

Abstract
Alcohol Use Disorder (AUD) is a common and chronic disorder with substantial effects on personal and public health1. The underlying pathophysiology is poorly understood but strong evidence suggests significant roles of both genetic and epigenetic components2. Given that alcohol affects many organ systems, we performed a cross-tissue and cross-phenotypic analysis of genome-wide methylomic variation in AUD using samples from 3 discovery, 4 replication, and 2 translational cohorts. We identified a differentially methylated region in the promoter of the proprotein convertase subtilisin/kexin 9 (PCSK9) gene that was associated with disease phenotypes. Biological validation showed that PCSK9 promoter methylation is conserved across tissues and positively correlated with expression. Replication in AUD datasets confirmed PCSK9 hypomethylation and a translational mouse model of AUD showed that alcohol exposure leads to PCSK9 downregulation. PCSK9 is primarily expressed in the liver and regulates low density lipoprotein cholesterol (LDL-C). Our finding of alcohol-induced epigenetic regulation of PCSK9 represents one of the underlying mechanisms between the well-known effects of alcohol on lipid metabolism and cardiovascular risk, with light alcohol use generally being protective while chronic heavy use has detrimental health outcomes.
Subject terms: epigenetics, LDL, cholesterol, methylation, cardiovascular, resting-state, PFC, liver disease
Introduction
Various pathways to the development of Alcohol Use Disorder (AUD) exist and include an interaction of environmental and genetic risk factors3. Identifying underlying genetic risk factors for AUD has been challenging due to small effect sizes, heterogeneity, and complex modes of inheritance2. However, the field of epigenetics in AUD is just developing4, and new technological advances are making it feasible to conduct epigenome-wide association studies (EWAS) of complex phenotypes using DNA methylation. Only a few EWAS for AUD exist, but they are limited by small sample sizes, low array-capture, tissue type, analysis strategy, and data interpretation5-11. Consequently, no universal DNA methylation loci for AUD have been identified; however, recent data suggest multiple loci for mild-moderate alcohol consumption12. Given that heavy alcohol consumption can cause significant alterations to multiple organs and tissue types, we carried out a systematic cross-tissue and cross-phenotypic analysis of methylomic variation in AUD. We used samples from independent cohorts involving postmortem brain, blood, and liver tissue as well as various clinical and imaging phenotypes with the goal of identifying disease-associated methylomic DNA variation (Fig. 1).
Figure 1. Methylomic profiling approach in AUD using three discovery and six replication data sets identified PCSK9 as main epigenetic target.

A schematic representation of cohorts investigated in this study broken into the discovery phase experiments (a,b,c) that identified PCSK9 association with alcohol use and the replication stage experiments (d-i) including biological target validation in animal models (h,i). Replication experiments were performed in multiple cohorts between blood and liver and data derived from publicly available datasets and direct investigation. For human blood, DNA from individuals who participated in the Grady Trauma Project (GTP) (n= 392) (d) was analyzed for CpG cg01444643 (hg38, chr1: 55039175) (PCSK9CpG1) PCSK9CpG1 was significantly associated with both the binary SCID37 lifetime alcohol abuse (β= -0.007 ± 0.004, F= 3.891, df= 2/325, P= 0.049) and the continuous KMSK Lifetime Alcohol scale38 (β= -0.001 ± 0, F= 4.944, df= 2/306, P= 0.027). In a second human blood cohort (e), we assessed PCSK9 DNA methylation levels with pyrosequencing at the PCSK9CpG1 and an adjacent CpG located at chr1: 55039185 (PCSK9CpG2) in human subjects (n= 90) aged 21-65 with a diagnosis of alcohol dependence and healthy volunteers (n= 62). We observed significantly lower DNA methylation at both CpGs (Student's t-test, PCSK9CpG1: Alcohol Abuse= 76.68 ± 0.05, No Alcohol Abuse= 78.6± 0.064, P= 0.0075; PCSK9CpG2 : Alcohol Abuse= 88.49 ± 0.025, No Alcohol Abuse= 89.28 ± 0.032, P= 0.028). In a liver cohort, we assessed PCSK9 DNA methylation levels at PCSK9CpG1 in DNA derived from individuals with normal livers (n= 34) or primary liver disease tissue arising in the setting of chronic hepatitis B (HBV) or C (HCV) viral infection, alcoholism (ETOH), and other causes (n= 66) (GSE60753) (f). PCSK9 DNA methylation was significantly elevated with alcohol-induced cirrhosis (Alcohol Cirrhosis= 0.54 ± 0.0023, Healthy= 0.49 ± 0.0015, P= 0.0021). Importantly, significant elevations were also observed with cirrhosis induced by hepatitis B (Hep B Cirrhosis= 0.55 ± 0.0079, Healthy= 0.49 ± 0.0015, P= 0.023) and C (Hep C Cirrhosis= 0.57 ± 0.0011, Healthy= 0.49 ± 0.0015, P= 1.2×10-9). Finally, PCSK9CpG1 DNA methylation levels assessed with pyrosequencing in liver tissue samples from liver transplant candidates with alcoholic cirrhosis (n= 50) and healthy controls (n= 47) (g). A significantly higher PCSK9 DNA methylation level was observed in alcoholic cirrhosis cases relative to controls (Student's t-test; Alcohol Abuse= 46.19 ± 1.07, No Alcohol abuse= 37.63 ± 0.89, P= 6.5×10-9). In translational models, mouse liver PCSK9 expression was significantly lower in the alcohol exposure group (h) (Student's t-test; Alcohol Exposure= 0.1 ± 0.011, No Alcohol Exposure= 1.02 ± 0.057, P= 0.0029). A translational rat model (i) was used to assess long-term effects of alcohol on PCSK9 expression in liver.
Materials and Methods
Subjects and samples
Subjects in the discovery and replication stages with alcohol dependence or abuse will be referred to as AUD.
I. Discovery stage genome-wide DNA methylation analysis in brain
Methylomic profiling
We used a step-wise, multiple-level approach across various tissue types and outcome for the initial discovery analysis. A graphic representation is shown in Fig. 1a, b, c.
Methylomic profiling discovery data set of postmortem prefrontal cortex tissue
Bulk brain derived DNA methylation profiles generated on Illumina Human Methylation 450 (HM450) bead chip microarrays from 46 postmortem prefrontal cortex (PFC) samples were downloaded from the Gene Expression Omnibus from GSE49393. The sample was comprised of 23 subjects with AUD (16 males and 7 females) and 23 age-matched healthy controls. Detailed methodology and sample description can be found elsewhere13. Fresh-frozen sections of Brodmann area 9 (BA9, mainly the dorsolateral PFC of the brain) postmortem brain tissues were obtained from the New South Wales Tissue Resource Centre (NSW TRC) at the University of Sydney and ethic approval was obtained from the Sydney Local Health Network and the University of Sydney. For each probe on the microarray, a linear model was performed to determine the association of DNA methylation with alcohol relative to control status, controlling for age, sex, and neuronal proportion as estimated by the Cell EpigenoType Specific (CETS) algorithm14. For this analysis, alcohol abuse and alcohol dependence were grouped together for case status. Correction for multiple testing was assessed using the False Discovery Rate method. Detailed sample demographics appear in Supplementary Table S1.
Neuronal and non-neuronal postmortem prefrontal cortical tissue National Institute of Child Health and Human Development (NICHD) brain bank sample
Postmortem cortical tissue from major depressive disorder (MDD) (n= 29) and matched control (n= 29) samples were obtained from the NICHD brain bank and DNA methylation profiles using HM450 arrays from FACs isolated neuronal and non-neuronal (glial) nuclei were obtained as previously described14. For the neuronal and glial fractions separately, a linear model was performed to determine the association of DNA methylation with alcohol relative to control status, controlling for age, sex, and postmortem interval for each probe on the microarray. Alcohol case status was based on reported alcohol abuse in the summary of health history provided by the NICHD brain bank. Correction for multiple testing was assessed using the False Discovery Rate method. Detailed sample demographics appear in Supplementary Table S2.
National Institute on Alcohol Abuse and Alcoholism (NIAAA) methylomic profiling discovery data set with resting-state magnetic resonance imaging (MRI) functional connectivity (FC)
A total of 68 right-handed individuals with a diagnosis of AUD and 72 healthy controls were recruited to the NIAAA at the National Institutes of Health (NIH), USA. Written informed consent to the study was obtained from all the subjects, which was approved by the Institutional Review Board (IRB) of the NIAAA and was in accordance with the Declaration of Helsinki and the NIH Combined Neuroscience IRB. Participants were compensated for their time. Blood was obtained from the 68 individuals with a diagnosis of AUD.
The Structured Clinical Interview for DSM-IV Axis I Disorders (SCID)15 was administered to all participants. A smoking questionnaire helped to determine the amount and frequency of a participant's cigarette use. The Alcohol Dependence Scale (ADS) was administered to determine the severity of alcohol dependence (AD)16. Exclusionary criteria included pregnancy, claustrophobia, and significant neurological or medical diagnoses. Participants were instructed not to consume any alcohol for 24 hours and no more than half a cup of caffeinated beverages 12 hours prior to each scanning visit. Participants could not participant if they had a positive alcohol breathalyzer or positive urine drug screen on the day of the scan. Controls were excluded if they met criteria for any current or past alcohol dependence. All subjects were required to be deemed physically healthy by a clinician. At the time of the MRI, AD participants could not be exhibiting severe symptoms of alcohol withdrawal, as determined by the Clinical Institutes Withdrawal Assessment-Alcohol revised (CIWA-Ar) score of greater than 817. Detailed sample demographics appear in Supplementary Table S3.
MRI data acquisition and pre-processing
Whole brain anatomical images and five minutes of closed-eyes resting-state fMRI were collected using 3T General Electric and 3T SIEMENS MRI scanners. High resolution T1-weighted 3-D structural scans were acquired for each subject using an MPRAGE sequence (128 axial slices, TR=1200ms TE=30ms, 256 × 256 matrix). Resting-state fMRI (rs-fMRI) datasets were collected using a single-shot gradient echo planar imaging pulse sequence with thirty-six axial slices acquired parallel to the anterior/posterior commissural line (TR=2000ms, TE=30ms, flip angle=90°, 3.75 mm × 3.75 mm × 3.8 mm voxels).
rs-fMRI data pre-processing
Using Functional MRI of the Brain (FMRIB)'s Software Library (www.fmrib.ox.ac.uk/fsl) we applied; 1) slice timing correction for interleaved acquisitions using Sinc interpolation with a Hanning windowing kernel; 2) motion correction using FMRIB's Linear Image Registration Tool (MCFLIRT)18; 3) non-brain removal using Brain Extraction Tool (BET)19; 4) spatial smoothing using a Gaussian kernel of full width at half maximum 5mm; 5) grandmean intensity normalization of the entire 4D dataset by a single multiplicative factor, the size of the voxels is 4×4×4 mm3; 6) high-pass temporal filtering (Gaussian-weighted least-squares straight line fitting, with sigma=50s); and 7) registration to high resolution structural MNI standard space images using the FMRIB's Linear Image Registration Tool (FLIRT)18, 20.
rs-fMRI connectivity analysis
We applied Probabilistic Independent Component Analysis (PICA)21 by using the Multivariate Exploratory Linear Decomposition into Independent Components (MELODIC) toolbox of the FSL package.
A temporal concatenation tool in MELODIC was used to derive group level components across all subjects. Pre-processed data were whitened and projected into a 36-dimensional subspace using PICA where the number of dimensions was estimated using the Laplace approximation to the Bayesian evidence of the model order21. The whitened observations were decomposed into sets of vectors which describe signal variation across the temporal domain (time-courses) and across the spatial domain (maps) by optimizing for non-Gaussian spatial source distributions using a fixed-point iteration technique22. These functional connectivity (FC) components maps were standardized into z statistic images via a normalized mixture model fit (thresholded at z > 7)21. Two criteria were used to remove biologically irrelevant components: 1) those representing known artifacts such as motion, and high-frequency noise; and 2) those with connectivity patterns not located mainly in gray matter. Networks of interest were identified as anatomically and functionally classical resting state networks (RSN)23 upon visual inspection.
The between-subject analysis was carried out using dual regression, a regression technique which back-reconstructs each group level component map at the individual subject level. Next, a non-parametric permutation test (FSL's randomize tool, with 5000 permutations), that utilizes a threshold-free cluster enhanced (TFCE) thresholding, was used to assess statistically significant differences in FC between the groups. To minimize the potential confounding influence of age, gender, ancestry informative marker (AIM) scores and scanner types in these results, these parameters were used as nuisance covariates for each network of interest. Finally, the resulting statistical maps were thresholded at P < 0.05 family-wise error (FWE) corrected for the main group effect. The Harvard-Oxford cortical and subcortical atlases incorporated in FSL were used to identify the anatomical regions of the resulting PICA maps. The FSL Cluster tools were used to report information about clusters in the selected maps. The functional connectivity maps were overlaid onto the mean standardized structural T1 1-mm MNI template and visualized using Mricrogl.
Peripheral blood Illumina HM450 data processing
Following removal of three subject samples with failed bisulfite conversion, raw Illumina HM450 microarray data was processed using the wateRmelon package in R24. Raw data was trimmed of probes failing quality assessment and a known list of 32323 cross reactive probes25, followed by scale based data correction for Illumina type I relative to type II probes. Methylated and unmethylated intensity values were then quantile normalized separately prior to the calculation of the β (beta) value based on following definition:
β value = (signal intensity of methylation-detection probe)/(signal intensity of methylation- detection probe ± signal intensity of non-methylation-detection probe ± 100).
Values were then adjusted by taking the residuals of a linear model of beta values as a function of sodium bisulfite modification batch.
II. PCSK9 Targeted Replication
Grady Trauma Project (GTP) blood replication sample
The subjects for this study were part of a larger investigation of genetic and environmental factors that predict response to stressful life events in a predominantly African American, urban population of low socioeconomic status26-28. Research participants were approached in the waiting rooms of primary care clinics of a large, public hospital while either waiting for their medical appointments or waiting with others who were scheduled for medical appointments. After the subjects provided written informed consent, they participated in a verbal interview and blood draw. This cohort was characterized by high rates of interpersonal violence and psychosocial stress; the majority of subjects reported at least one major trauma during their lifetime, and the number of traumatic experiences in childhood and adulthood predicted psychiatric symptom severity in adulthood28, 29. DNA methylation profiles from n = 392 African American individuals were downloaded from the Gene Expression Omnibus from GSE72680. Detailed sample demographics appear in Supplementary Table S5.
NIAAA blood samples of individuals with AUD for targeted plasma replication
Subjects (n= 90) with a diagnosis of alcohol dependence according to the Structured Clinical Interview for DSM-IV15 were recruited at the NIAAA alcohol treatment program. Participants were between 21–65 years old. Informed consent was obtained in accordance with the Declaration of Helsinki.
Most the alcohol dependent participants were recruited through local newspaper advertisements for the alcohol treatment program at the NIAAA at the NIH, Bethesda, Maryland. The participants consisted of men and women from a broad range of socioeconomic backgrounds ranging from executives to unemployed individuals. Participants underwent extensive clinical and physical examinations. Participants with a history of seizures, head trauma (defined as a period of unconsciousness exceeding 1h), or medical conditions requiring chronic medications were excluded from participation. Participants were literate in English and were not suffering from active psychotic symptoms or severe cognitive impairment. Participants were voluntarily admitted to the NIAAA inpatient unit in the NIH Clinical Center between February 2010 and May 2014. Once they were admitted, participants were detoxified from alcohol and participated in the alcohol treatment program.
Detailed sample demographics appear in Supplementary Table S6. Blood and plasma samples were obtained and DNA extracted using standard methods. Plasma samples were obtained from two IRB approved protocols. All the bloods were drawn around 0800 after overnight bed rest. Participants were nil per os (NPO) for approximately 10 hours prior to blood collection. Blood was collected in prechilled EDTA tubes which were promptly put into wet ice and taken to the laboratory to be processed. Samples were spun in a 4 °C centrifuge (Beckman-Coulter Allegra X-12R) at 1880 g for 10 minutes; the plasma was then aliquoted into NUNC cyrovials and frozen at -80 °C until assayed.
The first set of samples (n= 55) were alcohol treatment-seeking inpatient participants. Their blood samples were drawn after alcohol withdrawal within 5 days of their last drink of alcohol.
The second set of samples (n=35) were alcohol treatment-seeking inpatients who were also diagnosed with post-traumatic stress disorder (PTSD). After alcohol withdrawal and withdrawal from any other medications for up to 14 days after a participants last drink, the participants' blood samples were drawn.
In both sample sets, average drinks per day, drinking days, and heavy drinking days were recorded for the 90 days prior to inpatient admission using the Timeline Followback Questionnaire30. ADS was also assessed; this score can range from 0 to 47 and scores greater than 27 are classified as substantial or severe AD. Detailed sample demographics appear in Supplementary Table S6. Low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, total cholesterol, triglycerides, and total bilirubin were measured using standard procedures in milligrams per deciliter (mg/dL). Gamma-Glutamyltransferase (GGT) was measured using standard procedures in units per liter (U/L). Samples were collected on the date of admission to the NIAAA treatment program and processed by the NIH Clinical Center Department of Laboratory Medicine.
PCSK9 protein concentrations were determined in 90 subjects with AUD using Luminex technology (Luminex Corporation, Austin, TX, USA). A Human Magnetic Luminex Screening Assay was performed per the manufacturer's instructions (catalogue #LXSAHM, R&D Systems, Minneapolis, MN, USA), with plasma dilutions of 1:5. Data was analyzed using Milliplex Analyst software (EMD Millipore Corporation, Merck KGaA, Darmstadt, Germany).
Samples were assayed in duplicates. Mean CV for all 90 samples was 2.62 ± 2.39 (mean ± SD). Samples were randomized across four 96-well plates, and CV for two quality control samples run on all four plates were 0.069 and 0.065, respectively.
NIAAA AUD and healthy control blood samples for targeted pyrosequencing replication
Blood was obtained from 62 healthy controls that participated in studies at NIAAA as previously described13. Detailed sample demographics appear in Supplementary Table S6. Pyrosequencing was performed as detailed for these NIAAA healthy controls and 90 AUD subjects used in the plasma experiment described above.
Pyrosequencing was performed following techniques described by Kaminsky et al31. Sodium bisulfite conversion was carried out using EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA) according to the manufacturer's instructions on 500 ng of DNA from tested human tissues. Nested PCR amplifications were performed with a standard PCR protocol in 25 μl volume reactions containing 3-4 μl of sodium-bisulfite-treated DNA, 0.2 μM primers, and master mix containing Taq DNA polymerase (Sigma Aldrich, St. Louis, MO). Primer annealing temperatures for the outside and inside nested PCR were 58.6 and 61.9 degrees C, respectively. Outside primer sequences were as follows: Forward primer 5′- AGTAGTTAGTTGGTAAGGTTAGT-3′, Reverse primer 5′- ACCTTTTCAATATTTCCTAAATCCA -3′, Inside primer sequences were as follows: Forward primer 5′- GGTTAAGTTTAGAAGGTTGTTAGGT -3′, Reverse primer 5′- CCACCTTATCTCCTACTAACCAA -3′ with a biotin modification on the 5′ end. PCR amplicons were processed for pyrosequencing analysis per the manufacturer's standard protocol (Qiagen) using a PyroMark MD system (QIAGEN, Germantown, MD) with Pyro Q-CpG 1.0.9 software (QIAGEN) for CpG methylation quantification. The pyrosequencing primer sequences was 5′- AGGTTAAGGTTAGTGGAAAGAAT -3′. Only data passing internal quality checks for sodium bisulfite conversion efficiency, signal to noise ratio, and the observed vs. expected match of the predicted pyrogram peak profile using reference peaks were incorporated in subsequent analyses. Data generated derive from one technical replicate.
University of Minnesota Liver Tissue Cell Distribution System (LTCDS) liver samples
Normal human liver and pathologic human liver were obtained through the LTCDS (Minneapolis, Minnesota), which was funded by NIH Contract #HSN276200017C. The LTCDS is an NIH service contract to provide human liver tissue from regional centers to scientific investigators. These regional centers have active liver transplant programs. Human subject approval was obtained to provide portions of the resected livers for which the transplant was performed. We obtained liver tissue from transplant candidates with alcoholic cirrhosis (n= 50) and healthy controls (n= 50). Detailed sample demographics appear in Supplementary Table S7.
LTCDS human liver Illumina EPIC data processing
From the LTCDS data set, 2 control and 2 liver transplant tissues were excluded due to space limitations on the array. The remaining human alcohol-induced cirrhotic liver transplant tissues (n = 48) and controls (n = 48) were subjected to Illumina Methylation EPIC DNA methylation microarray analysis. Normalization of binary format .idat files for red and green channel intensities was carried out in R. Initial background correction was performed using the minfi R-package32 followed by individual red and green channel quantile normalization using the dasen method in wateRmelon24. After normalization, the beta value (β) is used to estimate the methylation level of each CpG locus using the ratio of intensities between methylated and unmethylated alleles (M/M + U=100) where M and U represent the methylated and unmethylated intensities respectively33.
LTCDS Targeted Pyrosequencing Replication Methods
DNA purification and extraction
DNA was extracted using a Maxwell Tissue Purification Kit with a Promega Maxwell 16 Forensic Instrument (Promega Corporation, Madison, WI). DNA extraction was performed according to manufacturer's instructions. DNA concentrations were verified to be of adequate concentration (> 95 ng/μl).
Real-Time Quantitative PCR
RNA was extracted using an Ambion Ribopure ™ RNA Purification Kit (Thermo Fisher Scientific, Rockville, MD) according to the manufacturer's instructions. 1 μg total RNA was reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen). Real-time quantitative PCR was run in ViiA™ 7 Real-Time PCR System with Using TaqMan Gene Expression Assays (Thermo Fisher). Gene expression was studied with TaqMan PCR assays (PCSK9; Hs00545399_m1). 18S gene was used as an endogenous control. Data were analyzed initially using ViiA™ 7 Software (Applied Biosystems). Gene expression was analyzed in liver tissue from transplant candidates with alcoholic cirrhosis (n= 24) and healthy controls (n= 24). The data analysis was performed using GraphPad prism 6.0 (GraphPad Software Inc., San Diego, CA, USA).
Sodium bisulfite pyrosequencing
Pyrosequencing was performed as detailed above. Methylation data was excluded for three healthy control samples due to experimental error (n= 47).
University of Florida liver sample
The samples used in this analysis were derived from cirrhotic (n= 66) and normal liver (n= 34) tissues from the University of Florida Shands Hospital by surgical resection as reported previously by Hlady et al34. Normal livers were derived from surgical patients for benign liver lesions. Data was downloaded from the Gene Expression Omnibus from GSE60753. Sample demographics appear in Supplementary Table S8.
III. Translational Animal Models
Mouse model
Mouse brain, blood, and liver tissues were analyzed for methylation, mRNA and protein expression using standard methodologies. Tissue was derived from mice that underwent the “NIAAA model” of chronic and binge ethanol feeding. Briefly, this model uses 10 days ad libitum oral feeding of the Lieber-DeCarli ethanol liquid diet plus a single-binge ethanol feeding. Samples were collected 8 hours after the single-binge feeding. Detailed information can be found elsewhere35. DNA of tissues was extracted using the Maxwell Tissue Purification Kit as described above.
Real-Time Quantitative PCR
Total RNA was extracted using an RNeasy mini-kit (Qiagen). 1 μg total RNA was reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen). Real-time quantitative PCR was run in ViiA™ 7 Real-Time PCR System with Using TaqMan Gene Expression Assays (Thermo Fisher). Gene expression was studied with TaqMan PCR assays (PCSK9; Mm01263610_m1). We used 18 S gene as an endogenous control. Data were analyzed initially using ViiA™ 7 Software (Applied Biosystems). The data analysis was performed using GraphPad prism 6.0 (GraphPad Software Inc., San Diego, CA, USA).
Western blot analysis
Liver homogenates were prepared in RIPA buffer (50 mM Tris; 1% NP40; 0.25% deoxycholic acid sodium salt; 150 mM NaCl; 1 mM EGTA) containing 1 mM Na3VO4 and a protease inhibitor cocktail (Sigma, St. Louis, MO). The protein concentrations were quantified using a detergent-compatible protein assay kit (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Aliquots of 50 μg of protein extract were denatured in Laemmli buffer containing 5% β-mercaptoethanol, and the samples were loaded and separated by gel electrophoresis on a 7% Bis-Tris gel (Invitrogen, Grand Island, NY). Samples were incubated with a primary antibody at 4°C overnight under shaking conditions. Immunoreactive bands were visualized on nitrocellulose membranes using alkaline-phosphatase-linked anti-rabbit antibodies and the ECF detection system with a PhosphorImager (GE Healthcare, Piscataway, NJ). Anti-PCSK9 antibody (ab125251) and beta-actin were purchased from Abcam (Cambridge, MA).
Rat model
Adult male Wistar rats (Charles River) received pre-vapor operant conditioning for 3 weeks and were first trained to self-administer alcohol using a modified sucrose-fading procedure in which 10% ethanol was added to a sweet solution. Sweeteners were gradually removed from the solution. Upon completion, animals could self-administer alcohol (10% ethanol and water solution) on a schedule of reinforcement. The case rats were then treated with a chronic intermittent exposure to alcohol in vapor chambers. Ethanol exposure lasted 14 hours and was followed by 10 hours off. During exposure, blood alcohol levels ranged between 150 and 250 mg%. This cycle was repeated for 20 days. Nondependent rats were not exposed to alcohol vapor.
DNA purification and extraction
DNA was extracted using a Maxwell Tissue Purification Kit as described above.
Real-Time Quantitative PCR
RNA was extracted using an Ambion Ribopure ™ RNA Purification Kit (Thermo Fisher Scientific, Rockville, MD) according to the manufacturer's instructions. 1 μg total RNA was reverse-transcribed using SuperScript® III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen). Real-time quantitative PCR was run in ViiA™ 7 Real-Time PCR System with Using TaqMan Gene Expression Assays (Thermo Fisher). Gene expression was studied with TaqMan PCR assays (PCSK9; Rn01416753_m1). 18S gene was used as an endogenous control. Data were analyzed initially using ViiA™ 7 Software (Applied Biosystems). The data analysis was performed using GraphPad prism 6.0 (GraphPad Software Inc., San Diego, CA, USA).
Results
Discovery strategy
For the discovery stage of our analysis, using the Illumina 450K HumanMethylation array platform, we conducted epigenome-wide association analyses of postmortem bulk brain tissue from prefrontal cortex (PFC) of individuals with AUD (n=23) and healthy controls (n=23) (Fig. 2a and Supplementary Table S1). In this phase, postmortem cortical brain tissue was also sorted into neuronal and non-neuronal cells from individuals with depression and alcohol use history (n=29) and healthy controls (n=29) (Fig. 2b,c and Supplementary Table S2). In addition, we performed an EWAS on neuroimaging network activation as a function of alcohol use with peripheral blood from individuals with AUD that previously underwent resting state functional connectivity (RSFC) analysis36 (n= 68) (Supplementary Table S3). Results of these 3 EWAS did yield various methylation sites that were associated with disease phenotypes (Fig. 2). To identify epigenetic consequences of systemic alcohol exposure further, we next conducted an overrepresentation analysis of functional brain imaging-associated peripheral blood DNA methylation with brain tissue-specific alcohol use associations. A total of 17 probes corresponding to 12 genes were significantly associated across the salience network (SN), executive control network (ECN), visual and motor networks. These probes were also significantly associated with alcohol status in the bulk PFC cohort. The most significant gene-associated probe cg01444643 (hg38, chr1: 55039175; PCSK9CpG1) was located in the promoter of the PCSK9 gene (β= -9.911 ± 3.034, F= 3.073, df= 5/43, P= 0.0021, Supplementary Table S4). We then performed the same overrepresentation analysis to assess for imaging network correlating peripheral blood DNA methylation and alcohol abuse associations in neuronal and non-neuronal nuclei from another postmortem brain cohort (Supplementary Table S2). In neurons, overrepresentation was observed in a visual network pathway different from that identified in the bulk PFC sample. Interestingly, only two networks were overrepresented in the non-neuronal fraction, which corresponded to the same SN and ECN networks identified in the bulk PFC analysis. A total of 86 loci were significantly associated across both the SN and ECN networks in peripheral blood as well as with alcohol abuse in non-neuronal nuclei. Of these loci only cg01444643 corresponding to PCSK9 (β= -0.026 ± 0.013, F= 6.23, df= 7/51, P= 0.048) and cg17845617 corresponding to Phosphatidylinositol-4-Phosphate 5-Kinase (PIP5K1C) were consistently identified in the National Institute of Child Health and Human Development (NICHD) and bulk brain cohorts (Supplementary Table S4). As the direction of association was consistent across discovery datasets for PCSK9, we targeted this locus for in-depth follow up.
Figure 2. Probe-wise association of alcohol use in the prefrontal cortex.
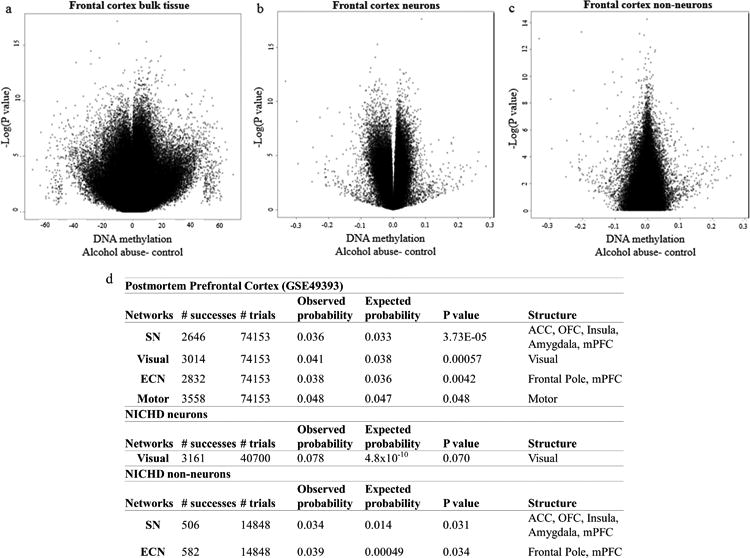
(a) Volcano plot depicting the negative natural log of the p-value of association of alcohol abuse (y-axis) as a function of the beta value (x-axis) from a linear model adjusting for age, sex, and neuronal proportion as estimated by the Cell EpigenoType Specific (CETS) algorithm. Data derived from frontal cortex bulk tissue from GEO dataset GSE49393. Three loci were significant after false discovery rate (FDR) based correction for multiple testing including cg00393248 (MYLK4; β= 9.32 ± 1.516, F= 10.061, df= 5/43, P= 2.23×10-7, FDRP= 0.038), cg19608003 (SLC44A4; β= -13.468 ± 2.011, F= 11.879, df= 5/43, P= 3.54×10-8, FDRP= 0.015), and cg19955284 (β= -19.036 ± 3.122, F= 9.902, df= 5/43, P= 2.64×10-7, FDRP= 0.038). (b) Volcano plot depicting the negative natural log of the p-value of association of alcohol abuse in FACs isolated NeuN positive neuronal nuclei from the NICHD cohort (y-axis) as a function of the beta value (x-axis) from a linear model adjusting for age, sex, race, and Body Mass Index (BMI). In neurons, only cg03982998 (LOC100130331; β= 0.087 ± 0.013, F= 10.487, df= 7/51, P= 2.3×10-8, FDR P= 0.0078) and cg06395265 (ZSWIM1; β= -0.049 ± 0.008, F= 8.967, df= 7/51, P= 2.36×10-7FDR P= 0.041) were significant after correction for multiple testing.(c) Volcano plot depicting the negative natural log of the p-value of association of alcohol abuse in FACs isolated NeuN negative non-neuronal nuclei from the NICHD cohort (y-axis) as a function of the beta value (x-axis) from a linear model adjusting for age, sex, race, and PMI. (d) A table depicting functional connectivity networks significantly over-represented among AUD associated probes from GEO dataset GSE49393 and isolated neuronal and glial nuclei from the NICHD cohort.Structures analyzed included the anterior cingulate cortex (ACC), orbitofrontal cortex (OFC), insula, amygdala, visual cortex, motor cortex, frontal pole and medial prefrontal cortex (mPFC).
PCSK9 methylation association with phenotype
To further investigate the association of PCSK9 methylation and alcohol use, we first analyzed peripheral blood DNA from human subjects who participated in the GTP (n= 392) (Supplementary Table S5) and found that lower methylation of cg01444643/PCSK9CpG1 was significantly associated with both the binary SCID37 lifetime alcohol abuse item (P= 0.049) and the continuous Kreek-McHugh-Schluger-Kellow (KMSK) Lifetime Alcohol scale38 (P=0.027) (Fig. 1d). In a second cohort, we assessed PCSK9 promoter methylation with pyrosequencing in individuals with AUD (n= 90) and healthy volunteers (n= 62) (Fig. 1e and Supplementary Table S6). Results showed significantly lower DNA methylation at PCSK9CpG1: (Student's t-test, PCSK9CpG1: Alcohol Abuse=76.68± 0.05, No Alcohol Abuse= 78.6± 0.064, P= 0.0075).
PCSK9 methylation cross-tissue validation
Although PCSK9 is mainly expressed in liver, using the EWAS approach, we also discovered it in brain and blood cohorts. However, it remains unknown to what degree there is conservation of methylation across tissues. Since blood, brain, and liver tissue was not available from the same individuals, we used a mouse model to assess PCSK9 DNA methylation across tissues using sodium bisulfite pyrosequencing of the syntenic PCSK9 CpG (chr4: 106464510). Brain DNA methylation exhibited a non-significant trend for correlation with that of blood (Rho= 0.51, P=0.064) and a significant correlation with liver methylation. Blood and liver methylation exhibited a non-significant trend for correlation (Fig. 3a,b,c). Blood methylation was significantly positively correlated with liver methylation in both mouse and human samples (Fig. 3c, Supplementary Fig. 1). These observations suggest that PCSK9 DNA methylation is relatively conserved across tissues, and warrants further replication in peripheral tissues as a biomarker of alcohol exposure.
Figure 3. Cross-tissue-specific PCSK9 methylation and association with gene expression and plasma levels.

a-c cross-tissue correlations: (a) Mouse syntenic PCSK9 DNA methylation in brain (y-axis) as a function of blood (x-axis) (Rho= 0.51, P= 0.064). (b) A scatterplot of mouse syntenic PCSK9 DNA methylation in brain (y-axis) as a function of liver (x-axis) (Rho= 0.62, P= 0.021). (c) A scatterplot of mouse syntenic PCSK9 DNA methylation in blood (y-axis) as a function of liver (x-axis) (Rho= 0.49, P= 0.087).
d-f methylation to expression correlations: (d) Mouse syntenic PCSK9 DNA methylation in blood (y-axis) as a function of liver-specific PCSK9 gene expression (x-axis) (Rho= 0.67, P= 0.0086). (e) Human PCSK9 DNA methylation in blood (y-axis) as a function of liver-specific PCSK9 gene expression from healthy controls (n= 47) (x-axis) (Rho= 0.41, P= 0.046). (f) PCSK9CpG1 DNA methylation (x-axis) as a function of plasma levels of PCSK9 (y-axis) in individuals with AUD (n= 90) (Rho = 0.31, P= 0.0027).
Functional relevance of PCSK9 methylation on expression
Next, we assessed the functional relevance of peripherally measured PCSK9 DNA methylation on levels of PCSK9 in the plasma and gene expression in liver. Using our mouse model where multiple types of tissues could be obtained from the same animal, we observed a significant positive correlation of syntenic PCSK9 DNA methylation and liver gene expression (Fig. 3d), which was consistent with observations in humans (Fig. 3e). Furthermore, in humans, we observed a significant correlation of PCSK9CpG1 methylation and PCSK9 plasma levels in AUD participants (Fig. 3f).
Effect of alcohol on PCSK9 methylation and expression
Since we consistently observed a decrease in PCSK9 DNA methylation with alcohol exposure, we next assessed liver PCSK9 gene expression and protein levels in the translational mouse model and in humans. Alcohol exposure was associated with significantly lower gene expression and protein levels of PCSK9 in the mouse model (Fig. 4a, b, c). In a cohort of human AUD liver transplant cases (n= 50) and healthy controls (n= 47) (Supplementary Table S7), a significantly lower PCSK9 gene expression level was observed in alcoholic cirrhosis cases relative to controls (Fig. 4e, Student's t-test; Alcohol Exposure= 0.3517 ± 0.125, No Alcohol Exposure= 0.9992 ± 0.1388, P< 0.01). Interestingly, we observed a marked increase in methylation in the human liver transplant cases, (Fig. 4d) which was contrary to our prediction of low methylation leading to low expression. The high methylation and low expression observed in liver tissue may be due to the direct toxic effects of alcohol. Therefore, a portion of the observed decreased liver PCSK9 expression may be accounted for by hepatocyte loss/toxicity in the assayed tissue. In fact, we found that mouse PCSK9 expression was negatively correlated with liver aspartate aminotransferase (AST) values (Supplementary Fig. 2). Furthermore, EWAS analysis of end-stage liver disease and control samples showed that estimated hepatocyte proportions were positively associated with human liver PCSK9 expression (Supplementary Fig. 3). Independent of acute or chronic alcohol-induced cell loss dynamics, the possibility remains that continued chronic heavy alcohol exposure may result in an increase in PCSK9 expression with time as we observed in a chronic rat model of alcohol exposure (Fig. 5, Supplementary Fig. 4), suggesting a need for a cell-type controlled longitudinal assessment of alcohol's effects on liver PCSK9 expression.
Figure 4. Alcohol exposure leads to lower PCSK9 expression in mice (a,b,c) and humans (d,e).

(a) PCSK9 mRNA expression in liver as a function of alcohol exposure status from mice that underwent the “NIAAA model of chronic and binge ethanol feeding”35 for 10-day chronic-plus-binge ethanol exposure. PCSK9 expression was significantly lower in the alcohol-exposed group (Student's t-test; Alcohol Exposure= 0.2443± 0.06553, No Alcohol Exposure= 1±0.07283, ****P<0.0001).
(b) PCSK9 protein expression in mouse liver was significantly higher in the control group compared to the case group that underwent the “NIAAA model of chronic and binge ethanol feeding”35 (Student's t-test; Alcohol Exposure= 0.2372± 0.01829, No Alcohol Exposure=0.4867 ± 0.0321, **P<0.01).
(c) Western blot of mouse liver PCSK9 levels as assessed by anti-PCSK9 antibody ab125251.
(d) Methylation analysis of PCSK9CpG1 in human alcohol-induced end-stage liver disease bulk tissue shows marked increase of methylation which might be due to general toxic effects of alcohol and end-stage organ disease. (Student's t-test; Alcohol Exposure= 46.19 ± 1.07, No Alcohol Exposure= 37.63 ± 0.8867, ****P< 0.0001).
(e) mRNA expression analysis of PCSK9 inhuman alcohol-induced end-stage liver disease bulk tissue reveals significantly decreased PCSK9 expression. (Student's t-test; Alcohol Exposure= 0.3517 ± 0.125, No Alcohol Exposure= 0.9992 ± 0.1388, **P< 0.01).
Figure 5. Model for PCSK9 interaction with alcohol.

The model shown describes different stages of alcohol exposure and subsequent PCSK9 methylation and expression findings. Consistent with mild use are our finding of alcohol exposure leading to hypomethylation (discovery data sets, Fig. 1a-e, plasma data set, Fig. 2f) with lower expression initially. Chronic alcohol use eventually leads to higher methylation and higher expression (rat data set, Supplementary Fig. 4) whereas alcohol liver toxicity (acute – NIAAA mouse model or chronic – liver transplant cases) leads to high methylation with ultimately low protein expression, consistent with end-stage liver disease (Fig. 4 and Supplementary Fig. 2, 5). Low expression and high methylation might also be affected by changes in cell type composition of tissue, as shown in end-stage liver disease tissue that varies greatly from healthy liver tissue on a global scale (Supplementary Fig. 3, 5). The early mild stage might be due to direct effects on transcription factor binding in the promoter regions (Supplementary Fig. 7), while later might be caused by liver toxicity effects and tissue composition changes (Supplementary Fig. 2, 3, 5).
Assessment of PCSK9 DNA methylation from diseased liver tissue must be interpreted with caution, as liver PCSK9 DNA methylation varies in a cell type specific manner. PCSK9CpG1 methylation exhibits significantly lower levels in mature hepatocytes relative to immature hepatocytes and other liver tissues (Supplementary Fig. 5). This suggests that liver damage and the significantly lower number of estimated hepatocyte proportions in the liver transplant cases (Supplementary Fig. 3c) may result in an observed higher PCSK9 DNA methylation relative to healthy controls. Indeed, we observed a significantly higher liver PCSK9 DNA methylation level in alcoholic cirrhosis cases (Fig. 4d: Student's t-test; Alcohol Abuse= 46.19 ± 1.07, No Alcohol abuse= 37.63 ± 0.89, P= 6.5×10-9). Within AUD subjects, a significant association of PCSK9CpG1 methylation and AST values was observed and further supports this hypothesis (Rho= 0.43, P= 0.0405). Significantly higher cirrhosis-associated PCSK9CpG1 DNA methylation was replicated in a second liver cohort derived from normal liver or cirrhotic liver arising from chronic hepatitis B or C viral infection, or alcoholism (Supplementary Table S8, Supplementary Table S9). It is important to note that significantly higher PCSK9 DNA methylation was observed across all cirrhosis categories (Supplementary Table S9), supporting the assessment that liver damage confounds PCSK9 DNA methylation assessment in the liver.
Biomarker for alcohol use / liver damage
Finally, building a statistical prediction model trained on PCSK9 DNA methylation status in AUD subjects and controls, we generated a prediction of high vs. low plasma PCSK9 levels using peripheral blood DNA methylation in an independent test set (Supplementary Fig. 6) documenting the possible use of PCSK9 methylation as a biomarker for alcohol exposure. This finding is important as plasma PCSK9 levels have been shown to act as predictors of treatment response to statin therapy39.
Discussion
We used a cross-tissue, cross-phenotypic, and translational approach to establish that alcohol exposure leads to methylation variation in the promoter region of PCSK9 affecting PCSK9 expression, which might contribute to regulation of low density lipoprotein cholesterol (LDL-C)40-42. Our finding is intriguing, given the direct action of alcohol on the liver which might modulate epigenetic regulation of PCSK9 expression via direct interference of transcription or alteration of transcription factor binding, such as sterol regulatory element-binding protein-2 (SREBP-2) and hepatocyte nuclear factor-1α (HNF1α) (Supplementary Fig. 7). This mechanism could explain the strong epidemiological evidence for the relationship between cardiovascular outcomes and alcohol use. Alcohol consumption and total mortality in cardiovascular disease (CVD) patients has been depicted to have a J-shaped relationship43, such that light to moderate drinking is associated with a reduction in CVD mortality risk while heavy drinking increases CVD mortality risk. PCSK9 methylation alteration may contribute to the downturn of this J-curve. The eventual upturn of risk in later stages of the disease might be due to chronic effects of alcohol on the liver and subsequent metabolic effects (Fig. 5). Our results and model suggest that PCSK9CpG1 could serve as an important biomarker in monitoring the effects of alcohol exposure.
Given the recent FDA approval of monoclonal antibodies as PCSK9 inhibitors in 2015 for patients with heterozygous familial hypercholesterolemia (HeFH) or clinical atherosclerotic cardiovascular disease44, as well as strong recent findings of RNA inhibitor (RNAi) compounds significantly influencing PCSK9 levels45, our finding suggests that the interaction of alcohol and PCSK9 regulation should be further studied in clinical populations. Although statins are the standard of care for hypercholesteremia, not all patients are able to tolerate statins, including patients with alcoholic liver disease. Understanding the relationship between alcohol and PCSK9 regulation may help determine an optimal treatment window for PCSK9 inhibitors in individuals with AUD (Fig. 5). However, given that alcohol dependence is most common in individuals with low socio-economic status46, access to currently expensive PCSK9 inhibitors may be difficult.
Interestingly, we identified methylomic variation of PCSK9 through an investigation of brain tissue and alcohol-associated neuroimaging endophenotypes. Although the role of PCSK9 function in the brain is not well understood, studies have suggested both direct and indirect effects on brain function. PCSK9 is directly involved in low density lipoprotein receptor (LDL-R) degradation as well as other apolipoprotein E-binding receptors47, 48 and plays a protective role in nervous system development49 by mediating cell differentiation47, 50. Indirectly, reports link its function to both decreasing neurite outgrowth through interference with LDL-R neurite induction51 and mediating neuronal apoptosis52 through a mechanism downstream of oxidated-LDL53. PCSK9 has also been implicated in Alzheimer's pathogenesis53-57 through its role in apoptosis53, 58-60 and disposal of non-acetylated β-site amyloid precursor protein (APP)- cleaving enzyme 1 (BACE1), the rate-limiting enzyme in the generation of the Alzheimer's disease amyloid β-peptide55. This is intriguing given recent findings that PCSK9 levels were significantly increased in cerebral spinal fluid (CSF) in patients with Alzheimer's disease30. Thus, future studies could investigate the effects of PCSK9 inhibitors in prevention of cognitive decline in Alzheimer's disease and vascular dementia.
It is likely that PCSK9 inhibitors will have pleiotropic effects that are unrelated to cholesterol regulation, such as effects on inflammation and immune function61, that might be relevant to brain physiology. Although the biological implications of PCSK9 regulation on brain function have not been fully characterized, an increased risk of neurocognitive adverse events in patients receiving PCSK9 inhibitors was recently reported62. However, new data from a large clinical trial showed no correlation between PCSK9 inhibitor treatment and neurocognitive side effects63. One potential explanation for the divergent findings of these studies might be unaccounted differences in alcohol exposure or other clinical characteristics.
While our data supports a dose dependent effect of alcohol exposure on PCSK9 levels (Fig. 5), an important limitation of our study is the lack of longitudinal data and controlled dose escalation of alcohol exposure. In addition, our sample cohorts might have been biased, as AUD is highly prevalent in disadvantaged populations of low socio-economic status46 who often have various other metabolomic risk factors. Another potential confounding factor may have been the presence of rare genetic PCSK9 variants influencing PCSK9 methylation patterns and plasma levels. Although PCSK9 was initially identified as a gain-of-function mutation in cholesterol metabolism in families with a history of familial hypercholesteremia42, loss-of-function PCSK9 missense mutations identified in certain ethnic populations were associated with lowered plasma LDL-C levels and significant protection from CVD48,64, 65. Presence of these rare PCSK9 variants may have potentially affected the PCSK9 expression levels measured in human plasma samples; however, given the low frequencies of such variants, it is unlikely that this had a major impact in our sample.
There are several technical limitations that should be carefully taken into account when interpreting EWAS results. We used array based assessment of genome wide methylation patterns in our study that could be affected by common or rare single nucleotide polymorphisms (SNPs). While we were unable to explore to what degree underlying genetic variation might have contributed to our findings, the role of genome-epigenome interaction is complex and warrants further study66. To address some of these concerns, we conducted comprehensive sequential replication analyses from independent cohorts, as well as biological validation of our main finding. The field of epigenetics is rapidly expanding and it is likely that future higher resolution capture might be able to provide more comprehensive coverage of CpG sites. In addition, as the costs of whole epigenome next-generation sequencing become more affordable, it might be feasible to assess DNA methylation variation more completely, including 5-hydroxymethyl-cytosine variation67. Epigenetic regulation in AUD is complex and most likely involves multiple genes. In this paper, we focused on biological validation of PCSK9. However, other intriguing candidates would include PIP5K1C, which has been implicated in pain signaling68, and Hepatocellular Carcinoma Associated Transcript 5 (HTA/HCCAT5) (Supplementary Table S4). These genes were consistently associated with disease phenotypes in our cross-tissue and cross-phenotypic discovery analysis of genome-wide methylomic variation in AUD.
Our findings could be biased by cell-type specificity of PCSK9 expression (Supplementary Fig. 3, 5), suggesting a need for a cell-type controlled assessment of alcohol's effects on PCSK9 expression in brain, liver, and blood. While the availability of brain tissue remains a challenge for most psychiatric phenotypes, we showed strong correlation of PCSK9 methylation across tissues (Fig. 3), further supporting the idea that use of peripheral biomarkers might be feasible in reflecting methylation status in other tissues. Future studies in AUD might take advantage of the possibility of liver biopsies to investigate cell-specific epigenetic changes longitudinally.
In summary, we provide evidence that alcohol exposure alters PCSK9 methylation and expression. Our data may shed light on novel mechanisms by which CVD risk can be altered. In addition, PCSK9 methylation might be an important biomarker that can track alcohol exposure and might predict treatment response, efficacy, and dosing regiments for individuals undergoing PCSK9 inhibitor therapy.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (NIH) intramural funding ZIA-AA000242 (Section on Clinical Genomics and Experimental Therapeutics; to FWL), Division of Intramural Clinical and Biological Research of the National Institute on Alcohol Abuse and Alcoholism (NIAAA).
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Grant BF, Goldstein RB, Saha TD, Chou SP, Jung J, Zhang H, et al. Epidemiology of DSM-5 alcohol use disorder: results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA psychiatry. 2015;72(8):757–766. doi: 10.1001/jamapsychiatry.2015.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tawa EA, Hall SD, Lohoff FW. Overview of the Genetics of Alcohol Use Disorder. Alcohol Alcohol. 2016 doi: 10.1093/alcalc/agw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ron D, Barak S. Molecular mechanisms underlying alcohol-drinking behaviours. Nature reviews Neuroscience. 2016;17(9):576–591. doi: 10.1038/nrn.2016.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nature reviews Neuroscience. 2011;12(11):623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Herman AI, Kranzler HR, Anton RF, Zhao H, Zheng W, et al. Array-based profiling of DNA methylation changes associated with alcohol dependence. Alcohol Clin Exp Res. 2013;37 Suppl 1:E108–115. doi: 10.1111/j.1530-0277.2012.01928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Wang F, Kranzler HR, Zhao H, Gelernter J. Profiling of childhood adversity-associated DNA methylation changes in alcoholic patients and healthy controls. PLoS One. 2013;8(6):e65648. doi: 10.1371/journal.pone.0065648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng JT, Wu LS, Lee CS, Hsu PW, Cheng AT. Integrative epigenetic profiling analysis identifies DNA methylation changes associated with chronic alcohol consumption. Comput Biol Med. 2015;64:299–306. doi: 10.1016/j.compbiomed.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Zhang R, Miao Q, Wang C, Zhao R, Li W, Haile CN, et al. Genome-wide DNA methylation analysis in alcohol dependence. Addict Biol. 2013;18(2):392–403. doi: 10.1111/adb.12037. [DOI] [PubMed] [Google Scholar]
- 9.Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SR. The impact of recent alcohol use on genome wide DNA methylation signatures. Frontiers in genetics. 2012;3:54. doi: 10.3389/fgene.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao R, Zhang R, Li W, Liao Y, Tang J, Miao Q, et al. Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia Pac Psychiatry. 2013;5(1):39–50. doi: 10.1111/appy.12010. [DOI] [PubMed] [Google Scholar]
- 11.Philibert RA, Penaluna B, White T, Shires S, Gunter T, Liesveld J, et al. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics. 2014;9(9):1212–1219. doi: 10.4161/epi.32252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2016 doi: 10.1038/mp.2016.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu H, Wang F, Liu Y, Yu Y, Gelernter J, Zhang H. Sex-biased methylome and transcriptome in human prefrontal cortex. Hum Mol Genet. 2014;23(5):1260–1270. doi: 10.1093/hmg/ddt516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8(3):290–302. doi: 10.4161/epi.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.First M, Spitzer RL, Gibbon M, Williams J. Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I), Clinician Version. American Psychiatric Press; Washington, D.C.: 1996. [Google Scholar]
- 16.Skinner HA, Horn JL Addiction Research Foundation of Ontario. Alcohol Dependence Scale (ADS) vol v, 38 leaves. Addiction Research Foundation; Toronto: 1984. [Google Scholar]
- 17.Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: The revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar) British Journal of Addiction. 1989;84:1353–1357. doi: 10.1111/j.1360-0443.1989.tb00737.x. [DOI] [PubMed] [Google Scholar]
- 18.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. NeuroImage. 2002;17(2):825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 19.Smith SM. Fast robust automated brain extraction. Human brain mapping. 2002;17(3):143–155. doi: 10.1002/hbm.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Medical image analysis. 2001;5(2):143–156. doi: 10.1016/s1361-8415(01)00036-6. [DOI] [PubMed] [Google Scholar]
- 21.Beckmann CF, Smith SM. Probabilistic independent component analysis for functional magnetic resonance imaging. IEEE transactions on medical imaging. 2004;23(2):137–152. doi: 10.1109/TMI.2003.822821. [DOI] [PubMed] [Google Scholar]
- 22.Hyvarinen A. Fast and robust fixed-point algorithms for independent component analysis. IEEE transactions on neural networks / a publication of the IEEE Neural Networks Council. 1999;10(3):626–634. doi: 10.1109/72.761722. [DOI] [PubMed] [Google Scholar]
- 23.Biswal BB, Mennes M, Zuo XN, Gohel S, Kelly C, Smith SM, et al. Toward discovery science of human brain function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(10):4734–4739. doi: 10.1073/pnas.0911855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pidsley R, CC YW, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation 450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gillespie CF, Bradley B, Mercer K, Smith AK, Conneely K, Gapen M, et al. Trauma exposure and stress-related disorders in inner city primary care patients. General Hospital Psychiatry. 2009;31(6):505–514. doi: 10.1016/j.genhosppsych.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470(7335):492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299(11):1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, et al. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Archives of general psychiatry. 2008;65(2):190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zimetti F, Caffarra P, Ronda N, Favari E, Adorni MP, Zanotti I, et al. Increased PCSK9 Cerebrospinal Fluid Concentrations in Alzheimer's Disease. J Alzheimers Dis. 2017;55(1):315–320. doi: 10.3233/JAD-160411. [DOI] [PubMed] [Google Scholar]
- 31.Kaminsky Z, Petronis A. Methylation SNaPshot: a method for the quantification of site-specific DNA methylation levels. Methods Mol Biol. 2009;507:241–255. doi: 10.1007/978-1-59745-522-0_18. [DOI] [PubMed] [Google Scholar]
- 32.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marabita F, Almgren M, Lindholm ME, Ruhrmann S, Fagerstrom-Billai F, Jagodic M, et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics. 2013;8(3):333–346. doi: 10.4161/epi.24008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hlady RA, Tiedemann RL, Puszyk W, Zendejas I, Roberts LR, Choi JH, et al. Epigenetic signatures of alcohol abuse and hepatitis infection during human hepatocarcinogenesis. Oncotarget. 2014;5(19):9425–9443. doi: 10.18632/oncotarget.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8(3):627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu X, Dutta N, Helton SG, Schwandt M, Yan J, Hodgkinson CA, et al. Resting-state functional connectivity and presynaptic monoamine signaling in Alcohol Dependence. Human brain mapping. 2015;36(12):4808–4818. doi: 10.1002/hbm.22951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.First M, Spitzer RL, Gibbon M, Williams J. Structured Clinical Interview for DSM-IV Axis I Disorders (SCID-I), Clinician Version. American Psychiatric Press; Washington, D.C.: 1996. [Google Scholar]
- 38.Kellogg SH, McHugh PF, Bell K, Schluger JH, Schluger RP, LaForge KS, et al. The Kreek-McHugh-Schluger-Kellogg scale: a new, rapid method for quantifying substance abuse and its possible applications. Drug Alcohol Depend. 2003;69(2):137–150. doi: 10.1016/s0376-8716(02)00308-3. [DOI] [PubMed] [Google Scholar]
- 39.Taylor BA, Panza G, Pescatello LS, Chipkin S, Gipe D, Shao W, et al. Serum PCSK9 Levels Distinguish Individuals Who Do Not Respond to High-Dose Statin Therapy with the Expected Reduction in LDL-C. Journal of lipids. 2014;2014:140723. doi: 10.1155/2014/140723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cariou B, Si-Tayeb K, Le May C. Role of PCSK9 beyond liver involvement. Current opinion in lipidology. 2015;26(3):155–161. doi: 10.1097/MOL.0000000000000180. [DOI] [PubMed] [Google Scholar]
- 41.Joseph L, Robinson JG. Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibition and the Future of Lipid Lowering Therapy. Progress in cardiovascular diseases. 2015;58(1):19–31. doi: 10.1016/j.pcad.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 43.Costanzo S, Di Castelnuovo A, Donati MB, Iacoviello L, de Gaetano G. Alcohol consumption and mortality in patients with cardiovascular disease: a meta-analysis. J Am Coll Cardiol. 2010;55(13):1339–1347. doi: 10.1016/j.jacc.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Hooper AJ, Burnett JR. Anti-PCSK9 therapies for the treatment of hypercholesterolemia. Expert Opin Biol Ther. 2013;13(3):429–435. doi: 10.1517/14712598.2012.748743. [DOI] [PubMed] [Google Scholar]
- 45.Fitzgerald K, White S, Borodovsky A, Bettencourt BR, Strahs A, Clausen V, et al. A Highly Durable RNAi Therapeutic Inhibitor of PCSK9. N Engl J Med. 2017;376(1):41–51. doi: 10.1056/NEJMoa1609243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Prev Chronic Dis. 2014;11:E206. doi: 10.5888/pcd11.140329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(3):928–933. doi: 10.1073/pnas.0335507100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1) PLoS One. 2013;8(5):e64145. doi: 10.1371/journal.pone.0064145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res. 2016;112(1):429–442. doi: 10.1093/cvr/cvw194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poirier S, Prat A, Marcinkiewicz E, Paquin J, Chitramuthu BP, Baranowski D, et al. Implication of the proprotein convertase NARC-1/PCSK9 in the development of the nervous system. J Neurochem. 2006;98(3):838–850. doi: 10.1111/j.1471-4159.2006.03928.x. [DOI] [PubMed] [Google Scholar]
- 51.Do HT, Bruelle C, Pham DD, Jauhiainen M, Eriksson O, Korhonen LT, et al. Nerve growth factor (NGF) and pro-NGF increase low-density lipoprotein (LDL) receptors in neuronal cells partly by different mechanisms: role of LDL in neurite outgrowth. J Neurochem. 2016;136(2):306–315. doi: 10.1111/jnc.13397. [DOI] [PubMed] [Google Scholar]
- 52.Kysenius K, Muggalla P, Matlik K, Arumae U, Huttunen HJ. PCSK9 regulates neuronal apoptosis by adjusting ApoER2 levels and signaling. Cellular and molecular life sciences : CMLS. 2012;69(11):1903–1916. doi: 10.1007/s00018-012-0977-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu Q, Tang ZH, Peng J, Liao L, Pan LH, Wu CY, et al. The dual behavior of PCSK9 in the regulation of apoptosis is crucial in Alzheimer's disease progression (Review) Biomedical reports. 2014;2(2):167–171. doi: 10.3892/br.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu M, Wu G, Baysarowich J, Kavana M, Addona GH, Bierilo KK, et al. PCSK9 is not involved in the degradation of LDL receptors and BACE1 in the adult mouse brain. J Lipid Res. 2010;51(9):2611–2618. doi: 10.1194/jlr.M006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9(9):916–922. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Banerjee Y, Santos RD, Al-Rasadi K, Rizzo M. Targeting PCSK9 for therapeutic gains: Have we addressed all the concerns? Atherosclerosis. 2016;248:62–75. doi: 10.1016/j.atherosclerosis.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 57.Xue-shan Z, Juan P, Qi W, Zhong R, Li-hong P, Zhi-han T, et al. Imbalanced cholesterol metabolism in Alzheimer's disease. Clin Chim Acta. 2016;456:107–114. doi: 10.1016/j.cca.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 58.Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS, Liu LS. PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem. 2012;359(1-2):347–358. doi: 10.1007/s11010-011-1028-6. [DOI] [PubMed] [Google Scholar]
- 59.Chiang LW, Grenier JM, Ettwiller L, Jenkins LP, Ficenec D, Martin J, et al. An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2814–2819. doi: 10.1073/pnas.051630598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bingham B, Shen R, Kotnis S, Lo CF, Ozenberger BA, Ghosh N, et al. Proapoptotic effects of NARC 1 (= PCSK9), the gene encoding a novel serine proteinase. Cytometry A. 2006;69(11):1123–1131. doi: 10.1002/cyto.a.20346. [DOI] [PubMed] [Google Scholar]
- 61.Bittner V. Pleiotropic Effects of PCSK9 (Proprotein Convertase Subtilisin/Kexin Type 9) Inhibitors? Circulation. 2016;134(22):1695–1696. doi: 10.1161/CIRCULATIONAHA.116.023687. [DOI] [PubMed] [Google Scholar]
- 62.Swiger KJ, Martin SS. PCSK9 inhibitors and neurocognitive adverse events: exploring the FDA directive and a proposal for N-of-1 trials. Drug Saf. 2015;38(6):519–526. doi: 10.1007/s40264-015-0296-6. [DOI] [PubMed] [Google Scholar]
- 63.Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N Engl J Med. 2017;376(18):1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 64.Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 65.Cohen JC, Boerwinkle E, Mosley TH, Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 66.Meaburn EL, Schalkwyk LC, Mill J. Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics : official journal of the DNA Methylation Society. 2010;5(7):578–582. doi: 10.4161/epi.5.7.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meaburn E, Schulz R. Next generation sequencing in epigenetics: insights and challenges. Semin Cell Dev Biol. 2012;23(2):192–199. doi: 10.1016/j.semcdb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 68.Wright BD, Loo L, Street SE, Ma A, Taylor-Blake B, Stashko MA, et al. The lipid kinase PIP5K1C regulates pain signaling and sensitization. Neuron. 2014;82(4):836–847. doi: 10.1016/j.neuron.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.