

Abstract
The biochemical, ionic, and signaling changes that occur within cardiomyocytes subjected to ischemia are exacerbated by reperfusion; however, the precise mechanisms mediating myocardial ischemia/reperfusion (I/R) injury have not been fully elucidated. The receptor for advanced glycation end-products (RAGE) regulates the cellular response to cardiac tissue damage in I/R, an effect potentially mediated by the binding of the RAGE cytoplasmic domain to the diaphanous-related formin, DIAPH1. The aim of this study was to investigate the role of DIAPH1 in the physiological response to experimental myocardial I/R in mice. After subjecting wild-type mice to experimental I/R, myocardial DIAPH1 expression was increased, an effect that was echoed following hypoxia/reoxygenation (H/R) in H9C2 and AC16 cells. Further, compared to wild-type mice, genetic deletion of Diaph1 reduced infarct size and improved contractile function after I/R. Silencing Diaph1 in H9C2 cells subjected to H/R downregulated actin polymerization and serum response factor-regulated gene expression. Importantly, these changes led to increased expression of sarcoplasmic reticulum Ca2 + ATPase and reduced expression of the sodium calcium exchanger. This work demonstrates that DIAPH1 is required for the myocardial response to I/R, and that targeting DIAPH1 may represent an adjunctive approach for myocardial salvage after acute infarction.
Abbreviations: DIAPH1, diaphanous 1; EGR1, early growth response 1; F actin, filamentous (F) actin; Fos, gene for c-Fos; G actin, globular (G) actin; GSK3B, glycogen synthase kinase 3 beta; Ex vivo I/R, ex vivo ischemia/reperfusion; H/R, hypoxia/reoxygenation; I/R, ischemia/reperfusion; LAD/reperfusion, left anterior descending coronary artery occlusion followed by reperfusion; LDH, lactate dehydrogenase; NCX1, sodium-calcium exchanger; PLN, phospholamban; Rac1, Ras-related C3 botulinum toxin substrate 1; ROCK2, Rho-associated protein kinase 2; ShRNA, short-hairpin RNA; SRF, serum response factor; Tagln, gene for Smooth Muscle Protein 22-Alpha; TTC, 2,3,5-triphenyl-2H-tetrazolium chloride; Slc8a1, gene for NCX1; SERCA2a, sarcoplasmic reticulum calcium ATPase 2a
Keywords: Formins, DIAPH1, Actin polymerization, Heart, Ischemia/reperfusion injury
Graphical Abstract
Highlights
-
•
DIAPH1 is a key mediator of ischemia/reperfusion injury in the heart.
-
•
Diaph1 deletion regulates the transcription factor, serum response factor, leading to modulation of calcium transporters in cardiomyocytes.
-
•
The effects of Diaph1 deletion are associated with improved contractile function after short-term ischemia/reperfusion injury, making it a potential adjunctive therapeutic target for treatment of evolving myocardial infarction.
Although reperfusion therapy, using thrombolytic agents, has become the mainstay for the treatment of patients with myocardial infarction, benefits of this therapy are reduced by multiple factors including reperfusion injury. The precise mechanisms for this effect are yet to be fully delineated. In this study we identify the formin DIAPH1 as a novel mediator of ischemia/reperfusion (I/R) injury in the heart. We show that Diaph1 deletion regulates the transcription factor, serum response factor, leading to modulation of calcium transporters in cardiomyocytes. We show that deletion of Diaph1 improved contractile function after I/R in the mouse heart, making DIAPH1 a potential adjunctive therapeutic target for the treatment of evolving myocardial infarction.
1. Introduction
The extent of myocardial tissue loss, as assessed by infarct size, is a key determinant of the prognosis of patients with acute myocardial infarction (Herlitz et al., 1984, Herlitz et al., 1987). Hence, limiting the infarct size is of paramount importance to ameliorate subsequent morbidity and mortality in affected patients (Pfeffer and Braunwald, 1990, St John Sutton et al., 1997, Miller et al., 1995, Miller et al., 1998, Gibbons et al., 2004). Given the magnitude of the problem (> 900,000 acute myocardial infarctions per year in the United States (Roger et al., 2012), the implications of developing effective cardioprotective therapies are immense, both from the perspective of public health and the financial burden of health care. Though reperfusion therapy using thrombolytic agents has become the mainstay for treatment of patients with myocardial infarction, the benefits of this therapy have been shown to be reduced by factors such as severity of ischemia, inadequate reflow, presence of residual stenosis, coronary reocclusion, and reperfusion injury (Sobel DPDaBE, 2001, Solomon et al., 2001). The biochemical, ionic, and signal transduction changes that occur within cardiomyocytes subjected to ischemia are exacerbated by reperfusion. Elucidation of the precise mechanisms mediating myocardial ischemia/reperfusion (I/R) injury is vital to accelerate the discovery of new and powerful adjunctive therapies to enhance the net benefit of reperfusion.
The signaling pathways involved in the response to myocardial I/R injury are complex. Intracellular signal transducers such as the Rho GTPases have been shown to modulate the response to I/R injury in mice (Shimokawa and Takeshita, 2005, Talukder et al., 2013, Xiang et al., 2011). Formins modulate Rho-GTPase signal transduction mechanisms (Narumiya et al., 2009) and regulate actin polymerization (Romero et al., 2004). The mammalian Diaphanous-related formin 1 (DIAPH1; also known as mDia1) is a canonical effector for Rho small GTP-binding proteins. When activated by GTP-bound RhoA, DIAPH1 relies upon its formin homology-2 domain to elongate linear actin filaments by adding actin monomers to their barbed ends (Romero et al., 2004). DIAPH1 also signals to the nucleus via the myocardin-related transcription factor/serum response factor (SRF) axis to effect gene expression (Mikhailov and Torrado, 2012). DIAPH1 interacts with the cytoplasmic domain of the receptor for advanced glycation end-products (RAGE) through its poly-proline-rich formin homology-1 domain. In response to RAGE ligand stimulation, DIAPH1 participates in signal transduction in smooth muscle cells (Toure et al., 2008), macrophages (Xu et al., 2010, Colucci-Guyon et al., 2005), immune cells (Shi et al., 2009), and transformed cells (Hudson et al., 2008). While RAGE is known to be an essential component of the in vivo cardiac response to I/R, knowledge of the role of DIAPH1 in such a response is unexplored.
To fill this knowledge gap, we subjected global Diaph1 knockout (Diaph1–/–) mice, deficient in DIAPH1 protein expression, and their wild-type (WT) littermates to I/R injury. We demonstrate that DIAPH1 expression was induced by I/R in murine hearts and mediated I/R injury through an F:G actin-regulated transcriptional mechanism that modulated calcium transporters, thereby leading to functional derangement in the I/R heart.
2. Materials and Methods
2.1. Animal Studies
All procedures were approved by the Institutional Animal Care and Use Committee of New York University Langone Medical Center (NYULMC) and conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH) (8th Edition, 2011, ISBN 10: 0-309-15400-6). The Animal Care and Use Program at NYULMC is in full compliance with NIH policy (NYULMC Compliance Number is A3435-01).
Homozygous Diaph1–/– mice were generated at the Van Andel Institute and obtained from Dr. Arthur Alberts (Peng et al., 2003). The mice were backcrossed 14 generations into C57BL/6J prior to any experimentation. Non-transgenic WT littermates were used as controls for in vivo and ex vivo I/R studies, as described below. Mouse studies were performed using male WT and Diaph1–/– mice aged 4–6 months as specified.
2.2. In Vivo I/R
Male mice aged approximately 4-6 months were anesthetized with an intraperitoneal injection of ketamine and xylazine (120 mg/kg body weight and 5 mg/kg body weight, respectively) and subjected to left anterior descending coronary artery occlusion followed by reperfusion (LAD/reperfusion) as previously described (Aleshin et al., 2008). The surgical procedures were performed by an operator naive to the mouse genotype. Briefly, the LAD was ligated for 30 min and then blood flow was restored. Sham animals were subjected to the surgical procedure without LAD occlusion. After 48 h, mice were anesthetized with isoflurane (4% to induce and 1.5–2% to maintain anesthesia, inhaled via a nose cone) for the duration of the procedure and 2-dimensional echocardiography was performed on a Vevo 2100 System (Visual Sonics, Toronto, Ontario, Canada). Echocardiographic images were analyzed by a researcher naive to the genotype. The left ventricular end-diastolic and end-systolic dimensions were measured and percent fractional shortening was calculated. Mice were then anesthetized with an intraperitoneal injection of ketamine and xylazine (100 mg/kg body weight and 10 mg/kg body weight, respectively) and the hearts were excised. A subset of hearts was used to assess area at risk and infarct area by 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) and Evan's blue staining. The remainders of the hearts were collected for biochemical analysis.
2.3. Ex Vivo I/R
Mice were anesthetized with an intraperitoneal injection of ketamine and xylazine (100 mg/kg body weight and 10 mg/kg body weight, respectively) and then hearts were isolated from WT and Diaph1–/– mice aged 4 months and perfused with Krebs-Henseleit buffer containing (in mM) the following: 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 5 glucose, 0.4 palmitate, 0.4 BSA, and 70 mU/l insulin, as previously described (Hwang et al., 2004). Left ventricular developed pressure (LVDP), left ventricular end diastolic pressure (LVEDP) and coronary perfusion pressure (PP) was continuously monitored using pressure transducer (ADInstruments). After an equilibration period of 30 min, global ischemia was performed for 30 min in the hearts, followed by 60 min of reperfusion (global I/R).
2.4. Cell Studies
Rat H9C2 cardiomyoblasts (ATCC, Manassas, VA) were cultured to assess the effects of in vitro I/R injury. Short-hairpin RNA (shRNA) particles (Sigma-Aldrich, St. Louis, MO) were used to silence Diaph1 (shDiaph1) according to the manufacturer’s instructions. Control H9C2 cells underwent the same protocol using control shRNA particles with a scrambled sequence (shScr). For subsequent experiments, shScr and shDiaph1 cells were placed in a hypoxia chamber (Biospherix, Lacona, NY) for 30 min of hypoxia (0.5% oxygen, 5% CO2) at 37 °C followed by 60 min of reoxygenation in 5% CO2 incubator (hypoxia/reoxygenation, H/R). Control cells were kept in a standard cell culture incubator (37 °C, 5% CO2) to simulate normoxia. All cell studies (including the hypoxia-reoxygenation studies) used DMEM supplemented with 10% FBS and 1 % penicillin-streptomycin. Cells were collected at the end of reoxygenation in ice cold PBS for further biochemical analysis. Lactate dehydrogenase (LDH) release into the media was measured to assess cardiomyocyte injury using a commercially available kit (Pointe Scientific, Canton, MI). The cells were treated with 100 nmol/l jasplakinolide, 1 μmol/l latrunculin B, or DMSO vehicle (Sigma-Aldrich) for 1 h prior to hypoxia when specified. Early growth response 1 (EGR1) was overexpressed in shScr and shDiaph1 cells using an adenoviral vector (Vector Biolabs, Malvern, PA). We subjected human AC16 cardiomyocytes (Davidson et al., 2005) (obtained as a gift from Dr. Mercy Davidson at Columbia University, New York, NY) to H/R when noted.
Primary cardiomyocytes were isolated from SHAM or LAD/reperfused adult mice hearts by a modified method as published by us and others (Das et al., 2005, Shang et al., 2010). In brief, hearts excised from anesthetized mice were subjected to Langendorff perfusion with Krebs medium containing calcium (2.5 mM) for 5 min, followed by perfusion with Calcium free Krebs medium (8–10 min). Hearts were then perfused with an enzymatic solution containing collagenase type II (0.35 mg/ml; Worthington, Freehold, NJ) and protease type XIV (0.01 mg/ml, Sigma) for 5–8 min. 50 μM Ca2 + was then added into the enzyme solution for perfusing the heart for another 5–10 min until the hearts became soft. The hearts were then removed, minced into small pieces and subjected to further serial enzymatic digestion at 37° C for ~ 3 min. Cardiomyocytes in the digests were collected by centrifugation at 500 rpm and the myocyte pellet was resuspended in storage medium (Ca2 + free Kreb's containing 1% BSA and Calcium (125 μM). Extracellular Ca2 + was incrementally added back to 500 μM over a period of 40 min. The rod shaped myocytes settled down immediately, whereas round ones and other cells were floating in the supernatant. By aspirating the supernatant and repeated washing, a rod-shaped myocyte population was obtained. Cardiomyocytes were incubated in Kreb's buffer containing 1 mM Ca2 + for the following study. Cell viability was assessed by using the commercially available CellTiter-FluorTM kit (Promega, Madison, WI, USA). Characterization of cardiomyocytes was done by staining with α-sarcomeric actinin antibody (Sigma, St. Louis, MO, USA) and FACS study.
2.5. Biochemical Analyses
Extracts from mouse hearts, H9C2 cells, and AC16 cells were obtained by homogenization in cell lysis buffer (Cell Signaling, Danvers, MA). Nuclear fractions were prepared using a commercially available kit (Thermo Scientific, Rockford, IL). Protein concentrations were determined using a DC Protein Assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were separated by SDS-PAGE and proteins were transferred to a nitrocellulose membrane (Life Technologies, Carlsbad, CA), which was probed with primary antibodies according to the manufacturer's instructions. We used primary antibodies (1:1000) as follows: mDia1 (DIAPH1), β-actin, ROCK2, nucleoporin p62 (BD Biosciences, San Jose, CA), pan-actin (Cytoskeleton, Denver, CO), GAPDH (Genetex, Irvine, CA), EGR1, SERCA2a (Santa Cruz, Dallas, TX), DIAPH1, myocardin, NCX1, calsequestrin, DIAPH2, histone 3, GAPDH (AbCam, Cambridge, UK), phosphorylated (ser16) and total phospholamban (EMD Millipore, Billerica, MA), GAPDH (Genetex, Irvine, CA), phosphorylated and total Rac1, RhoA, phosphorylated (ser9) and total GSK3β (Cell Signaling, Danvers, MA). We visualized and quantitated blots with a fluorescent western blot detection system (Licor, Lincoln, NE).
We measured the ratio of filamentous to globular (F:G) actin using a kit according to the manufacturer (Cytoskeleton, Denver, CO). Briefly, cell or tissue extracts were homogenized in F actin stabilization buffer, centrifuged to separate F and G actin, and then probed using Western blotting with a pan-actin antibody. In addition, we also used complimentary phalloidin/DNase I Staining method to determine F:G changes in H9C2 cells. Briefly, sub-confluent shScr and shDiaph1 H9C2 cells grown on gelatin-coated coverslips were placed in a hypoxia chamber (Biospherix, Lacona, NY) for either normoxia or 30 min of hypoxia (0.5% oxygen, 5% CO2) at 37° C followed by 60 min of reoxygenation (H/R). After these treatments, cells were washed with ice-cold PBS fixed in formaldehyde, permeabilized and stained in Alexa Fluor 647 Phalloidin and Deoxyribonuclease I Alexa Fluor 594 (Molecular Probes). Confocal microscopy was performed on a Zeiss LSM 510 Confocal Microscope, Single field images of 1024 × 1024 pixels were collected to visualise F-actin labelled with Alexa Fluor 647 Phalloidin and G-actin labelled with Alexa Fluor® 594 DNase I.
RNA was isolated from H9C2 cells using a commercially available kit (Qiagen, Valencia, CA) followed by reverse transcription (Bio-Rad) and RT-PCR using rat primers for Srf, Tagln (gene for Sm22), and Fos (the gene for c-Fos), with all values normalized to Ppia (Life Technologies).
2.6. Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the ChIP-IT kit (Active Motif, Carlsbad, CA). Briefly, mouse hearts subjected to LAD/reperfusion were crosslinked with formaldehyde in PBS for 15 min at room temperature and were processed as per instructions provided by the manufacturer to obtain chromatin. The chromatin was initially pre-cleared using protein-G beads and subsequently treated with SRF antibody (Abcam, Cambridge, UK) at a concentration of 1:200 for immunoprecipitation overnight. The immunoprecipitated ChIP product was washed, reverse crosslinked, treated with proteinase K, and eluted to obtain the ChIP enriched DNA. Simultaneously, IgG was used as a negative control to check the efficiency of the ChIP experiment. Wild Type heart lysates were incubated with negative control IgG for ChIP pulldown. 25 ng of ChIP enriched DNA was used to perform PCR for Egr1, Atp2a2, and Slc8a1. The PCR products were run on a 2% agarose gel and the amplicons were visualized using UV light.
2.7. Statistics
All values are presented as the mean ± standard error of the mean. Data were analyzed by unpaired two-tailed t-tests, and one-way or two-way ANOVAs to assess the difference between groups as specified. A probability value of ≤ 0.05 was considered significant.
3. Results
3.1. I/R Injury Increases DIAPH1 Expression
To evaluate the effects of I/R on DIAPH1 expression, we subjected male WT mice, aged 4-6 months, to left anterior descending coronary artery (LAD) occlusion for 30 min, followed by 48 h of reperfusion (LAD/reperfusion) or to sham surgery, and we measured DIAPH1 expression in whole cardiac tissue by immunoblotting using DIAPH1-specific antibodies. LAD/reperfused mouse hearts displayed a significant increase in DIAPH1 protein expression relative to sham controls (Fig. 1a). Thirty minutes of LAD occlusion followed by either 6, 24 or 48 h of reperfusion resulted in significant increases in DIAPH1 expression in mouse hearts compared with sham treatment, as assessed by Western blot analysis (P < 0.05; Fig. 1b). Cardiomyocytes isolated from WT mouse hearts after 48 h of reperfusion exhibited significant increases in Diaph1 gene expression compared to cardiomyocytes isolated from sham treated hearts (Fig. 1c.). To confirm these results in vitro, H9C2 rat cardiomyoblasts (Fig. 1d) and AC16 human cardiomyocytes (Supplementary Fig S1) were subjected to 30 min of hypoxia followed by 60 min of reoxygenation (hypoxia/reoxygenation, H/R), and both displayed elevated levels of DIAPH1 relative to cells maintained at normal oxygen levels.
Fig. 1.

I/R injury increased DIAPH1 protein levels. (a) Hearts from wild-type (WT) C57BL/6 mice were retrieved, and lysates were subjected to Western blot analysis for the detection of DIAPH1 epitopes in mice subject to sham procedure (48 h) or at 48 h of reperfusion. After probing with the primary antibody, blots were reprobed with anti-GAPDH IgG. Relative density units are reported; n = 5 mice/group. (a) DIAPH1/GAPDH expression is increased in WT mouse hearts after LAD/reperfusion (n = 8/group; *p < 0.05 vs. sham). (b) To probe for DIAPH1 changes at various time points during reperfusion, hearts were retrieved at 6 or 24, or 48 h after reperfusion or 48 h after sham procedure (identical anesthesia and surgery but without LAD occlusion) and lysates were subjected to Western blot analysis as in 1a. (n = 4/group; *p < 0.05 vs. sham). (c) mRNA expression of Diaph1 was increased in cardiomyocytes isolated from hearts after 48 h of recovery (n = 5/group; *p < 0.05 vs sham), (d) DIAPH1/GAPDH expression is increased in H9C2 cells after H/R (n = 5–8/group; *p < 0.05 vs. normoxia). Unpaired two-tailed t-tests were used; a p-value < 0.05 was considered significant.
3.2. Diaph1 Deletion Reduces Infarct Size and Damage After I/R
To determine potential roles for DIAPH1 in the heart after I/R-mediated injury, we subjected Diaph1–/– and WT mice to LAD/reperfusion. Infarct size was significantly lower in Diaph1–/– mice than in WT at 48 h, without differences in area at risk (Fig. 2a–b). At 48 h, Diaph1 deletion resulted in a significantly higher fractional shortening (Fig. 2c) and ejection fraction (Fig. 2d), as measured by echocardiography (Supplementary Fig S2), relative to WT mice after LAD/reperfusion (Fig. 2c). Other cardiac functional measures such as stroke volume were also improved in Diaph1–/– hearts (Table 1). At baseline, prior to LAD ligation, cardiac functional measures were similar in WT and Diaph1–/– hearts (Table 1). Isolated perfused Diaph1–/– mouse hearts subjected to global ischemia followed by reperfusion (ex vivo I/R) displayed significantly improved recovery of left ventricular developed pressure during reperfusion, relative to WT hearts (Fig. 2e). Left ventricular end diastolic pressure was attenuated in Diaph1–/– mouse hearts after ex vivo I/R compared to WT hearts (Table 2). In cultured H9C2 cells, the release of lactate dehydrogenase (LDH; a marker of injury induced by H/R), was significantly attenuated in short-hairpin (sh)Diaph1-silenced cells relative to control shRNA cells (shScr; Fig. 2f). These results demonstrate that genetic deletion of Diaph1 protects hearts from pathological and functional indices of I/R injury.
Fig. 2.

Diaph1 deletion reduced myocardial I/R injury. (a) Diaph1 deletion decreased infarct area (% of area at risk) in mouse hearts after LAD/reperfusion vs. WT (n = 10/group; *p < 0.05 vs. WT LAD) while (b) maintaining area at risk. (c) Diaph1 deletion improved fractional shortening after LAD/reperfusion (n = 10/group; *p < 0.05 vs. WT LAD). (d) Diaph1 deletion improved ejection fraction after LAD/reperfusion (n = 10/group; *p < 0.05 vs. WT LAD) (e) Left ventricular developed pressure (LVDP) recovery was improved in hearts isolated from Diaph1–/– mice compared to WT after ex vivo I/R (n = 6/group; *p < 0.05 vs. WT I/R). (f) LDH release was reduced in shDiaph1 cells after H/R compared to shScr (n = 15–16/group; *p < 0.05 vs. shScr H/R). Unpaired two-tailed t-tests were used; a p-value < 0.05 was considered significant.
Table 1.
Echocardiography measurements of cardiac function in LAD/reperfused hearts.
| Parameter | WT | Diaph1-/- | P value |
|---|---|---|---|
| Pre LAD/reperfusion | |||
| Heart rate (beats/min) | 556 ± 63 | 532 ± 46 | NS |
| LV ESD (mm) | 1.81 ± 0.1 | 1.78 ± 0.2 | NS |
| LV EDD (mm) | 2.8 ± 0.2 | 2.9 ± 0.3 | NS |
| LV mass (mg) | 98 ± 14 | 116 ± 18 | NS |
| Cardiac output (ml/min) | 10.6 ± 2.1 | 11. ± 1.9 | NS |
| Stroke volume (ml/min) | 19.8 ± 2.2 | 24.5 ± 3.6 | NS |
| LV thickness at end systole (mm) | 1.44 ± .32 | 1.54 ± 0.36 | NS |
| Post LAD/reperfusion (48 h after reperfusion) | |||
| Heart rate (beats/min) | 496 ± 28 | 502 ± 35 | NS |
| LV ESD (mm) | 3.6 ± 0.4 | 2.2 ± 0.3 | < 0.05 |
| LV EDD (mm) | 4.1 ± 0.5 | 3.3 ± 0.2 | < 0.05 |
| LV mass (mg) | 112 ± 19 | 121 ± 22 | NS |
| Cardiac output (ml/min) | 12.6 ± 1.5 | 17.8 ± 2.6 | < 0.05 |
| Stroke volume (ml/min) | 21.3 ± 4.8 | 36.4 ± 2.9 | < 0.05 |
| LV wall thickness at end systole(mm) | 1.38 ± 0.24 | 1.41 ± 0.19 | NS |
LVESD: left ventricular end systolic diameter, LVEDD: left ventricular end diastolic diameter, n = 10/group.
Table 2.
Cardiac function in ex vivo I/R hearts.
| Group | LVDP (mm Hg) | LVEDP (mmHg) | PP (mmHg) |
|---|---|---|---|
| Baseline | |||
| WT | 88.5 ± 6.2 | 5.6 ± 1.9 | 72.6 ± 12.1 |
| Diaph1-/- | 83.1 ± 5.6 | 5.2 ± 2.1 | 88.1 ± 19.5 |
| I/R | |||
| WT | 31.8 ± 3.9 | 28.6 ± 6.6 | 96.5 ± 18.6 |
| Diaph1-/- | 51.2 ± 6.8⁎ | 7.9 ± 2.9# | 82.7 ± 16.9 |
LVDP: left ventricular developed pressure, LVEDP: left ventricular end diastolic pressure, PP: coronary perfusion pressure. n = 6/group.
P < 0.05 vs WT for LVDP comparison in I/R conditions.
P < 0.05 vs WT for LVEDP comparison in I/R conditions.
3.3. Diaph1 Deletion Affects F:G Actin Ratio but Minimally Impacts RhoA/Rac1-regulated Signaling
Since formins modulate Rho-GTPase signal transduction (Narumiya et al., 2009) and regulate actin polymerization (Watanabe, 2013), we asked whether DIAPH1 mediates its effects in I/R via these mechanisms. RhoA directly stimulates DIAPH1, leading to direct interaction with profilin and stimulation of actin polymerization. Changes in ratio of filamentous (F) to globular (G) actin, a measure of actin polymerization, have been linked to modulation of signal transduction and transcriptional events in various cell types. Western blots (Fig. 3a) and phalloidin/DNase I staining method using confocal microscopy (Fig 3b) was used to investigate if DIAPH1 modulates changes in F:G actin. In the current study, the ratio of F:G actin was reduced in cultured shDiaph1 H9C2 cells after H/R compared to shScr cells (Fig. 3a,b), as well as in Diaph1–/– mouse hearts after ex vivo I/R (Fig. 3c). However, under normoxic conditions, Diaph1 silencing does not affect F:G actin (Supplementary Fig. S3). Although previous studies revealed that DIAPH1 mediates vascular injury in smooth muscle cells through alterations in Rho GTPases (Toure et al., 2012), we found no differences in Rac1 phosphorylation or in the expression of RhoA between shDiaph1 and shScr cells after H/R (Supplementary Fig. S4a–b). Diaph1 deletion had no effect on ROCK2 protein levels in H9C2 cells after H/R (Supplementary Figure S4c) or on GSK3β phosphorylation at Ser9 relative to WT mouse hearts after LAD/reperfusion (Supplementary Fig. S4d). We found no differences in DIAPH2 (also known as mDia2) protein levels between shScr and shDiaph1 H9C2 cells after H/R, demonstrating that DIAPH2 is not upregulated to compensate for the absence of DIAPH1 (Supplementary Fig. S4e). Thus, DIAPH1 exerts its effects via mechanisms distinct from Rho-mediated signaling to modulate myocardial I/R injury.
Fig. 3.

Diaph1 deletion inhibited actin polymerization and SRF activation after H/R. (a) F:G actin ratio was reduced in shDiaph1 H9C2 cells after H/R (n = 5/group; *p < 0.05 vs. shScr) and (b) representative confocal microscopy images indicating changes in F and G actin in shDiaph1 H9C2 cells after H/R. Single field images of 1024 × 1024 pixels were collected to visualize F-actin labelled with Alexa Fluor 647 Phalloidin and G-actin labelled with Alexa Fluor® 594 DNase I, Scale bar: 25 μm, (c) F:G actin ratio was reduced in hearts isolated from Diaph1–/– mice compared to WT after ex vivo I/R (n = 4–5/group; *p < 0.05 vs. WT I/R). (d) mRNA expression of Srf (n = 6–8/group) and target genes, Tagln and Fos (n = 16–17/group; normalized to Ppia), was decreased after H/R in shDiaph1 cells (*p < 0.05 vs. shScr). (e) mRNA expression of Srf (n = 6/group) and target genes, Tagln and Fos (n = 16/group; normalized to Ppia), was decreased in hearts isolated from Diaph1–/– mice compared to WT after ex vivo I/R (n = 4/group; *p < 0.05 vs. WT I/R) (f) The effects of pretreatment with LatB and Jas on expression of Srf (n = 6/group), Tagln (n = 9/group), and Fos (n = 9/group) in shScr H9C2 after H/R (*p < 0.05 vs. vehicle). (g) Nuclear myocardin/GAPDH was reduced in shDiaph1 H9C2 cells after H/R (n = 8/group; *p < 0.05 vs. shScr). (h) jasplakinolide (Jas) pretreatment increased LDH release in shDiaph1 H9C2 cells after H/R (n = 8/group; *p < 0.05 vs. shScr). Unpaired two-tailed t-tests (a, c, d, g) and one-way ANOVA (e, f) were used; a p-value < 0.05 was considered significant.
3.4. DIAPH1 Regulates Actin-mediated Transcriptional Changes After I/R Through Serum Response Factor
These unexpected results led us to consider primary roles for DIAPH1 in the regulation of actin polymerization and the stimulation of serum response factor (SRF)-regulated genes. We investigated mRNA levels of Srf and its target genes, Tagln (Sm22) and Fos (c-Fos) (Mack et al., 2001), and found that they were reduced in shDiaph1 cells relative to shScr cells after H/R (Fig. 3d). Similar reductions in mRNA levels of Srf and its target genes, Tagln and Fos were observed in Diaph1–/– mouse hearts after ex vivo I/R (Fig. 3e). Under normoxic conditions, Diaph1 silencing does not have an effect on the expression of these three genes (Supplementary Fig. S5).
To better understand the relationship between F:G actin changes and downstream SRF-driven transcriptional changes, we used the actin polymerization inhibitor latrunculin B, or the actin filament stabilizer jasplakinolide, 1 h prior to H/R in shScr H9C2 cells. As expected, latrunculin B reduced the ratio of F:G actin, and jasplakinolide significantly increased the F:G actin ratio (Supplementary Fig. S6). Since the transcription factor SRF regulates gene transcription downstream of actin polymerization, mRNA levels of Srf, Fos, and Tagln were measured in latrunculin B- and jasplakinolide-pretreated cells after H/R. Latrunculin B pretreatment reduced mRNA expression of these three genes, while jasplakinolide pretreatment increased mRNA expression in shScr H9C2 cells (Fig. 3f). Furthermore, the protein levels of myocardin, a coactivator of SRF, were significantly reduced in shDiaph1 H9C2 cells subjected to H/R (Fig. 3g). Treatment of shDiaph1 cells with jasplakinolide, known to increase F:G actin, significantly reduced LDH release during H/R (Fig. 3h). These data indicate that DIAPH1 is linked to SRF-regulated transcriptional changes during H/R, at least in part through mechanisms related to altered actin polymerization.
3.5. Early Growth Response 1 (EGR1) is Regulated Downstream of DIAPH1 in I/R Injury
EGR1 is a transcription factor that may be regulated by SRF (Kimura et al., 2014) and is increased in cardiac stress, including I/R (Bhindi et al., 2012). Nuclear EGR1 protein levels were significantly lower in Diaph1–/– mouse hearts after LAD/reperfusion relative to WT hearts (Fig. 4a) and in the nuclear fraction after H/R in shDiaph1 cells relative to shScr cells (Fig. 4b). ChIP assays revealed an interaction between SRF protein and the promoter region of the Egr1 gene in WT (but not Diaph1–/– hearts) subjected to LAD/reperfusion (Fig. 4c). These data link DIAPH1 to downstream regulation of SRF and EGR1.
Fig. 4.

Diaph1 deletion downregulated EGR1 expression. (a) Diaph1 deletion decreased nuclear EGR1/histone 3 after LAD/reperfusion in mouse hearts (n = 4/group; *p < 0.05 vs. WT LAD) and (b) Diaph1 silencing decreased nuclear EGR/nucleoporin p62 in H9C2 cells after H/R (n = 5/group; *p < 0.05 vs. shScr H/R). (c) ChIP studies revealed an interaction between SRF protein and the Egr1 promoter region in WT but not Diaph1–/– hearts following LAD/reperfusion (n = 3/group). Unpaired two-tailed t-tests were used; a p-value < 0.05 was considered significant.
3.6. Diaph1 Deletion Alters Regulation of Calcium-handling Proteins
Previous studies have suggested that EGR1 may play a role in the maintenance of calcium levels in the heart (Pacini et al., 2013, Wang et al., 2005). Impaired function of calcium transport proteins is a major mechanism of myocardial injury and poor functional recovery following I/R (Murphy and Steenbergen, 2008). Because Diaph1–/– hearts exhibited significantly greater cardiac functional recovery and reduced injury after I/R, we asked whether these outcomes were linked to altered expression of calcium transporter proteins in cardiomyocytes. The sarcoplasmic reticulum calcium ATPase (SERCA)2a shuttles Ca2 + from the cytosol to the lumen of the sarcoplasmic reticulum. It is a key regulator of intracellular Ca2 + in the cardiomyocyte and is inhibited by dephosphorylated phospholamban (PLN) (Periasamy and Huke, 2001). SERCA2a protein was more abundant in Diaph1–/– hearts from mice subjected to LAD/reperfusion relative to WT hearts (Fig. 5a) and in shDiaph1 H9C2 cells relative to shScr cells after H/R (Fig. 5b). However, under normoxic conditions, Diaph1 silencing did not impact SERCA2a expression in H9C2 cells (Supplementary Fig. S7a). Furthermore, phosphorylation of PLN at serine 16, which stimulates SERCA2a, was higher in shDiaph1 cells than in shScr cells after H/R (Fig. 5c). Expression of the sodium-calcium exchanger NCX1, which also regulates intracellular Ca2 + by removing Ca2 + from the cell in exchange for Na+ under normal physiological conditions (Philipson and Nicoll, 2000), was reduced in shDiaph1 cells compared with shScr cells (Fig. 5d). Under normoxic conditions, Diaph1 silencing did not significantly affect NCX1 expression or intracellular calcium in H9C2 cells (Supplementary Fig. S7b–c). In vivo, there was an interaction between SRF and the promoter region of the gene for NCX1, Slc8a1, in WT hearts (but not in Diaph1–/– hearts) from mice subjected to LAD/reperfusion (Fig. 5e).
Fig. 5.

Diaph1 deletion improved calcium dynamics after I/R. (a) SERCA2a/GAPDH protein levels were increased by Diaph1 deletion in both mouse hearts after LAD/reperfusion (n = 10/group; *p < 0.05 vs. WT LAD) and (b) H9C2 cells after H/R (n = 4/group; *p < 0.05 vs. shScr H/R). (c) Phosphorylated phospholamban (phospho/totalPLN; n = 3–4/group; *p < 0.05 vs. shScr H/R) was increased in shDiaph1 H9C2 cells after H/R. (d) NCX1/GAPDH protein expression was lower in shDiaph1 H9C2 cells after H/R (n = 3/group; *p < 0.05 vs. shScr H/R). (e) ChIP studies performed after LAD/reperfusion revealed an interaction between SRF protein and the promoter region for the NCX1 gene, Slc8a1, in WT but not Diaph1-/- hearts (n = 3/group). (f) Adenoviral Egr1 overexpression in shScr and shDiaph1 H9C2 cells suppressed the effect of DIAPH1 on SERCA2a/GAPDH protein expression after H/R (n = 12/group; *p < 0.05 vs. Ad-mock shScr H/R, #p < 0.05 vs. Ad-mock shDiaph1 H/R). Unpaired two-tailed t-tests (a–d) and a two-way ANOVA (e) were used, a p-value < 0.05 was considered significant.
Finally, to definitively link EGR1 to changes in intracellular calcium, we adenovirally overexpressed Egr1 in shDiaph1 H9C2 cells subjected to H/R. Unlike mock adenovirus, expression of Egr1 blocked the protective effect of Diaph1 deletion on SERCA2a (Fig. 5f). Taken together, these data demonstrate that DIAPH1 plays a role in calcium homeostasis through complex transcriptional mechanisms involving EGR1 and SRF and downstream regulation of Slc8a1.
4. Discussion
In the heart, the formins have been shown to play essential roles in development (Randall and Ehler, 2014). While Diaph1–/– mice do not display a pathological cardiac phenotype under normal physiological conditions, our data point to DIAPH1 as a mediator of I/R injury. Diaph1 deletion reduced infarct size and improved contractile function in mice following LAD/reperfusion, and Diaph1 gene silencing in H9C2 cells reduced H/R-induced LDH release via the following mechanisms. First, Diaph1 deletion suppressed actin polymerization and regulated transcriptional changes in cardiomyocytes, particularly through SRF and myocardin. Second, Diaph1 deletion led to increased SERCA2a expression, increased phosphorylated PLN, and reduced NCX1, in part via EGR1.
Reduced SERCA2a expression has been observed in heart failure patients (Smith, 2007), and mouse hearts with decreased SERCA2a expression are more susceptible to ischemic injury (Talukder et al., 2009). In our studies, increased SERCA2a expression and reduced infarct size after I/R were consistent with a key role for SERCA2a in the cardioprotection observed in Diaph1–/– mouse hearts. Previous studies have shown a major role for EGR1 in regulating SERCA2a protein expression. The expression of Atp2a2, the gene for SERCA2a, can be induced by reducing MK2-dependent EGR1 in mouse embryonic fibroblasts (Scharf et al., 2013). In rat neonatal cardiomyocytes, EGR1 binds to the 5’ regulatory region of the Atp2a2 gene (Arai et al., 2000). Furthermore, Atp2a2 gene expression is decreased in cardiomyocytes isolated from Egr1–/– mice (Pacini et al., 2013). In support of these earlier findings, our study shows for the first time that expression of calcium regulating proteins was mediated in part by DIAPH1 in cardiomyocytes through EGR1.
Interestingly, Diaph1 deletion reduces intracellular calcium not only by increasing SERCA2a protein levels, but also by decreasing NCX1, which is essential for maintaining calcium homeostasis in the cardiomyocyte (Blaustein and Lederer, 1999). Deletion of Slc8a1, the gene for NCX1, reduces infarct size and improves functional recovery after myocardial I/R in mice (Ohtsuka et al., 2004). Overexpression of Slc8a1 during heart failure led to diminished calcium stores in the sarcoplasmic reticulum and to subsequent contractile dysfunction (Bolck et al., 2004). Diaph1 deletion can both upregulate SERCA2a and downregulate NCX1, thus contributing to maintaining calcium homeostasis and reducing subsequent downstream events leading to I/R injury in the heart.
The effect of Diaph1 deletion on actin polymerization may be another mechanism for reduced injury. While the effects of actin polymerization on I/R-mediated injury have not been studied in cardiomyocytes, disruptions in cytoskeletal and actin-associated proteins, including desmin and tubulin, are associated with contractile dysfunction, hypertrophy, and subsequent heart failure (Schaper et al., 1991, Heling et al., 2000). Furthermore, I/R injury in endothelial cells contributes to F actin bundling, which can lead to apoptosis (van der Heijden et al., 2008). Actin polymerization requires ATP, which is depleted during I/R. One previous study showed that a model of ATP depletion in LLC-PK1 kidney proximal tubules leads to conversion of G actin to F actin (Atkinson et al., 2004). In the current study, we found that the F:G actin ratio was reduced in shDiaph1 cells after H/R. Hence, more G actin should be available to interact with myocardin and myocardin-related transcription factors, preventing their translocation to the nucleus and subsequent binding to the serum response element on SRF target genes (Mikhailov and Torrado, 2012). Indeed, it is suggested that the co-activators of SRF, rather than the SRF itself, determine the pathological outcomes of SRF-related gene transcription (Mikhailov and Torrado, 2012, Kontaraki et al., 2011, Kontaraki et al., 2007). Consistent with these studies, we observed a reduction in nuclear myocardin protein levels and reduced SRF-related gene transcription in shDiaph1 H9C2 cells after H/R.
Formins have not been studied in the context of cardiac I/R injury. Intriguingly, a recent report in Japanese subjects with dilated cardiomyopathy, which is associated with systolic dysfunction, uncovered a missense mutation in the gene encoding another formin family gene, formin homology 2 domain containing 3 (FHOD3). Functionally, the mutant was found to modulate actin filament assembly, highlighting potential links of the formins to cardiac injury (Arimura et al., 2013). In addition, we previously demonstrated key roles for DIAPH1 in vascular injury (Toure et al., 2012). Endothelial denudation injury to the femoral artery in mice upregulated DIAPH1 expression in vascular smooth muscle, and deletion of Diaph1 reduced pathological neointimal expansion after injury through alterations in Rac1 activation, AKT/GSK3β phosphorylation, and smooth muscle cell migration (Toure et al., 2012). We did not observe any changes in Rac1 signaling in H9C2 cells after H/R and GSK-3β phosphorylation in Diaph1–/– mice hearts after I/R, thereby highlighting the diverse cell-specific mechanisms by which DIAPH1 contributes to pathological responses to stress. It is noted, however, that further detailed studies using tissue specific Diaph1 knockout mice are required to determine the role of RhoA-driven signaling in DIAPH1-regulated myocardial I/R injury.
In summary, this work implicates DIAPH1 as a critical regulator mediating cardiac dysfunction during I/R. Through integration of a series of key events, DIAPH1 regulates SRF and EGR1 transcription and consequent regulation of calcium transporters (Fig. 6). Because deletion of Diaph1 protected against cardiac I/R injury, blockade of DIAPH1-driven mechanisms may reduce injury and slow progression towards heart failure after myocardial infarction.
Fig. 6.
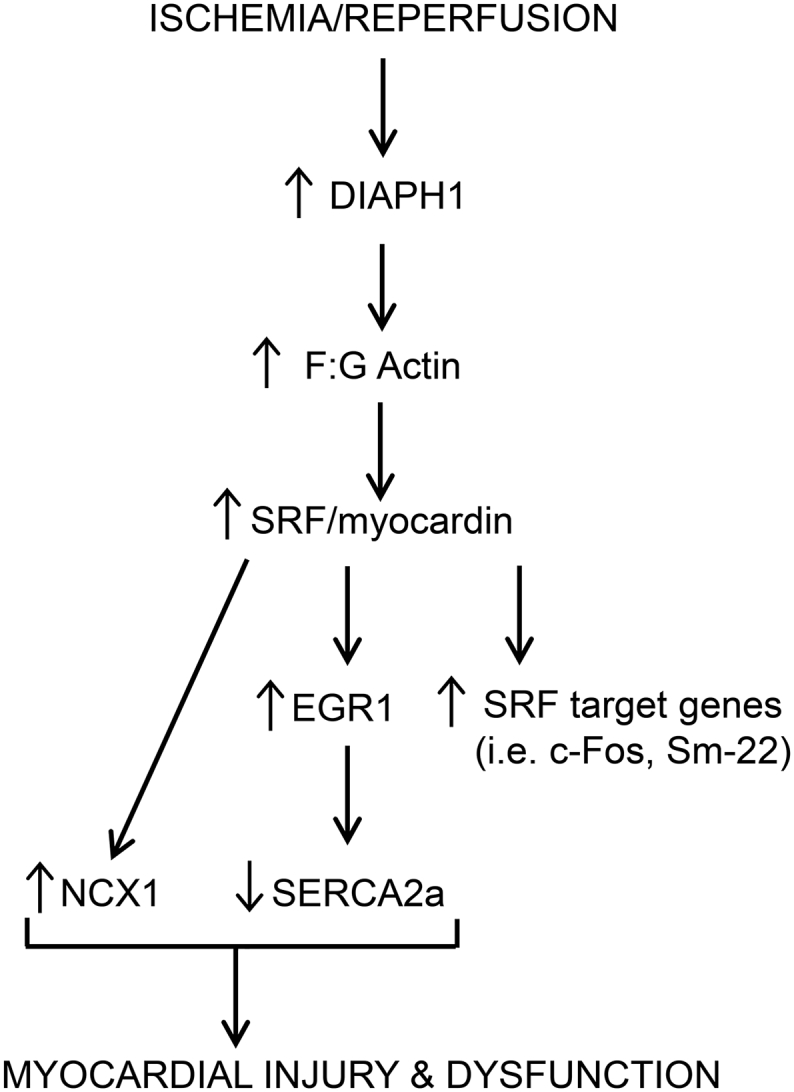
Proposed mechanism for the role of DIAPH1 in I/R injury. DIAPH1 is upregulated following I/R injury, which leads to an increase in actin polymerization. This allows for stimulation of SRF and myocardin, which results in elevated SRF-regulated gene transcription as well as increased EGR1. SRF also stimulates NCX1 expression. Downstream of EGR1, SERCA2a is reduced. The changes in NCX1 and SERCA2a expression contribute to injury and dysfunction in the myocardium.
Funding Sources
This work was supported by National Institutes of Health [F32HL110709-01, HL60901 and HL102022].
Conflicts of Interest
The authors declare no competing financial interests.
Author Contributions Statement
K.M.O. designed research, conducted research, analyzed data, and wrote the paper. R.A., Q.L., N.Q., D.T., G.S., L.W., H.Z., and J.F.A. conducted research and prepared critical reagents. R.R., A.S.A., and A.M.S. designed and directed research, analyzed data, and wrote the paper.
Acknowledgements
We thank Ms. Latoya Woods for assistance with manuscript and figure preparation. We thank Dr. Mercy Davidson from Columbia University for the generous gift of AC16 human cardiomyocytes.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2017.11.012.
Appendix A. Supplementary Data
Supplementary figures
References
- Aleshin A., Ananthakrishnan R., Li Q., Rosario R., Lu Y., Qu W. RAGE modulates myocardial injury consequent to LAD infarction via impact on JNK and STAT signaling in a murine model. Am. J. Physiol. Heart Circ. Physiol. 2008;294(4):H1823–32. doi: 10.1152/ajpheart.01210.2007. [DOI] [PubMed] [Google Scholar]
- Arai M., Yoguchi A., Takizawa T., Yokoyama T., Kanda T., Kurabayashi M. Mechanism of doxorubicin-induced inhibition of sarcoplasmic reticulum Ca(2 +)-ATPase gene transcription. Circ. Res. 2000;86(1):8–14. doi: 10.1161/01.res.86.1.8. [DOI] [PubMed] [Google Scholar]
- Arimura T., Takeya R., Ishikawa T., Yamano T., Matsuo A., Tatsumi T. Dilated cardiomyopathy-associated FHOD3 variant impairs the ability to induce activation of transcription factor serum response factor. Circ. J. 2013;77(12):2990–2996. doi: 10.1253/circj.cj-13-0255. [DOI] [PubMed] [Google Scholar]
- Atkinson S.J., Hosford M.A., Molitoris B.A. Mechanism of actin polymerization in cellular ATP depletion. J. Biol. Chem. 2004;279(7):5194–5199. doi: 10.1074/jbc.M306973200. [DOI] [PubMed] [Google Scholar]
- Bhindi R., Fahmy R.G., McMahon A.C., Khachigian L.M., Lowe H.C. Intracoronary delivery of DNAzymes targeting human EGR-1 reduces infarct size following myocardial ischaemia reperfusion. J. Pathol. 2012;227(2):157–164. doi: 10.1002/path.2991. [DOI] [PubMed] [Google Scholar]
- Blaustein M.P., Lederer W.J. Sodium/calcium exchange: its physiological implications. Physiol. Rev. 1999;79(3):763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Bolck B., Munch G., Mackenstein P., Hellmich M., Hirsch I., Reuter H. Na +/Ca2 + exchanger overexpression impairs frequency- and ouabain-dependent cell shortening in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2004;287(4):H1435–45. doi: 10.1152/ajpheart.00397.2003. [DOI] [PubMed] [Google Scholar]
- Colucci-Guyon E., Niedergang F., Wallar B.J., Peng J., Alberts A.S., Chavrier P. A role for mammalian diaphanous-related formins in complement receptor (CR3)-mediated phagocytosis in macrophages. Curr. Biol. 2005;15(22):2007–2012. doi: 10.1016/j.cub.2005.09.051. [DOI] [PubMed] [Google Scholar]
- Das A., Xi L., Kukreja R.C. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J. Biol. Chem. 2005;280(13):12944–12955. doi: 10.1074/jbc.M404706200. [DOI] [PubMed] [Google Scholar]
- Davidson M.M., Nesti C., Palenzuela L., Walker W.F., Hernandez E., Protas L. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005;39(1):133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Gibbons R.J., Valeti U.S., Araoz P.A., Jaffe A.S. The quantification of infarct size. J. Am. Coll. Cardiol. 2004;44(8):1533–1542. doi: 10.1016/j.jacc.2004.06.071. [DOI] [PubMed] [Google Scholar]
- van der Heijden M., Versteilen A.M., Sipkema P., van Nieuw Amerongen G.P., Musters R.J., Groeneveld A.B. Rho-kinase-dependent F-actin rearrangement is involved in the inhibition of PI3-kinase/Akt during ischemia-reperfusion-induced endothelial cell apoptosis. Apoptosis. 2008;13(3):404–412. doi: 10.1007/s10495-007-0173-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heling A., Zimmermann R., Kostin S., Maeno Y., Hein S., Devaux B. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ. Res. 2000;86(8):846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- Herlitz J., Waldenstrom J., Hjalmarson A. Relationship between the enzymatically estimated infarct size and clinical findings in acute myocardial infarction. Acta Med. Scand. 1984;215(1):21–32. doi: 10.1111/j.0954-6820.1984.tb04965.x. [DOI] [PubMed] [Google Scholar]
- Herlitz J., Hjalmarson A., Waldenstrom J. Five-year mortality rate in relation to enzyme-estimated infarct size in acute myocardial infarction. Am. Heart J. 1987;114(4 Pt 1):731–737. doi: 10.1016/0002-8703(87)90782-4. [DOI] [PubMed] [Google Scholar]
- Hudson B.I., Kalea A.Z., Del Mar Arriero M., Harja E., Boulanger E., D'Agati V. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008;283(49):34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y.C., Kaneko M., Bakr S., Liao H., Lu Y., Lewis E.R. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB J. 2004;18(11):1192–1199. doi: 10.1096/fj.03-1400com. [DOI] [PubMed] [Google Scholar]
- Kimura T.E., Duggirala A., Hindmarch C.C., Hewer R.C., Cui M.Z., Newby A.C. Inhibition of Egr1 expression underlies the anti-mitogenic effects of cAMP in vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2014;72:9–19. doi: 10.1016/j.yjmcc.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaraki J.E., Parthenakis F.I., Patrianakos A.P., Karalis I.K., Vardas P.E. Altered expression of early cardiac marker genes in circulating cells of patients with hypertrophic cardiomyopathy. Cardiovasc. Pathol. 2007;16(6):329–335. doi: 10.1016/j.carpath.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Kontaraki J.E., Marketou M.E., Zacharis E.A., Parthenakis F.I., Vardas P.E. Early cardiac gene transcript levels in peripheral blood mononuclear cells in patients with untreated essential hypertension. J. Hypertens. 2011;29(4):791–797. doi: 10.1097/HJH.0b013e3283424bc4. [DOI] [PubMed] [Google Scholar]
- Mack C.P., Somlyo A.V., Hautmann M., Somlyo A.P., Owens G.K. Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 2001;276(1):341–347. doi: 10.1074/jbc.M005505200. [DOI] [PubMed] [Google Scholar]
- Mikhailov A.T., Torrado M. In search of novel targets for heart disease: myocardin and myocardin-related transcriptional cofactors. Biochem. Res. Int. 2012;2012:973723. doi: 10.1155/2012/973723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T.D., Christian T.F., Hopfenspirger M.R., Hodge D.O., Gersh B.J., Gibbons R.J. Infarct size after acute myocardial infarction measured by quantitative tomographic 99mTc sestamibi imaging predicts subsequent mortality. Circulation. 1995;92(3):334–341. doi: 10.1161/01.cir.92.3.334. [DOI] [PubMed] [Google Scholar]
- Miller T.D., Hodge D.O., Sutton J.M., Grines C.L., O'Keefe J.H., DeWood M.A. Usefulness of technetium-99m sestamibi infarct size in predicting posthospital mortality following acute myocardial infarction. Am. J. Cardiol. 1998;81(12):1491–1493. doi: 10.1016/s0002-9149(98)00220-3. [DOI] [PubMed] [Google Scholar]
- Murphy E., Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008;88(2):581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S., Tanji M., Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28(1-2):65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- Ohtsuka M., Takano H., Suzuki M., Zou Y., Akazawa H., Tamagawa M. Role of Na +-Ca2 + exchanger in myocardial ischemia/reperfusion injury: evaluation using a heterozygous Na +-Ca2 + exchanger knockout mouse model. Biochem. Biophys. Res. Commun. 2004;314(3):849–853. doi: 10.1016/j.bbrc.2003.12.165. [DOI] [PubMed] [Google Scholar]
- Pacini L., Suffredini S., Ponti D., Coppini R., Frati G., Ragona G. Altered calcium regulation in isolated cardiomyocytes from Egr-1 knock-out mice. Can. J. Physiol. Pharmacol. 2013;91(12):1135–1142. doi: 10.1139/cjpp-2012-0419. [DOI] [PubMed] [Google Scholar]
- Peng J., Wallar B.J., Flanders A., Swiatek P.J., Alberts A.S. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 2003;13(7):534–545. doi: 10.1016/s0960-9822(03)00170-2. [DOI] [PubMed] [Google Scholar]
- Periasamy M., Huke S. SERCA pump level is a critical determinant of Ca(2 +)homeostasis and cardiac contractility. J. Mol. Cell. Cardiol. 2001;33(6):1053–1063. doi: 10.1006/jmcc.2001.1366. [DOI] [PubMed] [Google Scholar]
- Pfeffer M.A., Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- Philipson K.D., Nicoll D.A. Sodium-calcium exchange: a molecular perspective. Annu. Rev. Physiol. 2000;62:111–133. doi: 10.1146/annurev.physiol.62.1.111. [DOI] [PubMed] [Google Scholar]
- Randall T.S., Ehler E. A formin-g role during development and disease. Eur. J. Cell Biol. 2014;93(5-6):205–211. doi: 10.1016/j.ejcb.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Roger V.L., Go A.S., Lloyd-Jones D.M., Benjamin E.J., Berry J.D., Borden W.B. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero S., Le Clainche C., Didry D., Egile C., Pantaloni D., Carlier M.F. Formin is a processive motor that requires profilin to accelerate actin assembly and associated ATP hydrolysis. Cell. 2004;119(3):419–429. doi: 10.1016/j.cell.2004.09.039. [DOI] [PubMed] [Google Scholar]
- Schaper J., Froede R., Hein S., Buck A., Hashizume H., Speiser B. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation. 1991;83(2):504–514. doi: 10.1161/01.cir.83.2.504. [DOI] [PubMed] [Google Scholar]
- Scharf M., Neef S., Freund R., Geers-Knorr C., Franz-Wachtel M., Brandis A. Mitogen-activated protein kinase-activated protein kinases 2 and 3 regulate SERCA2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol. Cell. Biol. 2013;33(13):2586–2602. doi: 10.1128/MCB.01692-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L., Ananthakrishnan R., Li Q., Quadri N., Abdillahi M., Zhu Z. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS One. 2010;5(4):e10092. doi: 10.1371/journal.pone.0010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Zhang J., Mullin M., Dong B., Alberts A.S., Siminovitch K.A. The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. J. Immunol. 2009;182(6):3837–3845. doi: 10.4049/jimmunol.0803838. [DOI] [PubMed] [Google Scholar]
- Shimokawa H., Takeshita A. Rho-kinase is an important therapeutic target in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2005;25(9):1767–1775. doi: 10.1161/01.ATV.0000176193.83629.c8. [DOI] [PubMed] [Google Scholar]
- Smith G. Matters of the heart: the physiology of cardiac function and failure. Exp. Physiol. 2007;92(6):973–986. doi: 10.1113/expphysiol.2007.034314. [DOI] [PubMed] [Google Scholar]
- Sobel DPDaBE . Oxford, UK; Blackwell Science Inc: 2001. Challenges in Acute Coronary Syndromes. (320p) [Google Scholar]
- Solomon S.D., Glynn R.J., Greaves S., Ajani U., Rouleau J.L., Menapace F. Recovery of ventricular function after myocardial infarction in the reperfusion era: the healing and early afterload reducing therapy study. Ann. Intern. Med. 2001;134(6):451–458. doi: 10.7326/0003-4819-134-6-200103200-00009. [DOI] [PubMed] [Google Scholar]
- St John Sutton M., Pfeffer M.A., Moye L., Plappert T., Rouleau J.L., Lamas G. Cardiovascular death and left ventricular remodeling two years after myocardial infarction: baseline predictors and impact of long-term use of captopril: information from the Survival and Ventricular Enlargement (SAVE) trial. Circulation. 1997;96(10):3294–3299. doi: 10.1161/01.cir.96.10.3294. [DOI] [PubMed] [Google Scholar]
- Talukder M.A., Yang F., Nishijima Y., Chen C.A., Kalyanasundaram A., Periasamy M. Reduced SERCA2a converts sub-lethal myocardial injury to infarction and affects postischemic functional recovery. J. Mol. Cell. Cardiol. 2009;46(2):285–287. doi: 10.1016/j.yjmcc.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukder M.A., Elnakish M.T., Yang F., Nishijima Y., Alhaj M.A., Velayutham M. Cardiomyocyte-specific overexpression of an active form of Rac predisposes the heart to increased myocardial stunning and ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013;304(2):H294–302. doi: 10.1152/ajpheart.00367.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toure F., Zahm J.M., Garnotel R., Lambert E., Bonnet N., Schmidt A.M. Receptor for advanced glycation end-products (RAGE) modulates neutrophil adhesion and migration on glycoxidated extracellular matrix. Biochem. J. 2008;416(2):255–261. doi: 10.1042/BJ20080054. [DOI] [PubMed] [Google Scholar]
- Toure F., Fritz G., Li Q., Rai V., Daffu G., Zou Y.S. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ. Res. 2012;110(10):1279–1293. doi: 10.1161/CIRCRESAHA.111.262519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Dostanic S., Servant N., Chalifour L.E. Egr-1 negatively regulates expression of the sodium-calcium exchanger-1 in cardiomyocytes in vitro and in vivo. Cardiovasc. Res. 2005;65(1):187–194. doi: 10.1016/j.cardiores.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Watanabe N. Regulation of actin dynamics under physical stress: rapid actin filament regeneration by formin homology proteins and F- and G-actin homeostasis. Seikagaku. 2013;85(8):687–691. [PubMed] [Google Scholar]
- Xiang S.Y., Vanhoutte D., Del Re D.P., Purcell N.H., Ling H., Banerjee I. RhoA protects the mouse heart against ischemia/reperfusion injury. J. Clin. Invest. 2011;121(8):3269–3276. doi: 10.1172/JCI44371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Toure F., Qu W., Lin L., Song F., Shen X. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J. Biol. Chem. 2010;285(30):23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures