

Abstract
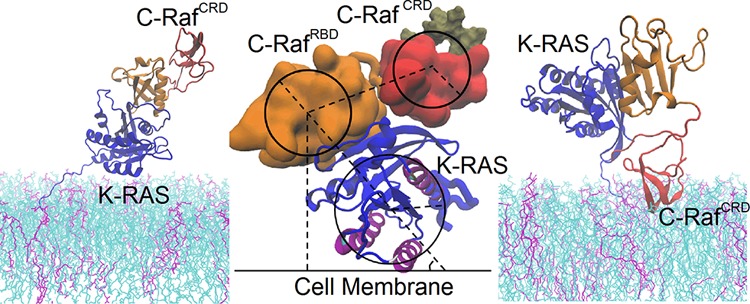
Association of Raf kinase with activated Ras triggers downstream signaling cascades toward regulating transcription in the cells’ nucleus. Dysregulation of Ras–Raf signaling stimulates cancers. We investigate the C-Raf RBD and CRD regions when bound to oncogenic K-Ras4B at the membrane. All-atom molecular dynamics simulations suggest that the membrane plays an integral role in regulating the configurational ensemble of the complex. Remarkably, the complex samples a few states dynamically, reflecting a competition between C-Raf CRD- and K-Ras4B- membrane interactions. This competition arises because the interaction between the RBD and K-Ras is strong while the linker between the RBD and CRD is short. Such a mechanism maintains a modest binding for the overall complex at the membrane and is expected to facilitate fast signaling processes. Competition of protein–membrane contacts is likely a common mechanism for other multiprotein complexes, if not multidomain proteins at membranes.
Short abstract
As two counterparts of a protein complex, K-Ras4B and C-Raf CRD interact with membrane in a competitive manner due to topological restriction, presenting two major dynamic membrane-associated states.
Introduction
The regulation of function of membrane peripheral protein complexes is achieved largely by the mutual interactions between the multiple proteins/protein domains involved, but may be heavily influenced also by the interactions the proteins make with the membrane. While the former can be characterized, in part, by experiments in solution, resolving the structure of a protein complex at a membrane remains a formidable challenge experimentally, given the nature of interactions involved. Recently, techniques such as cryoelectron microscopy, solution NMR with nanodiscs, and electron paramagnetic resonance (EPR) as well as fluorescence correlation methods of proteins bound to liposomes are advancing the characterization of the interfaces, of the orientation, and of the dynamics of peripheral membrane proteins at membranes.1−5 However, computational methods, such as dynamics simulations, are becoming particularly powerful in modeling the structures of proteins at membranes.6−9 In this study we apply this latter approach, specifically all-atom molecular dynamics (MD), to our knowledge for the first time to an oligomeric Ras–effector protein complex at a membrane.
Members of the Ras family of small GTPases are anchored to the intracellular leaflet of the plasma membrane and are a key regulator of cellular signal transduction: they convert signaling inputs from multiple transmembrane receptors to downstream activation, typically of kinases, eventually reaching and activating transcription factors in the cells’ nucleus.10 Signal transmission is achieved by the activation of Ras, converting it from Ras.GDP (inactive) to Ras.GTP (active) with the help of Guanine Nucleotide Exchange Factors, or GEF proteins. Activated Ras triggers downstream signaling through several pathways, including the Raf-MEK-ERK cascade. Oncogenic mutations, which usually result in permanently activated Ras and its persistent binding to Raf, lead to severe cellular dysfunction. Importantly, ∼20–30% of all human cancers harbor an oncogenic Ras mutation.11,12 Strategies that aim to disrupt the several steps which are required for Ras function, most notably drugs that interrupt the Raf–Ras interaction, are being developed in the recent five years and are expected to be promising therapies in cancer treatment.13−15
Downstream effectors such as Raf and PI3K interact with Ras.16 In the present investigation we studied the conformational and orientational dynamics of the C-RafRBD-CRD–K-Ras complex bound to a membrane model. That the Ras Binding Domain, or RBD (res. 56–132 of C-Raf), makes direct contacts with the membrane-bound Ras is well established, and structures for complexes of Raf with the K-Ras4B homologous Rap1A and H-Ras GTPase have been presented.17,18 There is as yet no study that clearly shows a direct interaction between Ras and the Cysteine Rich Domain, CRD (res. 138–187) which follows the RBD in the sequence of C-Raf, although a number of studies have indicated such interactions.19−22 In addition, the CRD was also identified as a membrane binding protein, especially for the lipid headgroup of phosphatidic acid (PA) and phosphatidylserine (PS).23−25 It has long been known that in most cases membrane localization is required for Ras activity. Specifically, the membrane helps to locally concentrate Ras proteins and likely directs Ras oligomerization, Ras cluster formation as well as association with other proteins, including Raf.26−28 Recent experiments as well as computer simulations have shown that the cell membrane determines the orientational preference of Ras relative to the membrane,4,29−31 which is predicted to have an effect on Raf–Ras recognition. Despite the many studies on the interactions of isolated Raf or Ras domains with the membrane, a biophysical-structural study of Raf–Ras as a protein complex at the membrane has not yet been reported, neither with experimental nor computational methods.
How are Raf–membrane, Ras–membrane, and Raf–Ras interactions integrated together in order to determine the structural features and function of the Raf–Ras complex at the intracellular membrane leaflet? In the present research, computational studies provide a powerful avenue for the detailed studies of the structure and dynamics of the C-RafRBD-CRD–K-Ras4B complex at the membrane.
Results
Configurations of C-Raf
We performed five independent all-atom MD simulations of a membrane-anchored complex of C-RafRBD-CRD–K-Ras4B for 1 μs each (Figure 1). Before this, we first examined the configurations of an isolated C-RafRBD-CRD (denoted C-Raf from now) in solution with three independent simulations of 1, 0.5, 0.5 μs each (Figure 2a). The configurations sampled a few major clusters (Figure 2b–c; Figure S1a). Overall, the CRD samples a wide configurational space relative to RBD, suggesting a large flexibility of the linker between the two domains. But it is noticeable that the CRD cannot reach the β-sheet surface (β2, β1, and β5) and the region (α1 and β2) of the RBD that is used for Ras binding. Therefore, none of these C-Raf configurations are likely to have major clashes with the binding of K-Ras. The program FPModeller32 independently predicts a similarly wide configurational space of C-Raf and similar excluded regions, by rotating residues in the linker (132–137) (Figure S1b). The interdomain linker is short and this makes it easy for the CRD less to access the distal α1−β2 region of the RBD. However, there is also a mismatch in surface electrostatic potential between these regions (of the α1−β2 region as well as the β2−β1−β5 region) with the CRD surface, making such configurations unfavorable (Figure 2c, lower). In contrast, interactions between the CRD and the RBD loops between β4 and α2, β3 and α1 as well as between β1 and β2 of the RBD are occasionally established (Figure S1c), which contribute to the higher population of configurations, denoted Cluster #1, Cluster #2, and Cluster #3 (Figure 2b).
Figure 1.
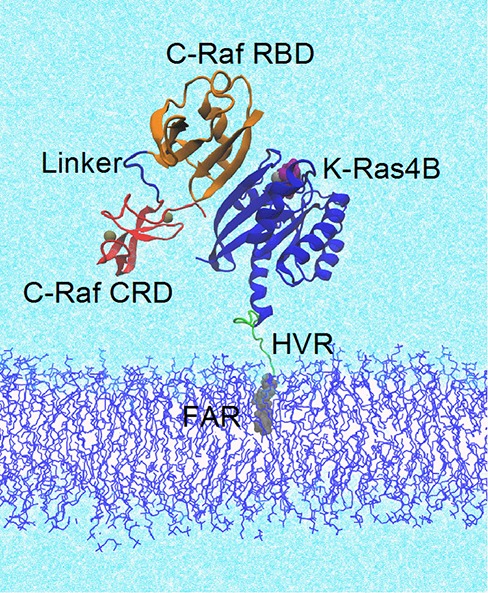
C-RafRBD-CRD–K-Ras4B complex at a mixed membrane consisting of 80% POPC and 20% POPS. The K-Ras4B protein consists of the globular, catalytic domain (CD, res. 1–166) and the largely unstructured hypervariable region, HVR (res. 167–185). C-Raf comprises the Ras Binding Domain, RBD and Cysteine Rich Domain, CRD, connected by a short linker (res. 132–137). Proteins shown as mainchain cartoon; K-Ras (blue); RBD (orange); CRD (red); small molecules/ions as space filling: farnesyl group (gray); GTP (purple); Zn and Mg (tan); shown as lines: linker region (blue); HVR (green), membrane (blue); water (light cyan).
Figure 2.

Configurations of C-RafRBD-CRD (C-Raf) in solution. (a) Starting structure. (b) Clustering of configurations with an RMSD cutoff distance of 5 Å, (c) representative configurations superimposed on RBD (pointing away from observer). Clusters #1–4 are shown (for other clusters, see Figure S1a). Surface electrostatic potential of domains (in configuration as seen in a), rotated by +90° around z.
When bound to membrane-anchored K-Ras4B, the configurational flexibility of C-Raf is largely preserved (Figure S2a–b). Configurations are comparable to the configurations of an isolated C-Raf, although the populations of the different configurations are changed to different extents (Figure S2c). In addition, the radius of gyration of the two domains of C-Raf is more extended when bound to membrane-anchored K-Ras4B (Figure S2d). These differences reflect the additional interactions of C-Raf with K-Ras4B as well as those of C-Raf CRD with the membrane.
Interaction of C-Raf with K-Ras4B
The C-Raf–K-Ras4B interactions are mostly confined to those that are known from the C-RafRBD–H-Ras crystal structure,18 and this interface is highly persistent in the simulations. The β-sheet interface is established between residues 65−70 of the RBD and residues 37−39 of K-Ras4B (Figure S3a). Outside these regions additional contacts K84:E31/D33, V88:I21/Y40, and R89:D38/S39/Y40 are seen between the RBD and K-Ras (Figure S3a). Overall, these detailed protein–protein contacts are in close agreement with the interactions seen in the experimental structure.18 There is indirect evidence suggesting that the switch regions (I or II) of Ras may directly interact with the CRD.21,22 Specifically, an NMR study reported interactions of the N-terminal region of Switch-I and the C-terminus of farnesylated H-Ras with the CRD.21 Our studies did not show the CRD in contact with the K-Ras switch-I region, because in our case it is already occupied by the tight binding RBD (Figure 3). The interactions between the CRD and the switch II region of Ras.GTP were also not observed in any of the five simulations (Figure S3b). To examine this issue, we performed one additional simulation (#6) by placing the CRD in the proximity of the switch II region. However, over the course of the simulation the CRD gradually moved away from this region (Figure S3c). Experimentally, the binding of the RBD or of the CRD to Ras was measured separately in the literature. We suggest that when RBD and CRD are linked, the CRD has a low potency for Ras binding at switch II, as the CRD needs to orientate itself, relative to the switch I bound RBD. This is only possible by configurations that were not seen/are unfavorable in sampling of the unbound Raf (distal α1−β2 region of the RBD, as discussed above). In a competition the RBD has a much greater advantage compared to the CRD for binding to Ras, given a Kd of 20 nM for the RBD versus an approximately 5.5 times weaker affinity for CRD (measured for H-Ras).33 Essentially, the CRD is engaged in a variety of competitive interactions among CRD-RBD, CRD-Ras, and CRD-membrane contacts, with the latter probably being the most favorable.
Figure 3.
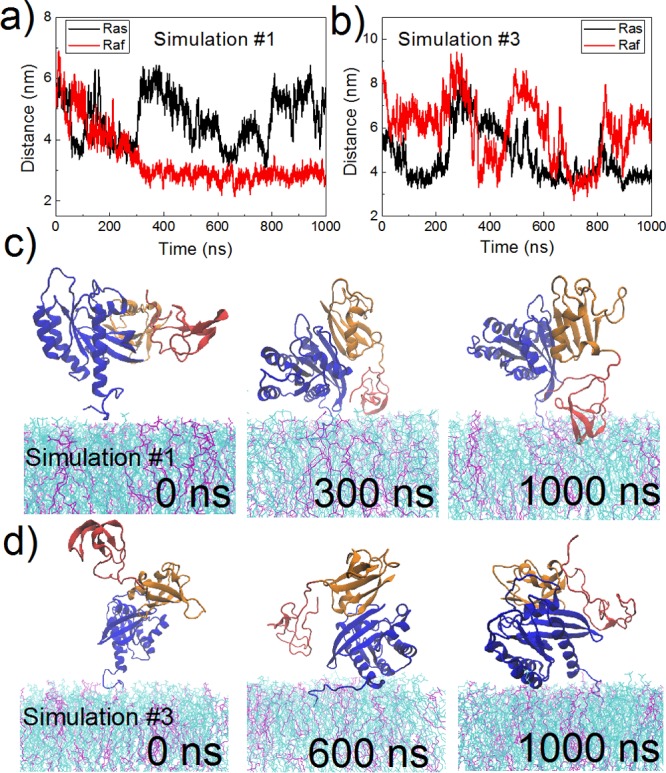
Membrane binding dynamics of the C-Raf–K-Ras4B complex. (a–b) Time evolution of the distance of the center of K-Ras4B CD2 (residues 87–166) or the center of C-Raf CRD to the membrane center. Snapshots taken at various time points from (c) simulation #1 and (d) simulation #3. Color scheme is the same as in Figure 1 except POPC is in cyan, and POPS is in purple.
The K-Ras4B Catalytic Domain and the C-Raf CRD Interact with the Membrane in a Dynamic Manner
Both the K-Ras4B catalytic domain and the C-Raf CRD region are able to contact the membrane in the simulations, but, when bound together as a protein–protein complex, do so in a dynamic way. For convenience of analysis, the K-Ras4B catalytic domain (CD) is further divided into two parts, i.e., CD1 or lobe1 containing residues 1–86 (containing P-loop, switch 1, switch 2, and the Raf RBD-binding interface of Ras), and CD2/lobe2 with residues 87–166 (containing helix 3, 4, and 5).34Figure 3 depicts the time evolution of the distance of the K-Ras4B CD2 or C-Raf CRD to the membrane center for simulation #1 and #3 (see Figure S4 for others). In simulation #1 (Figure 3a,c), the K-Ras4B catalytic domain binds to the membrane for the first time during the early 100 ns, but later it undergoes a few dissociation–association events. In comparison, the CRD gradually and spontaneously moves toward the membrane in the first 400 ns and remains bound to the membrane in the next 600 ns. In simulation #3 (Figure 3b,d), the K-Ras4B catalytic domain reaches the membrane first at ∼100 ns. However, along with the movement of the CRD toward the membrane at ∼350 ns, the K-Ras4B catalytic domain then moves away from the membrane. Later, the positions of both K-Ras4B and CRD undergo several fluctuations. Simulations #2, #4, and #5 show a range of similar scenarios (Figure S4). Overall, except for the CRD in simulation #1, perhaps, the membrane contacts of K-Ras4B or CRD are not highly persistent but are dynamic in nature (see discussion regarding convergence and optimal contacts below). This differs from previously reported situations when isolated K-Ras4B (and also K-Ras4A) are placed at a membrane with the same concentration of POPS lipid molecules, where K-Ras is mostly bound to the membrane, detaching less frequently.30,31
Interface of K-Ras4B–Membrane Interaction in the Presence of C-Raf
Residues involved in contacting the membrane are plotted in Figure 4a,b for K-Ras4B and C-Raf separately as a function of sequence and as averaged over the five simulations. For the catalytic domain of K-Ras4B, the membrane interacting residues belong mostly to helix 3 and helix 4, less so to helix 5 and a small loop segment between β2 and β3 (res. 43–50, loop 3). These interface residues establish the two dominant orientations of K-Ras at the membrane (O3 and O4V; numbering with reference to our previous study.31 see also Figure S5). In one orientation, helices 3 and 4 are bound to the membrane; in the other one, the loop 3, and partly helix 5 are the membrane interacting regions. For an isolated K-Ras, the O3 and O1 orientations (where β1-β3 associates with the membrane) are the dominant orientations.30,31 When bound to C-Raf, the O1 orientation is completely abolished, as the corresponding β1−β3 interface of K-Ras, also containing switch I, is now strongly bound to C-Raf. Therefore, O3 becomes the dominant orientation for K-Ras in complex with C-Raf at the membrane. Additionally, the membrane association of the CRD, in some cases, also brings the K-Ras loop 3 close to the membrane, resulting in the popularity of a variant of a previously characterized orientation, O4 (here denoted O4V). As shown in Figure 4c, a modest clustering of anionic POPS lipid molecules is observed under and around the C-Raf–K-Ras4B complex (especially under the GTPase). On average, 18% of the POPS are distributed within 2.5 nm of the center of K-Ras4B CD1 (accounting for ∼13.6% membrane area). Such clustering is expected to enhance the binding of K-Ras to the membrane.30,31 It should be noted that 1 μs presents an adequate amount of time for PS lipids to reorganize in the POPC/POPS bilayer under these conditions,30,35 although the simulations may not be completely converged for the more rapid reorientational transitions and some hysteresis could be present.
Figure 4.

Interfaces of the K-Ras4B–membrane and C-Raf–membrane interactions. (a) Frequency of K-Ras4B–membrane contacts (residues within 5 Å of membrane surface); position of helices 3–5 is indicated. Inset images show two major orientations of complex relative to the membrane. (b) Frequency of C-Raf–membrane contacts. The region, res. 143–150, is indicated in blue. Inset images: representative orientations of RBD and CRD at the membrane. (c) POPS distribution at the membrane. The distribution is normalized to make the maximum 100%. K-Ras4B CD1 is centered at (0, 0); the horizontal displacement between the center of mass of K-Ras4B CD1 and that of C-Raf RBD is aligned to the x-axis. The last 500 ns of the trajectories were used in these analyses.
Interface of the C-Raf:Membrane Interaction
As for C-Raf, only a few residues of the RBD (res. 56–132) can contact the membrane (Figure 4b), when bound to K-Ras. But the RBD–membrane contact frequency is rather low with the largest occupancy about 8.1% and 7.7% for E104 and H105, respectively. The most prominent membrane contacts involve residues in the CRD. Almost all of the residues in the CRD (res. 138–187) are able to contact the membrane more or less equally, except for a few residues hidden inside the folded conformation such as residues around V180 (Figure 4b). The CRD–membrane interaction is driven by both hydrophobic and by electrostatic contacts. The CRD can become partially buried into the membrane (most notably in simulation #1), with hydrophobic residues L147, L149, L159, and L160 largely buried into the membrane. The same region is also predicted as membrane inserted by the OPM (Orientations of Proteins in Membranes) Web server.36 Positively charged residues including R143, K144, K148, K157, and R164 also interact with the lipid bilayer, which is consistent with an experiment-based report that the 143-RKTFLKLA-150 segment of the CRD has a role in membrane binding.24 In addition, cation-π interactions established between F146, F158, and especially F151 and W187 and the nitrogen of the lipid headgroup (Figure S6a), as well as hydrogen bonding interactions between residues T145, Q156, and N161 (Figure S6b) also aid the membrane adhesion of the CRD.
Configurations of the C-Raf–K-Ras4B Complex at the Membrane
The configurations of the C-Raf–K-Ras4B complex with respect to the lipid bilayer are further characterized using several geometric parameters as shown in Figure 5. In Figure 5a, the distance of the K-Ras4B catalytic subdomain (CD2) to the membrane center is plotted versus the distance of the CRD to the membrane center, mapped over all five simulations. In one dominant conformation the CRD is away from the membrane and the K-Ras4B CD2 is bound to the membrane (6–8 nm and 3.5 nm, respectively). In contrast, another dominant conformation corresponds to the CRD approaching the membrane, with K-Ras CD2 pointing away from it (3–3.5 nm and 4.5–5.5 nm, respectively). Otherwise, there are instances when none of the domains are in contact with the membrane and times when both the K-Ras4B catalytic-domain and C-Raf CRD regions are close to the membrane (∼3.5 nm, ∼3.7 nm, respectively), although such instances are rare. Figure 5b plots another pair of parameters that characterize the relative position and orientation of the RBD and the K-Ras4B CD1 regions. Again, two orientations dominate: one with the RBD–K-Ras4B CD1 at ∼5.3 nm from the membrane center and with the RBD slanting upward (tilt angle centered at ∼50°); another represents a position tilted at around 95° and is centered at ∼4.2 nm. Unbound states can also be identified where the RBD–K-Ras4BCD1 is far away from the membrane and is almost parallel with the membrane surface. Overall, we classify the well populated configurations of the C-Raf–K-Ras4B complex at the membrane into four possible states based on the variables D1 and D2 (Figure 5a). Representative conformations for these four states are shown in Figure 5c. The first two states (CRD+/RAS- and CRD-/RAS+) represent the most dominant configurations for the complex. Moreover, there are large overlaps between different simulations (especially simulation #2 to #5, Figure S7). Thus, different states can interconvert to each other, suggesting an inherent dynamics of the protein complex unit with respect to the membrane.
Figure 5.

Configurations of the C-Raf–K-Ras4B complex at the membrane. The variables are defined as follows: D1, distance of K-Ras4B CD2, and D2, distance of CRD to membrane center, respectively. D, distance between the center of RBD–K-Ras4BCD1 and membrane center. V1, vector connecting the center of K-Ras4B CD1 to center of the RBD. θ, cross angle between vector V1 and normal to the membrane surface. K-Ras4B CD1, CD2, RBD, CRD in cyan, blue, orange, and red, respectively. (a) Contour maps (scaled to max.) with variables D1 versus D2. (b) D versus θ. (c) Representative configurations for the four possible states of the C-Raf–K-Ras4B complex. +/− denotes whether the domain binds the membrane or not.
Incompatibility of CRD (143-RKTFLKLAF-151) and K-Ras4B (Helix 3 and 4) in Making Membrane Contacts
It is apparent that the C-Raf CRD and K-Ras4B catalytic-domain contact the membrane mostly in a mutually exclusive manner. The non-cooperative mechanism is largely determined by the topology of C-Raf–K-Ras4B complex. As shown above, the most favorable membrane interaction interfaces are seen as residues 143-RKTFLKLAF-151 for the CRD and helix 3 and 4 for K-Ras4B, respectively (Figure 4a,b). However, when helix 3 and helix 4 of K-Ras4B contact the membrane, the RBD as the counterpart of K-Ras4B is moved away from the membrane with a slant angle at ∼50° (Figure 5b and Figure 6a). The calculated radius of gyration for the K-Ras catalytic domain, RBD, and CRD are 1.5, 1.2, and 1.1 nm respectively (the physical radius is even slightly larger). The distance between the center of the K-Ras catalytic domain and the C-Raf RBD is about 2.8 nm and between center of RBD and CRD is averaged at 2.9 nm. On the basis of the slant angle and the size of these domains, the RBD is at least 3.6 nm away from the membrane surface. Although the CRD has a considerable freedom to adopt multiple orientations relative to the RBD, none of them enable the CRD to reach the membrane without rotating the K-Ras domain, as the maximal length of the linker plus the CRD is estimated at 2.8 nm (2.9–1.2 + 1.1 nm, Figure 6a). Therefore, the most favorable membrane-associated state for CRD (143-RKTFLKLAF-151) and K-Ras4B (helix 3 and 4) cannot coexist. Figure S8 further discusses the opposite situation when CRD is bound to the membrane, yielding the same outcome. Overall, the C-Raf–K-Ras4B complex may either use 143-RKTFLKLAF-151 of CRD (state 1) or alternatively use helix 3 and helix 4 of K-Ras4B (state 2) to interact with the membrane. In state 3, both K-Ras4B and CRD are close to the membrane, but the complex is not using the 143-RKTFLKLAF-151 region of the CRD and helices 3, 4 of K-Ras4B together in membrane contact, and instead each use other, less favorable contacts with the membrane.
Figure 6.
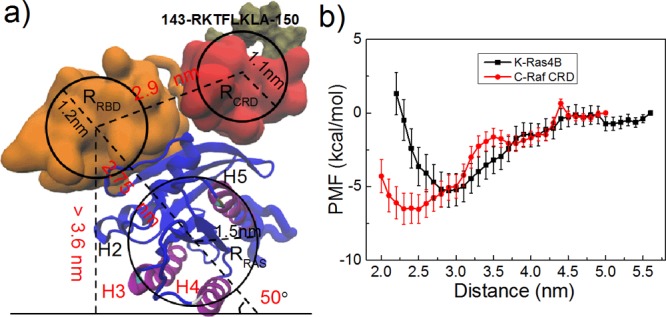
Tug of war between K-Ras4B and C-Raf CRD membrane interactions. (a) Schematic picture of steric/geometric limitations of the C-Raf–K-Ras4B complex in different states (here state 2; see Supporting Information, Figure S8 for state 1). (b) Potential of mean force, PMF, for K-Ras4B and CRD binding to the model membrane.
Free Energy of K-Ras4B and C-Raf CRD Membrane Adhesion
We estimate the binding affinity of monomeric C-Raf CRD and monomeric unlipidated K-Ras4B to the model membrane by calculating the potential of mean force (PMF) along a path of membrane (un)binding using the most favorable domain orientation in each case (Figure 6b, see Method). The free energy calculated for monomeric subdomains is not the exact free energy corresponding to state 1 and state 2 of the protein complex, but their calculation provides a rough free energy estimate for these states. The calculated PMF is −5.3 ± 1.1 kcal/mol for K-Ras4B and −6.5 ± 1.0 kcal/mol for the CRD binding to the membrane. Using microscale thermophoresis (MST) experiments, for a full-length unlipidated K-Ras4B binding to a membrane comprised of POPC and 5% PIP2 molecules, we measured Kd at 23.4 μM (ΔG = −6.31 kcal/Mol) at 298 K (manuscript in preparation). In the experiment of Gillette et al., a Kd was measured as 4.0 μM for lipidated K-Ras4B binding to nanodiscs mixed with DMPC (1,2-Dimyristoyl-sn-glycero-3-phosphorylcholine) and 30% DMPS (2,3-Bis(sulfanyl)propane-1-sulfonic acid).37 Thus, while difficult to simulate, lipid anchoring into the membrane via the farnesyl group likely has a favorable effect on the binding affinity of the catalytic domain to the membrane as well. No experimental Kd value for CRD binding to the membrane has been reported, but binding to liposomes suggests that the Kd should be at least 20 μM.31 The binding free energy predicted by the OPM server of −7.31 kcal/mol is larger than the value we estimate. Experimental measurements of the binding affinity of the CRD to the membrane are needed in the future. On the basis of our PMF calculation, the CRD could have a slight, but not an overwhelming advantage in membrane adhesion compared to the K-Ras4B catalytic domain.
Discussion
Structural preferences and dynamics of protein complexes at membranes are still a relatively new terrain for discovery. Several studies have examined the orientation and dynamics of isolated K-Ras at membranes,4,28−31 but there are as yet no simulations of the C-Raf–K-Ras complex at membranes. In this study, we performed multiple μs long all-atom MD simulation for the two-domain fragment C-RafRBD-CRD (denoted C-Raf) when bound to K-Ras4B at an anionic membrane. One might expect that additional interactions, such as possible direct interactions between the C-Raf CRD and K-Ras4B catalytic domains, would substantially increase this affinity via synergistic effects. In fact, this is frequently seen with cell signaling proteins, such as the interactions between WASP, Cdc42, and the membrane, which utilize multiple interactions in a cooperative manner to maximize the signaling output while minimizing output in the absence of coincident input signals.38,39 However, this is not the mechanism predicted by the calculations here. Importantly, our study predicts a novel competitive mechanism between membrane adhesion of the K-Ras4B catalytic domain and C-Raf CRD of the protein complex (Figure 5). We are able to rationalize the behavior of this system by considering both the geometric features of the domains within the complex (Figure 6a) as well as by the estimation of their binding affinity with the membrane (Figure 6b). While no experimental structural or biophysical data are available at present for a C-Raf RBD or CRD containing multidomain protein fragment binding to Ras, importantly, a relatively tight Ras–Raf RBD association was reported with a Kd of 20 nM.21 By contrast the Kd for both Ras catalytic domain-membrane and Raf CRD–membrane interactions are estimated at 10 to hundreds of μM from our simulations. Thus, membrane binding of the K-Ras4B catalytic domain and of the C-Raf CRD by themselves is only moderately strong, overall consistent with experimental data. Such a modest affinity is typically associated with cell signaling processes where interactions are kinetically labile, with the dissociation rate, koff ≈ Kd, and thus, such interactions are quick to separate and to switch off. An intrinsically modest affinity gives K-Ras more freedom to interact with multiple regulatory and effectors proteins. Even such interactions may enhance the binding affinity but not lead to “tightly frozen” protein–membrane complexes. Thus, we believe that the modest Raf- and K-Ras CD–membrane interactions likely have an important role in the regulation of the biological function of K-Ras4B and other protein–Ras complexes at membranes.
Our study implies that Raf–Ras interactions as well as the Ras CD- and CRD–membrane contacts are highly conserved (Figure S9). Specifically all K-, H and N-Ras are sequence invariant for the first 86 residues, parts of which comprise the contact region with the RBD. On the side of the RBD, res. 65–70 as well as K84, R89 are identical between B- and C-Raf. The region of residues 143–151 of the CRD primarily responsible for membrane interactions is also identical across many mammalian species (Figure S9a). The extent of conservation of Ras–membrane interaction sites is also high across the catalytic domains of Ras isoforms.40 Concerning the RBD-linker-CRD region of the protein, it is informative to put our simulation results in context of sequence conservation of this Raf protein segment and also with respect to cancer mutations (Figure S9b shows the alignment of C-Raf with A- and B-Raf homologues and discusses several mutations). Importantly, the linker region between the RBD and CRD is not changed in length, either between isoforms or between mammalian species (Figure S9a-b). The sequence differs between B-Raf and C-Raf in the linker with the change D132E and H133N (C-Raf numbering), but these changes are not dramatic. This suggests that the geometric restrictions we observe are likely conserved also between the Raf isoforms, including between the mammalian species.
A segment of residues 143-RKTFLKLAF-151 was found to be responsible for membrane binding of the CRD. Mutagenesis of R143Q or R143W as well as K144E suggested that the positive charge on these residues is critical for maintaining an autoinhibited state with the N-terminal of C-Raf (RBD-CRD) binding to the C-Raf kinase domain.41 Therefore, while RBD association with Ras.GTP is likely a major factor for releasing the self-inhibited state of C-Raf, the membrane binding of the 143–151 segment may be another positive factor for inducing or maintaining the open conformation of C-Raf. However, since we did not include the C-Raf kinase domain in our study, we cannot comment on whether the mutations have a stronger effect on weakening the autoinhibited state, compared to possibly weakening CRD–membrane binding.
It is accepted that Ras activity in cells requires its localization to the membrane. Ras clustering is an undisputed observation,26−28 but the role of catalytic domain contacts in Ras dimerization in solution and at the membrane is not yet clear. Recently, symmetric Ras–Ras dimers have been predicted through molecular docking, although they mainly represents a weak association (Kd ranges from 1 μM to greater than mM).42,43 A recent study of Ambrogio et al. showed that disruption of Ras dimerization attenuates the Ras signal pathway and oncogenic activity of mutant K-Ras.44 A similar inhibition was also observed when a designed monobody, NS1, was introduced into the cells. The monobody associates with the catalytic domain of Ras competitively against Ras dimerization.45 Both studies suggested that Ras dimerization occurs via helices 4 and 5. However, recent experiments showed, by contrast, that K-Ras4B lacks an intrinsic ability to dimerize at a supported lipid bilayer.46 This may indicate that some other factors in the cell, such as the Raf kinase domain or other proteins, may aid Ras dimerization. Still, as inference from our modeling and simulation study we notice that the predicted configurational state 2 of C-Raf–K-Ras4B does not match any of the predicted dimer forms of Ras (Figure 5c). But state 1 and of course, the non-membrane binding state 4 are suitable for the formation of Ras dimers, due to the exposure of helix 3, helix 4, and less so of helix 5 of K-Ras4B (Figure 5c) which are thought to comprise the dimer interface. However, in this case optimal K-Ras catalytic domain membrane binding and dimerization would also oppose one another. Nevertheless, CRD–membrane interactions may help to orient the GTPase at the membrane for improved dimerization kinetics, and depending on K-Ras vs C-Raf concentration, could stimulate K-Ras dimer formation. Formation of a K-Ras dimer could then bring the distal kinase domain of two Raf proteins sterically close to each other, hence stimulating also Raf dimerization. The activation of Raf probably involves a series of sequential processes (Ras.GTP binding through RBD, membrane binding of CRD, Ras dimerization, and Raf dimerization at distal kinase domain as well as possible Raf kinase domain membrane binding47), which will require in-depth studies. We are currently working on modeling possible higher order C-Raf–K-Ras complexes, but the added complexity is beyond the scope of this report.
Finally, the findings of our study also have general implications for cell signaling involving protein complexes at the cellular membrane. From other examples it is becoming clear that the membrane actively participates in the regulation of peripheral membrane protein function.1−9,48 However, the protein–membrane interactions typically synergize for protein localization at membranes. Alternatively, protein–membrane interactions can compete with protein–protein interactions. For example, the membrane adhesion of scaffold protein-Ste5 could release an autoinhibition between its two domains.49 Another example is the focal adhesion kinase, where both kinase and FERM domains can interact with PIP2 in membranes, leading to a separation of these two protein domains, then allowing the kinase domains to dimerize and activate.50 Here, we have found an example for a third type of mechanism involving a membrane peripheral protein complex: in this case, the domains are in competition with respect to their individual interactions with the membrane. Since their protein–protein interaction is stronger than the protein–lipid interactions, a “tug of war” is set up, yielding two configurations of the protein complex at the membrane. The study here reveals this mechanism at molecular level, further adding to the repertoire of signal processes by utilizing multidomain or multiprotein peripheral membrane protein complexes.
Method Summary
The model of C-RafRBD-CRD was built by connecting the crystal structures of the C-Raf RBD (PDB, 4G0N)18 and C-Raf CRD (PDB, 1FAQ)25 with the native linker. The C-RafRBD-CRD–K-Ras4B complex was further built by docking the modeled C-RafRBD-CRD structure and to the crystal structure of K-Ras4B (PDB, 4DSO), with reference to the crystal structure of H-Ras bound with the C-Raf RBD (PDB, 4G0N).18 The system was placed at a membrane containing 80% POPC (1-Palmitoyl-2-oleoylphosphatidylcholine) and 20% POPS (1-Palmitoyl-2-oleoylphosphatidylserine) (same ratio as in refs (4, 30, 31, 46)). Five independent simulations were performed each for 1 μs. Umbrella sampling simulations were performed to calculate the free energy of CRD and K-Ras4B binding to the membrane. The CHARMM36m force field was used in all simulations and energy calculations.51
Acknowledgments
This work is supported by NIGMS Grant R01GM112491 to the Buck lab; P.P. is supported by Cancer Prevention and Research Institute of Texas (CPRIT No. DP150093). We thank A. A. Gorfe for helpful discussion and the Ohio Super Computer center located in Columbus, Texas Advanced Computing Center (TACC), and the Extreme Science and Engineering Discovery Environment (XSEDE; Project MCB150054) for computational resources. Anton2 Computer time was provided by the Pittsburgh Supercomputing Center (PSC) through Grant R01GM116961 from the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00593.
Detailed methods, data analyses with associated figures and tables (PDF)
Author Contributions
Z.L.L. and M.B. conceived and designed the study with P.P contributing simulation #6; Z.L.L. and P.P. performed the simulations; Z.L.L., P.P. and M.B. analyzed the data; Z.L.L., P.P. and M.B. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Calvez P.; Schmidt T. F.; Cantin L.; Klinker K.; Salesse C. Phosphatidylserine allows observation of the calcium–myristoyl switch of recoverin and its preferential binding. J. Am. Chem. Soc. 2016, 138, 13533–13540. 10.1021/jacs.6b04218. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Sligar S. G. Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 2016, 23, 481–486. 10.1038/nsmb.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karandur D.; Nawrotek A.; Kuriyan J.; Cherfils J. Multiple interactions between an Arf/GEFcomplex and charged lipids determine activation kinetics on the membrane. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11416–11421. 10.1073/pnas.1707970114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhab-Jafari M. T.; Marshall C. B.; Smith M. J.; Gasmi-Seabrook G. M.; Stathopulos P. B.; Inagaki F.; Kay L. E.; Neel B. G.; Ikura M. Oncogenic and Rasopathy-associated K-Ras mutations relieve membrane-dependent occlusion of the effector binding site. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 6625–6630. 10.1073/pnas.1419895112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Lara Á; Thapa A.; Nyenhuis S. B.; Nyenhuis D. A.; Halder P.; Tietzel M.; Tittmann K.; Cafiso D. S.; Jahn R. PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. eLife 2016, 5, e15886. 10.7554/eLife.15886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorfe A. A.; Hanzal-Bayer M.; Abankwa D.; Hancock J. F.; McCammon J. A. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem. 2007, 50, 674–684. 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- Grauffel C.; Yang B.; He T.; Roberts M. F.; Gershenson A.; Reuter N. Cation−π interactions as lipid-specific anchors for phosphatidylinositol-specific phospholipase C. J. Am. Chem. Soc. 2013, 135, 5740–5750. 10.1021/ja312656v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckbosch S. M.; Wender P. A.; Pande V. S. Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun. 2017, 8, 6. 10.1038/s41467-016-0015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E.; Kalli A. C.; Yasuoka K.; Sansom M. S. P. Interactions of pleckstrin homology domains with membranes: adding back the bilayer via high-throughput molecular dynamics. Structure 2016, 24, 1421–1431. 10.1016/j.str.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu D. K.; Nissley D. V.; McCormick F. RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I. A.; Lewis P. D.; Mattos C. A. A Comprehensive survey of Ras mutation in cancer. Cancer Res. 2012, 72, 2457–2467. 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubbert S.; Shannon K.; Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Athuluri-Divakar S. K.; Vasquez-Del Carpio R.; Dutta K.; Baker S. J.; Cosenza S. C.; Basu I.; Gupta Y. K.; Reddy M. V.; Ueno L.; Hart J. R.; Vogt P. K.; Mulholland D.; Guha C.; Aggarwal A. K.; Reddy E. P. A small molecule Ras-mimetic disrupts Ras association with effector proteins to block signaling. Cell 2016, 165, 643–655. 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch M. E.; Kaplan A.; Chambers J. M.; Stokes M. E.; Bos P. H.; Zask A.; Zhang Y.; Sanchez-Martin M.; Badgley M. A.; Huang C. S.; Tran T. H.; Akkiraju H.; Brown L. M.; Nandakumar R.; Cremers S.; Yang W. S.; Tong L.; Olive K. P.; Ferrando A.; Stockwell B. R. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889. 10.1016/j.cell.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott H. R.; Owen D. Structures of Ras superfamily effector complexes: What have we learnt in two decades?. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 85–133. 10.3109/10409238.2014.999191. [DOI] [PubMed] [Google Scholar]
- Nassar N.; Horn G.; Herrmann C.; Scherer A.; McCormick F.; Wittinghofer A. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 1995, 375, 554–560. 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- Fetics S. K.; Guterres H.; Kearney B. M.; Buhrman G.; Ma B.; Nussinov R.; Mattos C. Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure 2015, 23, 505–516. 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brtva T. R.; Drugan J. K.; Ghosh S.; Terrell R. S.; Campbell-Burk S.; Bell R. M.; Der C. J. Two distinct Raf domains mediate interaction with Ras. J. Biol. Chem. 1995, 270, 9809–9812. 10.1074/jbc.270.17.9809. [DOI] [PubMed] [Google Scholar]
- Clark G. J.; Drugan J. K.; Terrell R. S.; Bradham C.; Der C. J.; Bell R. M.; Campbell S. Peptides containing a consensus Ras binding sequence from Raf-1 and the GTPase activating protein NF1 inhibit Ras function. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 1577–1581. 10.1073/pnas.93.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapar R.; Williams J. G.; Campbell S. L. NMR characterization of full-length farnesylated and non-farnesylated H-Ras and its implications for Raf activation. J. Mol. Biol. 2004, 343, 1391–408. 10.1016/j.jmb.2004.08.106. [DOI] [PubMed] [Google Scholar]
- Drugan J. K.; Khosravi-Far R.; White M. A.; Der C. J.; Sung Y. J.; Hwang Y. W.; Campbell S. L. Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J. Biol. Chem. 1996, 271, 233–237. 10.1074/jbc.271.1.233. [DOI] [PubMed] [Google Scholar]
- Hekman M.; Hamm H.; Villar A. V.; Bader B.; Kuhlmann J.; Nickel J.; Rapp U. R. Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J. Biol. Chem. 2002, 277, 24090–24102. 10.1074/jbc.M200576200. [DOI] [PubMed] [Google Scholar]
- Improta-Brears T.; Ghosh S. F.; Bell R. M. Mutational analysis of Raf-1 cysteine rich domain: requirement for a cluster of basic aminoacids for interaction with phosphatidylserine. Mol. Cell. Biochem. 1999, 198, 171–178. 10.1023/A:1006981411691. [DOI] [PubMed] [Google Scholar]
- Mott H. R.; Carpenter J. W.; Zhong S.; Ghosh S.; Bell R. M.; Campbell S. L. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 8312–8317. 10.1073/pnas.93.16.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abankwa D.; Gorfe A. A.; Inder K.; Hancock J. F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1130–1135. 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise K.; Kapoor S.; Denter M.; Nikolaus J.; Opitz N.; Koch S.; Triola G.; Herrmann A.; Waldmann H.; Winter R. Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. J. Am. Chem. Soc. 2011, 133, 880–887. 10.1021/ja107532q. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Prakash P.; Liang H.; Cho K. J.; Gorfe A. A.; Hancock J. F. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell 2017, 168, 239–251. 10.1016/j.cell.2016.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S.; Weise K.; Erlkamp M.; Triola G.; Waldmann H.; Winter R. The role of G-domain orientation and nucleotide state on the Ras isoform-specific membrane interaction. Eur. Biophys. J. 2012, 41, 801–813. 10.1007/s00249-012-0841-5. [DOI] [PubMed] [Google Scholar]
- Prakash P.; Zhou Y.; Liang H.; Hancock J. F.; Gorfe A. A. Oncogenic K-RAS binds to an anionic membrane in two distinct orientations: a molecular dynamics simulation. Biophys. J. 2016, 110, 1125–1138. 10.1016/j.bpj.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. L.; Buck M. Computational modeling reveals that signaling lipids modulate the orientation of K-Ras4A at the membrane reflecting protein topology. Structure 2017, 25, 679–689. 10.1016/j.str.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham E.; Chiang J.; Li I.; Shum W.; Truong K. A computational tool for designing FRET protein biosensors by rigid-body sampling of their conformational space. Structure 2007, 15, 515–523. 10.1016/j.str.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Xie W. Q.; Quest A. F.; Mabrouk G. M.; Strum J. C.; Bell R. M. The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J. Biol. Chem. 1994, 269, 10000–10007. [PubMed] [Google Scholar]
- Gorfe A. A.; Grant B. J.; McCammon J. A. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure 2008, 16, 885–896. 10.1016/j.str.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin B. S. Jr; Sodt A. J.; Cotten M. L.; Pastor R. W. The Curvature induction of surface-bound antimicrobial peptides piscidin 1 and piscidin 3 varies with lipid chain length. J. Membr. Biol. 2015, 248, 455–467. 10.1007/s00232-014-9733-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize M. A.; Lomize A. L.; Pogozheva I. D.; Mosberg H. I. OPM: Orientations of Proteins in Membranes database. Bioinformatics 2006, 22, 623–625. 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- Gillette W. K.; Esposito D.; Blanco M. A.; Alexander P.; Bindu L.; Bittner C.; Chertov O.; Frank P. H.; Grose C.; Jones J. E.; Meng Z.; Perkins S.; Van Q.; Ghirlando R.; Fivash M.; Nissley D. V.; McCormick F.; Holderfield M.; Stephen A. G. Farnesylated and methylated KRAS4b: high yield production of protein suitable for biophysical studies of prenylated protein-lipid interactions. Sci. Rep. 2015, 5, 15916. 10.1038/srep15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M.; Xu W.; Rosen M. K. A two-state allosteric model for autoinhibition rationalizes WASP signal integration and targeting. J. Mol. Biol. 2004, 338, 271–285. 10.1016/j.jmb.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Dueber J. E.; Yeh B. J.; Chak K.; Lim W. A. Reprogramming control of an allosteric signaling switch through modular recombination. Science 2003, 301, 1904–1908. 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- Prakash P.; Gorfe A. A. Membrane orientation dynamics of lipid-modified small GTPases. Small GTPases 2017, 8, 129–138. 10.1080/21541248.2016.1211067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler D. G.; Cutler R. E. Jr; Drugan J. K.; Campbell S.; Morrison D. K.; Cooper J. A. Identification of residues in the cysteine-rich domain of Raf-1 that control Ras binding and Raf-1 activity. J. Biol. Chem. 1998, 273, 21578–21584. 10.1074/jbc.273.34.21578. [DOI] [PubMed] [Google Scholar]
- Muratcioglu S.; Chavan T. S.; Freed B. C.; Jang H.; Khavrutskii L.; Freed R. N.; Dyba M. A.; Stefanisko K.; Tarasov S. G.; Gursoy A.; Keskin O.; Tarasova N. I.; Gaponenko V.; Nussinov R. GTP-dependent K-Ras dimerization. Structure 2015, 23, 1325–1335. 10.1016/j.str.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P.; Sayyed-Ahmad A.; Cho K. J.; Dolino D. M.; Chen W.; Li H.; Grant B. J.; Hancock J. F.; Gorfe A. A. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci. Rep. 2017, 7, 40109. 10.1038/srep40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrogio C.; Köhler J.; Zhou Z. W.; Wang H.; Paranal R.; Li J.; Capelletti M.; Caffarra C.; Li S.; Lv Q.; Gondi S.; Hunter J. C.; Lu J.; Chiarle R.; Santamaría D.; Westover K. D.; Jänne P. A.. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 2018, 10.1016/j.cell.2017.12.020 [DOI] [PubMed] [Google Scholar]
- Spencer-Smith R.; Koide A.; Zhou Y.; Eguchi R. R.; Sha F.; Gajwani P.; Santana D.; Gupta A.; Jacobs M.; Herrero-Garcia E.; Cobbert J.; Lavoie H.; Smith M.; Rajakulendran T.; Dowdell E.; Okur M. N.; Dementieva I.; Sicheri F.; Therrien M.; Hancock J. F.; Ikura M.; Koide S.; O’Bryan J. P. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. K.; Lee Y. K.; Denson J. P.; Gillette W. K.; Alvarez S.; Stephen A. G.; Groves J. T. K-Ras4B remains monomeric on membranes over a wide range of surface densities and lipid compositions. Biophys. J. 2018, 114, 137–145. 10.1016/j.bpj.2017.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baljuls A.; Mahr R.; Schwarzenau I.; Müller T.; Polzien L.; Hekman M.; Rapp U. R. Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J. Biol. Chem. 2011, 286, 16491–16503. 10.1074/jbc.M110.194167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timr S.; Pleskot R.; Kadlec J.; Kohagen M.; Magarkar A.; Jungwirth P. Membrane Binding of Recoverin: From Mechanistic Understanding to Biological Functionality. ACS Cent. Sci. 2017, 3, 868–874. 10.1021/acscentsci.7b00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan J. G.; Coyle S. M.; Rajan S.; Sidhu S. S.; Lim W. A. Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science 2012, 337, 1218–1222. 10.1126/science.1220683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog F. A.; Braun L.; Schoen I.; Vogel V. Structural insights how PIP2 imposes preferred binding orientations of FAK at lipid membranes. J. Phys. Chem. B 2017, 121, 3523–3535. 10.1021/acs.jpcb.6b09349. [DOI] [PubMed] [Google Scholar]
- Huang J.; Rauscher S.; Nawrocki G.; Ran T.; Feig M.; de Groot B. L.; Grubmüller H.; MacKerell A. D. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. 10.1038/nmeth.4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.