

Abstract
The lymphocyte toxicity assay (LTA) is a proposed surrogate marker of sulfonamide antibiotic hypersensitivity. In the LTA, peripheral blood mononuclear cells (PBMCs) undergo apoptosis more readily in hypersensitive versus tolerant patients when exposed to drug‐hydroxylamine metabolites in vitro. The purpose of this study was to identify key gene transcripts associated with increased cytotoxicity from sulfamethoxazole‐hydroxylamine in human PBMCs in the LTA. The LTA was performed on PBMCs of 10 patients hypersensitive to trimethoprim‐sulfamethoxazole (HS) and 10 drug‐tolerant controls (TOL), using two cytotoxicity assays: YO‐PRO (n = 20) and MTT (n = 12). mRNA expression profiles of PBMCs, enriched for CD8+ T cells, were compared between HS and TOL patients. Transcript expression was interrogated for correlation with % cytotoxicity from YO‐PRO and MTT assays. Correlated transcripts of interest were validated by qPCR. LTA results were not significantly different between HS and TOL patients, and no transcripts were found to be differentially expressed between the two groups. 96 transcripts were correlated with cytotoxicity by YO‐PRO (r = ±.63‐.75, FDR 0.188). Transcripts were selected for validation based on mechanistic plausibility and three were significantly over‐expressed by qPCR in high cytotoxicity patients: multi‐specific organic anion transporter C (ABCC5), mitoferrin‐1 (SLC25A37), and Porimin (TMEM123). These data identify novel transcripts that could contribute to sulfonamide‐hydroxylamine induced cytotoxicity. These include SLC25A37, encoding a mitochondrial iron transporter, ABCC5, encoding an arylamine drug transporter, and TMEM123, encoding a transmembrane protein that mediates cell death.
Keywords: cell death, hypersensitivity, idiosyncratic drug reactions, in vitro toxicity, reactive metabolites
Abbreviations
- ABCC5
ATP binding cassette subfamily C member 5
- CYB5A
cytochrome b 5
- CYB5R3
cytochrome b 5 reductase
- FDR
false discovery rate
- GO
gene ontology
- GSTM1
glutathione S‐transferase mu 1
- GSTT1
glutathione S‐transferase theta 1
- GUSB
glucuronidase beta
- HLA
human leukocyte antigen
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- HS
hypersensitivity, hypersensitive
- LTA
lymphocyte toxicity assay
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- NAT2
N‐acetyltransferase 2
- PBMC
peripheral blood mononuclear cell
- RPL37
ribosomal protein L37
- SLC25A28
solute carrier family 25 member 28
- SLC25A37
solute carrier family 25 member 37
- SMX‐HA
sulfamethoxazole‐hydroxylamine
- SMX
sulfamethoxazole
- TMEM123
transmembrane protein 123
- TMEM129
transmembrane protein 129
- TMP
trimethoprim
- TOL
tolerant
1. INTRODUCTION
Sulfonamide antibiotics, such as sulfamethoxazole (SMX) in combination with trimethoprim (TMP), are indicated for the treatment of urinary and respiratory tract infections and skin infections caused by methicillin‐resistant Staphylococcus aureus.1, 2, 3, 4 TMP‐SMX is also the drug of choice for the prevention and treatment of opportunistic fungal infections in immunocompromised patients, including those with AIDS.5, 6, 7, 8 Unfortunately, sulfonamide antibiotics, including potentiated sulfonamides as part of a folate synthesis combination, can cause idiosyncratic drug toxicity, also called sulfonamide hypersensitivity (HS),9 which is characterized by delayed onset of fever and pruritic skin rash after 5‐14 days of treatment. Less commonly, HS can progress to multiorgan involvement or severe bullous skin eruptions, such as Stevens‐Johnson syndrome and toxic epidermal necrolysis, with up to 30% mortality.10, 11
Sulfonamide HS occurs in both immunocompromised and immunocompetent populations with incidence rates of 20%‐57% in HIV‐infected patients and ~3% in the general population.12, 13, 14, 15, 16 In the former, the risk for this drug reaction appears to be acquired and is associated with advancing disease status.14, 17, 18, 19 In contrast, there is some evidence that sulfonamide HS in immunocompetent patients has a familial component,20, 21, 22 suggesting a genetic association.
A few polymorphic candidate genes have been investigated for associations with sulfonamide HS and focus predominantly on those involved with SMX biotransformation and detoxification. Genotypes for N‐acetyltransferase (NAT2), the primary SMX detoxification enzyme, have been associated previously with sulfonamide HS,23, 24 but in a larger, more recent study in our laboratory, we found no associations with HS despite resequencing of the entire NAT2 coding region.25 In that same population, polymorphism frequencies for two other sulfonamide detoxification genes, CYB5A and CYB5R3, were not significantly different between sulfonamide HS and tolerant patients.25 In another study, an increased risk for cutaneous drug eruptions overall was reported for patients with GSTM1 and GSTT1 null genotypes, but only 4 of 36 patients studied were hypersensitive to sulfonamide antibiotics.26 An association between sulfonamide‐induced skin eruptions and an HLA‐A haplotype (A30 B13 Cw6) was found in a Turkish population,27 but this has not been pursued in other ethnic groups. Finally, our group recently performed a genome‐wide association study in immunocompetent individuals, but did not identify any convincing genetic associations with sulfonamide HS,28 emphasizing the complex nature of this trait. Therefore, alternate approaches are needed to understand the mechanisms of risk for sulfonamide HS in the general population.
In both immunocompetent and HIV‐infected patients, sulfonamide HS has been associated with a surrogate marker identified by the lymphocyte toxicity assay (LTA). Results of this in vitro cytotoxicity assay show that peripheral blood mononuclear cells (PBMCs) from HS patients are more susceptible to apoptosis in the presence of sulfonamide metabolites, particularly SMX‐hydroxylamine (SMX‐HA), when compared to cells from sulfonamide‐tolerant patients. Interestingly, this enhanced cytotoxicity does not occur when cells are exposed to the parent drug (SMX) emphasizing the importance of drug bioactivation in the hypersensitivity reaction.29, 30, 31, 32 Furthermore, CD8+ T cells appear to be most susceptible to the LTA response, possibly due to lower intracellular antioxidant concentrations.33 The significance of this finding is unknown; however, T cells are the major effector cells in delayed‐type hypersensitivities.34 Cytotoxicity in the LTA has been measured by multiple methods, including trypan dye exclusion,22, 35 MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) conversion,29, 31, 32, 36, 37, 38 and YO‐PRO fluorescence.39
These findings have been interpreted as a “detoxification defect” in HS but not tolerant patients.30, 40, 41 Furthermore, this apparent defect occurs in family members of HS patients who have never received potentiated sulfonamides,21, 22, 41 which suggests a heritable component. However, the mechanism for this possible defect is not understood. Therefore, the purpose of this study was to identify key gene transcripts and pathways that are associated with increased cytotoxicity in the LTA in patients treated with potentiated sulfonamides.
2. MATERIALS AND METHODS
2.1. Subject recruitment
Patients with sulfonamide HS and controls tolerant of a course of potentiated sulfonamide antibiotics were identified in the electronic medical record and recruited through UW Health. Additional patients and controls were recruited among the UW‐Madison faculty, staff, and students. Medical records were searched electronically for a history of TMP‐SMX administration or for a diagnosis of sulfonamide HS, and then were reviewed using a structured abstraction form. The abstraction form included the following eligibility criteria: (1) administration of TMP‐SMX for at least 5 days prior to the adverse event9; (2) documentation of one or more new clinical signs after starting TMP‐SMX, including fever with or without eosinophilia, skin rash, increases in liver enzyme activities, hyperbilirubinemia, blood dyscrasias (anemia, leukopenia or thrombocytopenia), pneumonitis, myocarditis, aseptic meningitis, polyarthritis, acute interstitial nephritis, toxic epidermal necrolysis, or Stevens‐Johnson syndrome9; (3) lack of other clinical explanation for the adverse event; and (4) resolution of clinical signs with discontinuation of TMP‐SMX alone. Patients with only gastrointestinal symptoms such as nausea, vomiting, or diarrhea,9 or with acute anaphylactoid reactions,42, 43 were excluded. Because some forms of immunosuppression, in particular AIDS, lead to a high acquired risk of SMX hypersensitivity, apparently independent of genotype,44 immunocompromised patients, including those with HIV infection or undergoing immunosuppressive chemotherapy, were excluded. These criteria together were designed to yield a score of 6 or more, or “probable” adverse reaction, using the Naranjo Adverse Drug Reaction scale.45 Control patients (“tolerant;” TOL) must have been prescribed a course of TMP‐SMX at a standard therapeutic daily dosage for at least 10 days, with adequate follow‐up to indicate that the drug was taken and tolerated without adverse event. Clinical and demographic variables, including dosage, duration of treatment, and reason for TMP‐SMX prescription were also abstracted. Each case was adjudicated by a hospital pharmacist (WR) to ensure consistency and accuracy. This protocol was approved by the UW Health Sciences Institutional Review Board (Protocol # 2011‐0512).
2.2. Sample collection and processing
For each subject, 40 mL of heparinized blood were collected and PBMCs isolated using standard density centrifugation. CD8+ T cells were isolated from an aliquot of PBMCs (10.0 × 106) using an antibody‐based, negative selection magnetic bead system (MACS CD8+ T cell Isolation Kit; Miltenyi Biotec Inc., Auburn, CA). However, based on flow cytometry, the resultant cell population was not purely CD8+ T cells; therefore, these cells were considered CD8+ T cell‐enriched PBMCs. These enriched PBMCs were placed in RNAlater (Sigma‐Aldrich; St. Louis, MO) and stored at −80°C until RNA isolation. The remainder of the PBMCs was used immediately for the LTA. Unless otherwise noted, chemicals were obtained from Sigma‐Aldrich (St. Louis, MO).
2.3. Lymphocyte toxicity assay
Assays were performed in quadruplicate in tissue culture treated, 96‐well microtiter plates. PBMCs were suspended in cell media (Hank's balanced salt solution, 5% fetal bovine serum, 1% penicillin‐streptomycin solution, 15 mmol·L−1 HEPES) and incubated with 25‐800 μmol·L−1 SMX‐HA (Dalton Chemicals; Toronto, CA), and 1 mmol·L−1 SMX or vehicle as negative controls, at 37°C for 2 hours. To remove free drug, cells were washed twice in phosphate buffered saline (PBS), resuspended in 100 μL cell media, and incubated at 37°C for another 19 hours. Cytotoxicity was determined using YO‐PRO, an impermeant nucleic acid stain that identifies apoptotic or dead cells. YO‐PRO (final concentration 12 μmol·L−1) was added to each well and fluorescence was measured using a 96‐well plate reader (ex. 485, em. 530; Synergy 2; BioTek Instruments, Inc., Winooski, VT). To determine the total amount of cells in each well, cells were then lysed with the addition of 15 μL of 2% Triton X and incubated at room temperature for 1 hour; fluorescence was then remeasured. For each condition, % cytotoxicity was defined as pre‐Triton treatment fluorescence divided by the post‐Triton treatment value; values were normalized by subtracting the % cytotoxicity of the vehicle from that of each drug‐treated condition.
For a subset of samples where cell volumes were adequate, % cytotoxicity was also determined using the MTT detection method. After incubation of PBMC with SMX‐HA, SMX, or drug vehicle, and overnight incubation without drug as for the YO‐PRO assay, 10 μL of 12 nmol·L−1 MTT was added to each well and incubated at 37°C for 4 hours to allow generation of formazan.37 The formazan was solubilized with 100 μL 0.1 N HCl in isopropyl alcohol and absorbance measured at 570 nm. For each condition, % cell viability was defined as absorbance for drug‐exposed cells divided by the absorbance of cells treated with drug vehicle; % cytotoxicity was then calculated as 100% minus % viability.
2.4. RNA isolation and expression microarray of CD8+ T cell‐enriched PBMCs
CD8+ T cell‐enriched PBMCs were thawed and washed with 1 volume PBS. Samples were centrifuged at 4000 g at 4°C for 10 minutes and supernatant removed. RNA was isolated using the TRIzol/chloroform method: 1 mL TRIzol (ThermoFisher Scientific; Madison, WI) was added to the cell pellet and homogenized by repeated pipetting. Then 200 μg glycogen (ThermoFisher Scientific; Madison, WI) was added and genomic DNA sheared by passage through a 25 g needle. To each tube, 200 μL chloroform was added, shaken, and centrifuged at 16 100 g at 4°C for 5 minutes. The aqueous phase was transferred to a clean tube and 500 μL ice‐cold 100% isopropanol added. RNA was precipitated for 1 hour at −20°C. Tubes were centrifuged at 16 100 g at 4°C for 10 minutes, supernatant removed, and the pellet allowed to dry completely. RNA was then resuspended in nuclease free water. Genomic DNA contaminants were digested using TURBO DNA‐free (ThermoFisher Scientific; Madison, WI) and residual extraction reagents removed using RNeasy MinElute Cleanup (Qiagen; Hilden, Germany) according to manufacturers’ directions.
Prior to expression profiling, RNA quality was assessed on the Agilent 2100 Bioanalyzer (RNA 6000 Pico Kit; Agilent Technologies; Santa Clara, CA); samples with an RNA Integrity Number ≥8 were used for expression arrays.46 Microarray expression profiling was performed using the Human Transcriptome Array 2.0 (Affymetrix; Santa Clara, CA) on the Affymetrix GeneChip platform through the University of Wisconsin Biotechnology Center. The gene expression data were extracted using RMA‐Sketch in the Affymetrix Expression Console (version 1.14.46), and the exon data were extracted using Alt Splice Analysis in the Affymetrix Expression Console (version 1.14.46).
2.5. Validation of transcript expression with quantitative PCR (qPCR)
Expression levels of genes of interest identified by the microarray to be associated with % cytotoxicity were verified with qPCR. Reverse transcription with random oligomers (Superscript IV VILO cDNA Synthesis Kit; ThermoFisher Scientific; Madison, WI) was used to generate cDNA. Primers were designed for the target gene transcripts along with two reference genes (GUSB, HPRT1) and primer efficiencies determined for each using a 10‐fold dilution series of pooled cDNA (Table S1). SYBR Green (LightCycler 480 SYBR Green I Master; Roche Diagnostics; Indianapolis, IN) qPCR with melting curves was performed on the LightCycler 96 Instrument (Roche Diagnostics; Indianapolis, IN) with parameters adapted from manufacturer's directions (Table S2).
2.6. Statistical analyses
2.6.1. LTA analysis
Continuous data are presented as mean ± SD. For both YO‐PRO and MTT detection methods, mean % cytotoxicity at 800 μmol·L−1 SMX‐HA was compared between TOL and HS patients using a Student's t test. Correlation between YO‐PRO and MTT methods was assessed by comparing % cytotoxicity (at 800 μmol·L−1 SMX‐HA) using the Pearson's correlation coefficient. Analyses were performed with commercial statistical software (Prism 7; GraphPad Software Inc.; La Jolla, CA).
2.6.2. Microarray analysis
Microarray analyses were performed in R (www.r-project.org; version 3.3.2) and Bioconductor (bioconductor.org; version 3.4). Probe‐level‐data were aggregated to the gene level to improve power. After subtracting out the probe‐level mean across samples for each measurement, expression levels were aggregated by Entrez gene ID using mappings in the Bioconductor packages hta20transcriptcluster.db (version 8.5.0) and org.Hs.e.g.db (3.4.0). Probes that mapped to multiple genes or that did not appear in both database packages were excluded.
Three separate analyses were performed on gene‐level scores. The first assessed differential expression based on clinical status (TOL vs HS patients) using gene‐specific t tests. The second and third analyses assessed the correlation between gene expression and % cytotoxicity as determined by the YO‐PRO and MTT methods, respectively, using Fisher's z‐transformation. In all analyses, independent filtering 47 was used to remove the 50% of genes with the lowest variance, in an effort to increase power, and false discovery rates were computed using the Benjamini‐Hochberg adjustment after filtering. 48 Of the genes assessed, candidate genes previously hypothesized to be involved in sulfonamide HS pathogenesis (Table 1) were of particular interest. Transcripts significantly associated with % cytotoxicity by YO‐PRO were included in a pathway analysis using allez 49 (version 2.0.3), which grouped transcripts by gene ontology (GO) terms.
Table 1.
Candidate genes hypothesized to be involved in the pathogenesis of sulfonamide HS and evaluated by expression PBMC microarray, along with mechanistic rationale for each candidate
| Gene | Protein | Rationale | References |
|---|---|---|---|
| CYP1A2 | Cytochrome P450, family 1, subfamily A, polypeptide 2 | SMX and TMP biotransformation | 69, 70 |
| CYP2C8 | Cytochrome P450, family 2, subfamily C, polypeptide 8 | SMX biotransformation | 71 |
| CYP2C9 | Cytochrome P450, family 2, subfamily C, polypeptide 9 | SMX biotransformation | 69 |
| CYP2D6 | Cytochrome P450, family 2, subfamily D, polypeptide 6 | TMP biotransformation | 70 |
| CYP3A4 | Cytochrome P450, family 3, subfamily A, polypeptide 4 | SMX and TMP biotransformation | 69, 70 |
| GCLC | Glutamate‐cysteine ligase, catalytic subunit | Glutathione pathways for reactive drug metabolites | 72 |
| GCLM | Glutamate‐cysteine ligase, modifier subunit | Glutathione pathways | 72 |
| GSS | Glutathione synthetase | Glutathione pathways | 72 |
| GSTM1 | Glutathione S‐transferase mu 1 | Glutathione pathways | 72 |
| GSTP1 | Glutathione S‐transferase pi 1 | Glutathione pathways | 72 |
| GSTT1 | Glutathione S‐transferase tau 1 | Glutathione pathways | 72 |
| HLA‐A | Major histocompatibility complex, class I, A | Antigen presentation | Naisbitt et al. (2001), 74 |
| HLA‐B | Major histocompatibility complex, class I, B | Antigen presentation | Naisbitt et al. (2001), 74 |
| HLA‐C | Major histocompatibility complex, class I, C | Antigen presentation | Naisbitt et al. (2001), 74 |
| HLA‐DQA1 | Major histocompatibility complex, class II, DQ alpha 1 | Antigen presentation | Naisbitt et al. (2001), 74 |
| MARC1 | Mitochondrial amidoxime reducing component 1 | SMX biotransformation | 75 |
| MARC2 | Mitochondrial amidoxime reducing component 2 | SMX biotransformation | 75 |
| MPO | Myeloperoxidase | SMX biotransformation | 76 |
| NAT1 | N‐acetyltransferase 1 | SMX biotransformation | 77 |
| NAT2 | N‐acetyltransferase 2 | SMX biotransformation | 77 |
2.6.3. qPCR validation analysis
For selected target genes, ratios normalized to a pooled cDNA calibrator were generated using the LightCycler 96 software (Roche Diagnostics; Indianapolis, IN). The correlations between normalized expression ratios and % cytotoxicity were assessed by the Pearson's correlation coefficient in Prism (GraphPad Software Inc.; La Jolla, CA).
3. RESULTS
3.1. Patient population
Ten HS and 10 TOL patients were included in this study. Patient demographics are summarized in Table 2. Sulfonamide HS was characterized by cutaneous rash in all 10 HS patients, and one patient had a documented fever.
Table 2.
Demographic information for drug‐tolerant (TOL) and sulfonamide hypersensitive (HS) patients. Continuous data are presented as mean (range)
| TOL (n = 10) | HS (n = 10) | |
|---|---|---|
| Age at administration (yr) | 42 (17‐73) | 38 (21‐58) |
| Body weight (kg) | 81 (42‐123) | 60 (55‐68) |
| Total daily dose (mg/kg) | 14.2 (6.5‐23.2) | 16.5 (11.7‐28.4) |
| Gender | ||
| Female | 8 | 10 |
| Male | 2 | 0 |
| Race | ||
| Caucasian | 10 | 9 |
| African American | 0 | 1 |
| Indication for antibiotics | ||
| Urinary tract infection | 3 | 3 |
| Respiratory tract infection | 3 | 5 |
| Other soft tissue infection | 3 | 1 |
| Unknown/multiple | 1 | 1 |
3.2. Lymphocyte toxicity assays
The Lymphocyte toxicity assays (LTA) with YO‐PRO detection was performed for all 20 patients (10 TOL, 10 HS); the LTA with MTT detection was only available for 12 patients (6 TOL, 6 HS) because of limited PBMC availability. Unexpectedly, there was no statistically significant difference in % cytotoxicity between TOL and HS patients for either the YO‐PRO (41.5% ± 19.2% vs 38.1% ± 15.4%, P = .67; Figure 1A) or MTT (50.8% ± 21.3% vs 39.9% ± 34.2%, P = .52; Figure 1B) assays. Furthermore, % cytotoxicity for the two methods did not correlate across patients (r = .11, 95% CI: 0.50‐0.64, P = .74).
Figure 1.
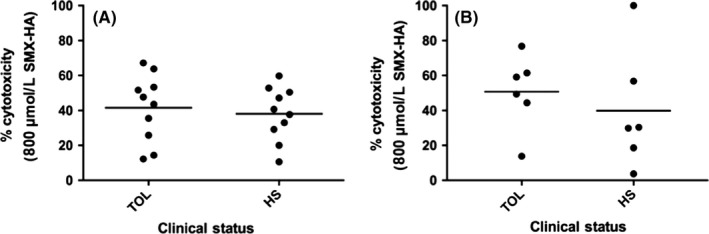
Relationship between clinical status and cytotoxicity from sulfamethoxazole‐hydroxylamine (SMX‐HA; 800 μmol·L−1) in human PBMCs as detected by the YO‐PRO and MTT methods. (A) Scatter plot of % cytotoxicity with YO‐PRO detection for SMX tolerant (TOL; n = 10) vs hypersensitive (HS; n = 10) patients, (P = .67 between groups); the horizontal line represents the mean for each group. (B) Scatter plot of % cytotoxicity with MTT detection for TOL (n = 6) versus HS (n = 6) patients, (P = .52 between groups); the horizontal line represents the mean for each group
3.3. Gene expression by clinical status and cytotoxicity
Expression data from in 20 patients (10 HS, 10 TOL) were included in the final comparison for YO‐PRO cytotoxicity, and in 12 patients (6 HS, 6 TOL) for MTT cytotoxicity. At a false discovery rate (FDR) of 0.05, no transcripts (candidate gene or otherwise) were detected to be significantly up‐ or down‐regulated in the HS group compared to the TOL group. The minimum FDR at which genes could be identified in this analysis was 0.96.
For cytotoxicity using YO‐PRO, 96 gene transcripts were significantly correlated with % cytotoxicity at an FDR of 0.188 (Figure 2; Table S3). No a priori candidate genes were among these transcripts. Of interest, expression of ABCC5, which encodes an organic anion pump and is known to transport arylamine‐containing drugs including methotrexate and leucovorin,50 was negatively correlated with YO‐PRO cytotoxicity (r = −.70). Expression of SLC25A37, which encodes a mitochondrial iron transporter, was also negatively correlated with YO‐PRO cytotoxicity (r = −.71). TMEM123, which encodes Porimin, a transmembrane cell protein associated with cell death, was positively correlated with YO‐PRO cytotoxicity (r = .72). Microarray data from this study are available in the GEO database (www.ncbi.nlm.nih.gov/geo; accession number GSE100443).
Figure 2.
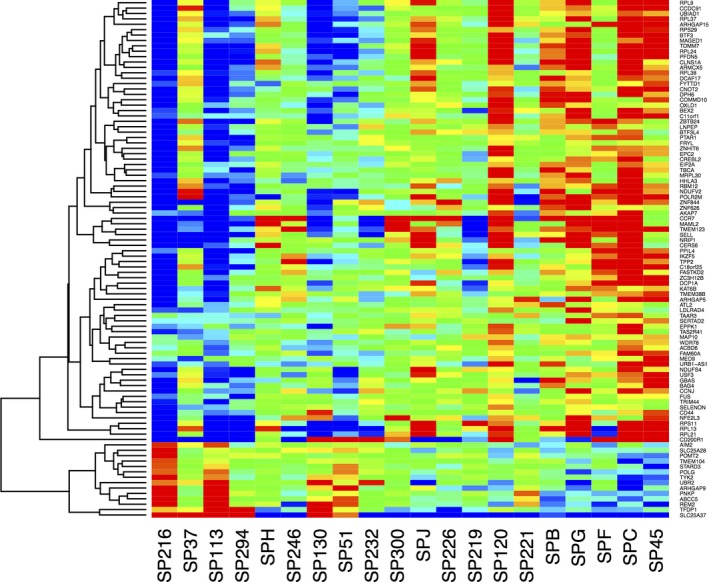
Heat map of expression of 96 transcripts correlated with SMX‐HA cytotoxicity in PBMCs from 20 patients using by the YO‐PRO method. The 20 samples are listed along the x‐axis in order of lowest (left) to highest (right) % cytotoxicity. Red indicates highest expression, blue lowest expression
Seven Gene Ontology (GO) terms were identified in the pathway analysis that contained more than one correlated transcript (Figure 3). Of interest, a group of eight ribosomal protein genes (RPL13, RPL21, RPL24, RPL37, RPL38, RPL9, RPS11, RPS29) was identified in four of the seven GO terms. All eight of these genes were positively correlated with cytotoxicity.
Figure 3.
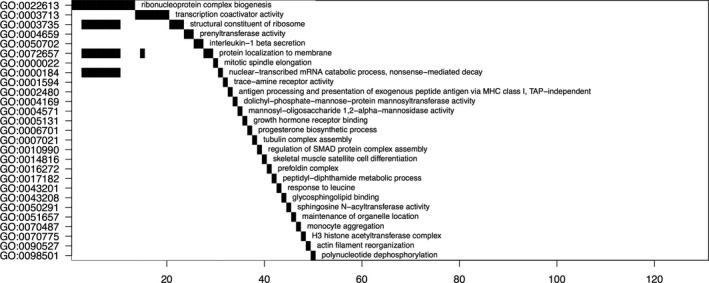
Waterfall plot of pathway analysis for 96 transcripts correlated with % cytotoxicity for PBMCs exposed to SMX‐HA in vitro and assessed by the YO‐PRO method of detection. Gene ontology (GO) terms are listed along the y‐axis, and numbers of transcripts identified for each term are indicated on the x‐axis
Transcripts were selected for validation by qPCR based on strength of correlation and mechanistic plausibility with SMX‐HA‐induced cytotoxicity (Table S1), including ABCC5, SLC25A37 because of the relationship between mitochondrial function and cell survival, TMEM123, and RPL37, which encodes ribosomal protein, large subunit 37, and was selected to represent the group of ribosomal proteins identified in the pathway analysis. The inverse correlation between ABCC5 expression and cytotoxicity in the array was confirmed by qPCR (r = −.49, P = .02; Figure 4). The negative correlation between SLC25A37 expression and % cytotoxicity approached statistical significance by qPCR (r = −.42, P = .067); when an extreme outlier (>3 SD above the mean) was removed the correlation (r = −.63, P = .0037; Figure 5) was comparable to that found in the array. The positive correlation between TMEM123 expression and cytotoxicity was also confirmed by qPCR (r = .53, P = .017; Figure 6). For RPL37 a weaker positive correlation was found between % cytotoxicity and RPL37 expression by qPCR that did not reach statistical significance (r = .32, P = .17; Figure 7).
Figure 4.
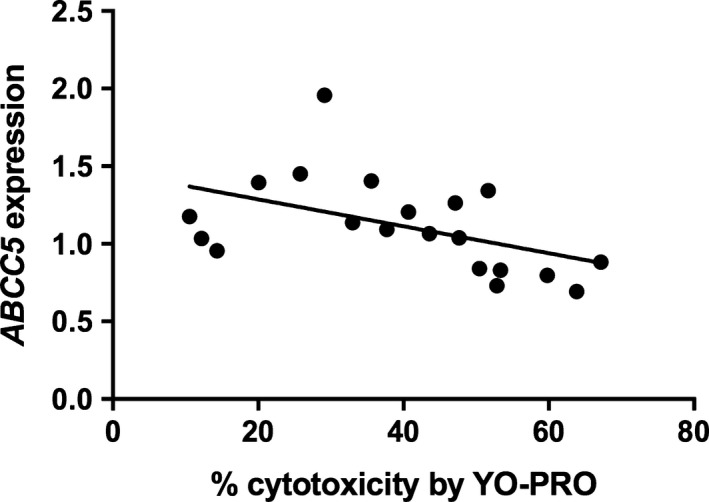
Correlation between % PBMC cytotoxicity by YO‐PRO detection and expression of ABCC5 by qPCR in PBMCs from 20 patients (Pearson's r = −.486, P = .030)
Figure 5.
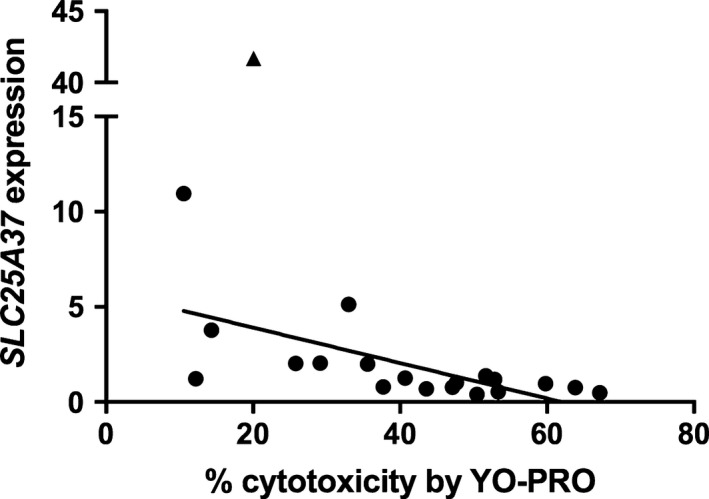
Correlation between % cytotoxicity by YO‐PRO detection and expression of SLC25A37 by qPCR (Pearson's r = −.632, P = .037) in PBMCs from 20 patients exposed in vitro to SMX‐HA (800 mmol·L−1), without inclusion of an extreme outlier (triangle)
Figure 6.
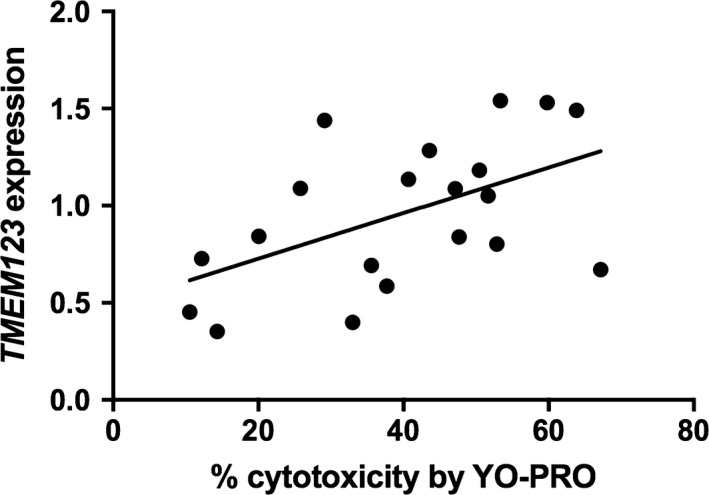
Correlation between % cytotoxicity by YO‐PRO detection and expression of TMEM123 by qPCR (Pearson's r = .526, P = .017)
Figure 7.
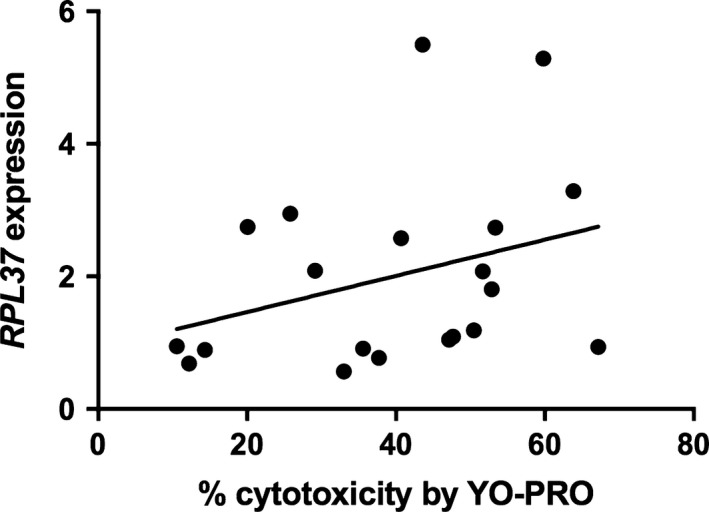
Correlation between % cytotoxicity by YO‐PRO detection and expression of RPL37 (Pearson's r = .322, P = .166) by qPCR
Using MTT cytotoxicity as the outcome, no transcripts were significantly correlated with % cytotoxicity when using independent filtering. However, without independent filtering, a correlation was found for a single transcript, TMEM129, which encodes a transmembrane protein that mediates cellular protein degradation (r = .92, with FDR <0.05). TMEM129 expression was also assessed by qPCR, but an association with % cytotoxicity could not be confirmed (r = .40, P = .19), possibly due to low power in just 12 patients.
4. DISCUSSION
The lymphocyte toxicity assay (LTA) has been proposed as an in vitro marker of sulfonamide HS.30, 40, 41 However, the mechanism underlying enhanced cytotoxicity of PBMCs to reactive SMX metabolites is not established. Therefore, we set out to define differences in gene expression between patients with high versus low susceptibility to SMX‐HA cytotoxicity in vitro, using two methods for evaluation of cytotoxicity. We chose YO‐PRO‐1 as our primary method, which is a DNA‐intercalating fluorophore that measures increased cell permeability is a marker of cellular apoptosis and death.51 We also used the classical MTT assay, which measures mitochondrial viability, in a subset of patients with adequate PBMCs for both assays.52
In our hands, there was poor correlation between cytotoxicity from SMX‐HA using the YO‐PRO versus MTT assay across patients. Part of this may due to small sample size. Additionally, we had inadequate PBMC numbers to perform the MTT assay on all patients. However, the lack of correlation could also be due to different assay outcomes (cell permeability versus mitochondrial function) and both assays have diagnostic weaknesses. As a vital dye, YO‐PRO is considered a conclusive test for cell death.53 However, false negatives can occur with cell death that does not involve increased membrane permeability 53 and false positives are possible with high expression of transporters for large cations.54 In the MTT assay, impaired mitochondrial reductase function in otherwise healthy cells will yield falsely low viability results.53 These factors may lead to discordant results between YO‐PRO and MTT assays.55
We used cytotoxicity of SMX‐HA at 800 μM for statistical comparisons of LTA results, since this metabolite concentration highlighted most of the variability among individuals. However, we did not detect increased cytotoxicity in HS patients compared to TOL patients at this concentration for either method, which was a concern given the results of previous studies.22, 32, 56 The reported specificity of the LTA is quite high (78%‐99%), but sensitivity varies greatly (41%‐100%).36, 41, 56, 57 In the relatively small numbers of patients recruited for expression arrays in our study, the lack of sensitivity reported by some groups could have led to poor discrimination between HA and TOL patients. Additionally, the performance of the LTA in drug HS appears to vary with the severity of clinical presentation. In patients with only an exanthemous rash, sensitivity is quite low (41%), but is much higher in patients with more serious systemic organ involvement (92%).56 Although patients with Stevens‐Johnson syndrome and toxic epidermal necrolysis were not excluded from this study, 9/10 patients in our population only exhibited a cutaneous rash and 1/10 exhibited a rash and fever. Thus it is possible that the LTA would have performed better if only patients with severe reactions were included. Alternately, these differences in apparent assay sensitivity may reflect problems not with the assay but with accurate phenotyping of patients with simple rash, which can have causes other than drug HS.
Although we did not find differences in cytotoxicity outcomes by clinical phenotype in our population, we were still interested in underlying gene expression patterns associated with the wide range of SMX‐HA cytotoxicities that we did observe. After independent filtering, which removes transcripts with low variance between samples, expression levels for 96 transcripts were significantly correlated with SMX‐HA cytotoxicity using the YO‐PRO method. These genes represent a wide range of cellular localization and functions, but none were a priori candidate genes known or hypothesized to metabolize sulfonamide antibiotics (Table 1). Since the lowest FDR was still relatively high (18.8%) in this exploratory study, we selected key transcripts with very high cytotoxicity correlations (RPL37, TMEM129) or plausible mechanistic significance (SLC25A37, ABCC5, TMEM123) for qPCR validation.
ABCC5 encodes a multispecific organic anion transporter; its expression was significantly and negatively correlated with SMX‐HA cytotoxicity in the microarray and by qPCR. This transcript is of interest because it mediates cellular efflux of methotrexate and leucovorin,50 which, like sulfonamide antibiotics, are arylamine drugs with therapeutic targets in folate metabolism. The ABCC5 gene is polymorphic 58 and variants have been associated with altered risk for toxicity to a variety of drugs including methotrexate,59 irinotecan 60 and azathioprine.61 These variants are thought to either modulate transporter affinity for the drug or drug metabolite or alter expression of the transporter itself. Therefore, decreased expression of ABCC5 observed in the present study could possibly lead to increased intracellular accumulation of SMX and its metabolites, contributing to increased cytotoxicity in vitro and possibly increased immunogenic drug adduct formation in vivo. This hypothesis merits further investigation.
SLC25A37 encodes mitoferrin 1, a solute carrier in the inner mitochondrial membrane that is known to transport iron. SLC25A37 expression was inversely correlated with SMX‐HA induced cytotoxicity in the microarray, and was confirmed with qPCR. SLC25A37 has been most studied relative to heme biosynthesis in erythroid cells, but its role in nonerythroid cells is not well‐characterized.62 Down‐regulation of mitoferrin 1 in lymphoid cells could plausibly lead to iron‐induced redox stress, and possible increased susceptibility to reactive xenobiotics such as SMX‐HA. Of note, SLC25A28, which encodes for mitoferrin 2, another mitochondrial iron transporter highly homologous to mitoferrin 1,63 was also inversely correlated with % cytotoxicity in the microarray (r = −.66, Table S3).
TMEM123, which encodes Porimin, was positively correlated with % cytotoxicity by YO‐PRO detection in the microarray and by qPCR. Porimin has been shown to increase membrane permeability and cell death in Jurkat (lymphoid) cells.64 This has direct relevance for the cytotoxicity observed in the in vitro LTA, but could also influence susceptibility to sulfonamide HS in vivo through increased cell death from relatively lower concentrations of reactive SMX metabolites, and enhanced “danger signals” to trigger a systemic immune response to cellular drug adducts.65 Like ABCC5, TMEM123 deserves further study relative to the risk of sulfonamide HS.
RPL37 was one of a group of 8 ribosomal proteins that were significantly and positively correlated with SMX‐HA cytotoxicity by YO‐PRO (r = .63‐.68). While this correlation could not be confirmed by qPCR (r = .32, P = .17), agreement between these two methodologies is affected by multiple factors and lack of concordance 66 does not necessarily invalidate microarray results. Although there is not a clear mechanistic relationship between upregulation of ribosomal proteins (which was assessed in an aliquot of PBMC that were not exposed to drug metabolite in vitro) and increased drug cytotoxicity, RPL37 overexpression has been shown to lead to increased cell death in vitro.67 Thus, the effects of up‐regulation of this and related genes on drug‐induced cytotoxicity deserved further study.
The major limitation of this study was its small sample size, which was limited in part by the expense of expression array analyses and by the logistics of prospective recruitment and adjudication of patients for fresh sample collection from a single hospital. We also only included patients with a well‐documented reaction, drug exposure, and history, further limiting the patient recruitment pool. However, our sample size was based on an a priori power calculation that 10 patients per group would provide 80% power to detect 2.5‐fold or greater differences in transcript expression.68 Thus, it is possible that there are differences in gene expression between HS and TOL patients that were not detectable by the current study design. We also used a lenient false positive discovery rate to detect novel transcripts that might lead to new mechanistic insights.
Another potential limitation of the study was the use of CD8+ enriched, but not pure, lymphocytes. While the LTA is traditionally performed on mixed PBMC, a previous study demonstrated that CD8+ T lymphocytes were the predominant cell type affected by SMX‐HA cytotoxicity.33 We chose to focus on the cell type with the highest toxicity phenotype for expression arrays, with the goal of providing a more homogenous background with less noise from different cell types. Negative selection for enrichment of CD8+ T cells provided adequate RNA for robust array analyses, but since cell yields were not pure, our results may have been clouded by expression patterns in other mononuclear leukocytes in the samples. Finally, certain clinical data, which could be of interest or introduce bias (severity of rash, brand of sulfonamide antibiotic), were not available for analysis.
In summary, although high SMX‐HA cytotoxicity toward patient PBMC has been previously attributed to a possible innate defect in drug detoxification,30, 40, 41 we found no evidence for an association between increased cytotoxicity and altered expression of genes known or hypothesized to be involved in sulfamethoxazole biotransformation. Instead, expression array analyses suggested other novel pathways that could contribute mechanistically to SMX‐HA cytotoxicity, and by extension, to sulfonamide HS in some populations. Novel genes of particular interest include ABCC5, which encodes an arylamine drug transporter, and TMEM123, which encodes a transmembrane protein that mediates cell death. ABCC5 in particular is polymorphic,58 and additional work is needed to determine whether this transporter mediates SMX or SMX‐HA efflux from cells, and whether this is affected by known ABCC5 polymorphisms.
DISCLOSURES
None declared.
AUTHORS’ CONTRIBUTIONS
Participated in research design: Reinhart, Rose, Trepanier; Conducted experiments: Reinhart, Rose, Liebenstein; Contributed new reagents or analytic tools: n/a; Performed data analysis: Reinhart, Panyard, Newton, Yee; Wrote or contributed to the writing of the manuscript: Reinhart, Panyard, Newton, Trepanier.
Supporting information
ACKNOWLEDGEMENTS
The authors thank the University of Wisconsin Gene Expression Center for providing Agilent Bioanalyzer and Affymetrix GeneChip services.
Reinhart JM, Rose W, Panyard DJ, et al. RNA expression profiling in sulfamethoxazole‐treated patients with a range of in vitro lymphocyte cytotoxicity phenotypes. Pharmacol Res Perspect. 2018;e00388 https://doi.org/10.1002/prp2.388
Funding Information
This work was supported by the National Institutes of Health [Grant R01 GM100784]. Dr. Reinhart was supported by the National Institutes of Health [Grant T32 OD010423]. Daniel Panyard was supported by the National Heart, Lung, and Blood Institute [Grant T32 HL083806]. Dr. Newton was supported by the National Institutes of Health [Grant U54 AI117924]. This work represents a portion of Dr. Reinhart's doctoral thesis.
REFERENCES
- 1. Avgeri SG, Matthaiou DK, Dimopoulos G, Grammatikos AP, Falagas ME. Therapeutic options for Burkholderia cepacia infections beyond co‐trimoxazole: a systematic review of the clinical evidence. Int J Antimicrob Agents. 2009;33:394‐404. [DOI] [PubMed] [Google Scholar]
- 2. Drekonja DM, Johnson JR. Urinary tract infections. Prim Care. 2008;35:345‐367. [DOI] [PubMed] [Google Scholar]
- 3. Drekonja DM, Traynor LM, DeCarolis DD, Crossley KB, Johnson JR. Treatment of non‐life‐threatening methicillin‐resistant Staphylococcus aureus infections with alternative antimicrobial agents: a 2‐year retrospective review. Diagn Microbiol Infect Dis. 2009;63:201‐207. [DOI] [PubMed] [Google Scholar]
- 4. Korbila IP, Manta KG, Siempos II, Dimopoulos G, Falagas ME. Penicillins vs trimethoprim‐based regimens for acute bacterial exacerbations of chronic bronchitis: meta‐analysis of randomized controlled trials. Can Fam Physician. 2009;55:60‐67. [PMC free article] [PubMed] [Google Scholar]
- 5. Gupta D, Zachariah A, Roppelt H, Patel AM, Gruber BL. Prophylactic antibiotic usage for Pneumocystis jirovecii pneumonia in patients with systemic lupus erythematosus on cyclophosphamide: a survey of US rheumatologists and the review of literature. J Clin Rheumatol. 2008;14:267‐272. [DOI] [PubMed] [Google Scholar]
- 6. Rodriguez M, Sifri CD, Fishman JA. Failure of low‐dose atovaquone prophylaxis against Pneumocystis jiroveci infection in transplant recipients. Clin Infect Dis. 2004;38:e76‐e78. [DOI] [PubMed] [Google Scholar]
- 7. Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140:255‐266. [DOI] [PubMed] [Google Scholar]
- 8. Thomas M, Rupali P, Woodhouse A, Ellis‐Pegler R. Good outcome with trimethoprim 10 mg/kg/day‐sulfamethoxazole 50 mg/kg/day for Pneumocystis jirovecii pneumonia in HIV infected patients. Scand J Infect Dis. 2009;41:862‐868. [DOI] [PubMed] [Google Scholar]
- 9. Cribb AE, Lee BL, Trepanier LA, Spielberg SP. Adverse reactions to sulphonamide and sulphonamide‐trimethoprim antimicrobials: clinical syndromes and pathogenesis. Adverse Drug React Toxicol Rev. 1996;15:9‐50. [PubMed] [Google Scholar]
- 10. Ortiz JE, Horn MS, Peterson HD. Toxic epidermal necrolysis‐case report and review of the literature. Ann Plast Surg. 1982;9:249‐253. [DOI] [PubMed] [Google Scholar]
- 11. Saka B, Kombaté K, Mouhari‐Toure A, Akakpo S, Tchangaï‐Walla K, Pitché P. Stevens‐Johnson syndrome and toxic epidermal necrolysis in a teaching hospital in Lomé, Togo: retrospective study of 89 cases. Med Trop (Mars). 2010;70:255‐258. [PubMed] [Google Scholar]
- 12. Bigby M. Drug‐induced cutaneous reactions. A report from the Boston Collaborative Drug Surveillance Program on 15,438 consecutive inpatients, 1975 to 1982. J Am Med Assoc. 1986;256:3358‐3363. [DOI] [PubMed] [Google Scholar]
- 13. Gordin FM, Simon GL, Wofsy CB, Mills J. Adverse Reactions to Trimethoprim‐Sulfamethoxazole in Patients with the Acquired Immunodeficiency Syndrome. Ann Intern Med. 1984;100:495‐499. [DOI] [PubMed] [Google Scholar]
- 14. Hennessy S, Strom BL, Berlin JA, Brennan PJ. Predicting cutaneous hypersensitivity reactions to cotrimoxazole in HIV‐infected individuals receiving primary Pneumocystis carinii pneumonia prophylaxis. J Gen Intern Med. 1995;10:380‐386. [DOI] [PubMed] [Google Scholar]
- 15. Medina I, Mills J, Leoung G, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. A controlled trial of trimethoprim‐sulfamethoxazole versus trimethoprim‐dapsone. N Engl J Med. 1990;323:776‐782. [DOI] [PubMed] [Google Scholar]
- 16. Walmsley SL, Khorasheh S, Singer J, Djurdjev O. A randomized trial of N‐acetylcysteine for prevention of trimethoprim‐sulfamethoxazole hypersensitivity reactions in Pneumocystis carinii pneumonia prophylaxis (CTN 057). J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19:498‐505. [DOI] [PubMed] [Google Scholar]
- 17. Carr A, Swanson C, Penny R, Cooper DA. Clinical and laboratory markers of hypersensitivity to trimethoprim‐sulfamethoxazole in patients with Pneumocystis carinii pneumonia and AIDS. J Infect Dis. 1993a;167:180‐185. [DOI] [PubMed] [Google Scholar]
- 18. Eliaszewicz M, Flahault A, Roujeau JC, Fillet AM. Prospective evaluation of risk factors of cutaneous drug reactions to sulfonamides in patients with AIDS. J Am Acad Dermatol. 2002;47:40‐46. [DOI] [PubMed] [Google Scholar]
- 19. Rabaud C, Charreau I, Izard S, et al.; Delta trial group . Adverse reactions to cotrimoxazole in HIV‐infected patients: predictive factors and subsequent HIV disease progression. Scand J Infect Dis. 2001;33:759‐764. [DOI] [PubMed] [Google Scholar]
- 20. Mohanasundaram J, Rajaram S, Mohanasundaram S. Familial tendency in hypersensitivity reactions to co‐trimoxazole. J Indian Med Assoc. 1998;96:21‐22. [PubMed] [Google Scholar]
- 21. Neuman MG, Malkiewicz IM, Shear NH. A novel lymphocyte toxicity assay to assess drug hypersensitivity syndromes. Clin Biochem. 2000;33:517‐524. [DOI] [PubMed] [Google Scholar]
- 22. Shear NH, Spielberg SP, Grant DM, Tang BK, Kalow W. Differences in metabolism of sulfonamides predisposing to idiosyncratic toxicity. Ann Intern Med. 1986;105:179‐184. [DOI] [PubMed] [Google Scholar]
- 23. Wolkenstein P, Carrière V, Charue D, et al. A slow acetylator genotype is a risk factor for sulphonamide‐induced toxic epidermal necrolysis and Stevens‐Johnson syndrome. Pharmacogenetics. 1995a;5:255‐258. [DOI] [PubMed] [Google Scholar]
- 24. Zielińska E, Niewiarowski W, Bodalski J. The arylamine N‐acetyltransferase (NAT2) polymorphism and the risk of adverse reactions to co‐trimoxazole in children. Eur J Clin Pharmacol. 1998;54:779‐785. [DOI] [PubMed] [Google Scholar]
- 25. Sacco JC, Abouraya M, Motsinger‐Reif A, Yale SH, McCarty CA, Trepanier LA. Evaluation of polymorphisms in the sulfonamide detoxification genes NAT2, CYB5A, and CYB5R3 in patients with sulfonamide hypersensitivity. Pharmacogenet Genomics. 2012;22:733‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ateş NA, Tursen U, Tamer L, et al. Glutathione S‐transferase polymorphisms in patients with drug eruption. Arch Dermatol Res. 2004;295:429‐433. [DOI] [PubMed] [Google Scholar]
- 27. Ozkaya‐Bayazit E, Bayazit H, Ozarmagan G. Drug related clinical pattern in fixed drug eruption. Eur J Dermatol. 2000;10:288‐291. [PubMed] [Google Scholar]
- 28. Reinhart JM, Motsinger‐Reif A, Dickey A, Yale S, Trepanier LA. Genome‐wide association study in immunocompetent patients with delayed hypersensitivity to sulfonamide antimicrobials. PLoS One. 2016;11:e0156000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carr A, Penny R, Cooper DA. Efficacy and safety of rechallenge with low‐dose trimethoprim‐sulphamethoxazole in previously hypersensitive HIV‐infected patients. AIDS. 1993;7:65‐71. [DOI] [PubMed] [Google Scholar]
- 30. Rieder MJ, Krause R, Bird IA. Time‐course of toxicity of reactive sulfonamide metabolites. Toxicology. 1995;95:141‐146. [DOI] [PubMed] [Google Scholar]
- 31. Rieder MJ, Uetrecht J, Shear NH, Spielberg SP. Synthesis and in vitro toxicity of hydroxylamine metabolites of sulfonamides. J Pharmacol Exp Ther. 1988;244:724‐728. [PubMed] [Google Scholar]
- 32. Rieder MJ, Uetrecht J, Shear NH, Cannon M, Miller M, Spielberg SP. Diagnosis of sulfonamide hypersensitivity reactions by in vitro rechallenge with hydroxylamine metabolites. Ann Intern Med. 1989;110:286‐289. [DOI] [PubMed] [Google Scholar]
- 33. Hess DA, Sisson ME, Suria H, et al. Cytotoxicity of sulfonamide reactive metabolites: apoptosis and selective toxicity of CD8(+) cells by the hydroxylamine of sulfamethoxazole. FASEB J. 1999;13:1688‐1698. [DOI] [PubMed] [Google Scholar]
- 34. Pichler WJ, Naisbitt DJ, Park BK. Immune pathomechanism of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127:S74‐S81. [DOI] [PubMed] [Google Scholar]
- 35. Shear NH, Spielberg SP. In vitro evaluation of a toxic metabolite of sulfadiazine. Can J Physiol Pharmacol. 1985;63:1370‐1372. [DOI] [PubMed] [Google Scholar]
- 36. Elzagallaai AA, Jahedmotlagh Z, Del Pozzo‐Magaña BR, et al. Predictive value of the lymphocyte toxicity assay in the diagnosis of drug hypersensitivity syndrome. Mol Diagn Ther. 2010;14:317‐322. [DOI] [PubMed] [Google Scholar]
- 37. Leeder JS, Cannon M, Nakhooda A, Spielberg SP. Drug metabolite toxicity assessed in human lymphocytes with a purified, reconstituted cytochrome P‐450 system. J Pharmacol Exp Ther. 1988;245:956‐962. [PubMed] [Google Scholar]
- 38. Reilly TP, Bellevue FH, Woster PM, Svensson CK. Comparison of the in vitro cytotoxicity of hydroxylamine metabolites of sulfamethoxazole and dapsone. Biochem Pharmacol. 1998;55:803‐810. [DOI] [PubMed] [Google Scholar]
- 39. Reilly TP, MacArthur RD, Farrough MJ, Crane LR, Woster PM, Svensson CK. Is hydroxylamine‐induced cytotoxicity a valid marker for hypersensitivity reactions to sulfamethoxazole in human immunodeficiency virus‐infected individuals? J Pharmacol Exp Ther. 1999;291:1356‐1364. [PubMed] [Google Scholar]
- 40. Westphal GA, Reich K, Schulz TG, Neumann C, Hallier E, Schnuch A. N‐acetyltransferase 1 and 2 polymorphisms in para‐substituted arylamine‐induced contact allergy. Br J Dermatol. 2000;142:1121‐1127. [DOI] [PubMed] [Google Scholar]
- 41. Wolkenstein P, Charue D, Laurent P, Revuz J, Roujeau JC, Bagot M. Metabolic predisposition to cutaneous adverse drug reactions. Role in toxic epidermal necrolysis caused by sulfonamides and anticonvulsants. Arch Dermatol. 1995;131:544‐551. [PubMed] [Google Scholar]
- 42. Gruchalla RS, Sullivan TJ. Detection of human IgE to sulfamethoxazole by skin testing with sulfamethoxazoyl‐poly‐L‐tyrosine. J Allergy Clin Immun. 1991;88:784‐792. [DOI] [PubMed] [Google Scholar]
- 43. Harle DG, Baldo BA, Wells JV. Drugs as allergens: detection and combining site specificities of IgE antibodies to sulfamethoxazole. Mol Immunol. 1988;25:1347‐1354. [DOI] [PubMed] [Google Scholar]
- 44. Pirmohamed M, Alfirevic A, Vilar J, et al. Association analysis of drug metabolizing enzyme gene polymorphisms in HIV‐positive patients with co‐trimoxazole hypersensitivity. Pharmacogenetics. 2000;10:705‐713. [DOI] [PubMed] [Google Scholar]
- 45. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239‐245. [DOI] [PubMed] [Google Scholar]
- 46. Fleige S, Pfaffl MW. RNA integrity and the effect on the real‐time qRT‐PCR performance. Mol Aspects Med. 2006;27:126‐139. [DOI] [PubMed] [Google Scholar]
- 47. Bourgon R, Gentleman R. Independent filtering increases detection power for high‐throughput experiments. Proc Natl Acad Sci USA. 2010;107:9546‐9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;157:289‐300. [Google Scholar]
- 49. Newton MA, Quintana FA, den Boon JA, Sengupta S, Ahlquist P. Random‐set methods identify distinct aspects of the enrichment signal in gene‐set analysis. Ann Appl Stat. 2007;1:85‐106. [Google Scholar]
- 50. Assaraf YG. The role of multidrug resistance efflux transporters in antifolate resistance and folate homeostasis. Drug Resist Update. 2006;9:227‐246. [DOI] [PubMed] [Google Scholar]
- 51. Plantin‐Carrenard E, Bringuier A, Derappe C, et al. A fluorescence microplate assay using Yopro‐1 to measure apoptosis: application to HL60 cells subjected to oxidative stress. Cell Biol Toxicol. 2003;19:121‐133. [DOI] [PubMed] [Google Scholar]
- 52. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55‐63. [DOI] [PubMed] [Google Scholar]
- 53. Méry B, Guy JB, Vallard A, et al. In vitro cell death determination for drug discovery: a landscape review of real issues. J Cell Death. 2017;. https://doi.org/10.1179/670717691251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Munns CH, Chung M‐K, Sanchez YE, Amzel LM, Caterina MJ. Role of the outer pore domain in transient receptor potential vanilloid 1 dynamic permeability to large cations. J Biol Chem. 2015;290:5707‐5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gruhlke MCH, Nicco C, Batteux F, Slusarenko AJ. The effects of allicin, a reactive sulfur species from garlic, on a selection of mammalian cell lines. Antioxidants. 2016;6: https://doi.org/10.3390/antiox6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Neuman MG, Shear NH, Malkiewicz IM, et al. Immunopathogenesis of hypersensitivity syndrome reactions to sulfonamides. Transl Res. 2007;149:243‐253. [DOI] [PubMed] [Google Scholar]
- 57. Riley RJ, Cribb AE, Spielberg SP. Glutathione transferase mu deficiency is not a marker for predisposition to sulphonamide toxicity. Biochem Pharmacol. 1991;42:696‐698. [DOI] [PubMed] [Google Scholar]
- 58. Saito S, Iida A, Sekine A, et al. Identification of 779 genetic variations in eight genes encoding members of the ATP‐binding cassette, subfamily C (ABCC/MRP/CFTR). J Hum Genet. 2002;47:147‐171. [DOI] [PubMed] [Google Scholar]
- 59. Zaruma‐Torres F, Lares‐Asseff I, Reyes‐Espinoza A, et al. Association of ABCB1, ABCC5 and xanthine oxidase genetic polymorphisms with methotrexate adverse reactions in Mexican pediatric patients with ALL. Drug Metab Pers Ther. 2015;30:195‐201. [DOI] [PubMed] [Google Scholar]
- 60. Chen S, Villeneuve L, Jonker D, et al. ABCC5 and ABCG1 polymorphisms predict irinotecan‐induced severe toxicity in metastatic colorectal cancer patients. Pharmacogenet Genomics. 2015;25:573‐583. [DOI] [PubMed] [Google Scholar]
- 61. Lee MN, Kang B, Choi SY, et al. Impact of genetic polymorphisms on 6‐thioguanine nucleotide levels and toxicity in pediatric patients with IBD treated with azathioprine. Inflamm Bowel Dis. 2015;21:2897‐2908. [DOI] [PubMed] [Google Scholar]
- 62. Mühlenhoff U, Hoffmann B, Richter N, et al. Compartmentalization of iron between mitochondria and the cytosol and its regulation. Eur J Cell Biol. 2015;94:292‐308. [DOI] [PubMed] [Google Scholar]
- 63. Haitina T, Lindblom J, Renström T, Fredriksson R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics. 2006;88:779‐790. [DOI] [PubMed] [Google Scholar]
- 64. Ma F, Zhang C, Prasad KV, Freeman GJ, Schlossman SF. Molecular cloning of Porimin, a novel cell surface receptor mediating oncotic cell death. Proc Natl Acad Sci USA. 2001;98:9778‐9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lavergne SN, Wang H, Callan HE, Park BK, Naisbitt DJ. “Danger” conditions increase sulfamethoxazole‐protein adduct formation in human antigen‐presenting cells. J Pharmacol Exp Ther. 2009;331:372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Morey JS, Ryan JC, Dolah FM. Microarray validation: factors influencing correlation between oligonucleotide microarrays and real‐time PCR. Biol Proc Online. 2006;8:175‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Daftuar L, Zhu Y, Jacq X, Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2‐p53‐MdmX network. PLoS ONE. 2013;8:e68667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pawitan Y, Michiels S, Koscielny S, Gusnanto A, Ploner A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics. 2005;21:3017‐3024. [DOI] [PubMed] [Google Scholar]
- 69. Cribb AE, Spielberg SP, Griffin GP. N4‐hydroxylation of sulfamethoxazole by cytochrome P450 of the cytochrome P4502C subfamily and reduction of sulfamethoxazole hydroxylamine in human and rat hepatic microsomes. Drug Metab Dispos. 1995;23:406‐414. [PubMed] [Google Scholar]
- 70. Damsten MC, de Vlieger JSB, Niessen WMA, Irth H, Vermeulen NPE, Commandeur JNM. Trimethoprim: novel reactive intermediates and bioactivation pathways by cytochrome p450s. Chem Res Toxicol. 2008;21:2181‐2187. [DOI] [PubMed] [Google Scholar]
- 71. Sanderson JP, Naisbitt DJ, Farrell J, et al. Sulfamethoxazole and its metabolite nitroso sulfamethoxazole stimulate dendritic cell costimulatory signaling. J Immunol. 2007;178:5533‐5542. [DOI] [PubMed] [Google Scholar]
- 72. Cribb AE, Miller M, Leeder JS, Hill J, Spielberg SP. Reactions of the nitroso and hydroxylamine metabolites of sulfamethoxazole with reduced glutathione. Implications for idiosyncratic toxicity. Drug Metab Dispos. 1991;19:900‐906. [PubMed] [Google Scholar]
- 73. Naisbitt DJ, Hough SJ, Gill HJ, Pirmohamed M, Kitteringham NR, Park BK. Cellular disposition of sulphamethoxazole and its metabolites: implications for hypersensitivity. Brit J Pharmacol. 1999; 126: 1393‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schnyder B, Burkhart C, Schnyder‐Frutig K, et al. Recognition of sulfamethoxazole and its Rreactive metabolites by drug‐specific CD4 + T cells from allergic individuals. J Immunol. 2000;164:6647‐6654. [DOI] [PubMed] [Google Scholar]
- 75. Ott G, Plitzko B, Krischkowski C, et al. Reduction of sulfamethoxazole hydroxylamine (SMX‐HA) by the mitochondrial amidoxime reducing component (mARC). Chem Res Toxicol. 2014;27:1687‐1695. [DOI] [PubMed] [Google Scholar]
- 76. Cribb AE, Miller M, Tesoro A, Spielberg SP. Peroxidase‐dependent oxidation of sulfonamides by monocytes and neutrophils from humans and dogs. Mol Pharmacol. 1990;38:44‐751. [PubMed] [Google Scholar]
- 77. Cribb AE, Nakamura H, Grant DM, Miller MA. Role of polymorphic and monomorphic human arylamine N‐acetyltransferases in determining sulfamethoxazole metabolism. Biochem Pharmacol. 1993;45:1277‐1282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials