

Abstract
Antibody-mediated rejection (ABMR) is implicated in 45% of renal allograft failure and 57% of late allograft dysfunction. Peritubular capillary C4d is a specific but insensitive marker of ABMR. The 2013 Banff Conference ABMR revised criteria included C4d-negative ABMR with evidence of endothelial-antibody interaction. We hypothesized that endothelial activation and lymphangiogenesis are increased with C4d-negative ABMR, and correlate with intra-graft T-regulatory cells (Tregs) and T-helper 17 (Th17).
Seventy-four renal transplant biopsies were selected to include: a) ABMR with C4d Banff scores ≥2 (n=35); b) variable microvascular injury (MI) and C4d score <2 (n=24); c) variable MI and C4d score=0 (n=15). Controls included normal pre-implantation donor kidneys (n=5). Immunohistochemistry for endothelial activation [P- and E- selectins (SEL)], lymphangiogenesis (D2-40), Tregs (FOXP3), and Th17 (STAT3) was performed. Microvessel and inflammatory infiltrate density was assessed morphometrically in interstitium and peritubular capillaries.
All transplants had significantly higher microvessel and lymph vessel density compared to normal. Increased expression of markers of endothelial activation predicted transplant glomerulopathy (P-SEL, p=0.003). Increased P-SEL and D2-40 were associated with longer interval from transplant to biopsy (p=0.005). All three markers were associated with increased interstitial fibrosis, tubular atrophy and graft failure (P-SEL, p<0.001; E-SEL, p=0.0011; D2-40, p=0.012). There was no association with the intragraft FOXP3/STAT3 ratio.
We conclude that endothelial activation and lymphangiogenesis could represent a late response to injury leading to fibrosis and progression of kidney damage, and are independent of the intragraft FOXP3/STAT3 ratio. Our findings support the therapeutic potential of specifically targeting endothelial activation.
Keywords: Kidney transplant, humoral rejection, microvascular injury, endothelial activation, selectin, transplant biopsy
1. INTRODUCTION
Antibody-mediated rejection (ABMR) has been implicated in 45% of renal allograft failure [1] and in 57% of new onset late allograft dysfunction [2]. Endothelial injury is one of the diagnostic pathologic features of ABMR. Chronic ABMR (CAMR) is the sequela of repeated/subclinical ABMR episodes with persistent endothelial injury and repair, leading to chronic endothelial remodeling with lamellation and deposition of newly formed basement membranes in peritubular capillaries and glomeruli, causing allograft dysfunction. Several mechanisms may contribute to endothelial injury during ABMR: complement-dependent antibody binding to endothelial surface antigens, endothelial activation by antibody alone [3], and complement-independent mechanisms mediated by natural killer cells [4]. Complement-dependent endothelial injury is detected in renal biopsies by C4d deposition in peritubular capillaries [5]. However, C4d has low sensitivity in routine biopsies [6] and in ABO donor group incompatible kidney transplants. Recent studies have shown that increased expression of endothelial cell transcripts predicted graft loss with more sensitivity than C4d alone [7, 8].
Activated endothelial cells increase expression of cell adhesion molecules. Selectins, transmembrane glycoproteins that are part of the cell adhesion molecule superfamily, mediate adhesion and rolling of leukocytes to the activated endothelium, the first step in leukocyte recruitment, through the mechanisms of chemokine-activated adhesion and extravasation. P-selectin (P-SEL) is stored in α-granules of platelets and in Weibel–Palade bodies of endothelial cells, and is translocated to the cell surface of activated endothelial cells and platelets. E-selectin (E-SEL) is not expressed under baseline conditions, except in skin microvessels, but is rapidly induced by inflammatory cytokines.
An additional mechanism which may contribute to ABMR is lymphatic neoangiogenesis. Lymph vessel density, assessed by D2-40 immunohistochemistry and morphometric analysis, is increased in areas with cellular infiltrates in renal biopsies with acute cellular rejection [9]. Lymphangiogenesis enhanced immune responses in corneal transplant rejection [10], and inhibition of lymphangiogenesis prolonged allograft survival after islet transplantation [11].
However, whether post-transplant lymphangiogenesis is beneficial or detrimental to the graft or whether this contributes to ABMR is still a matter of debate.
The aim of our study was to evaluate pathogenic markers of endothelial activation and lymphangiogenesis during ABMR and CAMR, and to correlate such markers with the progression of renal damage following humoral rejection. We hypothesized that upregulation of these markers is associated with pathophysiologic mechanisms of rejection, and with specific shifts in the intra-graft T-helper (Th) phenotype [regulatory T cells (Tregs) vs. Th17]. Further, we evaluated the ability of these markers to predict graft loss.
2. MATERIAL AND METHODS
Renal allograft biopsies performed for cause at Vanderbilt University Medical Center from 2007 to 2013 were retrospectively reviewed and cases with available tissue for immunohistochemical analysis, minimal glomerular number of 3 and availability of donor specific antibody (DSA) at time of biopsy were selected. Allograft biopsies were performed under ultrasound-guidance using a 16-gauge automated biopsy instrument. Tissue was examined at the biopsy site under a dissecting microscope, and allocated for light microscopy (LM), immunofluorescence (IF) and electron microscopy (EM) studies. Renal biopsies were processed by standard techniques for LM with multiple serial sections stained with hematoxylin and eosin, periodic acid-Schiff reagent, and PAS-methenamine silver, IF (stained for IgG, IgM, IgA, C3, C1q; Dako, Carpentaria, CA, and C4d, ABD Serotec-MorphoSys, Germany), and EM. C4d was performed by IF on frozen sections. The intensity of immunofluorescence staining was semiquantitatively graded on a scale from 0 to 3+ as follows: negative, 0; mild, 1+; moderate, 2+; and severe, 3+, with specification of staining localization. EM was done on all initial transplant biopsies (and as indicated on repeat biopsies) with available tissue and examined by a Philips FEI Morgagni transmission electron microscope. All biopsies were interpreted by experienced renal pathologists.
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded tissue for endothelial activation markers (P- and E- Selectins, Clone C34, Leica-Novacastra, Buffalo Grove, IL; Clone 16G4, Leica-Novacastra, Buffalo Grove, IL) lymphangiogenesis (D2-40, Clone D2-40, DAKO, Carpentaria, CA), Tregs [Forkhead box P3 (FOXP3), clone 236A/E7, eBiosciences, San Diego, CA)], TH17 [Signal transducer and activators of transcription (STAT3), clone 9D8, Abcam, Cambridge, MA)] and CD4 (Clone NCL-L-CD4-1F6, Leica Novacastra, Buffalo Grove, IL), on a Leica-Bond immunostainer. Sections for FOXP3 and STAT3 were sequentially stained with CD4 to confirm T-helper differentiation. Positive controls included human tonsil. Normal kidneys were used as negative controls. Controls stained as expected.
Microvessel and inflammatory infiltrate density of the cortical interstitium and peritubular capillaries were assessed morphometrically with ScanScope CS, Aperio (v11.2.0.780). Specifically, microvessel density was calculated using the microvessel density algorithm, and expressed as number of vessels per area (number of vessels/mm2). Tregs and Th17 were quantitated by the ratio of the positive cells to the biopsy area. Analysis was limited to peritubular capillaries for E-SEL and P-SEL. For D2-40, lymph vessels near interlobular arteries were not included in the analysis, as these are present in normal kidney cortex [9, 12].
Acute allograft dysfunction was defined as a rise in creatinine of ≥0.4 mg/dl over the baseline occurring during a one-month period. Delayed graft function (DGF) was defined as hemodialysis requirement in the first week post-transplant. Proteinuria was evaluated by spot urine protein/creatinine ratio (uPCR) (mg/mg), and a cut-off of ≥3.5 was used for definition of nephrotic-range proteinuria. Histologic findings were graded at time of biopsy by the Collaborative Clinical Trials in Transplantation (CCTT) classification [13], and subsequently reviewed and scored based on the 2007 Banff classification [14, 15] with 2013 update. Medical records were reviewed for demographic data, clinical presentation, parameters of renal function, and outcome.
Statistical analysis was performed using R version 3.0.2 (2013-09-25). Categorical variables were described with relative frequencies and percentages, and Chi-square test was used to analyze their relationship. Continuous variables were reported as mean±SD and/or median, and analyzed with Mann-Whitney U or Kruskal-Wallis tests as applicable. Survival curves were calculated by the Kaplan-Meier method and compared with log-rank test. Multivariable Cox proportional hazards models were used to evaluate the effect of risk factors on renal survival. Two-tailed p-values less than 0.05 were considered statistically significant. The study was approved by the Vanderbilt Institutional Review Board.
3. RESULTS
3.1. Patient characteristics
Seventy-four patients were selected from retrospective review of the renal pathology tissue archive at Vanderbilt University Medical Center from 2007 to 2013. Cases were included in the following groups based on C4d and DSA status: 1) Cases with diagnostic ABMR, defined by the presence of microvascular injury, positive donor-specific antibody (DSA), and C4d Banff score ≥2 (C4d+DSA+; n=35); 2) Cases with variable microvascular injury, positive DSA and C4d Banff score 0-1 (C4d−DSA+, n=24; 3) Cases with variable microvascular injury, negative DSA and C4d Banff score 0 (control, n=15). Pre-implantation wedge kidney biopsies of kidney donors (NL, n=5) were selected as normal controls. Additional biopsy diagnoses were recorded but not used for group classification. The clinical and demographic characteristics are shown in Table 1. Highly sensitized patients were present in the C4d+DSA+ group (n=5), and in the C4d−DSA+ group (n=2), and all received pre-operative intravenous immunoglobulin (IVIG). No highly sensitized patients were present in controls. All cases were donor blood group-compatible. In the C4d+DSA+ group, at biopsy, 22 patients (63%) were receiving triple immunosuppression (calcineurin inhibitor, mycophenolate mofetil and prednisone, or calcineurin inhibitor, sirolimus and prednisone), and 13 (37%) were on double immunosuppression (calcineurin inhibitor/prednisone, calcineurin inhibitor/mycophenolate mofetil, rapamycin/prednisone). In the C4d−DSA+ group, 20 (83%) were on triple immunosuppression and 4 (17%) were on double immunosuppression, while in the control group, 8 (53%) were on triple immunosuppression, and 7 (47%) were on double immunosuppression. The histopathologic findings at the time of renal biopsy are presented in Table 2. Briefly, there was a significant difference in the proportions of cases of TCMR, with the −C4d+DSA group having the highest proportion (Pearson’s Chi-square, p=0.0011). As expected by study group definitions, the differences in proportions were also significant for ABMR, CAMR, IFTA and Banff ptc and C4d scores across the three groups.
Table 1.
Clinical characteristics at time of renal biopsy
| C4d+DSA+ (n=35) |
C4d−DSA+ (n=24) |
Controls (n=15) |
|
|---|---|---|---|
| Age (years)(range) | 41(28.0-48.5) | 36.5 (27.5-46.2) | 39 (30.5-50.5) |
| Gender (M/F) (%) | 20/15 (57/43) | 18/6 (75/25) | 9/6 (60/40) |
| Race (C/AA/H) (%) | 19/15/1 (54/43/3) | 12/12/0 (50/50/0) | 12/3/0 (80/20/0) |
| Donor type | |||
| D/LR/LU (%) | 15/8/12a (43/23/34) | 14/3/7a (58/12/29) | 8/7/0a (53/47/0) |
| HLA compatibility (Incompatible/compatible) | 1/26 | 2/26 | 0.6 |
| HLA mismatches | |||
| A/B/DR≤3 | 16 | 16 | 9 |
| A/B/DR>3 | 19 | 8 | 6 |
| Time between transplant and biopsy (days) | 583 | 410 | 377 |
| Indication for biopsy [n (%)] | |||
| Increased creatinine | 21 (60) | 20 (84) | 10 (66) |
| Proteinuria | 1 (3) | 1 (4) | 1(7) |
| Increased creatinine and proteinuria | 13 (37) | 3 (12) | 3 (20) |
| DGF | 0 | 0 | 1(7) |
| Creatinine (mg/dl) | 4.49 | 5.78 | 4.10 |
| Proteinuria [n (%)] | |||
| Negative | 4 (11) | 2(8) | 5(33) |
| Sub-nephrotic | 25(72) | 20(84) | 8(53) |
| Nephrotic | 4 (11) | 0 | 1(7) |
| Not available | 2 (6) | 2 (8) | 1(7) |
M= male; F= female; C= Caucasian; AA= African-American; H=Hispanic; D=deceased donor; LR=living related donor; LU=living unrelated donor; DGF=delayed graft function.
p=0.037.
Table 2.
Histopathologic findings
| C4d+DSA+ (n=35) |
C4d−DSA+ (n=24) |
Controls (n=15) |
P-value | |
|---|---|---|---|---|
| Pathologic diagnosis [n (%)] | ||||
| TCMR or Borderline | 16 (46) | 21 (88) | 11 (73) | <0.003a |
| ABMR | 34(97) | 19 (79) | 0 (0) | <0.001b |
| CAMR | 13 (37) | 11 (46) | 0 (0) | 0.009b |
| CNIT | 1 (3) | 1 (4) | 1 (7) | 0.822 |
| DN | 2 (6) | 3 (12) | 1(7) | 0.627 |
| ATI | 4 (11) | 1 (4) | 4 (27) | 0.11 |
| PVN | 2 (6) | 0 (0) | 0 (0) | 0.318 |
| IFTA | 0 (0) | 0 (0) | 4 (27) | 0.001b |
| GN | 6 (17) | 3 (12) | 2 (13) | 0.87 |
| Banff Scores [n (%)] | ||||
| g | 0.437 | |||
| 0 | 6 (17) | 4 (17) | 6 (40) | |
| 1 | 10 (29) | 6 (25) | 5 (33) | |
| 2 | 9 (25) | 7 (29) | 3 (20) | |
| 3 | 10 (29) | 7 (29) | 1(7) | |
| i | 0.525 | |||
| 0 | 20 (57) | 9 (38) | 7 (47) | |
| 1 | 2 (6) | 3 (12) | 3 (20) | |
| 2 | 6 (17) | 4 (17) | 3 (20) | |
| 3 | 7 (20) | 8 (33) | 2 (13) | |
| t | 0.626 | |||
| 0 | 10 (29) | 4 (17) | 3 (20) | |
| 1 | 16 (46) | 10 (42) | 6 (40) | |
| 2 | 8 (23) | 9 (38) | 4 (27) | |
| 3 | 1 (3) | 1 (4) | 2 (13) | |
| v | 0.290 | |||
| 0 | 22 (63) | 17 (71) | 10 (67) | |
| 1 | 12 (34) | 3 (12) | 4 (27) | |
| 2 | 0 | 2 (8) | 1 (7) | |
| 3 | 1 (3) | 2 (8) | 0 | |
| cg | 0.210 | |||
| 0 | 19 (54) | 12 (50) | 8 (53) | |
| 1a | 6 (17) | 2 (8) | 5 (33) | |
| 1b | 10 (29) | 10 (42) | 2 (13) | |
| ci | 0.914 | |||
| 0 | 16 (46) | 10 (42) | 6 (40) | |
| 1 | 12 (34) | 8 (33) | 5 (33) | |
| 2 | 6 (17) | 6 (25) | 3 (20) | |
| 3 | 1 (3) | 0 | 1 (7) | |
| ct | 0.942 | |||
| 0 | 15 (43) | 10 (42) | 6 (40) | |
| 1 | 12 (34) | 8 (33) | 5 (33) | |
| 2 | 7 (20) | 6 (25) | 3 (20) | |
| 3 | 1 (3) | 0 | 1 (7) | |
| cv | 0.196 | |||
| 0 | 23 (66) | 16 (67) | 8 (53) | |
| 1 | 9 (26) | 6 (25) | 3 (20) | |
| 2 | 3 (9) | 2 (8) | 2 (13) | |
| 3 | 0 | 0 | 2 (13) | |
| ah | 0.431 | |||
| 0 | 20 (57) | 13 (54) | 8 (53) | |
| 1 | 14 (40) | 10 (42) | 5 (33) | |
| 2 | 0 | 1 (4) | 2 (13) | |
| 3 | 1 (3) | 0 | 0 | |
| mm | 0.08 | |||
| 0 | 23 (66) | 13 (54) | 12 (80) | |
| 1 | 9 (26) | 9 (38) | 1 (7) | |
| 2 | 3 (9) | 0 | 2 (13) | |
| 3 | 0 | 2 (8) | 0 | |
| ptc | 0.035b | |||
| 0 | 6 (17) | 2 (8) | 8 (53) | |
| 1 | 12 (34) | 12 (50) | 4 (27) | |
| 2 | 15 (43) | 9 (38) | 2 (13) | |
| 3 | 2 (6) | 1 (4) | 1 (7) | |
| ti | 0.350 | |||
| 0 | 10 (29) | 1 (4) | 4 (27) | |
| 1 | 8 (23) | 6 (25) | 4 (27) | |
| 2 | 7 (20) | 7 (29) | 4 (27) | |
| 3 | 10 (29) | 10 (42) | 3 (20) | |
| C4d | <0.001b | |||
| 0 | 0 | 20 (83) | 13 (87) | |
| 1 | 0 | 4 (17) | 2 (13) | |
| 2 | 8 (23) | 0 | 0 | |
| 3 | 27 (77) | 0 | 0 |
TCMR=T-cell mediated rejection; ABMR, antibody-mediated rejection; CAMR, chronic antibody-mediated rejection; CNIT, calcineurin-inhibitor toxicity; DN, diabetic nephropathy; ATI, acute tubular injury; PVN, polyomavirus nephropathy; IFTA, interstitial fibrosis-tubular atrophy; GN, glomerulonephritis.
There is a significant difference amongst groups in the proportions of cases of TCMR with the −C4d+DSA group showing the highest proportion (Pearson’s Chi-square, p= 0.0011).
Statistically different, as expected by study group definition.
3.2. Endothelial activation markers and lymphangiogenesis
E-SEL and P-SEL expressions were mostly limited to endothelium of peritubular capillaries, arteries and veins. There was no expression in glomerular capillary loops (Figure 1). C4d+DSA+, C4d−DSA+ and control groups had significantly higher microvessel density for P-SEL (p=0.035; p=0.0086; p=0.0036, respectively), and E-SEL (p=0.00040; p=0.00055; p=0.0010, respectively) compared to NL. However, there was no significant difference in expression of either P-SEL or E-SEL among C4d+DSA+, C4d−DSA+ and control (Figures 2a and 2b). Arteries and veins showed an increased trend in the proportion of E-SEL and P-SEL expression in all groups compared with NL, but this was not statistically significantly different (arteries: P-SEL, p=0.65; E-SEL, p=0.42; veins: P-SEL, p=0.10; E-SEL, p=0.055; all values vs. NL).
Figure 1.
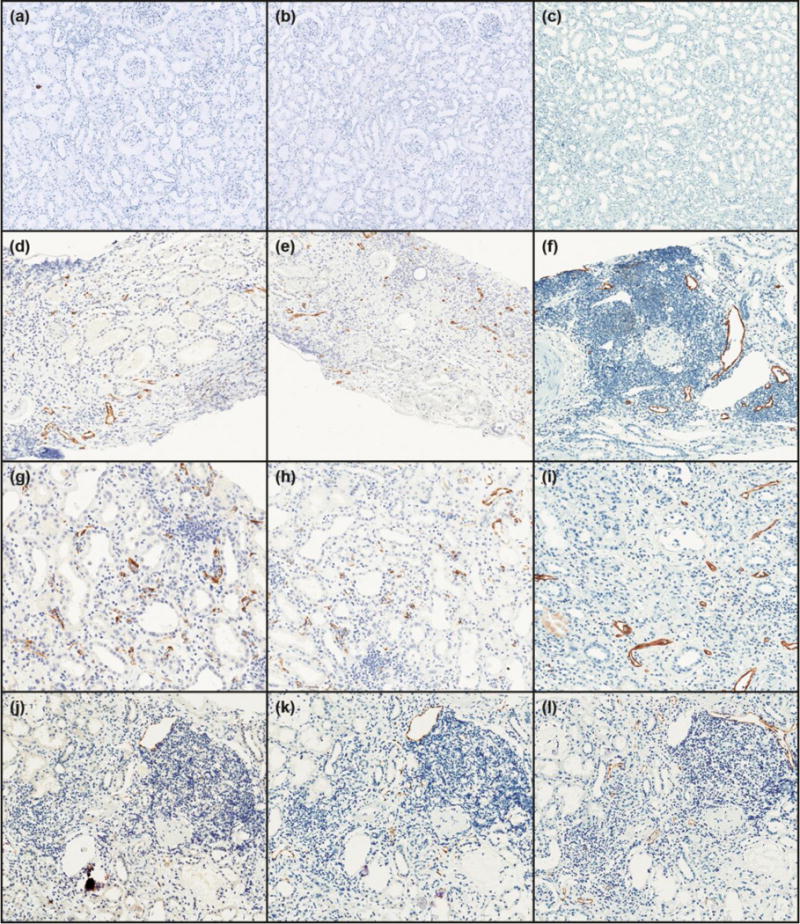
Expression of markers of endothelial activation and lymphangiogenesis. E-selectin, P-selectin and D2-40 in normal kidneys (a, b, and c, respectively; ×100), C4d+DSA+ (d, e, and f; ×100), C4d−DSA+ (g, h, and i; ×200), and control (C4d−, DSA−; CTR) (j, k and 1; ×200).
Figure 2.
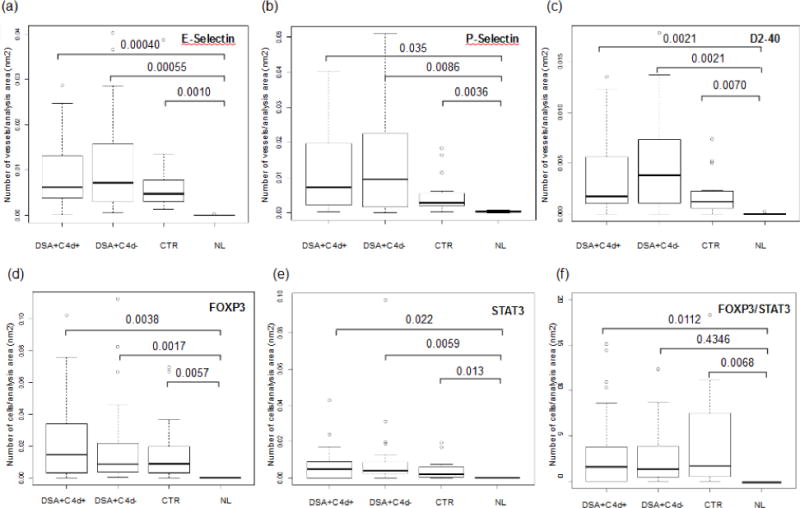
Expression of markers of endothelial activation and lymphangiogenesis (a, b and c) and T-helper subsets (d, e, and f) in different study groups (DSA+, C4d+, DSA+, C4d−), control (C4d−, DSA−; CTR) and normal kidneys (NL).
Lymph vessel density, evaluated by D2-40, was significantly higher in C4d+DSA+ (p=0.0021), C4d−DSA+ (p=0.0021) and control (p=0.0070) compared to NL. There was no significant difference in D2-40 expression among C4d+DSA+, C4d−DSA+ and control (Figure 2c). Lymph vessel density was higher around foci of interstitial lymphocytic infiltrate, as previously observed (Figure 1).
P-SEL, E-SEL and D2-40 expression was compared to demographic, clinical and histologic characteristics. Higher E-SEL and P-SEL expressions were significantly associated with African American race (E-SEL, p=0.014; P-SEL, p=0.010), and increased creatinine at biopsy (E-SEL, p=0.049; P-SEL, p=0.008). Higher E-SEL expression was significantly associated with deceased donor kidney transplant (p=0.038), while P-SEL and D2-40 were associated with longer interval from transplant to biopsy (p=0.005) (Table 3). There was no association for any of the three vascular markers with age, gender, number of human leukocyte antigen (HLA) mismatches, C4d and Banff microcirculation injury scores (i.e. g+ptc score), or DSA. However, increased expression of all three markers was associated with presence of interstitial fibrosis and tubular atrophy, compared to those with less expression and absence of fibrosis/atrophy (Banff scores ci and ct; P-SEL, p=0.007; E-SEL, p<0.001; D2-40, p=0.03) (Table 3) and later graft failure (P-SEL, p=0.000063; E-SEL, p=0.0011; D2-40, p=0.012) with P-SEL, E-SEL having the strongest association. Further, P-SEL was significantly associated with the presence of transplant glomerulopathy, a marker of CAMR (Table 3).
Table 3.
Comparison of E-SEL, P-SEL and D2-40 expression by histologic characteristics
| N | E-Sel (Microvessel density/mm2) |
P-Sel (Microvessel density/mm2) |
D2-40 (Microvessel density/mm2) |
|
|---|---|---|---|---|
| Microvascular inflammation (g+ptc Banff score) | ||||
| <2 | 21 | 0.0047 | 0.0029 | 0.0016 |
| ≥2 | 53 | 0.0069 | 0.0072 | 0.0032 |
| Interstitial inflammation and tubulitis | ||||
| 0 | 36 | 0.0059 | 0.0050 | 0.0017 |
| ≥ 1 | 38 | 0.0063 | 0.0094 | 0.0025 |
| Transplant glomerulopathy | ||||
| 0 | 39 | 0.0048 | 0.0038a | 0.0017 |
| 1a or 1b | 35 | 0.0080 | 0.012a | 0.0026 |
| Endarteritis | ||||
| 0 | 49 | 0.0067 | 0.0073 | 0.0023 |
| ≥ 1 | 25 | 0.0057 | 0.0031 | 0.0017 |
| Arteriosclerosis | ||||
| 0 | 47 | 0.0062 | 0.0062 | 0.0020 |
| ≥ 1 | 27 | 0.0068 | 0.0095 | 0.0026 |
| Interstitial fibrosis (ci)/tubular atrophy (ct) scores | ||||
| 0 | 32 | 0.0039b | 0.023c | 0.0014d |
| ≥ 1 | 42 | 0.0083b | 0.012c | 0.0037d |
| C4d Banff score | ||||
| 0-1 | 39 | 0.0062 | 0.0049 | 0.0021 |
| 2-3 | 35 | 0.0062 | 0.0072 | 0.0017 |
P-selectin, p=0.003
E-selectin, p<0.001
P-selectin, p=0.007
D2-40, p=0.03
3.3. Intragraft T-helper subsets
FOXP3 and STAT3 were significantly elevated in all groups compared to NL (FOXP3, p=0.0038; p=0.0017; p=0.0057, and STAT3, p=0.022; p=0.0059; p=0.013, C4d+DSA+, C4d−DSA+, and control vs NL, respectively) (Figure 2d and 2e), while their distribution was similar among the study groups. The FOXP3/STAT3 ratio was significantly increased in C4d+DSA+ and control compared to NL (p=0.011 and 0.0068, respectively) (Figure 2f), and only numerically increased in C4d−DSA+ compared to controls (p=0.43). Increased E-SEL expression was associated with increased FOXP3 (p=0.033) and STAT3 (p=0.0060). Expression of P-SEL was associated with STAT3 (p=0.026) but not with FOXP3 (p=0.089). Expression of P-SEL, E-SEL and D2-40 was not associated with any change in the intragraft FOXP3/STAT ratio. When adjusting for creatinine, donor type and Banff chronicity scores (ci and ct), increased P-SEL, E-SEL and D2-40 were not significantly associated with increased graft failure (Cox regression, HR= 1.029). Although there was a trend for decreased graft survival in the C4d+DSA+ cases compared to control and C4d−DSA+ cases, the difference was not statistically significant (Figure 3) (mean follow-up 30 months, range 2-88, log-rank p=0.136).
Figure 3.

Kaplan-Meier analysis of renal survival by group.
4. DISCUSSION
After the first three decades of kidney transplantation and the subsequent development of powerful and effective anti-T cell immunosuppressive agents, focus has shifted to the role of alloantibodies and humoral rejection in long term renal allograft survival. Class I or II antibodies directed to HLA antigens expressed on the endothelial cell surface and capable of complement fixation and activation are found in a substantial fraction of renal allograft recipients [16] and are associated with graft loss. Endothelial cells can also directly interact with allogeneic T-cells through major histocompatibility complex (MHC) antigens and associated surface co-stimulatory and adhesion proteins [17]. Microvascular injury and peritubular capillary deposition of C4d, a degradation product of complement factor C4, are markers of humoral response [5] and correlate with the presence of DSA [18, 19]. A recent microarray study by Sis et al. [8] showed altered endothelial gene expression in antibody-mediated rejection. The endothelium is thus the key in alloantibody-mediated injury, and persistent/recurrent, smoldering antibody-mediated injury has been associated with transplant glomerulopathy. This injury manifests with endothelial remodeling, lamellation and deposition of newly formed basement membrane in peritubular capillaries and glomeruli, leading to progressive graft loss.
In this study we evaluated the role of markers of endothelial activation and lymphangiogenesis, E-selectin, P-selectin, and D2-40, in progression of renal damage and prediction of graft loss in renal biopsies with ABMR and positive DSA with and without C4d positivity, and compared to cases without DSA or C4d deposition. We showed that in biopsies done for cause, endothelial activation and lymphatic neo-formation were significantly increased in cases with ABMR with and without C4d, and even in cases without DSA, compared to normal controls. E- and P-selectin were expressed largely in peritubular capillaries, arteries and veins and not in glomerular capillaries, while D2-40 predominantly marked newly formed lymphatic vessels clustering around lymphoid aggregates, as previously demonstrated [9]. This increase was not specifically associated with features of acute/active microvascular injury, such as microvascular inflammation, DSA or C4d positivity, and did not differ among the study groups. Lymphangiogenesis was associated with longer interval from transplant to biopsy and more interstitial fibrosis and tubular atrophy, and/or with transplant glomerulopathy, both features of established and irreversible disease. Further, expression of markers of endothelial activation was associated with subsequent graft loss.
Based on these results, we speculate that endothelial activation represents a late response to injury leading to fibrosis and progression of kidney damage. Alternatively, endothelial activation and lymphangiogenesis could represent a non-specific or compensatory response to ensuing fibrosis and end-organ damage in a final attempt to reverse loss of graft function.
Although lymphangiogenesis was mostly localized around lymphoid clusters, no association was seen with Banff scores of interstitial inflammation and tubulitis in non-scarred parenchyma (Banff “i” and “t” scores). Thus, lymphangiogenesis was not associated with active T-cell mediated rejection, but rather was part of late damage.
An important role of selectins in the pathophysiology of graft rejection has been suggested by previous studies in animal models. The degree of arterial intimal thickening significantly correlated with endothelial expression of P-selectin in a rat model of cardiac allograft vasculopathy, the morphologic correlate of chronic allograft rejection [20]. Grafts from mice lacking E-, P-, and L- selectins showed longer survival compared with wild-type grafts, and less chronic vascular rejection in coronary arteries [21]. In an established animal model of cardiac allograft vasculopathy and chronic rejection, mice lacking fucosyltransferase-VII, an enzyme essential for biosynthesis of selectin ligands, exhibited increased long-term graft survival with minimal vasculopathy compared with wild-type controls [22]. In a rat kidney allograft model, the selectin inhibitor bimosiamose significantly prolonged kidney graft survival with associated decreased infiltration of CD4+ and CD8+ lymphocytes and macrophages, reduced mRNA levels for interleukin (IL)-1β, IL-3, IL-6, IL-10, tumor necrosis factor (TNF)-α, interferon (IFN)-γ and intragraft expression of P-selectin glycoprotein ligand-1, CX3CL1, CCL19, CCL20, and CCL2 [23]. These findings are in agreement with the results of the current study, and support a significant role of selectins in the development of chronic allograft rejection.
CD4+ CD25+ FOXP3+ Tregs and Th17 are T-helper subsets involved in the mechanisms of tolerance and inflammation in the graft. Tregs and Th17 can develop from the same precursor CD4+CD25− cells. The presence of IL-6 in combination with transforming growth factor-beta preferentially induces Th17 development [24]. In addition, Tregs can undergo conversion to Th17 [25], suggesting that the balance of pro- and anti-rejection conditions may depend on the cytokine microenvironment within the graft. Our study shows that FOXP3 and STAT3 were significantly elevated in all groups compared to normal, and were significantly associated with increased E-selectin, while P-selectin was associated with STAT3 but not with FOXP3 infiltrating cells. The FOXP3/STAT3 ratio was significantly increased in DSA+C4D+ and control transplant without C4d or DSA compared to normal, but was not significantly increased in C4d−DSA+ compared to normal. However, increased expression of endothelial activation markers and lymphangiogenesis was independent of the intragraft FOXP3/STAT3 ratio. Endothelial cells can directly activate CD4+ T-helper subsets through the expression of class I and II MHC molecules, which in turn are recognized by T-cell receptor and CD4 co-receptor [26–28]. Endothelial cells also express intercellular adhesion molecule (ICAM)-1 (CD54) and lymphocyte function-associated antigen (LFA)-3 (CD58), which bind to LFA1 (CD11a/18) and LFA2 (CD2) on T-cells [29], inducing proliferation, and trans-endothelial migration. Further, under inflammatory conditions, human microvascular endothelial cells are able to selectively amplify both Th17 and Tregs, the former by an IL-6/STAT-3-dependent mechanism, the latter via contact–CD54–dependent interaction [30]. These findings corroborate the results of our study, highlighting the central role of endothelial cells in initiating and/or perpetuating mechanisms of alloimmune inflammatory response during rejection.
A rather intriguing finding emerging from this study is the presence of endothelial activation in the C4d–DSA– (control) group. The significance of this finding in the absence of ABMR by current diagnostic criteria is unclear. Microvascular injury has been described in DSA-negative patients [31], and thus, it has high diagnostic specificity only in presence of C4d positivity and/or circulating DSA. However, it is also possible that current tecniques may not detect all DSA, particularly those directed to endothelium. Conversely, glomerulitis has been reported in 21% of biopsies with TCMR, 10% of biopsies with borderline change [32] and 47% of C4d negative, DSA negative biopsies with TCMR diagnosis [33]. For this reason, the Banff 2013 meeting report suggested that peritubular capillaritis in presence of TCMR should not be considered as evidence of ABMR unless concurrent glomerulitis is also present [15]. Whether additional testing could elucidate antibody-related injury in some of these patients remains to be further investigated.
In summary, our study shows that kidney allografts have significantly increased expression of markers of endothelial activation and lymphangiogenesis compared to normal kidneys, irrespective of the humoral immunologic status expressed by C4d, DSA or both. These endothelial activation markers are associated with increased intragraft FOXP3 and STAT3 infiltrating cells, are expressed in late stages of disease and are associated with morphologic features of established and irreversible disease, such as transplant glomerulopathy and interstitial fibrosis/tubular atrophy. These findings suggest that endothelial activation and lymphangiogenesis may play a role in the development of chronic rejection, and could potentially represent prognostic tissue markers of irreversible organ damage. Furthermore, these findings support the therapeutic potential of new treatment strategies targeting mechanisms of endothelial activation and lymphangiogenesis to prevent graft loss due to immune-activated mechanisms.
Acknowledgments
The authors would like to thank Mr. John Bobbitt for his technical assistance with digital images, and Marcela Brissova, Ph.D. in the Islet Procurement and Analysis Core of the Vanderbilt Diabetes Research and Training Center (DRTC) for her support with the ScanScope Aperio image analysis.
FINANCIAL SUPPORT
This project was supported by the Vanderbilt Transplant Center and the CTSA award No. UL1TR000445 from the National Center for Advancing Translational Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare that they have no relevant financial interests.
References
- 1.Sellares J, de Freitas DG, Mengel M, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant. 2012;12:388–99. doi: 10.1111/j.1600-6143.2011.03840.x. [DOI] [PubMed] [Google Scholar]
- 2.Gaston RS, Cecka JM, Kasiske BL, et al. Evidence for antibody-mediated injury as a major determinant of late kidney allograft failure. Transplantation. 2010;90:68–74. doi: 10.1097/TP.0b013e3181e065de. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Reed EF. Effect of antibodies on endothelium. Am J Transplant. 2009;9:2459–65. doi: 10.1111/j.1600-6143.2009.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirohashi T, Chase CM, Della Pelle P, et al. A novel pathway of chronic allograft rejection mediated by NK cells and alloantibody. Am J Transplant. 2012;12:313–21. doi: 10.1111/j.1600-6143.2011.03836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feucht HE, Schneeberger H, Hillebrand G, et al. Capillary deposition of C4d complement fragment and early renal graft loss. Kidney Int. 1993;43:1333–8. doi: 10.1038/ki.1993.187. [DOI] [PubMed] [Google Scholar]
- 6.Cohen D, Colvin RB, Daha MR, et al. Pros and cons for C4d as a biomarker. Kidney Int. 2012;81:628–39. doi: 10.1038/ki.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sellares J, Reeve J, Loupy A, et al. Molecular diagnosis of antibody-mediated rejection in human kidney transplants. Am J Transplant. 2013;13:971–83. doi: 10.1111/ajt.12150. [DOI] [PubMed] [Google Scholar]
- 8.Sis B, Jhangri GS, Bunnag S, et al. Endothelial gene expression in kidney transplants with alloantibody indicates antibody-mediated damage despite lack of C4d staining. Am J Transplant. 2009;9:2312–23. doi: 10.1111/j.1600-6143.2009.02761.x. [DOI] [PubMed] [Google Scholar]
- 9.Stuht S, Gwinner W, Franz I, et al. Lymphatic neoangiogenesis in human renal allografts: results from sequential protocol biopsies. Am J Transplant. 2007;7:377–84. doi: 10.1111/j.1600-6143.2006.01638.x. [DOI] [PubMed] [Google Scholar]
- 10.Dietrich T, Bock F, Yuen D, et al. Cutting edge: lymphatic vessels, not blood vessels, primarily mediate immune rejections after transplantation. J Immunol. 2010;184:535–9. doi: 10.4049/jimmunol.0903180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin N, Zhang N, Xu J, et al. Targeting lymphangiogenesis after islet transplantation prolongs islet allograft survival. Transplantation. 2011;92:25–30. doi: 10.1097/TP.0b013e31821d2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonsib SM. Renal lymphatics, and lymphatic involvement in sinus vein invasive (pT3b) clear cell renal cell carcinoma: a study of 40 cases. Mod Pathol. 2006;19:746–53. doi: 10.1038/modpathol.3800589. [DOI] [PubMed] [Google Scholar]
- 13.Colvin RB, Cohen AH, Saiontz C, et al. Evaluation of pathologic criteria for acute renal allograft rejection: reproducibility, sensitivity, and clinical correlation. J Am Soc Nephrol. 1997;8:1930–41. doi: 10.1681/ASN.V8121930. [DOI] [PubMed] [Google Scholar]
- 14.Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753–60. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 15.Haas M, Sis B, Racusen LC, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant. 2014;14:272–83. doi: 10.1111/ajt.12590. [DOI] [PubMed] [Google Scholar]
- 16.Hourmant M, Cesbron-Gautier A, Terasaki PI, et al. Frequency and clinical implications of development of donor-specific and non-donor-specific HLA antibodies after kidney transplantation. J Am Soc Nephrol. 2005;16:2804–12. doi: 10.1681/ASN.2004121130. [DOI] [PubMed] [Google Scholar]
- 17.Rose ML. Endothelial cells as antigen-presenting cells: role in human transplant rejection. Cell Mol Life Sci. 1998;54:965–78. doi: 10.1007/s000180050226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trpkov K, Campbell P, Pazderka F, et al. Pathologic features of acute renal allograft rejection associated with donor-specific antibody, Analysis using the Banff grading schema. Transplantation. 1996;61:1586–92. doi: 10.1097/00007890-199606150-00007. [DOI] [PubMed] [Google Scholar]
- 19.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–56. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 20.Koskinen PK, Lemstrom KB. Adhesion molecule P-selectin and vascular cell adhesion molecule-1 in enhanced heart allograft arteriosclerosis in the rat. Circulation. 1997;95:191–6. doi: 10.1161/01.cir.95.1.191. [DOI] [PubMed] [Google Scholar]
- 21.Izawa A, Ueno T, Jurewicz M, et al. Importance of donor- and recipient-derived selectins in cardiac allograft rejection. J Am Soc Nephrol. 2007;18:2929–36. doi: 10.1681/ASN.2006111261. [DOI] [PubMed] [Google Scholar]
- 22.Sarraj B, Ye J, Akl AI, et al. Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection. Proc Natl Acad Sci U S A. 2014;111:12145–50. doi: 10.1073/pnas.1303676111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langer R, Wang M, Stepkowski SM, et al. Selectin inhibitor bimosiamose prolongs survival of kidney allografts by reduction in intragraft production of cytokines and chemokines. J Am Soc Nephrol. 2004;15:2893–901. doi: 10.1097/01.ASN.0000142425.23036.AC. [DOI] [PubMed] [Google Scholar]
- 24.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25– Foxp3− T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–9. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 26.Savage CO, Hughes CC, Pepinsky RB, et al. Endothelial cell lymphocyte function-associated antigen-3 and an unidentified ligand act in concert to provide costimulation to human peripheral blood CD4+ T cells. Cell Immunol. 1991;137:150–63. doi: 10.1016/0008-8749(91)90065-j. [DOI] [PubMed] [Google Scholar]
- 27.Adams PW, Lee HS, Waldman WJ, et al. Alloantigenicity of human endothelial cells. 1. Frequency and phenotype of human T helper lymphocytes that can react to allogeneic endothelial cells. J Immunol. 1992;148:3753–60. [PubMed] [Google Scholar]
- 28.Page C, Thompson C, Yacoub M, Rose M. Human endothelial stimulation of allogeneic T cells via a CTLA-4 independent pathway. Transpl Immunol. 1994;2:342–7. doi: 10.1016/0966-3274(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 29.Murakami K, Ma W, Fuleihan R, Pober JS. Human endothelial cells augment early CD40 ligand expression in activated CD4+ T cells through LFA-3-mediated stabilization of mRNA. J Immunol. 1999;163:2667–73. [PubMed] [Google Scholar]
- 30.Taflin C, Favier B, Baudhuin J, et al. Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A. 2011;108:2891–6. doi: 10.1073/pnas.1011811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JJ, Balasubramanian R, Michaelides G, et al. The clinical spectrum of de novo donor-specific antibodies in pediatric renal transplant recipients. Am J Transplant. 2014;14:2350–8. doi: 10.1111/ajt.12859. [DOI] [PubMed] [Google Scholar]
- 32.Batal I, Lunz JG, 3rd, Aggarwal N, et al. A critical appraisal of methods to grade transplant glomerulitis in renal allograft biopsies. Am J Transplant. 2010;10:2442–52. doi: 10.1111/j.1600-6143.2010.03261.x. [DOI] [PubMed] [Google Scholar]
- 33.Randhawa P. T-cell-mediated rejection of the kidney in the era of donor-specific antibodies: diagnostic challenges and clinical significance. Curr Opin Organ Transplant. 2015;20:325–32. doi: 10.1097/MOT.0000000000000189. [DOI] [PubMed] [Google Scholar]