

Abstract
Multiple system atrophy (MSA) is a complex and multifactorial neurodegenerative disease, and its pathogenesis remains uncertain. Patients with MSA or spinocerebellar ataxia (SCA) show overlapping clinical phenotypes. Previous studies have reported that intermediate or long CAG expansions in SCA genes have been associated with other neurodegenerative disease. In this study, we screened for the number of CAG repeats in ATXN1, 2 and 3 in 200 patients with MSA and 314 healthy controls to evaluate possible associations between (CAG)n in these three polyQ-related genes and MSA. Our findings indicated that longer repeat lengths in ATXN2 were associated with increased risk for MSA in Chinese individuals. No relationship was observed between CAG repeat length in the three examined genes and age at onset (AO) of MSA.
Introduction
Multiple system atrophy (MSA) is an adult-onset neurodegenerative disease characterized by various combinations of autonomic failure, cerebellar ataxia and Parkinsonism1,2. MSA is classified into two subtypes, MSA with predominant cerebellar ataxia (MSA-C) and MSA with predominant Parkinsonism (MSA-P)2.
Spinocerebellar ataxias (SCAs) are a group of neurodegenerative diseases characterized by cerebellar dysfunction that may be accompanied by other neurological abnormalities3,4. Patients with MSA have similar symptoms to those with SCA, including prominent ataxia, dysmetria and eye movement anomalies3,4. Several studies have suggested links among neurodegenerative diseases, their causative genes and clinical syndromes characterized by overlapped phenotypes5–7. For instance, patients diagnosed with MSA carry expanded CAG repeats in ATXN38. Furthermore, research has revealed a strong effect of ATXN2 expansion on Parkinson disease (PD) and the MSA-P subtype9, and intermediate-expansion within ATXN1 or ATXN2 has been associated with an increased risk for amyotrophic lateral sclerosis (ALS)10. In animal and cell models of ALS, ataxin2 interacts with TDP-43 and forms characteristic cytoplasmic aggregates in neurons, suggesting a relationship between ATXN2 and TDP-43 toxicity10,11.
The specific molecular mechanisms underlying the pathogenesis of MSA remain unclear. However, several genes other than COQ2 have been implicated in MSA, including SNCA, SHC2 and ATXN29,12,13. From this perspective, we aimed to test the hypothesis that CAG repeat sizes for the genes involved in SCA (namely, ATXN1, 2 and 3) might play a role as risk factors for MSA and affect age at onset (AO) of the disease.
Materials and Methods
Subjects and samples
A total of 200 patients (128 males, 72 females; 35–72 years of age, with a mean age of 53.4 ± 7.6 years) who satisfied consensus criteria for MSA were recruited from the outpatient neurology clinic of Xiangya Hospital, Central South University, Hunan, China2. Clinical stage was evaluated using the Unified Multiple System Atrophy Rating Scale (UMSARS). There were 148 patients with MSA-C and 52 patients with MSA-P. This study also included 314 healthy controls with no history of neurodegenerative disease or other diseases.
The control and patient groups were matched with respect to age (age range, 30–74 years; mean age, 53.5 ± 7.6 years), sex ratio (203 males, 111 females), and region of residence. All patients and controls were from the Han Chinese population. The study was approved by the Ethics Committee of Xiangya Hospital, and written informed consent was obtained from all participants.
DNA analysis and genotype classification
Genomic DNA was extracted from peripheral blood using standard phenol-chloroform extraction procedures14. Genotyping of ATXN1, 2 and 3 was determined by polymerase chain reaction (PCR) amplification of CAG tracts in combination with capillary electrophoresis, using GeneMarker software (SoftGenetics). At the SCA1, SCA2 and SCA3/MJD loci, the allele containing the larger repeat was designated the ‘long’ allele, and the other allele was regarded as the ‘short’ allele. The short and long alleles were considered separately in statistical models. With respect to the multimodal or skewed distributions in Fig. 1, repeats in long alleles of ATXN2 and ATXN3 were classified as short, short-medium, medium or long in accordance with the approach described in previous studies15,16. For ATXN1, since the size of the long allele exhibited a nearly normal distribution, we divided long alleles into two groups, short (≤29 repeats) and long (å 29 repeats), based on mean repeat size.
Figure 1.
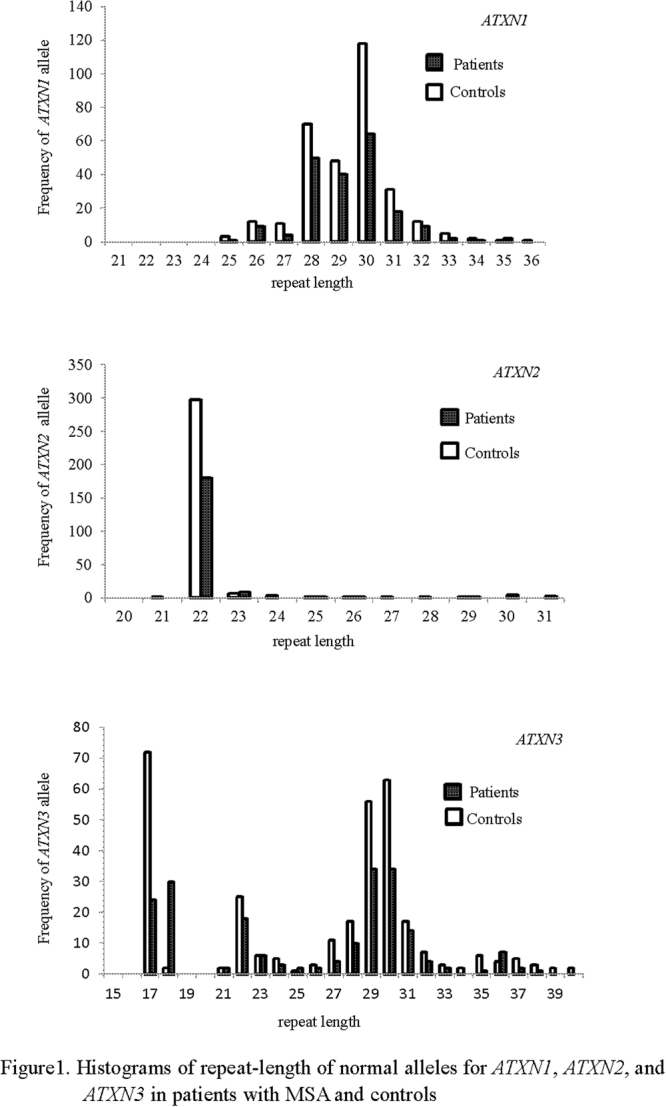
Histograms showing repeat lengths for the long allele in ATXN1, ATXN2, and ATXN3 in patients with MSA and controls.
Statistical analysis
Differences in age and sex between the patients with MSA and the controls were assessed using a t-test and a chi-square test. Descriptive statistics are expressed as mean ± standard deviation (Table 1). Associations between size of (CAG)n and risk for MSA were determined via logistic regression, adjusting for age and sex. We used one-way factorial analysis of variance (ANOVA) or the Kruskal-Wallis test to investigate the association between (CAG)n size and AO for the patients with MSA. The Mann-Whitney U test or t-tests were used to test for differences in repeat length between patients with MSA-C and patients with MSA-P. A two-tailed P-value ≤ 0.05 was regarded as significant.
Table 1.
Demographic data and mean (CAG)n repeats for patients with MSA and controls.
| Variable | MSA | Controls |
|---|---|---|
| number | 200 | 314 |
| males (%) | 128 (64.0) | 203 (64.6) |
| mean age/age at onset ± SD | 53.4 ± 7.4 | 53.5 ± 7.6 |
| age range | 35–72 | 30–74 |
| (CAG)n in the long allele | ||
| ATXN1: | ||
| mean ± SD | 29.3 ± 1.6 | 29.4 ± 1.6 |
| range | 25–35 | 25–36 |
| ATXN2: | ||
| mean ± SD | 22.4 ± 1.6 | 22.1 ± 0.7 |
| range | 22–31 | 21–29 |
| ATXN3: | ||
| mean ± SD | 25.6 ± 5.9 | 26.2 ± 6.1 |
| range | 17–38 | 17–40 |
SD = standard deviation.
Results
Table 1 summarizes demographic data for all 514 participants. No pathological (CAG)n expansion in the three SCA genes was detected (in either patients or controls). Distributions of (CAG)n size are shown in Fig. 1. In ATXN1, the mean size was 29.3 ± 1.6, ranging 25–35 repeats in patients and 29.4 ± 1.6 in controls, ranging 25–36 repeats. In ATXN2, the mean size was 22.4 ± 1.6, ranging 22–31 repeats in patients and in controls 22.1 ± 0.7, ranging 22–29 repeats; 8 patients and 3 controls carried long expansions (Supplementary Table S2). In ATXN3, the mean size was 25.6 ± 5.9, ranging 17–38 repeats in patients and in controls 26.2 ± 6.1, ranging 17–40 repeats.
For the ATXN2 locus, there was a significant difference in the distribution of CAG repeats between patients and controls (P = 0.011, OR = 1.253, 95% CI = [1.052–1.492]) (Table 2). The number of CAG repeats in ATXN1 and ATXN3 did not significantly differ between patients with MSA and controls. There were no significant correlations between AO for MSA and repeat length in ATXN1, 2 or 3 (Table 3). In addition, the distributions of (CAG)n size in any of these three genes did not significantly differ between patients with MSA-C and patients with MSA-P (Supplementary Table S1).
Table 2.
Comparison of (CAG)n sizes for polyQ-related genes in patients with MSA and controls.
| Locus | n | P-value | OR | 95% CI | |
|---|---|---|---|---|---|
| ATXN1 | patients | 200 | 0.793 | 0.985 | 0.879–1.104 |
| controls | 314 | ||||
| ATXN2 | patients | 200 | 0.011 | 1.253 | 1.052–1.492 |
| controls | 314 | ||||
| ATXN3 | patients | 200 | 0.207 | 0.981 | 0.952–1.011 |
| controls | 314 | ||||
P-values were calculated via logistic regression using SPSS 18.0.
Table 3.
Effects of (CAG)n size in polyQ-related genes on age at onset for patients with MSA.
| Locus | Group (CAGs) | n | Age at onset | P-value |
|---|---|---|---|---|
| ATXN1 | short | 104 | 52.5 ± 7.2 | 0.072 |
| long | 96 | 54.3 ± 7.5 | ||
| ATXN2 | short-medium | 181 | 53.4 ± 7.2 | 0.842 |
| medium | 11 | 54.1 ± 8.5 | ||
| long | 8 | 51.6 ± 10.4 | ||
| ATXN3 | short-medium | 85 | 53.3 ± 7.2 | 0.898 |
| long | 115 | 53.4 ± 7.5 |
*Group: Long alleles of ATXN1, ATXN2, and ATXN3 were divided into several groups as follows:
ATXN1: short: ≤29 CAGs; long: >29 CAGs.
ATXN2: short: <22 CAGs; short-medium: 22 CAGs; medium: 23–26 CAGs; long: 27–32 CAGs.
ATXN3: short: <19 CAGs; short-medium: 19–25 CAGs; long: 26–40 CAGs.
P-value estimated by one-way ANOVA or Kruskal-Wallis test.
Discussion
None of our patients exhibited pathogenic expansion in any of the three examined polyQ-related genes, indicating that such expansion may not be a causative factor for MSA. Nevertheless, we found a significant association between CAG repeat sizes in ATXN2 and risk for MSA.
The most common size (over 95%) of the (CAG)n in SCA2 is either 22 or 23 (range, 14–32)17–20. Ataxin2, which is encoded by ATXN2, is localized to the rough endoplasmic reticulum and plays a critical role in mRNA processing21. In the pathogenesis of SCA2, polyQ expansion of ataxin2 confers a gain-of-function mutation that induces neuronal impairment and triggers the disease phenotype21. Ataxin2 is also closely related to other neurodegenerative diseases, such as ALS, PD and spinocerebellar ataxia type 3 (SCA3/MJD)22–25. Functional studies have proven that ataxin2 interacts with TDP-43 via joint mRNA binding, aggravating TDP-43 toxicity and thereby further increasing the risk of developing ALS22. In a yeast model, ataxin2 was shown to be a modifier of α-synuclein biotoxicity in specific molecular pathways and a predictive nodal point in the α-synuclein network6. As an mRNA-related translation factor, ataxin2 has been associated with α-synuclein toxicity in neurons of patients with PD6. The neuropathological hallmark of MSA is the presence of glial cytoplasmic inclusions (GCIs) containing α-synuclein; given this characteristic, MSA can be regarded as a synucleinopathy, together with PD and Lewy body dementia (DLB)26–28. We can speculate that ATXN2 increases the risk for MSA by perturbing mRNA metabolism and translation and thereby influencing α-synuclein biotoxicity.
An association was found between MSA and CAG repeat sizes in ATXN2 but not CAG repeat size in ATXN1 or ATXN3, implying that ATXN2 may play a role as a risk factor for MSA (at least) in the Chinese population; however, no modifying effects of repeat lengths at SCA1, SCA2 or SCA3/MJD loci on AO of MSA were observed, possibly due to sample size. The small number of ATXN2 long-expansion, over 26 CAGs, found in patients and controls (due to their rarity in the general population) is, of course, a limitation that cannot be easily overcome. Further studies in different ethnic populations and a larger sample size are needed to confirm the present findings.
In terms of mechanisms for neurodegeneration, it would be important to shed some light on a possible pathogenic interaction between ataxin2 and α-synuclein in MSA. The genetic association between ATXN2 and MSA may contribute to a more comprehensive understanding of neurodegenerative disorders and to foster new therapies for such diseases.
Ethics statement
This study was approved by the Ethics Committee of Xiangya Hospital of Central South University in China (equivalent to an institutional review board), and all methods were performed in accordance with approved guidelines. Written informed consent was obtained from all participants.
Electronic supplementary material
Acknowledgements
We are grateful to all of the subjects for their participation in our study. This study was supported by the National Natural Science Foundation of China (Nos 81471156, and 81771231 to Hong Jiang, Nos 81130021 and 81430023 to Beisha Tang), the National Key Research and Development Program of China (Nos 2016YFC0901504 and 2016YFC0905100 to Hong Jiang), the National Natural Science Foundation of China (No. 31401135 to Rong Qiu), and Clinical Research Funds of Xiangya Hospital (No. 2014L03 to Hong Jiang).
Author Contributions
Research project: Conception: Xin Zhou, Chunrong Wang, Huirong Peng and Hong Jiang; Organization: Xin Zhou, Puzhi Wang, Xaocan Hou, Tianjiao Li and Wei Ye; Execution: Xin Zhou, Chunrong Wang, Dongxue Ding and Xuan Hou. Statistical analysis: Design: Xin Zhou, Chunrong Wang and Hong Jiang; Execution: Xin Zhou, Chunrong Wang, Zhao Chen, Yun Peng and Huihua Yang; Review and Critique: Dongxue Ding, Hong Jiang, Kun Xia and Rong Qiu. Manuscript: Writing of the first draft: Xin Zhou; Review and critique: Xin Zhou, Chunrong Wang, Hong Jiang, Beisha Tang and Jorge Sequeiros.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22290-0.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fanciulli A, Wenning GK. Multiple-system atrophy. The New England journal of medicine. 2015;372:249–263. doi: 10.1056/NEJMra1311488. [DOI] [PubMed] [Google Scholar]
- 2.Gilman S, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. The Lancet. Neurology. 2004;3:291–304. doi: 10.1016/S1474-4422(04)00737-9. [DOI] [PubMed] [Google Scholar]
- 4.Tan EK, Ashizawa T. Genetic testing in spinocerebellar ataxias: defining a clinical role. Archives of neurology. 2001;58:191–195. doi: 10.1001/archneur.58.2.191. [DOI] [PubMed] [Google Scholar]
- 5.Chen, K. H., Lin, C. H. & Wu, R. M. Psychotic-affective symptoms and multiple system atrophy expand phenotypes of spinocerebellar ataxia type 2. BMJ case reports2012, 10.1136/bcr.10.2011.5061 (2012). [DOI] [PMC free article] [PubMed]
- 6.Khurana V, et al. Genome-Scale Networks Link Neurodegenerative Disease Genes to alpha-Synuclein through Specific Molecular Pathways. Cell systems. 2017;4:157–170.e114. doi: 10.1016/j.cels.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou, X. et al. Association of TNF-alpha rs1799964 and IL-1beta rs16944 polymorphisms with multiple system atrophy in Chinese Han population. The International journal of neuroscience, 1–4, 10.1080/00207454.2017.1418346 (2018). [DOI] [PubMed]
- 8.Nirenberg MJ, Libien J, Vonsattel JP, Fahn S. Multiple system atrophy in a patient with the spinocerebellar ataxia 3 gene mutation. Movement disorders: official journal of the Movement Disorder Society. 2007;22:251–254. doi: 10.1002/mds.21231. [DOI] [PubMed] [Google Scholar]
- 9.Kim JM, et al. Importance of low-range CAG expansion and CAA interruption in SCA2 Parkinsonism. Archives of neurology. 2007;64:1510–1518. doi: 10.1001/archneur.64.10.1510. [DOI] [PubMed] [Google Scholar]
- 10.Conforti FL, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology. 2012;79:2315–2320. doi: 10.1212/WNL.0b013e318278b618. [DOI] [PubMed] [Google Scholar]
- 11.Van Damme P, et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76:2066–2072. doi: 10.1212/WNL.0b013e31821f445b. [DOI] [PubMed] [Google Scholar]
- 12.Gwinn K, et al. Clinical features, with video documentation, of the original familial lewy body parkinsonism caused by alpha-synuclein triplication (Iowa kindred) Movement disorders: official journal of the Movement Disorder Society. 2011;26:2134–2136. doi: 10.1002/mds.23776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki H, et al. Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2)gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Molecular brain. 2011;4:24. doi: 10.1186/1756-6606-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tezenas du Montcel S, et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain: a journal of neurology. 2014;137:2444–2455. doi: 10.1093/brain/awu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, et al. (CAG)n loci as genetic modifiers of age-at-onset in patients with Machado-Joseph disease from mainland China. Brain: a journal of neurology. 2016;139:e41. doi: 10.1093/brain/aww087. [DOI] [PubMed] [Google Scholar]
- 17.Yagishita S, Inoue M. Clinicopathology of spinocerebellar degeneration: its correlation to the unstable CAG repeat of the affected gene. Pathology international. 1997;47:1–15. doi: 10.1111/j.1440-1827.1997.tb04429.x. [DOI] [PubMed] [Google Scholar]
- 18.Riess O, et al. SCA2 trinucleotide expansion in German SCA patients. Neurogenetics. 1997;1:59–64. doi: 10.1007/s100480050009. [DOI] [PubMed] [Google Scholar]
- 19.Sanpei K, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nature genetics. 1996;14:277–284. doi: 10.1038/ng1196-277. [DOI] [PubMed] [Google Scholar]
- 20.Pulst SM, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nature genetics. 1996;14:269–276. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 21.Albrecht M, Golatta M, Wullner U, Lengauer T. Structural and functional analysis of ataxin-2 and ataxin-3. European journal of biochemistry. 2004;271:3155–3170. doi: 10.1111/j.1432-1033.2004.04245.x. [DOI] [PubMed] [Google Scholar]
- 22.Elden AC, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nkiliza A, et al. RNA-binding disturbances as a continuum from spinocerebellar ataxia type 2 to Parkinson disease. Neurobiology of disease. 2016;96:312–322. doi: 10.1016/j.nbd.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Ding, D. et al. ATXN2 polymorphism modulates age at onset in Machado-Joseph disease. Brain: a journal of neurology, 10.1093/brain/aww176 (2016). [DOI] [PubMed]
- 25.Chen Z, et al. Ubiquitin-related network underlain by (CAG)n loci modulate age at onset in Machado-Joseph disease. Brain: a journal of neurology. 2017;140:e25. doi: 10.1093/brain/awx028. [DOI] [PubMed] [Google Scholar]
- 26.Farrer M, Gwinn-Hardy K, Hutton M, Hardy J. The genetics of disorders with synuclein pathology and parkinsonism. Human molecular genetics. 1999;8:1901–1905. doi: 10.1093/hmg/8.10.1901. [DOI] [PubMed] [Google Scholar]
- 27.Galvin JE, Lee VM, Trojanowski JQ. Synucleinopathies: clinical and pathological implications. Archives of neurology. 2001;58:186–190. doi: 10.1001/archneur.58.2.186. [DOI] [PubMed] [Google Scholar]
- 28.Sun ZF, et al. Increase of the plasma alpha-synuclein levels in patients with multiple system atrophy. Movement disorders: official journal of the Movement Disorder Society. 2014;29:375–379. doi: 10.1002/mds.25688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.