

Abstract
As geroscience research extends into the role of epigenetics in aging and age-related disease, researchers are being confronted with unfamiliar molecular techniques and data analysis methods that can be difficult to integrate into their work. In this review, we focus on the analysis of DNA modifications, namely cytosine methylation and hydroxymethylation, through next-generation sequencing methods. While older techniques for modification analysis performed relative quantitation across regions of the genome or examined average genome levels, these analyses lack the desired specificity, rigor, and genomic coverage to firmly establish the nature of genomic methylation patterns and their response to aging. With recent methodological advances, such as whole genome bisulfite sequencing (WGBS), bisulfite oligonucleotide capture sequencing (BOCS), and bisulfite amplicon sequencing (BSAS), cytosine modifications can now be readily analyzed with base-specific, absolute quantitation at both cytosine-guanine dinucleotide (CG) and non-CG sites throughout the genome or within specific regions of interest by next-generation sequencing. Additional advances, such as oxidative bisulfite conversion to differentiate methylation from hydroxymethylation and analysis of limited input/single-cells, have great promise for continuing to expand epigenomic capabilities. This review provides a background on DNA modifications, the current state-of-the-art for sequencing methods, bioinformatics tools for converting these large data sets into biological insights, and perspectives on future directions for the field.
Keywords: Epigenetics, Methods, DNA methylation
DNA modifications and aging
Epigenetic changes as a regulator of the aging process have been an area of research interest for a number of decades (Waddington 1940), even prior to the discovery of DNA methylation (Hotchkiss 1948). The roles of DNA methylation in regulation of genome organization and gene expression have progressed from solely being a mechanism of permanent gene inactivation to one in which DNA methylation is also a dynamic gene regulator (Schubeler 2015), though the degree of dynamism is a matter of debate (Bestor et al. 2015). The concept of non-sequence-based changes to the genome and how their accumulation with advancing age may adversely affect lifespan and cellular function have obvious relevance to geroscience research (Lopez-Otin et al. 2013; Kennedy et al. 2014). Early efforts sought to examine total levels of DNA methylation and resulted in a theory of genomic hypomethylation (decreased methylation levels across the genome) with aging (Vanyushin et al. 1973; Wilson and Jones 1983). While the hypomethylation hypothesis is often referenced as dogma (Zampieri et al. 2015; Sen et al. 2016), the general consensus has shifted to rejecting this hypothesis based on data from modern quantitative techniques (Unnikrishnan et al. 2017a). Thus, the field has moved toward identifying specific genomic sites and regions with differential methylation in animal models (Maegawa et al. 2010) and humans (Rakyan et al. 2010). A number of recent reports have provided the first comprehensive analyses of altered DNA methylation with aging in a range of mouse tissues (Cole et al. 2017; Hahn et al. 2017; Masser et al. 2017b; Petkovich et al. 2017; Stubbs et al. 2017). Human studies have also advanced by examining age-affected tissues (e.g., Zykovich et al. 2014) and through the development of a variety of chronological aging “clocks” (Hannum et al. 2013; Horvath 2013; Weidner et al. 2014; Chen et al. 2016). While these clocks have reproducible predictive validity for chronological age and potentially mortality (Marioni et al. 2015), their relevance to biological aging processes remains to be determined. This is particularly evident given the counterintuitive results from recent reports regarding the relationship between “methylation age” and tissue function (Marioni et al. 2016; Kim et al. 2017; Kozlenkov et al. 2017; Simpkin et al. 2017). Overall, there is clearly a need for epigenetic studies of aging. This review is focused on the epigenetic domain of DNA modification—namely cytosine modifications. Given the demonstrated importance of the genetics of aging (Jeck et al. 2012), a similar effort is needed to understand how the epigenome changes in response to the various stimuli encountered throughout the lifecycle. Further review of the current state of scientific findings in the epigenomics of aging is beyond the scope of this review, but several recent publications provide an overview of findings in different models and organ systems (Valdes et al. 2013; Benayoun et al. 2015; Jones et al. 2015; Sen et al. 2016). The purpose of this review is to examine the technical approaches that can be used in these studies to best meet individual experimental goals and educate geroscience researchers in designing and interpreting epigenomic studies.
Nature and regulation of DNA modifications
Epigenetic mechanisms are classically defined as chromatin structure and DNA modifications (Allis and Jenuwein 2016). While chromatin structure and histone modifications are an area of intense interest for aging research (Benayoun et al. 2015), the focus of this review is on DNA modifications due to their regulation of chromatin (Martinowich et al. 2003), the long-lasting nature of DNA modifications (Bird 2002), and because these modifications to the genome can be passed to daughter cells (Holliday 2006). The primary DNA modifications are methylation (mC) and hydroxymethylation (hmC) of cytosine at the 5′ position of the pyrimidine ring (Globisch et al. 2010). Hydroxymethylation is especially abundant in brain tissues as compared to other tissues/organs (Kriaucionis and Heintz 2009; Lister et al. 2013). Much rarer modifications of formylcytosine (fC) and carboxylcytosine (caC) exist as well (He et al. 2011; Ito et al. 2011) (Fig. 1a). Non-cytosine modifications, though rarer still, are also beginning to be investigated (Yao et al. 2017).
Fig. 1.
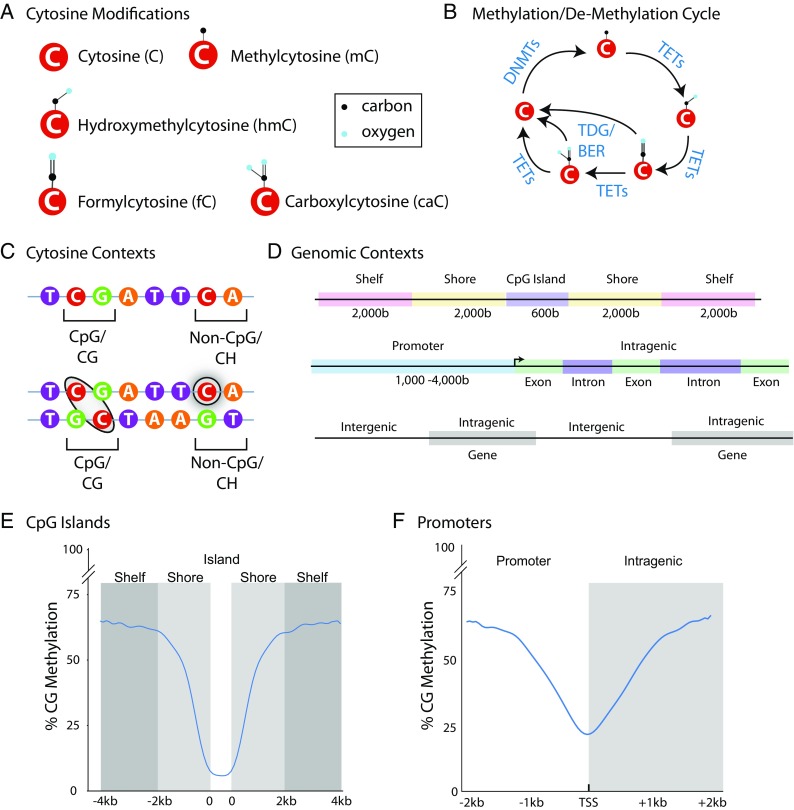
Fundamental principles of DNA modifications. a Cytosine bases exist in an unmodified form and with methyl, hydroxymethyl, formyl, and carboxy additions at the five positions of the pyrimidine ring. b Over the last decade, advances in the understanding of modification regulation have characterized the cycle of modification addition and oxidation. DNA methyltransferases (DNMTs) add methyl groups in which Tet methylcytosine dioxygenases (TETs) sequentially oxidize modifications back to an unmodified cytosine or include base excision repair (BER) through thymine DNA glycosylase (TDG). c Cytosines are present in palindromic CG contexts with methylation paralleled between the two strands while CH (where H is A, C, or T) modifications are by nature strand specific. d Changes in DNA modifications can occur across different forms of genomic elements with differing functional outcomes. CpG islands are areas of high CG density that are flanked by shore and shelf regions upstream and downstream. Similarly, methylation of promoter regions has traditionally been a focus but intragenic regions, either in exons or introns are an area of growing interest and play a role in gene expression regulation. e CG methylation is typically low over CG islands but higher in shores and shelves. f CG methylation across promoters often varies greatly across a relatively narrow region with lowest methylation typically observed around the transcription start site (TSS), even in un-expressed genes
DNA methyltransferases (DNMTs) maintain methylation patterns in CG contexts during cell division (DNMT1) and de novo methylate cytosines (DNMT3a and DNMT3b) in response to stimuli (Okano et al. 1999). Tet methylcytosine dioxygenases (TETs) oxidize mC into hmC, as well as fC and caC before the cycle is completed back to unmodified C (He et al. 2011) (Fig. 1b). This final stage of the cycle can also occur through thymine DNA glycosylase (TDG) and base excision repair (BER) (Maiti and Drohat 2011). Modifications occur on cytosines in both CG and CH (H being C, A, or T) dinucleotide contexts. Importantly, mCG is mirrored across the DNA strands because of the palindrome nature of the CG dinucleotide motif. On the other hand mCH is, by its nature, strand-specific (Fig. 1c) (He and Ecker 2015). Historically, CG methylation has been the focus of investigation. However, methylation of CH sites and hydroxymethylation of CG sites are prevalent in several organ systems, particularly the central nervous system (Lister et al. 2013; Kinde et al. 2015). The high levels of hmCG and mCH in the brain stand in contrast to other organs (Nestor et al. 2012) and suggest the possibility that the epigenomics of the aging brain is substantially different from other tissues (Masser et al. 2017a).
CG dinucleotides occur far less frequently (< 1% of dinucleotide pairs) than any other dinucleotide pairs (Bird 1980; Karlin and Mrázek 1997) and are clustered together in regions termed CG islands (Lander et al. 2001). Definitions for the regions around CG islands have also been established (Fig. 1d). The context of cytosine modification—promoter vs intergenic, intergenic vs intragenic—can result in different functional outcomes. For example, mCG in promoter regions typically represses gene expression whereas promoter hmCG levels are greater in highly expressed genes (Chapman et al. 2015). Within gene bodies, both mCG and hmCH are positively associated with gene expression (Lister et al. 2013; Lou et al. 2014). Promoter mCH is inversely correlated to gene expression and gene body mCH varies in its relationship to gene expression, being either repressive or associated with increased expression (Lister et al. 2013; Kinde et al. 2015; Lister and Mukamel 2015). The relationship of hmCH to gene expression is unknown, although it does appear that hmCH levels are extremely low (Hadad et al. 2016).
Although mC and hmC are recognized by common DNA binding proteins, they also have modification-specific binders (Spruijt et al. 2013), thereby providing a potential mechanism for the differential effects on gene expression between mC and hmC (Xu et al. 2011). Additionally, TET enzymes may prefer CG sites (Hu et al. 2013) which raises the intriguing possibility that mCH may be resistant to oxidation and elimination. Our understanding of the effects of mC/hmC on gene expression and genome organization with aging is in its infancy. A critical task for the field is to generate the data needed to test how, or whether, these general concepts apply to epigenomics of aging. From a geroscience perspective, it is essential that we clarify and expand our understanding of aging-associated alterations of epigenetic processes to determine their contribution to health and disease.
Historical methods for analysis of DNA modifications
The focus of this review is on current, next-generation sequencing (NGS) approaches to analyzing DNA modifications that provide base-specific, absolute quantitation. The differentiating factor of the historical methods from the current generation of NGS methods is that now absolute quantitation of modifications can be performed in a base-specific manner for either the whole genome or at least significant portions of it. However, a brief perspective of the approaches that lead to the current NGS methods is worthwhile. As the epigenomic field developed, the first assays were for total levels of cytosine modifications in the genome. High-performance liquid chromatography (HPLC) was one of the first methods to detect global methylation levels through column chromatography of hydrolyzed DNA (Reddy and Reddy 1990). Samples were then compared to a set of standards with known methylation levels (Reddy and Reddy 1990; Fuke et al. 2004). Updates to this method include replacing UV detection with mass spectrometry (Song et al. 2005). For more simple assays, enzyme-linked immunosorbent assays (ELISA) use antibodies that are specific for mC or hmC with an in vitro generated standard curve to measure the global levels of methylation in a given sample (Kalani et al. 2014). Challenges with this method are that many of the standards used in ELISAs contain different CG densities than mammalian genomes, resulting in inaccurate quantitation of methylation levels (Hadad et al. 2016) and modifications at CG and CH sites cannot be differentiated. The same antibodies developed for ELISAs can be used for immunohistochemistry (IHC) (Almeida et al. 2012), but great care must be taken as IHC is generally not quantitative and spurious conclusions can be drawn from simple densitometry of tissue sections. Another commonly proposed surrogate measure for total genomic methylation levels is pyrosequencing of repetitive elements (Yang et al. 2004). Repetitive elements constitute a large portion of the genome (de Koning et al. 2011), and analysis by relatively simple pyrosequencing approaches is a potentially facile approach to estimating genome-wide methylation. While methylation of repetitive elements is of biological interest in and of themselves, care should be taken to not simplistically assume they are an accurate marker of genome-wide methylation. Methylation levels vary greatly over the genome (Lister et al. 2013), and there is, in fact, very little evidence to support a strong relationship between repeat element methylation level and other genomic regions (Blueprint_consortium 2016; Hadad et al. 2016; Crary-Dooley et al. 2017).
All of these traditional methylation analysis approaches have been applied to a number of aging research studies but with varying degrees of limitations. Many of the findings have not been recapitulated across studies (see Unnikrishnan et al. 2017a), especially given the large magnitude of changes often presented. At times, the technical limitations of these methods have resulted in nonsensical findings like the sum of methylation and hydroxymethylation being greater than 100% (Mei et al. 2015). This argues for better analytical methods and analysis procedures that can reproducibly quantify DNA modifications. Preventing progress to more quantitative methods were the barriers of the cost of generating large amounts of sequencing data and the difficulty in differentiating modified and unmodified cytosines.
Affinity enrichment NGS
With the advent of high throughput sequencing (Shendure and Ji 2008), now commonly known as NGS, generating large amounts of accurate sequencing data become economical. This led initially to development of affinity-based methods in which modified DNA was “pulled” out from unmethylated DNA by immunoprecipitation in a manner similar to chromatin immunoprecipitation sequencing (ChIP-Seq) (Furey 2012). Methylated DNA immunoprecipitation (meDIP-Seq) (Weber et al. 2005; Jorgensen et al. 2006), methyl-binding domain sequencing (MBD-Seq) (Rauch and Pfeifer 2005; Serre et al. 2010), and a number of variations of these techniques (see Lister and Ecker 2009; Laird 2010) were developed. However, the limitation to any affinity-based approach is that the specific methylation sites (or hydroxymethylation; Tan et al. 2013) cannot be determined, only a general genomic region of methylation. While this was a significant advance, the primary limitations of these approaches, relative quantitation across regions and lack of base-specific data analyses, diminish the insight that could be garnered from such approaches.
Base-specificity of DNA modifications
Base-specificity in DNA modification analysis is critical for four principle reasons: (1) levels of DNA modifications demonstrate large differences over narrow genomic regions, (2) age-related changes appear to occur in a base-specific manner, (3) base-specificity is required to differentiate CG from CH methylation, and (4) with base-specific absolute quantitation, meta-analysis and comparisons between studies can be readily performed. Starting with the first point, it has become apparent across a large number of studies that methylation levels differ dramatically over relatively short genomic distances (e.g., Edgar et al. 2014; Zhao et al. 2014; Schultz et al. 2015). This is especially true at transcription start sites (TSSs) and at CpG islands which tend to be minimally methylated. Methylation levels then rise dramatically upstream and downstream of these genomic features. For example, taking base-specific whole genome bisulfite sequencing data from mouse prefrontal cortex, nadirs of methylation are evident at CpG islands and TSSs (Fig. 1e, f). Therefore, averages over regions that are 1–10 kb wide provide limited insight into the true pattern of methylation. Furthermore, a number of reports have described base-specific changes in methylation with aging (Maegawa et al. 2010; Mangold et al. 2017a; Petkovich et al. 2017; Stubbs et al. 2017). Moreover, given the growing importance of examining methylation at both CG and CH sites, site-specific quantitation is required to differentiate between these two. We have previously demonstrated in the mouse hippocampus, for example, the majority of changes in methylation with aging are in the CH context, which cannot be differentiated from CG by meDIP-Seq and MBD-Seq techniques outlined above (Masser et al. 2017a). Lastly, absolute quantitation in a base-specific manner allows data from multiple studies to be combined to increase power, test reproducibility, identify biomarkers, and compare between species (Horvath 2013; Masser et al. 2017a; Petkovich et al. 2017; Stubbs et al. 2017). Taken together, these points provide strong justification that to fully understand differences in DNA modifications with aging, base-specific resolution is required.
The concept of examining base-specific methylation levels is not new. In the past, this has generally been performed on small genomic regions (< 100 bp) at specific genomic loci through bisulfite conversion and pyrosequencing or Sanger sequencing. Bisulfite conversion works through deamination of unmodified cytosines to uracils which are then copied as thymines during subsequent amplification (Frommer et al. 1992; Clark et al. 1994). Methylated and hydroxymethylated cytosines are protected from this conversion. Thus, modification status is turned into a base difference that can easily be read by sequencing (Fig. 2a). The methylation level in the sample is determined by collecting a number of sequencing reads over the region and counting the number of cytosines and thymines at a specific site. Methylation level equals C/C + T (Fig. 2b). Sequencing was traditionally performed by pyrosequencing (Dupont et al. 2004) or standard Sanger sequencing (Parrish et al. 2012). These methods have been used in a wide range of studies and perform comparably (Reed et al. 2010). However, they provide coverage over only a small region, usually just a few cytosines. Additionally, pyrosequencing requires multiple sequencing primers and Sanger sequencing traditionally required cloning of PCR products, a laborious task (Zhang et al. 2009). Nevertheless, these techniques have been used to great effect to generate base-specific analysis of selected regions and changes in methylation, but not hydroxymethylation, with aging (Noer et al. 2007; Maegawa et al. 2010; King et al. 2012).
Fig. 2.

Bisulfite conversion and quantitation of mC. a Bisulfite sequencing approaches are based on the concept that methylation, or hydroxymethylation, prevents the deamidation of cytosine to uracil by bisulfite. With PCR amplification, uracils are copied as thymines. The difference in methylation, or hydroxymethylation, status of a cytosine can then be read through sequencing as a base difference (C vs T). b Quantitation of methylation level is achieved by sequencing over a genomic region many times. The number of times a cytosine in the reference sequence is read as a thymine or cytosine is counted. The methylation level is then computed for each cytosine as C/C + T for that site. The greater the number of times that site is sequenced, the more accurate the methylation quantitation
Bisulfite sequencing methods
As stated above, developments in NGS allowed rapid and efficient generation of massive amounts of high-quality sequencing data and bisulfite conversion approaches make base-specific quantitation possible. In a natural progression, these techniques were combined into a new generation of methods. Whole-genome bisulfite sequencing (WGBS) was first introduced to quantify total genome methylation levels in the plant Arabidopsis (Cokus et al. 2008; Lister et al. 2008) but was rapidly adapted for use in mammalian genomes (Lister et al. 2009). For WGBS, genomic DNA libraries are created and subsequently bisulfite converted, sequenced, and mapped back to the reference genome. Subsequent versions of WGBS perform library construction after bisulfite conversion (Miura et al. 2012; Khanna et al. 2013) (Fig. 3a) or use alternative transposase-mediated library construction (Adey and Shendure 2012). The benefits of WGBS are obvious; it provides the most comprehensive, base-specific, absolute quantitation of DNA methylation at potentially all cytosines in a genome. The challenge with WGBS is the amount of sequencing data needed. Humans and standard rodent laboratory animals have genomes of ~ 3 billion bases (3 Gb) per DNA stand. Sufficient sequencing depth (5–15×) over any given base is needed to accurately quantify methylation (Ziller et al. 2015) and thus across the whole genome requires > 25 Gb of sequencing per sample, after accounting for losses due to quality control and alignment efficiency. This makes WGBS studies very large undertakings and not reaching needed sequencing depths can impair identification of statistically significant group differences. NGS costs have come down in recent years, but the depth requirements as detailed by the scientific community have increased. Therefore, more sequencing is required to meet expectations of the community for the validity of the quantitative data. These factors make WGBS an expensive approach and the analytical methods required to process the data daunting, especially when including detailed analysis of all cytosine contexts (CG and CH), which can increase the number of analyzed cytosines by orders of magnitude. One approach to decreasing the scale of WGBS is to perform low coverage sequencing and eliminate base-specific analyses. With this approach, patterns of methylation can be examined by collapsing genomic elements, e.g., promoters, CpG islands (Hadad et al. 2016). This is very useful for getting initial insights into the total levels and general patterns of DNA modifications but goes against the goal of determining base-specific differences with aging. Thus, a number of approaches have been developed to target sequencing to only a portion of the genome and thus greatly reduce the amount of sequencing required.
Fig. 3.
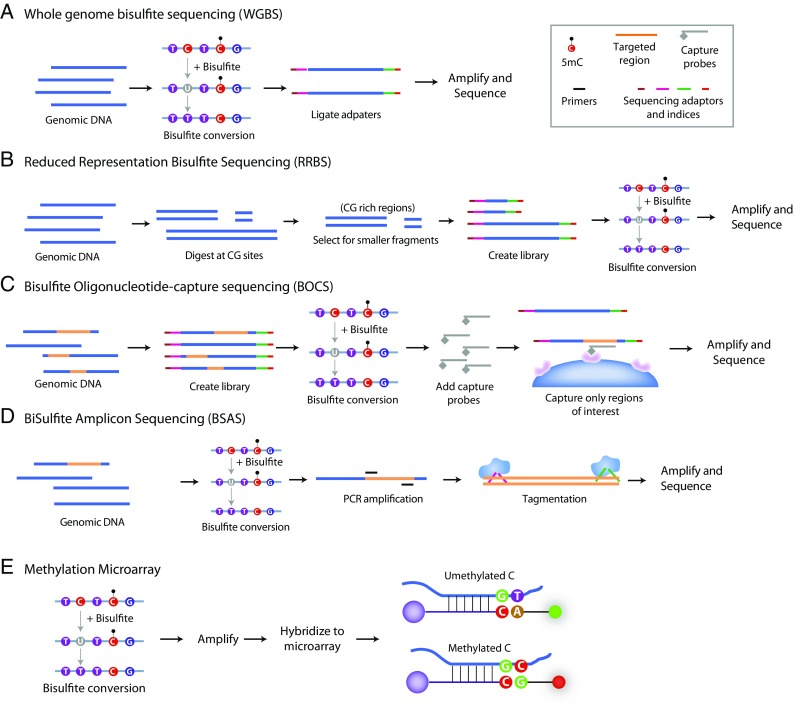
Methods for base-specific cytosine modification quantitation. a Whole-genome bisulfite sequencing (WGBS) in general principle consists of bisulfite modification of genomic DNA and then creation of a sequencing library. Variants may switch the order of bisulfite conversion and library preparation. This approach gives quantitation across the genome but requires very large amounts of sequencing. b A long-standing approach to the analysis of large portions of the genome, but without the expense of WGBS is reduced representation bisulfite sequencing (RRBS). Genomic DNA is digested at CG sites, and then, the resulting smaller molecular weight fragments are isolated. These fragments are made into a sequencing library and bisulfite converted prior to sequencing. This approach focuses the data on CG rich regions of the genome. c Bisulfite oligonucleotide capture sequencing (BOCS) is analogous to exome sequencing techniques. In this approach, a whole genome library is made and then bisulfite converted. Genomic regions of interest are then captured with oligonucleotide probes greatly enriching for regions of interest and thereby decreasing the amount of sequencing required. d Often, analysis of a specific genomic loci is desired, e.g., a gene of interest. With bisulfite amplicon sequencing (BSAS), the specific regions are amplified from bisulfite converted DNA and then made into a sequencing library. In effect, the PCR amplification greatly enriches for the region of interest. e While not a sequencing approach, one of the most common approaches to methylation quantitation is with a microarray format. This is a modification of SNP microarrays where probes are designed to detect a converted (T) or unconverted (C) at a CG site. Genomic DNA is bisulfite converted and amplified prior to hybridization to a microarray were the two different “alleles” have different reported colors. This is a high throughput approach, but a microarray must be available for the CG sites of interest and the species being examined. Currently, only human microarrays are commercially available
Reduced representation bisulfite sequencing—RRBS/eRRBS
The first sequencing approach to reducing the amount of the genome covered utilized the use of restriction enzymes. This was built on prior work that used combinations of methyl-sensitive and insensitive restriction enzymes (Cedar et al. 1979; Reilly et al. 1982). Reduced representation bisulfite sequencing (RRBS) uses the restriction enzyme MspI to cleave CCGG motifs (Meissner et al. 2005) (Fig. 3b). Libraries are then made and only lower weight fragments (40–200 bp) (Gu et al. 2011) are selected. This enriches for CG-rich regions like CpG islands, and promoter regions containing CpG islands. However, coverage over other genome elements like CpG island shores and some enhancer elements is limited (Gu et al. 2011). With refinements to the method, enhanced RRBS (ERRBS) coverage in some of the less CG dense regions was improved (Akalin et al. 2012b; Garrett-Bakelman et al. 2015). The primary limitation to these methods is the dependence on the location of restriction sites in the genome and inability to tune coverage to regions of interest. Additionally, the size selection step is critical to which genomic regions are analyzed which is why there are often very limited overlapping sites between different RRBS datasets (Stubbs et al. 2017).
Oligonucleotide capture
Selecting regions of interest for sequencing is not a new concept. For a number of years, what is popularly termed exome sequencing has been used in genetics to focus sequencing on protein coding regions of the genome (Mamanova et al. 2010). An analogous approach can be applied to DNA modification analyses. Combining bisulfite sequencing with oligonucleotide capture offers the potential to focus analyses on genomic regions of interest while maintaining base-specific, absolute quantitation (Fig. 3c). We term these approaches as bisulfite oligonucleotide capture sequencing (BOCS). Several very similar approaches have been developed for assessing the human (Wang et al. 2011; Ivanov et al. 2013; Allum et al. 2015; Li et al. 2015), mouse (Hing et al. 2015; Li et al. 2015), and rat (Masser et al. 2016) genomes. In all of these approaches, capture of targeted regions is by complimentary oligonucleotide probes. Slight differences in the oligonucleotides and whether capture is performed before or after bisulfite conversion are the main differences between these methods. These approaches, which provide base-specific absolute quantitation over ~ 80–200 Mb of relevant genomic regulatory regions, are poised to be a widely employed approach for aging research studies. Recently, we have described the use of this approach for geroscience research (Hadad et al. 2017a; Masser et al. 2017c).
Generation of the oligonucleotide probes by an individual lab would be impractical and a variety of commercially available BOCS approaches are available. Roche, Agilent, and Illumina offer probesets for humans and mice. Regardless of the specific capture approach/supplier used, the ability to tune the regions of the genome being examined (Ziller et al. 2016) and the relatively straightforward nature of building probesets for other species (Masser et al. 2016) make this a promising approach for the future.
Amplicon sequencing
While both whole genome and BOCS approaches are well suited to discovery studies, often experimental goals only require analysis of a specific genomic locus or set of loci. Highly focused sequencing decreases the cost per sample and increases throughput. Additionally, very high sequencing depth is easily achieved when only small amounts of (< 1 Mb) of the genome are being tested. For hypothesis testing experiments, several approaches have been developed that still take advantage of the power of bisulfite conversion and NGS. To achieve this, PCR primers can be designed to amplify bisulfite converted DNA, and the resultant amplicons are used to generate sequencing libraries. We term these approaches bisulfite amplicon sequencing (BSAS) (Masser et al. 2013; Masser et al. 2015) (Fig. 3d). Several other BSAS variations include using restriction enzymes (Varley and Mitra 2010), use of microdroplets for the PCR (Komori et al. 2011), further evolutions of the library preparation (Bernstein et al. 2015; Bhat et al. 2016), and the addition of analyzing hydroxymethylation (Chen et al. 2017). These approaches provide base-specific absolute quantitation in up to 96 samples of 1–10 kb targeted regions, making this method highly valuable for hypothesis-driven aging research. Additionally, this approach is much less expensive compared to whole genome or genome-wide capture techniques making it accessible to more laboratories. Recent aging studies have employed this technique demonstrating its usefulness in any tissue or cells with multiple targets of interest (Franzen et al. 2017; Mangold et al. 2017a; Unnikrishnan et al. 2017b).
The primary technical hurdle to amplicon based approaches is the design and validation of the PCR primers. They must be designed against bisulfite converted genomic sequence and generally avoid having CG sites in the primer binding region (Masser et al. 2015). In some cases, such as highly rich CG regions, primer design can be difficult and may preclude analysis of a specific locus.
Methylation microarrays
The primary alternate approach to methylation analysis that does not use NGS is high density microarrays (Bibikova et al. 2009; Bibikova et al. 2011). These micorarrays are adaptations of SNP microarrays widely used in the genetics field, coupled with bisulfite conversion. This method works through bisulfite conversion of the sample DNA and hybridization to oligonucleotide probes on a microarray slide. The detection probe used is either complimentary to C—if the base was methylated and protected from bisulfite conversion, or complimentary to A—when the cytosine was unmodified and not protected from bisulfite conversion (Fig. 3e). Different fluorophores are conjugated to the different detection probes allowing discrimination. Microarray slides are scanned and automated software computes a beta (β) value equivalent to methylation percentage. Recently, a new generation of these microarrays which cover 850,000 CG sites in the human genome have been released (Moran et al. 2016), although there are questions about the reproducibility of this platform as compared to prior microarrays (Logue et al. 2017). The advantages of this method are the relatively simple workflow, comparatively low cost per sample, and automated data analysis. This method is also amenable to high throughput analysis of large numbers of human samples (Chen et al. 2016). The primary limitations of methylation microarrays are that they are available only from one company (Illumina) and only for the human genome. As well, these arrays have been designed only against CG sites and cover far fewer sites than can be obtained through WGBS or BOCS. Nonetheless, this has been a valuable method for clinical geroscience studies and has seen wide utility in the field (Christensen et al. 2009).
Comparison of sequencing methods
As a researcher new to the study of DNA modifications, it would be entirely reasonable to be left overwhelmed by the variety and complexity of all the methods now available for DNA methylation analysis. Analytical performance of some of these methods has been compared (Harris et al. 2010; Blueprint_consortium 2016). Among focused analyses, bisulfite amplicon sequencing approaches performed better than pyrosequencing or methylation microarrays (Blueprint_consortium 2016). In-depth comparisons of WGBS, BOCS, and RRBS have not been performed although this is desperately needed by the field. Both BOCS (Li et al. 2015; Masser et al. 2016) and RRBS (Gu et al. 2011) have been compared in a limited manner to WGBS, though more in-depth comparisons are needed. It has been suggested that post-bisulfite library construction approaches may be better (Olova et al. 2017). If all bisulfite sequencing methods are considered technically sound, the choice of methods is a balance of the degree of genome coverage needed scientifically and practical reality of the resources available. Considering genomic coverage requirements, sample throughput, and resources available (Fig. 4), the current range of technologies can meet most needs.
Fig. 4.

Sample throughput and cost as a function of genomic coverage. Experimental design for DNA modification analysis requires consideration of a range of scientific and practical factors. a Genomic coverage as a function of sample throughput. With greater coverage of the genome, as a general principle, sample throughput decreases. b Cost per sample increases as a function of genomic coverage. Costs vary on the number of samples performed and the specific circumstances of a study, but generally, the more genome analyzed the greater the cost as more sequencing is performed. Note: costs are estimated at a 15× sequencing depth and the microarray technology is denoted with dashed lines as the calculations of genomic coverage are inherently different as this is not a sequencing approach
Bioinformatics
So far, we have considered the methods for collection of methylation data, but this is only the first part of any study. Analysis of bisulfite sequencing data and the subsequent tertiary analysis are a complex topic. While a full discussion is beyond the scope of this review, recent publications provide greater detail (Bock 2012; Krueger et al. 2012; Baubec and Akalin 2016; Shafi et al. 2017). Nonetheless, the basic steps of sequencing data analysis consist of the following: quality control, alignment, quantitation, differential methylation determination, and tertiary analysis where patterns of methylation are integrated with other forms of annotation such as genomic features, other epigenomic data (e.g., chromatin and enhancers), gene expression, and pathways.
Quality control uses many of the same tools available for genomic and transcriptomic studies and consist of filtering out low-quality sequencing reads, contaminating adaptor sequences, etc. The principle quality control step specific to bisulfite sequencing is positive controls for complete bisulfite conversion of unmethylated cytosines to uracils. Estimation of conversion efficiencies was previously performed using non-CG sites as it was assumed that non-CG methylation did not exist, and therefore, any methylation signal at non-CG sites was failed conversion (Sun et al. 2015). This is clearly unwise given our current understanding of non-CG methylation as a common and biological relevant phenomenon. Therefore, the current best practice for conversion controls is exogenous unmethylated spike-in sequences such as PhiX that are known to be unmethylated. When included at the beginning of workflow for each sample, conversions rates can be calculated and corrections can be applied on a sample-by-sample basis.
Alignment of bisulfite sequencing data also differs from standard sequencing alignments. Bisulfite conversion has created an ambiguous base at cytosines that can now be either C or T. Bismark is generally the most frequently used aligner for bisulfite sequencing data (Krueger and Andrews 2011), though other sequence aligners have been reported including BSMAP (Xi and Li 2009), BS Seeker (Chen et al. 2010), and others (Bock 2012). Bisulfite sequencing alignment is computationally intensive and can take days and weeks of processing time. Some new approaches seek to utilize computer hardware differently to greatly speed bisulfite sequencing alignments (Klus et al. 2012; Koster and Rahmann 2014; Manconi et al. 2014). With quality controlled and aligned sequencing data, quantitation of methylation levels is the next stage of analysis. It should be noted that sometimes an approach of “calling” whether methylation is present or not is employed at this stage. We have found this to be counter-productive as it oversimplifies and creates missing data. Rather, treating methylation as a continuous variable from 0 to 1 is the most useful approach to employ.
Most studies are performed to determine differential methylation between conditions. The primary decisions at this stage are whether to analyze differential methylation in a base-specific or regional fashion and whether or not to examine CH methylation. Based on our experience in brain aging research (Hadad et al. 2016; Hadad et al. 2017c; Mangold et al. 2017b; Masser et al. 2017a), we would advocate for base-specific and inclusion of CH analysis. To provide visualization of the advantage of base-specific analysis, we include an example comparing methylation between two different brain regions at an MHCI promoter by BSAS (Mangold et al. 2017a). This promoter region contains a number of CG and CH sites, as well as a range of transcription factor motifs (Fig. 5a). Examining CG methylation with base resolution a pattern of lower methylation nearer the TSS is evident with significantly higher methylation in the cortex at CGs in the promoter region than in the cerebellum (Fig. 5b). Similarly, differences in CH methylation between cortex and cerebellum throughout the promoter region and into the intragenic regions are evident (Fig. 5c). Clearly, not examining CH data would have missed a number of differences in the methylation pattern between these brain regions. If quantitation is performed in a regional manner (1 kb region), higher cortical CG methylation (Fig. 5d) and higher cerebellar CH methylation (Fig. 5e) are evident. However, all resolutions as to where the specific differences are and their relationship to introns, exons, promoters, and transcription factors (Fig. 5a) are lost. It seems reasonable that if sufficient sequence depth is achieved, that base-specific analysis is superior to region-based analyses. The primary reason to perform regional analyses is when sequencing depth is limited and combining data across a region provides for more data.
Fig. 5.
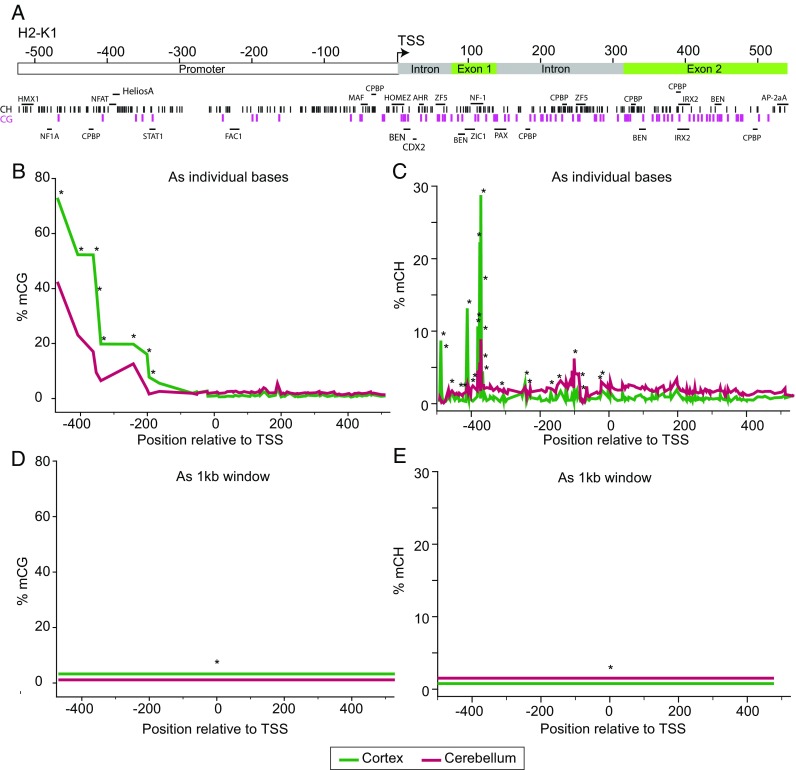
Base-specific versus region-based quantitation. For computational simplicity, methylation analysis is sometimes performed as regions. This signal compression comes with distinct disadvantages. a In this example from Mangold et al. (2017a), a region ± 500 bp of the mouse H2-K1 promoter containing a number of genomic features (promoter, intron, exon, and transcription factor sites) and CG (purple tics) and CH sites (black, tics) was examined by BSAS. CG (b) and CH (c) methylation demonstrated distinct, base-specific, profiles across the region examined. Differences in methylation levels between the cerebral cortex and cerebellum were found at a number of sites (*p < 0.05, t test, B-H MTC, n = 8/group). If these values are compressed into a region analysis across the 1 kb, differences are still evident in CG (d) and CH (e) methylation but the magnitude of differences are compressed and the specificity for the differentially methylated sites to genomic features is lost
For base-specific quantitation, a number of different software packages are available including MethylKit (Akalin et al. 2012a), DSS (Park and Wu 2016),BSmooth (Hansen et al. 2012), and Limma (Pacheco et al. 2011). In-lab developed R or Python scripts are also commonly performed, requiring development of programming skills. This does require commitment of time to training but opens up many new possibilities in data analysis (Tippmann 2015). Whether as a base-specific or regional analyses, the outcome is a listing of differentially methylated cytosines (DMCs) or regions (DMRs). Interpreting the genomic localization and potentially relationships to gene expression is the next step of analysis—typically referred to a tertiary analysis.
Tertiary analysis requires a review of its own but is an important aspect of experimental design and output. For a brief summary, it is best to think of integration with genomic feature data, other epigenetic data, and gene expression data. For genomic features, this often takes the form of determining if changes are more or less likely than chance to occur in a certain feature such as a promoter, intron, and CG island. A number of examples exist from recent studies (Cole et al. 2017; Dozmorov 2017; Hadad et al. 2017b; Masser et al. 2017a; Wang et al. 2017). For integration with other epigenomic data, the process is similar but the comparison is with other epigenomic data such as ChIP-Seq. This requires data from the same tissue and ideally from the same sex and age. Most directly, this can be viewed as comparing two sets of genomic locations to determine where they overlap and if there are statistically significant over- or under-representations (Dozmorov 2017). Comparing methylation with gene expression possesses a unique problem given the base-specific data to compare to transcript count data. The field is working on packages for aiding in this analysis but accepted solutions have not yet been developed. Suffice it to say that simple correlations of region methylation to transcript count may not fully reveal the epigenetic control of a region. The best available approach is to utilize data visualization such as by methylPipe (Kishore et al. 2015).
Other modifications
With the discovery of the abundance and specific functional roles for alternate cytosine modifications (for reviews, see Moore et al. 2013; Shi et al. 2017), new methods have emerged in order to distinguish these modifications (hmC, fmC, caC) from mC (Raiber et al. 2017). hmC is of the greatest interest given its prevalence and potential regulation with aging (Szulwach et al. 2011). Because traditional bisulfite sequencing cannot differentiate between mC and hmC, methylation in a bisulfite sequencing experiment is really the sum of methylation and hydroxymethylation. When methods that distinguish mC from hmC are used up to a quarter of what was previously assumed to be methylation is actually hydroxymethylation depending on the tissue (Fig. 6a). This is further supported by the base-specific regulation of mC and hmC over small genomic regions (Fig. 6b).
Fig. 6.
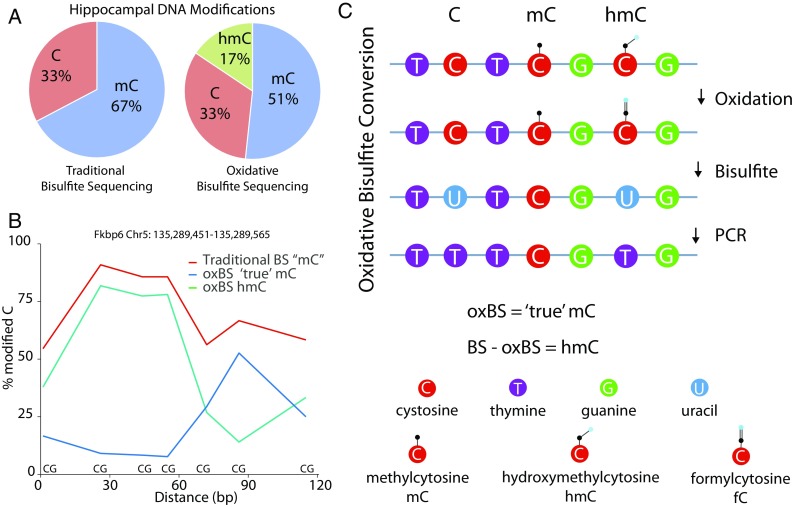
Hydroxymethylation analysis. Most DNA modification analysis studies use bisulfite sequencing approaches that do not differentiate between methylation and hydroxymethylation. However, hydroxymethylation is abundant in a number of tissues. For example, using data from Hadad et al. (2016) who examined the mouse hippocampus by oxidative bisulfite sequencing that differentiates between methylation and hydroxymethylation, it is clear (a) that one fourth of what would normally be called as methylation is in fact hydroxymethylation. Further demonstrating this point, within a relatively small region (~ 120 bp of Fkbp6; b), the levels of methylation and hydroxymethylation are clearly regulated in a base-specific fashion. Hydroxymethylation can be differentiated from methylation by an oxidation step (c) that de-protects hydroxymethylation and allows a specific methylation quantitation. When combined with bisulfite sequencing, a subtractive approach can be used to quantify methylation and hydroxymethylation individually
There are new methods that have been developed to distinguish between mC and hmC including Ox-BS (Booth et al. 2014) and TAB-seq (Yu et al. 2012). Oxidative bisulfite sequencing (Ox-BS) includes an oxidation step to convert hmC to fmC (Fig. 6c) which is then subject to bisulfite conversion. Parallel runs of standard bisulfite sequencing and oxidative bisulfite sequencing respectively give combined mC + hmC and mC only data, respectively. hmC is then determined by subtraction. Importantly, Ox-BS as a conversion chemistry has been adapted to analysis of focused genomic regions (Chen et al. 2017). TAB-Seq uses glycosylation of 5-hmC and then bisulfite conversion and sequencing DNA libraries to quantify methylation levels (Booth et al. 2012; Yu et al. 2012; Booth et al. 2013). RedBS-seq was developed to measure levels of fmC through reducing the fmC base back to hmC in order for quantify by sequencing, much like the Ox-BS method (Booth et al. 2014). caC can be quantified using CAB-seq, where EDC is used to chemically modify the 5-caC to prevent it from converting during bisulfite conversion, and levels quantified by sequencing (Lu et al. 2013). An alternative is methylation assisted bisulfite sequencing (MAB-Seq) which can be used to determine, in a base-by-base fashion combined caC and fC (Neri et al. 2016).
Single cell/cell-type analyses
While the field has experienced rapid advances in epigenomic analysis techniques, method development continues, especially in the context of analyzing singe cells, single cell types, or very small samples. Several groups including those lead by Reik (Angermueller et al. 2016; Clark et al. 2016; Clark et al. 2017), Vijg (Gravina et al. 2015; Gravina et al. 2016; Yu et al. 2017), Adey (Mulqueen et al. 2017), and Eckardt (Luo et al. 2017) have also demonstrated that methylation patterns can be generated from single cells. However, the principle caveat to these methods is that only a small amount of the genome is analyzed (often around 5%) and the same genomic regions are not analyzed in each cell, making cell-to-cell comparisons of specific loci difficult. Clearly, there is a great need for these methods but they require additional development to provide comparisons of individual single cells. Currently, the best option for dealing with cellular heterogeneity in complex tissues is through existing methods for isolation of specific cell types that can then be analyzed (Mo et al. 2015; Mo et al. 2016).
An alternate approach to the question of cellular heterogeneity is through the use of algorithms that “correct” for cellular heterogeneity. This can either be based on reference patterns of cell-type specific methylation (Houseman et al. 2012) or without reference cell data (Zou et al. 2014). Comparisons of these methods have been performed (Kaushal et al. 2017; Teschendorff and Relton 2017) with recent data casting doubt on the accuracy of these correction methods (Zheng et al. 2017). Given the still limited data on the DNA modification profiles of different cell types, and their response to aging the accuracy of these methods, it needs to be fully validated before they can see widespread usage in aging studies.
Alternatives to conversion-based sequencing
As discussed above, the current standard for analysis of DNA modifications is to use chemical conversion approaches to change modification status to a base difference that can be read with current next-generation sequencing approaches. This has strengths and weaknesses including controlling for conversion efficiency and accurately aligning altered sequences to their reference genome. An alternative approach may be to use nanopore sequencers to directly read different modifications from the DNA (Burgess 2017; Schatz 2017). Nanopore sequencing uses pores through which nucleic acid strands are “pulled” and the ionic pattern reveals the nucleotide sequence, including modifications (Jain et al. 2017; Shendure et al. 2017). These technologies are still in the developmental phase but show great promise (Rand et al. 2017). With continued technical development, they may be employed to aging research making direct analysis of DNA modification patterns from un-manipulated DNA possible. An exciting possibility of this single-molecule approach would be that individual cells could potentially be interrogated, without the need for extensive amplification (and the issues this can cause) but this still requires additional method development (Simpson et al. 2017).
Future directions
Employing the above described technologies offers great promise to understand the nature and patterns of DNA modifications as well as how they respond to aging. In addition to performing these discovery studies, the greatest challenge for the field is transitioning from much needed epigenome phenotyping studies to interventional studies that seek to prevent or cause age-related patterns of DNA modifications to determine the function. A major difficulty is that simplistic manipulation of DNMT or TET enzyme activity, similar to those typically performed in “mechanistic” studies through pharmacological or genetic manipulation, does not seem appropriate here. As demonstrated by studies described above, changes in DNA modification are a complex set of hyper- and hypomethylation at specific genomic loci across the genome. The functional role of these patterns will almost certainly not be mechanistically unraveled by simply increasing or decreasing overall enzymatic activity and/or expression. An alternative approach would be to enlist the current generation of genome editing tools (Kim and Kim 2014) to modify the epigenome at specific genomic locations. Recent reviews provide a good overview of this potential paradigm shift (Voigt and Reinberg 2013; Thakore et al. 2016; Stricker et al. 2017; Willyard 2017). Briefly, the concept is to use tools like CRISPR (Vojta et al. 2016), zinc fingers (Kungulovski et al. 2015) or transcription activator-like effector (TALES) (Maeder et al. 2013) to drive fusion proteins with DNMTs or TETs to a specific location or locations in the genome to alter modification status. This offers great promise to move epigenomic patterns to more mechanistic investigations (Stricker et al. 2017). These approaches still require more development and the concerns about the specificity of these targeting mechanisms will need to be addressed. Nonetheless, a pathway from discovery to intervention is now feasible and should be able to, in time, address many of the fundamental questions in epigenomics of aging.
Conclusion
This review is intended to provide an overview and primer on analysis of DNA modifications in geroscience studies, especially for those new to critically analyzing or performing these studies. With previous hypotheses of the epigenetic contribution to aging already being refuted (Unnikrishnan et al. 2017a) and new era of understanding the epigenetics of aging is likely upon us (Lopez-Leon and Goya 2017). With a full understanding of how these new analytical techniques work, their strengths and limitation, and possible future directions, geroscientists will be equipped to understand and participate in these studies.
Acknowledgements
The authors thank Donald Dunn for assistance with figure generation.
Funding information
This work was supported the Donald W. Reynolds Foundation, the Oklahoma Nathan Shock Center of Excellence in the Biology of Aging Targeted DNA Methylation and Mitochondrial Heteroplasmy Core (P30AG050911), the National Institute on Aging (R01AG026607, F31AG038285, T32AG052363, K99AG051661), the National Eye Institute (R01EY021716, R21EY024520, T32EY023202), and the Oklahoma Center for Advancement of Science and Technology (HR14-174).
References
- Adey A, Shendure J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012;22:1139–1143. doi: 10.1101/gr.136242.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012;8:e1002781. doi: 10.1371/journal.pgen.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Allum F, et al. Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants. Nat Commun. 2015;6:7211. doi: 10.1038/ncomms8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida RD, et al. Semi-quantitative immunohistochemical detection of 5-hydroxymethyl-cytosine reveals conservation of its tissue distribution between amphibians and mammals. Epigenetics. 2012;7:137–140. doi: 10.4161/epi.7.2.18949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angermueller C, et al. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13:229–232. doi: 10.1038/nmeth.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T, Akalin A. Genome-wide analysis of DNA methylation patterns by high-throughput sequencing. In: Aransay AM, Lavín Trueba JL, editors. Field guidelines for genetic experimental designs in high-throughput sequencing. Cham: Springer International Publishing; 2016. pp. 197–221. [Google Scholar]
- Benayoun BA, et al. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16:593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DL, et al. The BisPCR(2) method for targeted bisulfite sequencing. Epigenetics Chromatin. 2015;8:27. doi: 10.1186/s13072-015-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH, et al. Notes on the role of dynamic DNA methylation in mammalian development. Proc Natl Acad Sci U S A. 2015;112:6796–6799. doi: 10.1073/pnas.1415301111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, et al. DNA methylation detection at single base resolution using targeted next generation bisulfite sequencing and cross validation using capillary sequencing. Gene. 2016;594:259–267. doi: 10.1016/j.gene.2016.09.019. [DOI] [PubMed] [Google Scholar]
- Bibikova M, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- Bibikova M, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8:1499–1504. doi: 10.1093/nar/8.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- Blueprint_consortium Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat Biotechnol. 2016;34:726–737. doi: 10.1038/nbt.3605. [DOI] [PubMed] [Google Scholar]
- Bock C. Analysing and interpreting DNA methylation data. Nat Rev Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- Booth MJ, et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- Booth MJ, et al. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat Protoc. 2013;8:1841–1851. doi: 10.1038/nprot.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth MJ, et al. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat Chem. 2014;6:435–440. doi: 10.1038/nchem.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ. Epigenetics: rich pore methods for DNA methylation detection. Nat Rev Genet. 2017;18:209. doi: 10.1038/nrg.2017.18. [DOI] [PubMed] [Google Scholar]
- Cedar H, et al. Direct detection of methylated cytosine in DNA by use of the restriction enzyme MspI. Nucleic Acids Res. 1979;6:2125–2132. doi: 10.1093/nar/6.6.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CG, et al. TET-catalyzed 5-hydroxymethylcytosine regulates gene expression in differentiating colonocytes and colon cancer. Sci Rep. 2015;5:17568. doi: 10.1038/srep17568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, et al. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BH, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 2016;8:1844–1865. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GG, et al. Medium throughput bisulfite sequencing for accurate detection of 5-methylcytosine and 5-hydroxymethylcytosine. BMC Genomics. 2017;18:96. doi: 10.1186/s12864-017-3489-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BC, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602. doi: 10.1371/journal.pgen.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, et al. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, et al. Single-cell epigenomics: powerful new methods for understanding gene regulation and cell identity. Genome Biol. 2016;17:72. doi: 10.1186/s13059-016-0944-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ, et al. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq) Nat Protoc. 2017;12:534–547. doi: 10.1038/nprot.2016.187. [DOI] [PubMed] [Google Scholar]
- Cokus SJ, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JJ, et al. Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol. 2017;18:58. doi: 10.1186/s13059-017-1185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary-Dooley FK, et al. A comparison of existing global DNA methylation assays to low-coverage whole-genome bisulfite sequencing for epidemiological studies. Epigenetics. 2017;12:206–214. doi: 10.1080/15592294.2016.1276680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning AP, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7:e1002384. doi: 10.1371/journal.pgen.1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozmorov MG. Epigenomic annotation-based interpretation of genomic data: from enrichment analysis to machine learning. Bioinformatics. 2017;33:3323–3330. doi: 10.1093/bioinformatics/btx414. [DOI] [PubMed] [Google Scholar]
- Dupont JM, et al. De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem. 2004;333:119–127. doi: 10.1016/j.ab.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Edgar R, et al. Meta-analysis of human methylomes reveals stably methylated sequences surrounding CpG islands associated with high gene expression. Epigenetics Chromatin. 2014;7:28. doi: 10.1186/1756-8935-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen J, et al. Senescence-associated DNA methylation is stochastically acquired in subpopulations of mesenchymal stem cells. Aging Cell. 2017;16:183–191. doi: 10.1111/acel.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, et al. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuke C, et al. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann Hum Genet. 2004;68:196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Bakelman FE et al. (2015) Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J Vis Exp:e52246 [DOI] [PMC free article] [PubMed]
- Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina S, et al. Single-cell, locus-specific bisulfite sequencing (SLBS) for direct detection of epimutations in DNA methylation patterns. Nucleic Acids Res. 2015;43:e93. doi: 10.1093/nar/gkv366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina S, et al. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol. 2016;17:150. doi: 10.1186/s13059-016-1011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, et al. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6:468–481. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- Hadad N, et al. Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenetics Chromatin. 2016;9:30. doi: 10.1186/s13072-016-0080-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadad N et al (2017a) Caloric-restriction attenuates age-associated alterations in CG and non-CG methylation in the old brain. BioRxv. 10.1101/175810
- Hadad N et al. (2017b) Caloric restriction mitigates age-associated hippocampal differential CG and non-CG methylation. bioRxiv [DOI] [PMC free article] [PubMed]
- Hadad N et al (2017c) Caloric-restriction attenuates age-associated alterations in CG and non-CG methylation in the old brain. BioRxv: 10.1101/175810
- Hahn O, et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017;18:56. doi: 10.1186/s13059-017-1187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, et al. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RA, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Ecker JR. Non-CG methylation in the human genome. Annu Rev Genomics Hum Genet. 2015;16:55–77. doi: 10.1146/annurev-genom-090413-025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hing B, et al. Adaptation of the targeted capture methyl-Seq platform for the mouse genome identifies novel tissue-specific DNA methylation patterns of genes involved in neurodevelopment. Epigenetics. 2015;10:581–596. doi: 10.1080/15592294.2015.1045179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. Epigenetics: a historical overview. Epigenetics. 2006;1:76–80. doi: 10.4161/epi.1.2.2762. [DOI] [PubMed] [Google Scholar]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. 1948;175:315–332. [PubMed] [Google Scholar]
- Houseman EA, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Ito S, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov M, et al. In-solution hybrid capture of bisulfite-converted DNA for targeted bisulfite sequencing of 174 ADME genes. Nucleic Acids Res. 2013;41:e72. doi: 10.1093/nar/gks1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M et al. (2017) Nanopore sequencing and assembly of a human genome with ultra-long reads. bioRxiv [DOI] [PMC free article] [PubMed]
- Jeck WR, et al. Review: a meta-analysis of GWAS and age-associated diseases. Aging Cell. 2012;11:727–731. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MJ et al. (2015) DNA methylation and healthy human aging. Aging Cell [DOI] [PMC free article] [PubMed]
- Jorgensen HF, et al. Engineering a high-affinity methyl-CpG-binding protein. Nucleic Acids Res. 2006;34:e96. doi: 10.1093/nar/gkl527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalani A, et al. Nutri-epigenetics ameliorates blood-brain barrier damage and neurodegeneration in hyperhomocysteinemia: role of folic acid. J Mol Neurosci. 2014;52:202–215. doi: 10.1007/s12031-013-0122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Mrázek J. Compositional differences within and between eukaryotic genomes. Proc Natl Acad Sci. 1997;94:10227–10232. doi: 10.1073/pnas.94.19.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal A, et al. Comparison of different cell type correction methods for genome-scale epigenetics studies. BMC Bioinformatics. 2017;18:216. doi: 10.1186/s12859-017-1611-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159:709–713. doi: 10.1016/j.cell.2014.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna A et al (2013) EpiGnome[trade] methyl-Seq kit: a novel post-bisulfite conversion library prep method for methylation analysis. Nat Methods 10
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Kim S, et al. The frailty index outperforms DNA methylation age and its derivatives as an indicator of biological age. Geroscience. 2017;39:83–92. doi: 10.1007/s11357-017-9960-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinde B, et al. Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A. 2015;112:6800–6806. doi: 10.1073/pnas.1411269112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, et al. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age (Dordr) 2012;34:1405–1419. doi: 10.1007/s11357-011-9315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishore K, et al. methylPipe and compEpiTools: a suite of R packages for the integrative analysis of epigenomics data. BMC Bioinformatics. 2015;16:313. doi: 10.1186/s12859-015-0742-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P, et al. BarraCUDA—a fast short read sequence aligner using graphics processing units. BMC Res Notes. 2012;5:27. doi: 10.1186/1756-0500-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori HK, et al. Application of microdroplet PCR for large-scale targeted bisulfite sequencing. Genome Res. 2011;21:1738–1745. doi: 10.1101/gr.116863.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J, Rahmann S. Massively parallel read mapping on GPUs with the q-group index and PEANUT. PeerJ. 2014;2:e606. doi: 10.7717/peerj.606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlenkov A et al (2017) DNA methylation profiling of human prefrontal cortex neurons in heroin users shows significant difference between genomic contexts of hyper- and hypomethylation and a younger epigenetic age. Genes (Basel) 8 [DOI] [PMC free article] [PubMed]
- Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, et al. DNA methylome analysis using short bisulfite sequencing data. Nat Methods. 2012;9:145–151. doi: 10.1038/nmeth.1828. [DOI] [PubMed] [Google Scholar]
- Kungulovski G, et al. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics Chromatin. 2015;8:12. doi: 10.1186/s13072-015-0002-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Li Q, et al. Post-conversion targeted capture of modified cytosines in mammalian and plant genomes. Nucleic Acids Res. 2015;43:e81. doi: 10.1093/nar/gkv244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Ecker JR. Finding the fifth base: genome-wide sequencing of cytosine methylation. Genome Res. 2009;19:959–966. doi: 10.1101/gr.083451.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA. Turning over DNA methylation in the mind. Front Neurosci. 2015;9:252. doi: 10.3389/fnins.2015.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logue MW et al. (2017) The correlation of methylation levels measured using Illumina 450K and EPIC BeadChips in blood samples. Epigenomics [DOI] [PMC free article] [PubMed]
- Lopez-Leon M, Goya RG. The emerging view of aging as a reversible epigenetic process. Gerontology. 2017;63:426–431. doi: 10.1159/000477209. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, et al. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou S, et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 2014;15:408. doi: 10.1186/s13059-014-0408-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, et al. Chemical modification-assisted bisulfite sequencing (CAB-Seq) for 5-carboxylcytosine detection in DNA. J Am Chem Soc. 2013;135:9315–9317. doi: 10.1021/ja4044856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357:600–604. doi: 10.1126/science.aan3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat Biotechnol. 2013;31:1137–1142. doi: 10.1038/nbt.2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010;20:332–340. doi: 10.1101/gr.096826.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamanova L, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- Manconi A, et al. GPU-BSM: a GPU-based tool to map bisulfite-treated reads. PLoS One. 2014;9:e97277. doi: 10.1371/journal.pone.0097277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold CA, et al. CNS-wide sexually dimorphic induction of the major histocompatibility complex 1 pathway with aging. J Gerontol A Biol Sci Med Sci. 2017;72:16–29. doi: 10.1093/gerona/glv232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold CA, et al. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J Neuroinflammation. 2017;14:141. doi: 10.1186/s12974-017-0920-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16:25. doi: 10.1186/s13059-015-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE et al. (2016) The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int J Epidemiol [DOI] [PMC free article] [PubMed]
- Martinowich K, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Masser DR, et al. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenetics Chromatin. 2013;6:33. doi: 10.1186/1756-8935-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR et al. (2015) Targeted DNA methylation analysis by next-generation sequencing. J Vis Exp [DOI] [PMC free article] [PubMed]
- Masser DR, et al. Bisulfite oligonucleotide-capture sequencing for targeted base- and strand-specific absolute 5-methylcytosine quantitation. Age (Dordr) 2016;38:49. doi: 10.1007/s11357-016-9914-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser DR et al. (2017a) Sexually divergent DNA methylation patterns with hippocampal aging. Aging Cell [DOI] [PMC free article] [PubMed]
- Masser DR et al. (2017b) Sexually divergent DNA methylation programs with hippocampal aging. bioRxiv 161752 [DOI] [PMC free article] [PubMed]
- Masser DR et al. (2017c) Sexually divergent DNA methylation patterns with hippocampal aging. Aging Cell:Preprint from BioRxv. doi: 10.1101/161752 [DOI] [PMC free article] [PubMed]
- Mei Y, et al. Aging-associated formaldehyde-induced norepinephrine deficiency contributes to age-related memory decline. Aging Cell. 2015;14:659–668. doi: 10.1111/acel.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–5877. doi: 10.1093/nar/gki901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura F, et al. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136. doi: 10.1093/nar/gks454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron. 2015;86:1369–1384. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, et al. Epigenomic landscapes of retinal rods and cones. elife. 2016;5:e11613. doi: 10.7554/eLife.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LD, et al. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. doi: 10.1038/npp.2012.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, et al. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016;8:389–399. doi: 10.2217/epi.15.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulqueen RM et al. (2017) Scalable and efficient single-cell DNA methylation sequencing by combinatorial indexing. bioRxiv
- Neri F, et al. Methylation-assisted bisulfite sequencing to simultaneously map 5fC and 5caC on a genome-wide scale for DNA demethylation analysis. Nat Protoc. 2016;11:1191–1205. doi: 10.1038/nprot.2016.063. [DOI] [PubMed] [Google Scholar]
- Nestor CE, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–477. doi: 10.1101/gr.126417.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noer A, et al. Dynamics of adipogenic promoter DNA methylation during clonal culture of human adipose stem cells to senescence. BMC Cell Biol. 2007;8:18. doi: 10.1186/1471-2121-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/S0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Olova N et al. (2017) Comparison of whole-genome bisulfite sequencing library preparation strategies identifies sources of biases affecting DNA methylation data. bioRxiv [DOI] [PMC free article] [PubMed]
- Pacheco SE, et al. Integrative DNA methylation and gene expression analyses identify DNA packaging and epigenetic regulatory genes associated with low motility sperm. PLoS One. 2011;6:e20280. doi: 10.1371/journal.pone.0020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Wu H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics. 2016;32:1446–1453. doi: 10.1093/bioinformatics/btw026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish RR et al. (2012) Direct bisulfite sequencing for examination of DNA methylation with gene and nucleotide resolution from brain tissues. Curr Protoc Neurosci Chapter 7:Unit 7 24 [DOI] [PMC free article] [PubMed]
- Petkovich DA, et al. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 2017;25:954–960 e956. doi: 10.1016/j.cmet.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiber E-A, et al. Mapping and elucidating the function of modified bases in DNA. Nat Rev Chem. 2017;1:0069. doi: 10.1038/s41570-017-0069. [DOI] [Google Scholar]
- Rakyan VK, et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand AC, et al. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods. 2017;14:411–413. doi: 10.1038/nmeth.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch T, Pfeifer GP. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab Investig. 2005;85:1172–1180. doi: 10.1038/labinvest.3700311. [DOI] [PubMed] [Google Scholar]
- Reddy PM, Reddy PR. Effect of prolactin on DNA methylation in the liver and kidney of rat. Mol Cell Biochem. 1990;95:43–47. doi: 10.1007/BF00219528. [DOI] [PubMed] [Google Scholar]
- Reed K, et al. Comparison of bisulfite sequencing PCR with pyrosequencing for measuring differences in DNA methylation. Anal Biochem. 2010;397:96–106. doi: 10.1016/j.ab.2009.10.021. [DOI] [PubMed] [Google Scholar]
- Reilly JG, et al. DNA methylation in mouse cells in culture as measured by restriction enzymes. Biochim Biophys Acta. 1982;697:53–59. doi: 10.1016/0167-4781(82)90044-6. [DOI] [PubMed] [Google Scholar]
- Schatz MC. Nanopore sequencing meets epigenetics. Nat Methods. 2017;14:347–348. doi: 10.1038/nmeth.4240. [DOI] [PubMed] [Google Scholar]
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- Schultz MD, et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015;523:212–216. doi: 10.1038/nature14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, et al. Epigenetic mechanisms of longevity and aging. Cell. 2016;166:822–839. doi: 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, et al. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafi A et al. (2017) A survey of the approaches for identifying differential methylation using bisulfite sequencing data. Brief Bioinform [DOI] [PMC free article] [PubMed]
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Shendure J, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550:345–353. doi: 10.1038/nature24286. [DOI] [PubMed] [Google Scholar]
- Shi DQ, et al. New insights into 5hmC DNA modification: generation, distribution and function. Front Genet. 2017;8:100. doi: 10.3389/fgene.2017.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkin AJ, et al. Are objective measures of physical capability related to accelerated epigenetic age? Findings from a British birth cohort. BMJ Open. 2017;7:e016708. doi: 10.1136/bmjopen-2017-016708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, et al. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods. 2017;14:407–410. doi: 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- Song L, et al. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal Chem. 2005;77:504–510. doi: 10.1021/ac0489420. [DOI] [PubMed] [Google Scholar]
- Spruijt CG, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Stricker SH, et al. From profiles to function in epigenomics. Nat Rev Genet. 2017;18:51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- Stubbs TM, et al. Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 2017;18:68. doi: 10.1186/s13059-017-1203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, et al. Base resolution methylome profiling: considerations in platform selection, data preprocessing and analysis. Epigenomics. 2015;7:813–828. doi: 10.2217/epi.15.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, et al. Genome-wide comparison of DNA hydroxymethylation in mouse embryonic stem cells and neural progenitor cells by a new comparative hMeDIP-seq method. Nucleic Acids Res. 2013;41:e84. doi: 10.1093/nar/gkt091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Relton CL (2017) Statistical and integrative system-level analysis of DNA methylation data. Nat Rev Genet [DOI] [PubMed]
- Thakore PI, et al. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods. 2016;13:127–137. doi: 10.1038/nmeth.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippmann S. Programming tools: adventures with R. Nature. 2015;517:109–110. doi: 10.1038/517109a. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan A et al. (2017a) Revisiting the genomic hypomethylation hypothesis of aging. Annals of the New York Acedemy of Science In press [DOI] [PMC free article] [PubMed]
- Unnikrishnan A, et al. Role of DNA methylation in the dietary restriction mediated cellular memory. Geroscience. 2017;39:331–345. doi: 10.1007/s11357-017-9976-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdes AM, et al. Omics technologies and the study of human ageing. Nat Rev Genet. 2013;14:601–607. doi: 10.1038/nrg3553. [DOI] [PubMed] [Google Scholar]
- Vanyushin BF, et al. The 5-methylcytosine in DNA of rats. Tissue and age specificity and the changes induced by hydrocortisone and other agents. Gerontologia. 1973;19:138–152. doi: 10.1159/000211967. [DOI] [PubMed] [Google Scholar]
- Varley KE, Mitra RD. Bisulfite patch PCR enables multiplexed sequencing of promoter methylation across cancer samples. Genome Res. 2010;20:1279–1287. doi: 10.1101/gr.101212.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P, Reinberg D. Epigenome editing. Nat Biotechnol. 2013;31:1097–1099. doi: 10.1038/nbt.2756. [DOI] [PubMed] [Google Scholar]
- Vojta A, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–5628. doi: 10.1093/nar/gkw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. Organisers & genes. Cambridge: The University Press; 1940. [Google Scholar]
- Wang J, et al. High resolution profiling of human exon methylation by liquid hybridization capture-based bisulfite sequencing. BMC Genomics. 2011;12:597. doi: 10.1186/1471-2164-12-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, et al. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017;18:57. doi: 10.1186/s13059-017-1186-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet. 2005;37:853–862. doi: 10.1038/ng1598. [DOI] [PubMed] [Google Scholar]
- Weidner CI, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15:R24. doi: 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willyard C. The epigenome editors: how tools such as CRISPR offer new details about epigenetics. Nat Med. 2017;23:900–903. doi: 10.1038/nm0817-900. [DOI] [PubMed] [Google Scholar]
- Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AS, et al. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, et al. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat Commun. 2017;8:1122. doi: 10.1038/s41467-017-01195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B et al. (2017) Genome-wide, single-cell DNA methylomics reveals increased non-CpG methylation during human oocyte maturation. Stem Cell Rep [DOI] [PMC free article] [PubMed]
- Zampieri M, et al. Reconfiguration of DNA methylation in aging. Mech Ageing Dev. 2015;151:60–70. doi: 10.1016/j.mad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. DNA methylation analysis by bisulfite conversion, cloning, and sequencing of individual clones. Methods Mol Biol. 2009;507:177–187. doi: 10.1007/978-1-59745-522-0_14. [DOI] [PubMed] [Google Scholar]
- Zhao L, et al. The dynamics of DNA methylation fidelity during mouse embryonic stem cell self-renewal and differentiation. Genome Res. 2014;24:1296–1307. doi: 10.1101/gr.163147.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SC, et al. Correcting for cell-type heterogeneity in epigenome-wide association studies: revisiting previous analyses. Nat Methods. 2017;14:216–217. doi: 10.1038/nmeth.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, et al. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat Methods. 2015;12:230–232. doi: 10.1038/nmeth.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, et al. Targeted bisulfite sequencing of the dynamic DNA methylome. Epigenetics Chromatin. 2016;9:55. doi: 10.1186/s13072-016-0105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, et al. Epigenome-wide association studies without the need for cell-type composition. Nat Methods. 2014;11:309–311. doi: 10.1038/nmeth.2815. [DOI] [PubMed] [Google Scholar]
- Zykovich A, et al. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell. 2014;13:360–366. doi: 10.1111/acel.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]