and male FAT-ATTAC mice (10 to 12 weeks old) were used for fasting/refeeding studies with their age-matched WT controls, respectively. For FAT-ATTAC mice, the study was performed 3 days post dimerizer compound AP20187 (Ariad) administration as described (25). Blood samples (30 µl) were collected in heparinized capillaries from tail tip at specified time points. Samples were centrifuged and aliquots of plasma were frozen at −20°C.
Plasma and bile measurements
Blood samples (30 µl) from tail tip were collected in heparinized capillaries from male WT and age-matched ob/ob mice (12 to 30 weeks old) and male rats (10 weeks old). Samples were centrifuged and aliquots of plasma were frozen at −20°C. Bile samples were collected from gallbladders from male WT, female WT and age-matched ob/ob mice (28 to 32 weeks old). Plasma uridine and uric acid, and bile uridine levels were quantified by HPLC-MS/MS at UT South-western Medical Center Mouse Metabolic Phenotyping Core. Plasma glucose was measured by an oxidase-peroxidase assay (Sigma) as described (44). Plasma leptin levels were measured using an ELISA kit (Millipore).
Effect of uridine and glucose on body temperature
Male WT (15 to 35 weeks old) and age-matched ob/ob mice were used for body temperature measurements through an IPTT-300 transponder implanted longitudinally above the shoulder of mouse (Bio Medic Data Systems). To study the thermal effect of uridine, 0.1 g/ml uridine (Sigma) in PBS was administrated to mice (1 g/kg body weight) via intraperitoneal injection or oral gavage. To study the thermal effect of glucose, 0.25 g/ml glucose (Sigma) in H2O was administrated orally to mice (2.5 g/kg body weight). To study the combined effect of glucose and uridine, glucose-uridine solution (0.25 g/ml glucose and 0.1 g/ml uridine in H2O) was administrated orally at the same dose as the study for glucose alone. All the treatment and temperature measurements were performed at ambient room temperature (23° to 25°C), if not specified. A refrigerated incubator at 29°C (Powers Scientific) was used to achieve a near-thermoneutral environment.
[3H]Uridine clearance study
[5-3H]Uridine (25 µCi, PerkinElmer) in 250 µl PBS was administrated to male C57BL/6 mice (15 to 35 weeks old) via tail vein injection or oral gavage. This dose (0.274 µg uridine per mouse) was lower than that for thermal effects of uridine (1 g/kg body weight), such that no drop in body temperature was triggered in the clearance study. Plasma from tail tip was sampled from intact mouse before its bile was harvested from gallbladder. Plasma (5 µl) and bile (2 µl) were used for radioactivity measurements. To collect blood from portal vein, mice were anesthetized with isoflurane after plasma had been first sampled from its tail tip.
Indirect calorimetry and NMR
VO2 (ml/hr/kg), VCO2 (ml/hr/kg), and RER (VCO2/VO2, dimensionless) were calculated using an open-flow, indirect calorimeter in the UTSW Metabolic Phenotyping Core. Two male C57BL/6 mice (10 to 13 weeks old, chow fed) were studied at a time such that the variables were recorded every 6 min for 2 hours. After 15 min acclimation in the test chambers, three recordings were performed before injection (−13 min, −7 min, and −1 min) from which the base line of VO2 and VCO2 were calculated for each mouse. One minute after the third recording, the mouse was taken out of the test chamber to receive an intraperitoneal injection of uridine (1 g/kg bodyweight) or PBS, such that the time 0 indicated when the injection was performed. This paired study was repeated for five times, and the results were expressed as mean ± SEM. The administration of uridine or PBS required opening the test chamber, which caused a sudden drop in the calculated rates of O2 consumption and CO2 production in the first recording, 5 min post injection for each mouse. Accurate determination of body composition in intact mouse was conducted using nuclear magnetic resonance (Bruckner Minispec mq10).
Tolerance tests
For glucose tolerance tests, glucose (0.25 g/ml in H2O) was gavaged (2.5 g/kg body weight) orally to male WT mice and age-matched ob/ob mice (15 to 35 weeks old) after fasted for 4 hours. To test parenteral effect of uridine, uridine (0.1 g/ml in PBS) was administrated via intraperitoneal injection(1 g/kg body weight) 15 min before glucose tolerance tests. To test enteral effect of uridine, uridine was dissolved together with glucose in H2O and the glucose-uridine solution (0.25 g/ml glucose and 0.1 g/ml uridine in H2O) was used for oral gavage at the same dose as glucose tolerance test.
N-(phosphonacetyl)-l-aspartate (PALA) and streptozotocin (STZ) treatments
PALA (100 mg/ml in 0.9% saline) was administrated via intraperitoneal injection at 62.5 mg/kg body weight to ob/ob (10 to 35 weeks old) or male WT mice (16 to 20 weeks old on HFD for 30 days). Food was removed after PALA injection up to 24 hours to monitor response in body temperature and plasma glucose. PALA was synthesized at UT Southwestern Medical Center as described (45) with purity greater than 95% as assessed by nuclear magnetic resonance. STZ (Sigma) was dissolved freshly in ice-cold sodium citrate buffer (0.1 M, pH 4.5) and administrated via intraperitoneal injection to male C57BL/6 mice (10 to 15 weeks old) at 120 µg/g body weight as described previously (46). Body weight and plasma glucose levels were monitored up to 7 weeks post STZ treatment, and the mice were euthanized for plasma, bile, and tissue collection after 24 hours of fasting.
RNA isolation and qPCR
Male C57BL/6 mice (15 to 25 weeks old) fed or fasted of 24 hours were euthanized and tissues were harvested and immediately frozen in liquid nitrogen. Total RNA was isolated using NucleoSpin RNA II mini columns (Macherey-Nagel) from 50 to 100 mg tissue and total RNA (100 ng to 1 µg) was used for reverse transcription with iScript kit (Bio-Rad). cDNA samples were then diluted tenfold with ddH2O and stored at −20°C for qPCR (Roche). Primers are shown in table S2. 18S RNA served as an internal control.
Statistical analysis
Results are reported as mean ± SEM. Statistical analysis of the data was performed with two-tailed Student t test or two-way ANOVA, as specified. Paired t test was performed for samples collected from the same mouse during a time course study. A P value < 0.05 was considered as significant. Statistical software consisted of Microsoft Excel and GraphPad Prism 6.04.
Supplementary Material
Acknowledgments
We thank D. Li, W. Tan (Division of Cardiology, UT Southwestern Medical Center), M.-Y. Wang (Touchstone Diabetes Center, UT Southwestern Medical Center), and S. Shah (Metabolic Phenotyping Core, UT Southwestern Medical Center) for excellent technical assistance with animal studies. Supported by American Diabetes Association postdoctoral fellowship 7-08-MN-53 (Y.D.), American Heart Association scientist development grant 14SDG18440002 (Z.V.W.), NIH grants R01-DK55758 and R01-DK099110 (P.E.S.), NIH grant P01DK088761 (P.E.S., J.K.E., J.D.H.), NIH grant R01-DK101578 (S.K.), Robert A. Welch Foundation grant I-1422 (J.D.B.), and American Heart Association grant 14SFRN20740000 and NIH grants HL-120732, HL-100401, and HL-126012 (J.A.H.).
Footnotes
Author contributions: Y.D. conceived the study, designed and performed the experiments, and analyzed data; Z.V.W. helped with uridine clearance study and tolerance tests; R.G. performed uridine analysis with HPLC-MS/MS; Y.A. performed the studies using the FAT-ATTAC mice; K.M.C. performed studies using the Agpat2 KO mice; C.Z. helped with gallbladder and tissue collection; Q.L. and J.D.B. synthesized PALA; J.Y. and S.K. provided the human samples and contributed to the discussion; J.K.E. and J.D.H. contributed to the discussion; J.A.H. contributed to the discussion and edited the manuscript; P.E.S. conceived the study; and Y.D. and P.E.S. wrote the manuscript with suggestions from all authors.
The authors have no conflicting financial interests.
REFERENCES AND NOTES
- 1.Connolly GP, Duley JA. Uridine and its nucleotides: Biological actions, therapeutic potentials. Trends Pharmacol. Sci. 1999;20:218–225. doi: 10.1016/S0165-6147(99)01298-5. [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto T, et al. Biochemistry of uridine in plasma. Clin. Chim. Acta. 2011;412:1712–1724. doi: 10.1016/j.cca.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Pizzorno G, et al. Homeostatic control of uridine and the role of uridine phosphorylase: A biological and clinical update. Biochim. Biophys. Acta. 2002;1587:133–144. doi: 10.1016/S0925-4439(02)00076-5. [DOI] [PubMed] [Google Scholar]
- 4.Gasser T, Moyer JD, Handschumacher RE. Novel single-pass exchange of circulating uridine in rat liver. Science. 1981;213:777–778. doi: 10.1126/science.7256279. [DOI] [PubMed] [Google Scholar]
- 5.Liu MP, Beigelman L, Levy E, Handschumacher RE, Pizzorno G. Discrete roles of hepatocytes and nonparenchymal cells in uridine catabolism as a component of its homeostasis. Am. J. Physiol. 1998;274:G1018–G1023. doi: 10.1152/ajpgi.1998.274.6.G1018. [DOI] [PubMed] [Google Scholar]
- 6.Connolly GP, Simmonds HA, Duley JA. Pyrimidines and CNS regulation. Trends Pharmacol. Sci. 1996;17:106–107. doi: 10.1016/0165-6147(96)20001-X. [DOI] [PubMed] [Google Scholar]
- 7.Urasaki Y, Pizzorno G, Le TT. Chronic uridine administration induces fatty liver and pre-diabetic conditions in mice. PLOS ONE. 2016;11:e0146994. doi: 10.1371/journal.pone.0146994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters GJ, et al. Uridine-induced hypothermia in mice and rats in relation to plasma and tissue levels of uridine and its metabolites. Cancer Chemother. Pharmacol. 1987;20:101–108. doi: 10.1007/BF00253962. [DOI] [PubMed] [Google Scholar]
- 9.Peters GJ, et al. Effect of pyrimidine nucleosides on body temperatures of man and rabbit in relation to pharmacokinetic data. Pharm. Res. 1987;4:113–119. doi: 10.1023/A:1016410817898. [DOI] [PubMed] [Google Scholar]
- 10.Steculorum SM, et al. Hypothalamic UDP increases in obesity and promotes feeding via P2Y6-dependent activation of AgRP neurons. Cell. 2015;162:1404–1417. doi: 10.1016/j.cell.2015.08.032. [DOI] [PubMed] [Google Scholar]
- 11.Lee SH, Paz-Filho G, Mastronardi C, Licinio J, Wong ML. Is increased antidepressant exposure a contributory factor to the obesity pandemic? Transl. Psychiatry. 2016;6:e759. doi: 10.1038/tp.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung RT, Shetty PS, James WP, Barrand MA, Callingham BA. Reduced thermogenesis in obesity. Nature. 1979;279:322–323. doi: 10.1038/279322a0. [DOI] [PubMed] [Google Scholar]
- 13.Gavrilova O, et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 1999;96:14623–14628. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins KD, Stark GR. Aspartate transcarbamylase. Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J. Biol. Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 15.Jones ME. Regulation of pyrimidine and arginine biosynthesis in mammals. Adv. Enzyme Regul. 1970;9:19–49. doi: 10.1016/S0065-2571(71)80036-5. [DOI] [PubMed] [Google Scholar]
- 16.Mayfield KP, Hong EJ, Carney KM, D’Alecy LG. Potential adaptations to acute hypoxia: Hct, stress proteins, and set point for temperature regulation. Am. J. Physiol. 1994;266:R1615–R1622. doi: 10.1152/ajpregu.1994.266.5.R1615. [DOI] [PubMed] [Google Scholar]
- 17.Simonson DC, DeFronzo RA. Indirect calorimetry: Methodological and interpretative problems. Am. J. Physiol. 1990;258:E399–E412. doi: 10.1152/ajpendo.1990.258.3.E399. [DOI] [PubMed] [Google Scholar]
- 18.Ramos-Jiménez A, et al. The respiratory exchange ratio is associated with fitness indicators both in trained and untrained men: A possible application for people with reduced exercise tolerance. Clin. Med. Circ. Respirat. Pulm. Med. 2008;2:1–9. doi: 10.4137/ccrpm.s449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JG, et al. Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat. Neurosci. 2014;17:908–910. doi: 10.1038/nn.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landsberg L, Young JB. Caloric intake and sympathetic nervous system activity. Implications for blood pressure regulation and thermogenesis. J. Clin. Hypertens. 1986;2:166–171. [PubMed] [Google Scholar]
- 21.Begriche K, et al. Beta-aminoisobutyric acid prevents diet-induced obesity in mice with partial leptin deficiency. Obesity. 2008;16:2053–2067. doi: 10.1038/oby.2008.337. [DOI] [PubMed] [Google Scholar]
- 22.Moyer JD, Oliver JT, Handschumacher RE. Salvage of circulating pyrimidine nucleosides in the rat. Cancer Res. 1981;41:3010–3017. [PubMed] [Google Scholar]
- 23.Tseng J, Barelkovski J, Gurpide E. Rates of formation of blood-borne uridine and cytidine in dogs. Am. J. Physiol. 1971;221:869–878. doi: 10.1152/ajplegacy.1971.221.3.869. [DOI] [PubMed] [Google Scholar]
- 24.Grem JL, King SA, O’Dwyer PJ, Leyland-Jones B. Biochemistry and clinical activity of N-(phosphonacetyl)-L-aspartate: A review. Cancer Res. 1988;48:4441–4454. [PubMed] [Google Scholar]
- 25.Pajvani UB, et al. Fat apoptosis through targeted activation of caspase 8: A new mouse model of inducible and reversible lipoatrophy. Nat. Med. 2005;11:797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- 26.Cortés VA, et al. Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 2009;9:165–176. doi: 10.1016/.jcmet.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenny GP, Jay O. Thermometry, calorimetry, and mean body temperature during heat stress. Compr. Physiol. 2013;3:1689–1719. doi: 10.1002/cphy.c130011. [DOI] [PubMed] [Google Scholar]
- 28.Landsberg L, Young JB. Diet-induced changes in sympathoadrenal activity: Implications for thermogenesis. Life Sci. 1981;28:1801–1819. doi: 10.1016/0024-3205(81)90352-0. [DOI] [PubMed] [Google Scholar]
- 29.Boyer JL. Bile formation and secretion. Compr. Physiol. 2013;3:1035–1078. doi: 10.1002/cphy.c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol. Med. 2015;21:702–714. doi: 10.1016/j.molmed.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Mackinnon AM, Deller DJ. Purine nucleotide biosynthesis in gastrointestinal mucosa. Biochim. Biophys. Acta. 1973;319:1–4. doi: 10.1016/0005-2787(73)90034-8. [DOI] [PubMed] [Google Scholar]
- 32.He Y, Sanderson IR, Walker WA. Uptake, transport and metabolism of exogenous nucleosides in intestinal epithelial cell cultures. J. Nutr. 1994;124:1942–1949. doi: 10.1093/jn/124.10.1942. [DOI] [PubMed] [Google Scholar]
- 33.Cass CE, Baldwin SA, Young J. xPharm: The Comprehensive Pharmacology Reference. Elsevier; 2011. pp. 1–4. [DOI] [Google Scholar]
- 34.Molina-Arcas M, Pastor-Anglada M. In: Pharmacogenomics of Human Drug Transporters: Clinical Impacts. Ishikawa T, Kim RB, König J, editors. Wiley; 2013. pp. 243–270. [DOI] [Google Scholar]
- 35.Pinilla-Macua I, Casado FJ, Pastor-Anglada M. Structural determinants for rCNT2 sorting to the plasma membrane of polarized and non-polarized cells. Biochem. J. 2012;442:517–525. doi: 10.1042/BJ20110605. [DOI] [PubMed] [Google Scholar]
- 36.Pinilla-Macua I, Claudio-Montero A, Pastor-Anglada M. rCNT2 extracellular cysteines, Cys615 and Cys649, are important for maturation and sorting to the plasma membrane. FEBS Lett. 2014;588:4382–4389. doi: 10.1016/j.febslet.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 37.Errasti-Murugarren E, Molina-Arcas M, Casado FJ, Pastor-Anglada M. A splice variant of the SLC28A3 gene encodes a novel human concentrative nucleoside transporter-3 (hCNT3) protein localized in the endoplasmic reticulum. FASEB J. 2009;23:172–182. doi: 10.1096/fj.08-113902. [DOI] [PubMed] [Google Scholar]
- 38.Domingo P, et al. Uridine metabolism in HIV-1-infected patients: Effect of infection, of antiretroviral therapy and of HIV-1/ART-associated lipodystrophy syndrome. PLOS ONE. 2010;5:e13896. doi: 10.1371/journal.pone.0013896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L. A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature. 1998;393:684–688. doi: 10.1038/31474. [DOI] [PubMed] [Google Scholar]
- 40.Froy O. Metabolism and circadian rhythms—implications for obesity. Endocr. Rev. 2010;31:1–24. doi: 10.1210/er.2009-0014. [DOI] [PubMed] [Google Scholar]
- 41.Eckel-Mahan KL, et al. Coordination of the transcriptome and metabolome by the circadian clock. Proc. Natl. Acad. Sci. U.S.A. 2012;109:5541–5546. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larder R, O’Rahilly S. Shedding pounds after going under the knife: Guts over glory-why diets fail. Nat. Med. 2012;18:666–667. doi: 10.1038/nm.2747. [DOI] [PubMed] [Google Scholar]
- 43.Kenny GP, Sigal RJ, McGinn R. Body temperature regulation in diabetes. Temperature. 2016;3:119–145. doi: 10.1080/23328940.2015.1131506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang ZV, et al. PANIC-ATTAC: A mouse model for inducible and reversible beta-cell ablation. Diabetes. 2008;57:2137–2148. doi: 10.2337/db07-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris AD, Cordi AA. A new, efficient, two step procedure for the preparation of the antineoplastic agent sparfosic acid. Synth. Commun. 1997;27:1259–1266. doi: 10.1080/00397919708003363. [DOI] [Google Scholar]
- 46.Ye R, et al. Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes β-cell regeneration. eLife. 2014;3 doi: 10.7554/eLife.03851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshino J, et al. Diurnal variation in insulin sensitivity of glucose metabolism is associated with diurnal variations in whole-body and cellular fatty acid metabolism in metabolically normal women. J. Clin. Endocrinol. Metab. 2014;99:E1666–E1670. doi: 10.1210/jc.2014-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.