

Abstract
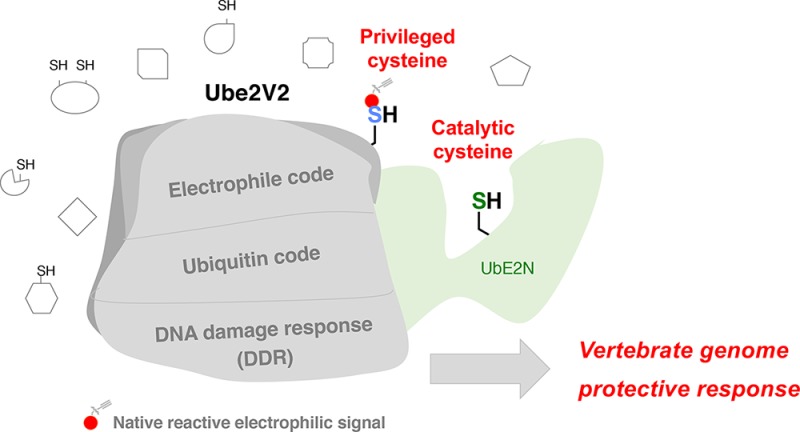
Posttranslational modifications (PTMs) are the lingua franca of cellular communication. Most PTMs are enzyme-orchestrated. However, the reemergence of electrophilic drugs has ushered mining of unconventional/non-enzyme-catalyzed electrophile-signaling pathways. Despite the latest impetus toward harnessing kinetically and functionally privileged cysteines for electrophilic drug design, identifying these sensors remains challenging. Herein, we designed “G-REX”—a technique that allows controlled release of reactive electrophiles in vivo. Mitigating toxicity/off-target effects associated with uncontrolled bolus exposure, G-REX tagged first-responding innate cysteines that bind electrophiles under true kcat/Km conditions. G-REX identified two allosteric ubiquitin-conjugating proteins—Ube2V1/Ube2V2—sharing a novel privileged-sensor-cysteine. This non-enzyme-catalyzed-PTM triggered responses specific to each protein. Thus, G-REX is an unbiased method to identify novel functional cysteines. Contrasting conventional active-site/off-active-site cysteine-modifications that regulate target activity, modification of Ube2V2 allosterically hyperactivated its enzymatically active binding-partner Ube2N, promoting K63-linked client ubiquitination and stimulating H2AX-dependent DNA damage response. This work establishes Ube2V2 as a Rosetta-stone bridging redox and ubiquitin codes to guard genome integrity.
Short abstract
G-REX and T-REX double tap strategy identifies privileged electrophile responsivity of Ube2V2 to promote genome protection.
Through a phenomenal research effort we now understand much about complex post-translational regulation in cell signaling. Approximately 10% of the genome is involved in phosphorylation1 and ubiquitination:2,3 a complex series of “codes” specific to both healthy and disease states is overseen by this suite of enzymes. Gaining a clearer comprehension of these paradigmatic signaling pathways has impacted several aspects of human health, including prophylaxis, diagnosis, drug design, and personal medicine. Recent years have witnessed success of kinome-targeting pharmaceuticals,4 and an intense pursuit of drug discovery is now also aimed at proteins/pathways involving ubiquitination.5,6 No approved drugs currently target ubiquitin (Ub) conjugation/deconjugation,7 but the proteasome—a molecular machine intrinsically linked to the ubiquitin pathways—is a bona fide drug target.6
Against the backdrop of these exquisite enzyme-regulated signaling subsystems, the cell has also harnessed reactive small-molecule signaling mediators to fine-tune responses. In this paradigm, reactive oxygen or electrophilic species (ROS/RES) directly modify a specific signal-sensing protein, preempting decision-making.8−11 Because ROS and RES exist at low levels during signaling, sensor residues on redox-responsive proteins are likely “kinetically privileged”, i.e., inherently tuned to rapidly react with specific ROS/RES12 with rapid second-order rate constants (high kcat/Km). Unlike phosphorylation, ubiquitination is dominated by reactive thiol chemistry: Ub-conjugation proceeds through multiple enzyme-bound Ub-thioester intermediates. These conjugating enzymes are ROS-sensitive.13 Deubiquitinating/deSUMOylating enzymes (DUBs/SENPs) are mostly thiol-active proteases; many DUBs and SENPs are indeed targets of ROS. Many of these ROS adducts involve direct modification of the active-site cysteine residue that is privileged due to its low pKa and low kinetic barrier to reaction with ROS. In most cases, free thiol ushers regain in enzymatic activity.
The Ub-proteasome pathway is RES-sensitive, although this is more nuanced than ROS-sensing. Many natural electrophiles, including prostaglandins,14 4-hydroxynonenal (HNE),15 and dietary isothiocyanates16 affect Ub-modification events (semi)-specifically. Several RES target active-site or other important cysteines on Ub-activating (E1), -conjugating (E2), and -ligating (E3, HECT-type) enzymes. Regardless of the site, most RES-modifications are irreversible—an attribute of RES signaling that may improve efficacy/confer different latency/longevity relative to ROS signaling.12
With the resurgence of electrophilic pharmacophores17 and the search for novel drug-targeting mechanisms,18 privileged RES-sensing residues and the proteins/pathways they control have come to the fore of disease treatment.19 Our recent work indicates that (i) there are specific subsets of orthogonal ROS- and RES-sensing cysteines,20 meaning specific RES-sensors could be ideal foundations for electrophilic drug discovery;21 and (ii) privileged electrophile sensors may neither need to be active-site nucleophiles nor present in proteins with any specific chemical function. Hence RES-sensor identification could offer a pipeline to regulate pathway flux and modulate undruggable proteins without the difficulties associated with targeting active-site residues.22
Identification of bona fide sensor cysteines is difficult.23 An extensive series of innovative work has been done to identify electrophile-sensor proteins in cells, and some work has studied model organisms. There are two main strategies. The first is bolus exposure of cells with reactive electrophiles, followed by affinity capture of modified proteins and MS.24 This strategy excitingly identifies a huge number of targets but is dominated by mass action/hypermodification and is often prone to artifacts caused by end-point toxicity and perturbation of innate redox balance. A second strategy uses competitive profiling of a specific set of reactive residues.25 This innovative strategy is very powerful and more sensitive than bolus-dosing.
While much of our current understanding in redox biology has been derived using these pioneering methods, some key limitations remain to be addressed.21 For instance, both of the above methods administer an excess of electrophile from outside of cells/animals; thus nuances of low-stoichiometry on-target RES-modifications that drive phenotypically dominant redox responses at a specific time are often lost. Indeed, RES permeation into the cell, interaction with cellular redox machinery (e.g., glutathione), and buildup of metabolites are complex, time-dependent processes, rendering bulk RES-exposure from outside of cells a far-from-controlled environment. This effect is magnified in whole organisms where phenotypic outputs from bulk exposure are a function of complex pharmacokinetics as well as amalgamation of on-target and off-target responses elicited by uncontrolled RES-exposure. Furthermore, as the competitive profiling method measures loss of labeling by the proxy (e.g., iodoacetamide), conclusions from the indirect measure of RES-modification may be confounded by off-target and/or secondary modifications/functional coupling: selective labeling of minor isoforms/complexes in low abundance may also be missed.
G-REX: An Unbiased Method To Profile Privileged Innate Electrophile-Sensor Proteins
To gain a new ability to directly assay downstream ramifications of these nonenzymatic redox-modification events at single-protein-target resolution and obtain mechanistic information about precision RES-signaling, we recently developed “T-REX” (targetable reactive electrophiles and oxidants)26 (Figure S1A–B). While T-REX has provided the previously inaccessible ability to directly read-out functional consequences of target-specific RES-modifications in vivo (cells, worms, and fish) in the backdrop of an otherwise unperturbed proteome, it has the following limitations: (1) T-REX relies on ectopic overexpression of Halo-fusion proteins, in which HaloTag is fused to individual select targets; (2) one thus has to have some prior knowledge about which targets could potentially bear functionally responsive cysteines [i.e., privileged first responders (PFRs)]. To that end, potential PFRs for a medium-throughput T-REX screen need to be cherry-picked from previously published hits obtained from profiling methods built on bulk-RES-introduction from outside the cell; and (3) T-REX is a one-target-at-a-time, low/medium-throughput gel-based screen.
To strive for a functional high-throughput screen at the whole-genome scale to target-ID PFRs, we here devised a new platform, “G-REX”, that captures PFR-cysteines directly in vivo, at a specific user-defined time (Figure 1A). Unlike T-REX, G-REX (1) requires no reliance on ectopic overexpression of the target protein of interest (i.e., HaloTag ectopically expressed alone in cells with no transgene fused to it serves as an anchor to a cell/fish/worm-permeable bioinert photocaged RES-precursor that can liberate controlled amount of RES such as HNE on demand); (2) G-REX is thus “casting a net” for innate PFRs at the whole-genome scale in a high-throughput manner, at the user-defined time/dose/duration/locale; (3) in that capacity, G-REX is the first method to deliver a native RES signal of choice in situ; and (4) it selectively captures targets specifically under RES-limited conditions (thus targets likely represent those that undergo low-stoichiometric RES-modifications, i.e., likely to be PFRs), as opposed to identification of targets labeled by RES following bulk introduction of reactive signals from outside of cells/animals, which often is nondiscriminating in all parameters (time, locale, target, and extent of RES-occupancy on individual targets).
Figure 1.

G-REX identifies endogenous privileged first responder (PFR)-cysteines, including two novel sensors, Ube2V1 and Ube2V2, through its capability to genome-wide target-ID endogenous PFRs under electrophile-limited conditions. (A) General setup for G-REX. Treatment of HEK293T cells ectopically expressing HaloTag results in specific binding of the inert photocaged RES-precursor [e.g., Ht-PreHNE(alkyne)]. (In dotted box is the ribbon model of Halo bound to Ht-PreHNE(alkyne), chemical structure of which is shown in Figure S1B). Any unbound probe is washed out. Upon low-energy light exposure of cells (see methods section), this Halo–Ht-PreHNE(alkyne) complex releases a stoichiometric amount of HNE(alkyne) (t1/2 < 1–2 min) (red dot) within the microenvironment of Halo, enabling substoichiometric covalent tagging of native PFRs to HNE. Indicated established pulldown-proteomics analysis permits HT-target-ID genome-wide. (B) (Left) The indicated recombinant protein (12 μM) was treated with HNE-alkyne (12 μM) at 37 °C and aliquots were removed as a function of indicated time and diluted into Click reaction mixture containing Cy5-azide (reporting on HNEylation) and subsequently analyzed by Sypro-Ruby (total protein). (Right) Plot of total unlabeled protein remaining as a function of time based on band-intensity quantitation. Also see Figure S3A. (C) An illustrative model of N-terminal HaloTagged Ube2V2 complexed to Ube2N with the photocaged-precursor, Ht-PreHNE, bound at Halo. (See Figure S1A for general T-REX setup.) Ribbon structure is adapted from PDB: 1J7D. The newly discovered HNE-responsive C69 (this work) within Ube2V2, and the catalytic cysteine (C87) within Ube2N, are indicated. (D) MS/MS spectrum of a triply charged ion at m/z 795.75653+ identifying an HNE alkyne modified peptide: IYSLKVECGPKYPEAPPSVR in which Cys69 of the UBE2 V2 protein digested by ArgC is modified by HNE alkyne with Xcorr score at 2.20 by Proteome Discoverer database search. The MS/MS spectrum was manually inspected and confirmed. The relative low Xcorr score is apparently due to the labile nature of the HNE alkyne which readily loses the HNE alkyne molecule from Cys69 residue during the CID fragmentation. This observation was supported by the two dominant ions (m/z 745.353+ and m/z 1117.372+) in the MS/MS spectrum representing the native targeted peptide (after loss of HNE alkyne). The inset shows the expanded view of MS survey scan for the precursor ion of the HNE-alkyne-modified peptide (m/z 795.75653+) with observed mass accuracy at 1.04 ppm. Also see Tables S1–S2. (E) HEK293T cells were transfected with the indicated plasmids, treated with Ht-PreHNE, and either exposed to light, or not irradiated. Normalized lysates from these two sets of cells were treated with either TEV-protease or buffer alone, respectively. Lysates were then subject to Click coupling with Cy5-azide, and analyzed by in-gel fluorescence for Cy5 signal. “1, 2, and N” respectively designate Ube2V1, Ube2V2, and Ube2N. Refer to Figure S4A for Cy5 gel and blots in full-view. (F) Similar to E but cells were cotransfected with either empty vector (−) or a plasmid of the same vector expressing HA-Ube2N (+). Region of interest in Cy5 gel is marked by a red rectangle. Refer to Figure S4B for Cy5 gel and blots in full-view. (G) Similar to F. Refer to Figure S4C for Cy5 gel and blots in full-view. M designates molecular weight marker lane in all gels/blots in this and all figures elsewhere.
Specifically herein, G-REX identifies PFR-cysteines via instantaneous release of a minimal amount of specific endogenous electrophile [e.g., 4-hydroxynonenal (HNE)—a known signaling messenger that is cytotoxic and mutagenic at high concentrations8−10] in a cell (Figure 1A and Figure S1B). We chose HNE as a well-established model for native bioactive signaling electrophiles housing cysteine-reactive Michael-acceptor motifs prevalent in many successful covalent drugs.21 Ultimately different electrophile chemotypes are anticipated to have differential fingerprint patterns of privileged sensors. Because the amount of electrophile is low and release is rapid (t1/2 < 1–2 min), only the most sensitive cysteines can react before HNE metabolism (true kcat/Km-type conditions). Under the conditions deployed, based on the calculated intracellular concentration of ectopic Halo and photo-uncaging efficiency of the Ht-PreHNE probe bound to Halo, ∼5 μM of HNE is released to the cell during G-REX (Supporting Information, Figure S1C). We compared time-dependent HNE-labeling of the proteome following either G-REX or bulk HNE (5 μM) exposure to live HEK293T cells ectopically expressing HaloTag construct. These experiments revealed that in situ HNE release by G-REX afforded a larger degree of proteome labeling in the initial times relative to exogenous HNE treatment (Figure S1D). Time required for HNE permeation into cells may partly explain these differences.
The G-REX system—when directly coupled to our T-REX single-protein-redox targeting26 (Figure S1A)—presents a previously inaccessible two-in-one capability, simultaneously enabling (1) proteome-wide profiling, and (2) target-specific functional validations of novel sensors and phenotypically dominant responses specifically triggered as a direct result of low-occupancy on-target RES-modifications under electrophile-limited conditions in situ (Figure 1A and Figure S1A–B). Using this G-REX–T-REX double-tap strategy, we identified a novel privileged cysteine of conserved importance present in two proteins that acts as a redox–Ub signaling shunt modulating two disparate signaling pathways.
Gel-based analysis coupled with streptavidin blot verified successful labeling of proteins from G-REX (Figure 1A and Figure S1E–F). Many redox-sensing proteins, such as Keap1, are unusually cysteine (Cys) rich. These proteins likely use mass action to improve their odds of being HNEylated and trigger downstream signaling. In addition, for Keap1, many Cys’s are functional sensors.27 Our recent findings make a strong case that sensing ability is not necessarily correlated with the number of Cys’s: we have found some HNE-sensing proteins/enzymes—such as Akt320 and small heat shock protein26—contain unique sensor Cys’s and rely mostly upon kinetic privilege to sense endogenous RES such as HNE. To increase the odds of finding highly reactive yet cysteine-poor sensors, we focused on the low-molecular-weight (LMW) protein pool (Figure S1G). A 15–25-kDa region of the resultant gel (Figure S1G) was thus cut and HNE-labeled proteins were identified by MS. The top hit from this pool was Ube2V2 (Mms228)—a Ub-conjugating protein-variant with a poorly understood role in DNA damage. Human Ube2V2 bears only one cysteine (C69) (Figure S2). A homologous protein, Ube2V1 (Uev129) was the fourth highest confidence hit (Figure S1H and Table S1). Ube2V1 bears three cysteines, one of which (C94) is analogous to C69 in Ube2V2 (Figure S2).30 Although neither of these proteins is reportedly redox-sensitive, remarkably, five other hits in the top 10 [peptidyl-prolyl cis–trans isomerase,31 ADP-ribosylation factor-332 and -4,32 nucleoside diphosphate kinase,33 and cofilin-134] were known HNE-sensors. This evidence suggests that G-REX is a sensitive method to identify first-responding sensor proteins. It also gave us further impetus to validate Ube2V1 and Ube2V2 as novel low-MW PFRs, without undertaking standard parallel MS analyses on samples not treated with light or Ht-PreHNE, which could be used to eliminate false-positive hits.
G-REX Identifies a Novel Conserved Cysteine Present in Two Privileged Sensors in Humans, Ube2V1 and Ube2V2
Because Ube2V2 that contains only one cysteine (C69) was the top hit and it was also found together with a homologous protein Ube2V1 containing a similar cysteine, we hypothesized that Ube2V2(C69)/[Ube2V1(C94)] (Figure S2A) are privileged sensors. Alignments of human Ube2V1 and Ube2V2 with other vertebrate counterparts showed that C69 and C94 are both conserved from humans to yeast (Figure S2B–D). Neither cysteine is implicated in electrophile sensing. The analogous cysteine in Saccharomyces cerevisiae Ube2V2 (Mms2) is also not required for activity.35 The longest isoform of human Ube2V1 has two other cysteines, one of which is not conserved beyond chimpanzees, whereas the other is conserved to frogs (Figure S2B). Interestingly, both Ube-2 V1 and -2V2 modulate ubiquitination activity of Ube2N (a low confidence hit in our G-REX data). Ube2N is an established Ub-conjugating E2 enzyme that assembles K63-linked ubiquitin, unanchored, or anchored on target proteins.28,29 As opposed to proteasomal targeting, K63-linked ubiquitination plays a role in cell signaling, including NF-κB signaling29,36 and the DNA damage response (DDR).37−39 Intriguingly, Ube2V2/Ube2V1 both lack a catalytic cysteine required for E2-activity, whereas Ube2N houses a catalytically essential nucleophilic Cys(C87) required for E2-catalytic function. Indeed, Ube2N(C87) has also proven susceptible to nitro-furan-derived electrophiles and acrylonitrile sulfonates.40 However, direct comparison of the efficiency of HNEylation of Ube2V2 and Ube2N in vitro (Figure 1B) unambiguously demonstrated that HNE sensing is unique to Ube2V2. By contrast, Ube2V2(C69S)—which we predicted to be a mutant unable to undergo HNEylation but otherwise similar to wild-type (wt)-Ube2V2—lost sensing (Figure S3A–B). Circular dichroism analyses showed that both Ube2V2 wt and C69S adopted a similar secondary structure (Figure S3C). The in vitro ubiquitination assays validated that these recombinantly purified wt and C69S Ube2V2 were functional (Figure S3D). HNEylation of Ube2V2 also did not significantly perturb/destabilize the protein’s fold (Figure S3E). Our findings from G-REX—together with these in vitro analyses—thus raise the tantalizing possibility that Ube2V1/2 serve as novel signaling shunts bridging the human redoxome and ubiquitome, enabling “signal exchange” between a native redox-linked signal (HNE) and a canonical enzyme-catalyzed signal (Ub).
The in-cell-relevance of target- and site(C69)-specific HNE-sensing within Ube2V2 was verified using our single-protein redox-targeting method, T-REX, in live cells (Figure S1A–B),26 followed by LC-MS/MS on Ube2V2 enriched from extracts (Figure 1C–D and Table S2). These data underscore the capability of a G-REX–T-REX coupled platform to identify bona fide HNE-sensors at the proteome scale, and subsequently interrogate the consequences of on-target electrophilic modification under conditions that mimic endogenous signaling. From our recent unbiased screens, approximately 10% of proteins can react with HNE under T-REX.20,26 Because T-REX is built on a quasi-intramolecular delivery,41 the efficiency of this targeting process is not affected by protein expression.20,26,41 Regardless, expression of Ube2N and Ube2V2/Ube2V1 proteins was similar in our experiments (Figure 1E–F, Figure S4A–B). T-REX-targeted delivery efficiencies26 independently measured for Ube2V2, Ube2V1, and Ube2N were 15 ± 6%, 5 ± 2%, and <2%, respectively. Delivery to Ube2V1/2 V2 was unaffected by Ube2N-co-overexpression (Figure 1C,F–G and Figure S4B–D). Ube2V1(C94S) and Ube2V2(C69S) both showed significantly reduced targeting relative to wt counterparts (Figure 1G and Figure S4C–E). Furthermore, affinity capture of any proteins nonspecifically adducted by adventitious HNE released during T-REX-targeted delivery to Ube2V2 showed that there was no detectable HNEylation of Ube2N (Figure S4F). This finding is consistent with Ube2N being a low-confidence hit in G-REX (Figure S1G and Table S1) as well as our in vitro data on HNE conjugation (Figure 1B).
Aside from documenting the power of G-REX to identify novel HNE sensors, the finding that Ube2V1/2 are privileged HNE-sensors is significant for several reasons. First, the relative HNE-sensitivity of these similar proteins does not correlate with the number of Cys’s they contain. Second, in this series, HNE-sensing ability does not correlate with enzymatic function: catalytically inactive E2-variants (Ube2V1/2) are much better HNE-sensors than catalytically active E2-conjugating-enzyme (Ube2N). Third, although HNE released under G-REX labeled many proteins, it has been shown that only 1–8% of free HNE in cells labels proteins.42 Thus, the 15% Ube2V2(C69)-specific labeling above represents a significant enrichment over these background proteins—a result consistent with there being no labeling of the C69S mutant.
Target- and Cys-Site-Specific RES-Sensing by Ube2V1 and Ube2V2 Triggers Specific Pathway Activation
We next evaluated functional responses brought about by on-target HNEylation of Ube2V1 and Ube2V2 separately. Ube2N–Ube2V2 heterodimer (Figure 1C) is implicated to be important in DNA damage response (DDR), whereas Ube2N–Ube2V1 regulates NF-κB inflammatory signaling.29,36,43 Consistent with previous data, subsequent to T-REX-targeted HNEylation of Ube2V1 (with coexpression of Ube2N), a 3-fold upregulation in NF-κB-signaling was measured relative to all controls. Replicating this experiment with Ube2V2 in place of Ube2V1 did not result in NF-κB-pathway activation (Figure 2A), confirming that HNE-induced NF-κB signaling upregulation was Ube2V1-specific. These data further demonstrate that G-REX can identify functional first responders.
Figure 2.

T-REX-assisted Ube2V2(C69)-specific HNEylation elicits K63-linked-polyubiquitination that is lost in the C69S mutant. (A) HNEylation of Ube2V1 (specifically of C94), but not Ube2V2, selectively elicits upregulation in NF-κB-signaling (mean ± s.e.m, n ≥ 2 independent biological replicates). (B) HEK293T cells expressing wt-Halo-(FLAG)-Ube2V2 or the C69S mutant were treated with the DUB-inhibitor PR619 and high-molecular-weight (HMW) polyubiquitylated-Ube2V2 was assessed by western blot. (C) HEK293T cells expressing wt-Halo-(FLAG)-Ube2V2 (or the C69S mutant) and HA-Ubiquitin were exposed to the indicated conditions. Halo-(FLAG)-Ube2V2 from these cells was enriched by FLAG-immunoprecipitation (IP), and resulting samples were analyzed using the indicated antibodies. Left panel = “Input lanes”; Right panel = “IP-samples”. (D) Similar experiment to C, except nonenriched whole cell lysates were analyzed using the indicated antibodies, and an additional sample from the use of no-alkyne-variant of Ht-PreHNE but otherwise treated under identical conditions was also analyzed. (E) Similar experiment to C except global Ub pools were precipitated using HA IP (right panel) (there is no change in Ub since Ub is being IP-ed).
By contrast to the established position of Ube2V1 in NF-κB-directed inflammatory signaling, the precise biological mechanisms underlying the role of Ube2V2 in DDR are unclear. To elucidate how Ube2V2’s HNE-sensing function fits into the DDR program, we first studied the ramifications of Ube2V2-specific HNEylation relative to Ube2V2(C69S). To this end, we also validated that both wt and mutant proteins ectopically expressed are stable (Figure S3F). Ube2N also coprecipitated with Ube2V2(C69S) as efficiently as wt-Ube2V2 (Figure S5A–B). PR-619—a deubiquitinating-enzyme inhibitor (one of the few inhibitor classes that elevate K63-linked Ub-pools39,44)—was able to promote ubiquitination of Ube2V2(C69S) as efficiently as wt-Ube2V2 (Figure 2B). These data are consistent with the results from our in vitro ubiquitination assays (Figure S3D).
Ube2V2–Ube2N-heterodimer catalyzes the synthesis of noncanonical K63-linked polyubiquitin chains with various functional roles in cell signaling.28,40,43,45,46 We found that Ube2V2-specific HNEylation enabled by T-REX, did not alter K63-linked total cellular Ub pools (Figure 2C, “input”), whereas treatment with PR-619 promoted elevation of K63-ubiquitination as expected (Figure 2B). In addition, there was also no effect on pan- (Figure S5B–C, compare “input” lanes) or K48-linked- (Figure 2D) cellular poly-Ub-pools as a consequence of Ube2V2-specific HNEylation. Two different proteasome inhibitors—bortezomib and MG132—elevated pan-Ub-pools as expected (Figure S5C, “input”; Figure S5D, dotted box in “input” and Figure S7A, “input”).
Unexpectedly, however, upon assessment of Ube2V2 enriched subsequent to T-REX, we discovered that C69-selective-HNEylation is accompanied by Ube2V2-specific-ubiquitination [Figure 2C,E; Figure S5B,C, IP-panels, and S5E]. Importantly, the effect was suppressed in Ube2V2(C69S), indicating that HNEylation of Ube2V2 is a trigger for its selective ubiquitination (Figure 2C,E and Figures S5B,E and S6A). A band (∼150 kDa) was observed in IP samples for both wt and C69S mutant upon T-REX (e.g., Figure 2C, top blot in IP). However, as we show below this has no bearing on downstream signaling as C69S mutant is hypomorphic for all downstream events.
To further substantiate that Ube2V2–C69-specific HNEylation promotes Ube2N activity, we measured the extent of K63-chain elongation in vitro using recombinant proteins. We varied the percentage of HNEylated-Ube2V2 balanced with non-HNEylated-Ube2V2 to keep the overall concentration the same (Figure S3G). Consistent with the observations in cells above, the rate of K63-linked Ub chain formation increased with increasing HNEylated-Ube2V2 present in the assay.
We next ascertained the nature of the Ub-linkages formed selectively on Ube2V2. Bortezomib and MG132 had no effect on the amount of ubiquitinated-Ube2V2 formed upon targeted-HNEylation [Figure S5C–D (treated/untreated conditions within IP-panel and IP-lanes, respectively)]. Thus, the high-molecular weight (HMW)-ubiquitinated Ube2V2 was not primed for proteasomal degradation, and hence unlikely contains K48-linked-Ub—the canonical proteasome-targeting signal. As a corollary, we found very little upregulation in K48-linked-Ub post targeted-HNEylation (Figure 2D). By contrast, K63-linked ubiquitination of Ube2V2 was significantly upregulated upon T-REX, only in cells expressing wt, and not in cells expressing HNE-sensing-defective-C69S mutant (Figure 2C, Figure S6B). This outcome was further verified using affinity capture of K63-linked-Ub using a tandem Ub-binding protein (TUBE) that showed an increase in Ube2V2 only upon T-REX (Figure S6C). Overexpression of HA-Ub(K63R) reduced the amount of Ube2V2(wt) in the polyubiquitin pool (Figure S6D).
Since Ube2N is an established E2-Ub-conjugating-enzyme promoting K63-linked ubiquitination,28,40,43,45,46 we hypothesized that Ube2N was responsible for elevated Ube2V2-ubiquitination. Overexpression of Ube2N had no significant effect on HNEylation-driven Ube2V2-polyubiquitination (Figure 3A). Ube2V2-HNEylation also did not alter Ube2N-levels (Figure 1F–G and Figure S4B–D). This is not unexpected since RNF447/RNF8/16837,48,49—Ub-E3 ligases—are also required for Ube2N-Ub discharge; thus factors other than Ube2N could limit this process, rendering overexpression of Ube2N alone ineffective. However, we consistently found on Ube2N that had been enriched by IP of wt-Ube2V2 from native cells, a minor band of MW equivalent to monoubiquitinated-Ube2N (i.e., Ub-Ube2N). The same band was detected when Ube2N bearing either a T7 (detected using rabbit-secondary-HRP; Figure S6E) or an HA tag (detected using a rat-HRP conjugated primary; Figures S5B and S7A) was used, ruling out nonspecific binding. Intriguingly, this band was selectively lost only when T-REX was carried out in wt-Ube2V2, but not in the HNE-insensitive C69S-hypomorph-expressing cells [Figure 3B–D; Figure S5B]. This putative monoubiquitinated-Ube2N band was removed upon addition of hydroxylamine to the loading buffer (Figure 3D, “IP”),50 and depleted by proteasome-inhibitor treatment (conditions known to deplete labile/exchangeable Ub-pool) (Figures S5C and S7A).51 These results are characteristic of a nonamide linkage between Ube2N and Ub, likely the active Ub-thioester bond that serves as a Ub-donor to downstream targets during the catalytic cycle. In addition, no HMW-ubiquitination of Ube2N was observed under both native and SDS-/sonication-induced denatured conditions (Figures S5C and S7A).
Figure 3.

Ube2V2(C69)-specific HNEylation functionally impacts the monoubiquitinated state of Ube2N. (A) HEK293T cells ectopically expressing wt-Halo-(FLAG)-Ube2V2 and HA-Ubiquitin were cotransfected with either empty vector (EV) or a plasmid of the same backbone expressing HA-Ube2N and analyzed for HMW band of HaloUbe2V2 (i.e., Ube2V2-polyUbiquitin) by indicated antibodies. (B) HEK293T cells transfected with the indicated plasmids were subjected to T-REX conditions against controls followed by immunoprecipitation using FLAG resin (Input: top panel; IP: panel on right). Eluates and inputs were analyzed by western blot using indicated antibodies. Levels of Ube2N/Ube2N-monoUb bound to Ube2V2 were analyzed by western blot. See Figure 3C for quantitation. (C) Quantitation of the relative amount of mono-Ub-Ube2N bound to Halo-Ube2V2 enriched from cells subjected to T-REX against controls. See representative blots, for instance, in Figure 3B,D and Figures S5B and S6E (mean ± s.d., n = 3 independent sets of biological replicates at different passages). (D) HEK293T cells transfected with the indicated plasmids were subjected to T-REX conditions against indicated controls. Levels of Ube2N/Ube2N-monoUb bound to Halo-(FLAG)-Ube2V2 were analyzed by western blot subsequent to enrichment using anti-FLAG-beads (IP: right panel). Half of precipitated fractions were treated with NH2OH (conditions known to hydrolyze thioester bonds; detailed in SI methods) and analyzed separately. All samples were exposed to light in this experiment.
Loss of the active intermediate, mono-Ub-Ube2N, coupled with the upregulation in Ube2V2-ubiquitination, is consistent with HNEylation of Ube2V2 stimulating Ub-release from Ube2N. It further suggests that Ube2V2 is likely a target of Ube2N itself. To evaluate this hypothesis, we prepared cell lines expressing different shRNAs targeting Ube2N. Two of these shRNAs gave >50% reduction in Ube2N levels (#16, #17) relative to both wt-lines and lines expressing nontargeted shRNAs, and a third shRNA (#18) gave weaker knockdown (Figure S7B–C). Ube2N-knockdown lines did not show perturbation in poly-Ub-pools [Figure S7B, Anti-Ub (endogenous) blot]. Lines with higher knockdown-efficiencies (#16, #17) showed significantly reduced HMW-ubiquitination of Ube2V2 following T-REX-assisted HNEylation (Figure 4A, Figure S7D–E). Line #18 showed weak suppression of polyubiquitination, consistent with Ube2N-dose-dependent regulation of Ube2V2-polyubiquitination (Figure 4A–B). These data—in conjunction with Ube2V2(C69S) hypomorphism—establish that Ube2V2(C69)-specific electrophilic modification stimulates Ube2N-enzymatic activity.
Figure 4.

HNEylation of Ube2V2 upregulates γ-H2AX and decreases DNA synthesis: these phenotypes depend on both C69 and Ube2N. (A) Ube2N knockdown lines #16, 17, 18, and control-knockdown line were transfected with Halo-(FLAG)-Ube2V2 and HA-Ubiquitin, then subjected to T-REX against no-“Ht-PreHNE”-controls. HA-Ubiquitin was immunoprecipitated, and inputs (top panels) and elutions (lower panels) were analyzed by western blot using indicated antibodies. See Figure 4B for quantitation [n = 3 independent biological replicates at different passages except shUbe2N-#17 (n = 1)]. (B) Quantitation of data from A. (C) SILAC workflow used to identify proteins that bind preferentially to HNEylated-Ube2V2/Ube2N complex (upper left) and graphical depiction of hits (bottom right). (D) HEK293T cells coexpressing Myc-MCM6 and either wt-Halo-(FLAG)-Ube2V2 or C69S-Halo-(FLAG)-Ube2V2 were subjected to T-REX conditions against “no Ht-PreHNE”-controls. 3-h-Post light exposure, cells were lysed and analyzed by western blot using the indicated antibodies. (E) Similar experiment to D, except cells were transfected with Halo-(Flag)-Ube2V2 (wt- or C69S mutant) and HA-Ubiquitin, and lysates were analyzed for endogenous PCNA-ubquitination using sandwich ELISA [binding: anti-HA(Ub); detection: anti-PCNA (endogenous) antibody] as detailed in SI methods. [mean ± s.d., two independent replicates were performed. N = 3 for each set of cells transfected with either wt- or C69S-Halo-(FLAG)-Ube2V2, under individual experimental conditions as indicated. (F) HEK293T cells were transfected with Halo-(FLAG)-Ube2V2 (or the C69S mutant), then exposed to T-REX conditions. The EdU/BrdU-dual-pulse DNA-labeling (detailed in SI methods) was measured and levels of second pulse (BrdU) from EdU-positive-only cells were quantified and displayed. [mean ± s.e.m., for wt-Halo-(FLAG)-Ube2V2, n = 339 (T-REX), n = 375 (Light alone), n = 300 (Ht-PreHNE alone), n = 465 (DMSO), n = 280 (mitomycin C), for C69S- Halo-(FLAG)-Ube2V2, n = 266 (T-REX), n = 212 (Light alone), n = 283 (Ht-PreHNE alone), n = 312 (DMSO), n = 305 (mitomycin C)]. See Figure S8B for representative images for data in panel F. (G) HEK293T cells were transfected with Halo-(FLAG)-Ube2V2 (or the C69S mutant), then exposed to T-REX conditions. Levels of γ-H2AX were assessed by immunofluorescence (detailed in SI methods) as a function of time post light exposure. [mean ± s.e.m., for wt-Halo-(FLAG)-Ube2V2, n = 648 (0 h), n = 624 (1 h), n = 634 (3 h), n = 571 (6 h), n = 542 (18 h); for C69S- Halo-(FLAG)-Ube2V2, n = 585 (0 h), n = 615 (1 h), n = 644 (3 h), n = 649 (6 h), n = 646 (18 h)]. (H) Ube2N knockdown lines #16 (shUbe2N-16) and control-knockdown line (shLacZ-D11) were transfected with wt-Halo-(FLAG)-Ube2V2, then exposed to T-REX conditions. Levels of γ-H2AX were assessed by immunofluorescence at the similar peak hour (3 h) as in F [mean ± s.e.m., for shUbe2N-#16, n = 379 (T-REX), n = 297 (Light alone), n = 342 (Ht-PreHNE alone), n = 402 (DMSO), n = 241 (Mitomycin C); for shLacZ-D11, n = 434 (T-REX), n = 390 (Light alone), n = 483 (Ht-PreHNE alone), n = 434 (DMSO), n = 445 (Mitomycin C)].
Ube2V2-(C69)HNEylation-Controlled Ube2N Stimulation Drives DDR Signaling
We further tracked the physiologic ramifications of this HNEylation event by mapping, at the proteome-scale, perturbations in Ube2V2-interactome in response to Ube2V2-specific electrophilic modification using SILAC–T-REX. In other words, we were motivated to profile altered associations as a result of Ube2V2-specific HNEylation. Targets identified in this experimental setup consist of Ube2V2-HNEylation-dependent associating partner(s) of Ube2V2/Ube2N or other protein(s) strongly associated with Ube2V2. SILAC—in contrast to standard pulldown-proteomics—was employed to eliminate false-positives and bias toward abundant targets, and avoid missing low-affinity interactions.52 Briefly, T-REX was executed independently in cells cultured in heavy or light arginine/lysine, expressing either HaloTagged-wt-Ube2V2 (heavy) or -(C69S) mutant (light). A 1:1 mixture of these cells was lysed, IP-ed for Ube2V2, and the heavy/light ratio was analyzed following trypsin-digest and LC-MS/MS (Figure 4C, Table S3). Ub was significantly enriched in the heavy (wt) fraction, consistent with enhanced Ube2V2-polyubiquitination upon HNEylation. The heavy fraction was also enriched in Ube2N-binding proteins known to be involved in DDR: p5353 and H2A.54−56 We also found MCM657—a protein previously unknown to be regulated by Ube2V2. Altogether these data indicate that HNEylation of Ube2V2 promotes Ube2V2/Ube2N to bind client proteins with higher affinity, offering an elegant explanation for the observed loss of Ube2N-monoUb accompanying Ube2V2(C69)-specific HNEylation (Figure 3B–C; Figures S5C, S6E, and S7A), and consequent increased ubiquitination of downstream targets.
Our hypothesis that “Ube2V2-specific HNEylation stimulates Ube2V2-dependent ubiquitination” would predict increased ubiquitination of proteins enriched in the SILAC–T-REX. We co-overexpressed MCM6 with either wt-Halo-Ube2V2 or the HNE-sensing-defective C69S mutant. Upregulated ubiquitination of MCM6 upon T-REX was only observed in cells expressing wt-Ube2V2 (Figure 4D). Thus, Ube2V2-specific HNEylation promotes ubiquitination of MCM6, validating that T-REX coupled with SILAC can identify novel regulatory intersections.
We also detected by ELISA increased ubiquitination of endogenous PCNA (Figure 4E), a known downstream target of Ube2N58 and a low confidence hit in our SILAC data. Indeed, K63-linked Ube2N-dependent PCNA-ubiquitination is required for fork stall during genotoxic stress.59 We thus hypothesized that the elevation of ubiquitination of PCNA and other DDR proteins, such as MCM6, may bypass DNA damage control and cause fork stalling. We used a dual-pulse assay, which involves sequential, timed pulsing with two orthogonal DNA-labeling agents (EdU, followed by BrdU) that can be detected orthogonally by fluorescence imaging (Figure S8A). This assay is very accurate because DNA-synthesis is measured over a defined time range, allowing a good estimate of synthesis-rate to be determined. In cells expressing wt-Ube2V2, the extent of DNA-synthesis stall upon T-REX was similar to that observed under mitomycin-C treatment, whereas corresponding cells expressing Ube2V2(C69S) exhibited a normal DNA-synthesis rate post T-REX (Figure 4F and Figure S8A–B). This DNA-synthesis stall did not occur in Ube2N-depleted cells (Figure S8C).
Slowing of the replication fork has been shown to upregulate γ-H2AX formation at the point of replication.59,60 We thus proceeded to assay for changes in γ-H2AX following T-REX-directed Ube2V2(C69)-HNEylation. Indeed, an approximately 2-fold increase in cellular γ-H2AX was observed following Ube2V2(C69)-targeted-HNEylation (Figure 4G; Figure S8D–F). γ-H2AX upregulation was transient: increased γ-H2AX could be measured up to 18-h post light-induced HNE targeting, but γ-H2AX reduced to basal after 24 h (Figure S8E). This behavior is consistent with a signaling/preconditioning response. Mitomycin-C upregulated γ-H2AX-upregulation by 3–4-fold (Figure 4H; Figure S8A,C),61,62 validating the assay and showing that the effect of HNEylation is biologically—as well as statistically—significant. γ-H2AX-upregulation required both Ube2V2(C69) and Ube2N because (1) no significant γ-H2AX upregulation was measured in cells expressing Ube2V2(C69S) mutant at any point over the time course (18-h) post T-REX (Figures 4G and S8E); and (2) notably, wt-Ube2V2-specific HNEylation was amorphic for γ-H2AX-upregulation in Ube2N-depleted background (Figure 4H). However, both backgrounds hypomorphic for T-REX-specific γ-H2AX-upregulation [i.e., Ube2V2(C69S) mutant-expressing cells and cells deficient of Ube2N] still upregulated γ-H2AX upon mitomycin-C treatment (Figure 4G–H; Figure S8D–F).
DDR Preconditioning Occurs in Zebrafish Embryos
The role of γ-H2AX in DNA-damage checkpoint remains enigmatic, although evidence exists that H2AX is required to initiate several DNA-damage checkpoints. In light of the reported increase in ubiquitination of other proteins during DNA damage (such as PCNA), we postulated that HNEylation, specifically of Ube2V2, and subsequent increase in ubiquitination of downstream targets of Ube2N, like PCNA, retards the replication fork and ultimately promotes DDR-like responses. Although preconditioning is a common mechanism whereby RES-signals elicit beneficial cytoprotective responses to defend against or adapt to cellular stress, triggering of DDR is not one of the pathways known to function through this mechanism.
We were thus compelled to draw functional relevance of this intriguing signal exchange mechanism to a whole-vertebrate system. We chose zebrafish (Danio rerio).63 Our phylogenetic analysis indicated that zebrafish Ube2V2 possess a cysteine analogous to C69 in humans (Figure S2C,D). We first demonstrated that ectopic human Ube2V2 senses bioactive electrophiles such as HNE in zebrafish. We expressed human HaloTagged-Ube2V2 in zebrafish by injecting embryos at the one-cell stage with in vitro transcribed mRNA, incubated embryos with Ht-PreHNE (1 μM) for 1 day in the dark, then washed away excess probe, exposed the live specimens to light (5 min, 365 nm at 0.3 mW/cm2). A portion of these samples was subjected to biotin-mediated enrichment of in situ HNEylated proteins. The remaining portion was incubated for 3 h post light exposure. Subsequently the fish were fixed and γ-H2AX levels were measured using whole-mount immunofluorescence.
Ube2V2 was significantly HNEylated following T-REX in zebrafish (Figure 5A). Targeted HNEylation led to detectable levels of HMW HNEylated-Ube2V2 that was polyubiquitinated (Figure S9A). As in cell culture, global ubiquitin pools were unchanged (Figures 2D, 3A, and 4A). Both the above results are consistent with our sequence alignments that showed conservation of the sensor cysteine (C69) from humans to fish (Figure S2C,D).
Figure 5.

Ube2V2-Specific HNEylation regulates γ-H2AX levels in zebrafish. (A) Casper zebrafish embryos were injected with mRNA-encoding Halo-(FLAG)-Ube2V2 at the 1–4 cell stage. Once injection was complete, eggs were either exposed to Ht-PreHNE or DMSO. After 24-h-incubation, fish were washed and exposed to light. After dechorionation and deyolking at 4 °C, embryos were lysed, biotin was attached using Click chemistry by biotin-azide, and lysates were ethanol precipitated. After resolubilization, biotinylated [i.e., HNE(alkyne)-modified] proteins were pulled down using streptavidin and analyzed by western blot. (See Figure S1A for workflow; Clicking with biotin-azide.) (B) Similar experiment to A, but at 3-h postlight-exposure, fish were fixed, permeabilized, and analyzed by whole-mount immunofluorescence using indicated antibodies. Scale bars, 100 μm. (C) Quantitation of images in B [mean ± s.e.m., n = 69 (T-REX), n = 74 (Light alone), n = 64 (Ht-PreHNE alone), n = 69 (DMSO)].
Furthermore, T-REX-treated fish selectively showed upregulation in γ-H2AX (Figure 5B–C). On the other hand, treatment of embryos with mitomycin-C led to a high level of γ-H2AX-expression relative to untreated fish, but these fish were severely deformed or died during treatment (Figure 5B–C; Figure S9B). Although latencies and other confounding factors make the comparison between mitomycin-C treatment and T-REX indirect, this result at least suggests that RES-targeting of Ube2V2 could constitute a noninvasive method to prime DDR, without eliciting severe genotoxic stress.
Conclusion
In sum, we have devised a regimen to identify functional electrophile-sensor proteins and surgically interrogate their specific responses in the backdrop of an otherwise largely unperturbed cell or animal. Previous studies to model electrophilic signaling subsystems have made use of global HNE flooding. Here we take a different track: we configure a “low-occupancy modification” setting, in which a brief “pulse” of HNE is delivered in situ, such that only first responders (regardless of expression level) are tagged. This approach offers a much-needed user-controlled protocol to tag, and (when coupled to T-REX) road-test functional cysteines. Importantly, G-REX uses direct affinity capture of modified cysteines, as opposed to current strategies that detect loss of labeling. Thus, G-REX minimizes false positives associated with off-target reactivity. The G-REX regimen could readily be applied to target-ID of a specific electrophile; to identify new RES-sensors for drug discovery; or extended to identification of off-target proteins hit by electrophilic drugs and linking these specific events to phenotypes, such as drug-induced hepatotoxicity.
We validated G-REX by investigating the in vitro and in cell-specific HNEylation effects of our highest confidence novel hits. In vitro, Ube2V2 showed highly efficient conjugating efficiency to HNE and stimulated activity of the cognate E2, Ube2N. In cells, remarkably, mono-HNEylation of either privileged sensor was sufficient to elicit hyper-stimulation of Ube2V1- or Ube2V2-specific downstream signaling. The outcomes from the chemically and kinetically competent Ube2V2-point mutant that cannot sense HNE (in vitro or cells) strikingly support this functional sensing and signal propagation. These data support an HNE-shunt mechanism, whereby flux through the ubiquitin signaling node, Ube2N, is elevated by HNEylation of its allosteric binding-partner, Ube2V2.
We propose this new signaling currency exchange to be a functional pathway that operates as a surveillance and maintenance mechanism for DDR64 in response to a transient rise in cellular electrophile flux. These data indicate that redox signaling—HNEylation of one regulatory protein (at a site with no “expected” reactivity)—can affect ubiquitin signaling via a third-party enzyme containing a catalytic cysteine (Ube2N). Given that defects in DDR65 are a common source of heritable disease, it is likely that exquisite regulation by small-molecule electrophiles on specific pathways could be mined for drug discovery and biomedical benefits.
Acknowledgments
Professors Todd Evans (Weill Cornell Medicine) and Joseph Fetcho (Cornell University) for early guidance and zebrafish techniques transfer; Nikki Gilbert and Brian Miller from the Fetcho laboratory for assistance with fish husbandry and breeding; Dr. Xuyu Liu for assistance with cell-based protein stability assays; Alexandra Van Hall-Beauvais for providing recombinant purified His6-Halo protein; Professor Brian Crane’s laboratory for the use of CD spectrometer; zebrafish husbandry and microinjection/imaging facility [NIH R01 NS026593, J. Fetcho (Cornell University)], Cornell NMR facility [NSF MRI: CHE-1531632, Y.A.], and Cornell Imaging Center [NIH 1S10RR025502, R. M. Williams (Cornell University)] for instrumentation. Instrumentation, personnel, and supplies are fully or partly supported by NIH New Innovator (1DP2GM114850), Beckman Young Investigator, NSF CAREER (CHE-1351400), Office of Naval Research (ONR) Young Investigator (specific to work involving zebrafish), and Sloan Fellowship programs (FG-2016-6379) (to Y.A.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00556.
Author Contributions
Y.Z. performed all the cell-based and in vitro biochemical assays. M.J.C.L. performed zebrafish microinjection and M.J.C.L. and Y.Z. collaborated on imaging and biochemical assays involving zebrafish. Y.W. assisted Y.Z. in small-molecule synthesis. S.Z. assisted in LC-MS/MS data analysis. Y.A., Y.Z., and M.J.C.L. analyzed the data. Y.A. wrote the paper with assistance from M.J.C.L. and Y.Z.
The authors declare the following competing financial interest(s): Provisional patent for the G-REX method in filing stages (by Cornell University).
Supplementary Material
References
- Pearlman S. M.; Serber Z.; Ferrell J. E. Jr. A mechanism for the evolution of phosphorylation sites. Cell 2011, 147 (4), 934–46. 10.1016/j.cell.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R.; Rape M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- Wilkinson K. D.; Ventii K. H.; Friedrich K. L.; Mullally J. E. The ubiquitin signal: assembly, recognition and termination. Symposium on ubiquitin and signaling. EMBO Rep. 2005, 6 (9), 815–820. 10.1038/sj.embor.7400506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrant M. K.; Cole P. A. The chemical biology of protein phosphorylation. Annu. Rev. Biochem. 2009, 78, 797–825. 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love K. R.; Catic A.; Schlieker C.; Ploegh H. L. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat. Chem. Biol. 2007, 3 (11), 697–705. 10.1038/nchembio.2007.43. [DOI] [PubMed] [Google Scholar]
- Salami J.; Crews C. M. Waste disposal—An attractive strategy for cancer therapy. Science 2017, 355 (6330), 1163–1167. 10.1126/science.aam7340. [DOI] [PubMed] [Google Scholar]
- Hoeller D.; Dikic I. Targeting the ubiquitin system in cancer therapy. Nature 2009, 458, 438–444. 10.1038/nature07960. [DOI] [PubMed] [Google Scholar]
- Schopfer F. J.; Cipollina C.; Freeman B. A. Formation and signaling actions of electrophilic lipids. Chem. Rev. 2011, 111 (10), 5997–6021. 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Systems Analysis of Protein Modification and Cellular Responses Induced by Electrophile Stress. Acc. Chem. Res. 2010, 43 (5), 673–683. 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J.; Aye Y. The Die Is Cast: Precision Electrophilic Modifications Contribute to Cellular Decision Making. Chem. Res. Toxicol. 2016, 29 (10), 1575–1582. 10.1021/acs.chemrestox.6b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer T. F.; Garcia F. J.; Onak C. S.; Carroll K. S.; Chang C. J. Chemical approaches to discovery and study of sources and targets of hydrogen peroxide redox signaling through NADPH oxidase proteins. Annu. Rev. Biochem. 2015, 84, 765–790. 10.1146/annurev-biochem-060614-034018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Poganik J. R.; Ghosh S.; Aye Y. Subcellular Redox Targeting: Bridging in Vitro and in Vivo Chemical Biology. ACS Chem. Biol. 2017, 12 (3), 586–600. 10.1021/acschembio.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-G.; Baek K.; Soetandyo N.; Ye Y. Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat. Commun. 2013, 4, 1568. 10.1038/ncomms2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullally J. E.; Moos P. J.; Edes K.; Fitzpatrick F. A. Cyclopentenone prostaglandins of the J series inhibit the ubiquitin isopeptidase activity of the proteasome pathway. J. Biol. Chem. 2001, 276 (32), 30366–30373. 10.1074/jbc.M102198200. [DOI] [PubMed] [Google Scholar]
- Okada K.; Wangpoengtrakul C.; Osawa T.; Toyokuni S.; Tanaka K.; Uchida K. 4-hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress - Identification of proteasomes as target molecules. J. Biol. Chem. 1999, 274 (34), 23787–23793. 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- Keum Y. S.Regulation of the Keap1/Nrf2 system by chemopreventive sulforaphane: implications of posttranslational modifications. In Nutrition and Physical Activity in Aging, Obesity, and Cancer; Surh Y. J.; Song Y. S.; Han J. Y.; Jun T. W.; Na H. K., Eds.; Blackwell Science Publishing: Oxford, 2011; Vol. 1229, pp 184–189. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Rask-Andersen M.; Almen M. S.; Schioth H. B. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery 2011, 10 (8), 579–590. 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- Blewett M. M.; Xie J.; Zaro B. W.; Backus K. M.; Altman A.; Teijaro J. R.; Cravatt B. F. Chemical proteomic map of dimethyl fumarate–sensitive cysteines in primary human T cells. Sci. Signaling 2016, 9 (445), rs10. 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Parvez S.; Zhao Y.; Surya S. L.; Wang Y.; Zhang S.; Aye Y. Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat. Chem. Biol. 2017, 13, 333–338. 10.1038/nchembio.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J.; Aye Y. Privileged electrophile sensors: a resource for covalent drug development. Cell Chemical Biology 2017, 24 (7), 787–800. 10.1016/j.chembiol.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews C. M. Targeting the undruggable proteome: the small molecules of my dreams. Chem. Biol. 2010, 17 (6), 551–555. 10.1016/j.chembiol.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S. M.; Gladyshev V. N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287 (7), 4419–4425. 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Carroll K. S.; Liebler D. C. The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteomics 2016, 15 (1), 1–11. 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Weerapana E.; Blewett M. M.; Cravatt B. F. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 2014, 11 (1), 79–85. 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Long M. J. C.; Lin H.-Y.; Zhao Y.; Haegele J. A.; Pham V. N.; Lee D. K.; Aye Y. T-REX on-demand redox targeting in live cells. Nat. Protoc. 2016, 11, 2328–2356. 10.1038/nprot.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Fu Y.; Li J. Y.; Long M. J. C.; Lin H. Y.; Lee D. K.; Hu G. S.; Aye Y. Substoichiometric Hydroxynonenylation of a Single Protein Recapitulates Whole-Cell-Stimulated Antioxidant Response. J. Am. Chem. Soc. 2015, 137 (1), 10–13. 10.1021/ja5084249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann R. M.; Pickart C. M. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 1999, 96 (5), 645–653. 10.1016/S0092-8674(00)80575-9. [DOI] [PubMed] [Google Scholar]
- Deng L.; Wang C.; Spencer E.; Yang L.; Braun A.; You J.; Slaughter C.; Pickart C.; Chen Z. J. Activation of the NF-kB Kinase Complex by TRAF6 Requires a Dimeric Ubiquitin-Conjugating Enzyme Complex and a Unique Polyubiquitin Chain. Cell 2000, 103 (2), 351–361. 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Michelle C.; Vourc’h P.; Mignon L.; Andres C. R. What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor?. J. Mol. Evol. 2009, 68, 616–628. 10.1007/s00239-009-9225-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluise C. D.; Rose K.; Boiani M.; Reyzer M. L.; Manna J. D.; Tallman K.; Porter N. A.; Marnett L. J. Peptidyl-prolyl cis/trans-Isomerase A1 (Pin1) Is a Target for Modification by Lipid Electrophiles. Chem. Res. Toxicol. 2013, 26, 270–279. 10.1021/tx300449g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacko B. K.; Wall S. B.; Kramer P. A.; Ravi S.; Mitchell T.; Johnson M. S.; Wilson L.; Barnes S.; Landar A.; Darley-Usmar V. M. Pleiotropic effects of 4-hydroxynonenal on oxidative burst and phagocytosis in neutrophils. Redox Biol. 2016, 9, 57–66. 10.1016/j.redox.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano J. i.; Nagata M.; Okamura S.; Shiraya T.; Mitsui T. Identification of Oxidatively Modified Proteins in Salt-Stressed Arabidopsis: A Carbonyl-Targeted Proteomics Approach. Plant Cell Physiol. 2014, 55, 1233–1244. 10.1093/pcp/pcu072. [DOI] [PubMed] [Google Scholar]
- Chavez J.; Chung W.-G.; Miranda C. L.; Singhal M.; Stevens J. F.; Maier C. S. Site-Specific Protein Adducts of 4-Hydroxy-2(E)-Nonenal in Human THP-1 Monocytic Cells: Protein Carbonylation Is Diminished by Ascorbic Acid. Chem. Res. Toxicol. 2010, 23, 37–47. 10.1021/tx9002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann R. M.; Pickart C. M. In Vitro Assembly and Recognition of Lys-63 Polyubiquitin Chains. J. Biol. Chem. 2001, 276, 27936–27943. 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- Iwai K. Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol. 2012, 22, 355–364. 10.1016/j.tcb.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Mailand N.; Bekker-Jensen S.; Faustrup H.; Melander F.; Bartek J.; Lukas C.; Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Doil C.; Mailand N.; Bekker-Jensen S.; Menard P.; Larsen D. H.; Pepperkok R.; Ellenberg J.; Panier S.; Durocher D.; Bartek J.; Lukas J.; Lukas C. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- Silva G. M.; Finley D.; Vogel C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct. Mol. Biol. 2015, 22, 116–123. 10.1038/nsmb.2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge C. D.; Edwards R. A.; Markin C. J.; McDonald D.; Pulvino M.; Huen M. S. Y.; Zhao J.; Spyracopoulos L.; Hendzel M. J.; Glover J. N. M. Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem. Biol. 2015, 10, 1718–1728. 10.1021/acschembio.5b00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H. Y.; Haegele J. A.; Disare M. T.; Lin Q. S.; Aye Y. A Generalizable Platform for Interrogating Target- and Signal-Specific Consequences of Electrophilic Modifications in Redox-Dependent Cell Signaling. J. Am. Chem. Soc. 2015, 137 (19), 6232–6244. 10.1021/ja5132648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich O.; Huser H.; Ehrlich W.; Grune T. Intracellular metabolism of 4-hydroxynonenal in primary cultures of rabbit synovial fibroblasts. Free Radical Biol. Med. 1997, 22 (7), 1153–1157. 10.1016/S0891-5849(96)00496-0. [DOI] [PubMed] [Google Scholar]
- Andersen P. L.; Zhou H.; Pastushok L.; Moraes T.; McKenna S.; Ziola B.; Ellison M. J.; Dixit V. M.; Xiao W. Distinct regulation of Ubc13 functions by the two ubiquitin-conjugating enzyme variants Mms2 and Uev1A. J. Cell Biol. 2005, 170, 745–755. 10.1083/jcb.200502113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun M.; Kramer H. B.; Willems L. I.; McDermott J. L.; Leach C. A.; Goldenberg S. J.; Kumar K. G.; Konietzny R.; Fischer R.; Kogan E.; Mackeen M. M.; McGouran J.; Khoronenkova S. V.; Parsons J. L.; Dianov G. L.; Nicholson B.; Kessler B. M. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011, 18 (11), 1401–1412. 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Xia Z.-P.; Sun L.; Chen X.; Pineda G.; Jiang X.; Adhikari A.; Zeng W.; Chen Z. J. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009, 461, 114–119. 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broomfield S.; Chow B. L.; Xiao W. MMS2, encoding a ubiquitin-conjugating-enzyme-like protein, is a member of the yeast error-free postreplication repair pathway. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 5678–5683. 10.1073/pnas.95.10.5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branigan E.; Plechanovová A.; Jaffray E. G.; Naismith J. H.; Hay R. T. Structural basis for the RING-catalyzed synthesis of K63-linked ubiquitin chains. Nat. Struct. Mol. Biol. 2015, 22, 597–602. 10.1038/nsmb.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S. J.; Edwards R. A.; Leung C. C. Y.; Neculai D.; Hodge C. D.; Dhe-Paganon S.; Glover J. N. M. Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J. Biol. Chem. 2012, 287, 23900–23910. 10.1074/jbc.M112.359653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge C. D.; Ismail I. H.; Edwards R. A.; Hura G. L.; Xiao A. T.; Tainer J. A.; Hendzel M. J.; Glover J. N. M. RNF8 E3 Ubiquitin Ligase Stimulates Ubc13 E2 Conjugating Activity That Is Essential for DNA Double Strand Break Signaling and BRCA1 Tumor Suppressor Recruitment. J. Biol. Chem. 2016, 291 (18), 9396–9410. 10.1074/jbc.M116.715698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart C. M.; Rose I. A. Mechanism of ubiquitin carboxyl-terminal hydrolase. Borohydride and hydroxylamine inactivate in the presence of ubiquitin. J. Biol. Chem. 1986, 261 (22), 210–217. [PubMed] [Google Scholar]
- Xu Q.; Farah M.; Webster J. M.; Wojcikiewicz R. J. H. Bortezomib rapidly suppresses ubiquitin thiolesterification to ubiquitin-conjugating enzymes and inhibits ubiquitination of histones and type I inositol 1,4,5-trisphosphate receptor. Mol. Cancer Ther. 2004, 3 (10), 1263–1269. [PubMed] [Google Scholar]
- Mann M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7 (12), 952–958. 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- Topisirovic I.; Gutierrez G. J.; Chen M.; Appella E.; Borden K. L.; Ronai Z. A. Control of p53 multimerization by Ubc13 is JNK-regulated. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (31), 12676–12681. 10.1073/pnas.0900596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T.; Tashiro S.; Kakino A.; Shima H.; Jacob N.; Amunugama R.; Yoder K.; Izumi S.; Kuraoka I.; Tanaka K.; Kimura H.; Ikura M.; Nishikubo S.; Ito T.; Muto A.; Miyagawa K.; Takeda S.; Fishel R.; Igarashi K.; Kamiya K. DNA Damage-Dependent Acetylation and Ubiquitination of H2AX Enhances Chromatin Dynamics. Mol. Cell. Biol. 2007, 27, 7028–7040. 10.1128/MCB.00579-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F.; Sixma T. K. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 2014, 21 (4), 308–316. 10.1038/nsmb.2792. [DOI] [PubMed] [Google Scholar]
- Wang B.; Elledge S. J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (52), 20759–20763. 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell S. P.; Dutta A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002, 71, 333–374. 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- Hoege C.; Pfander B.; Moldovan G. L.; Pyrowolakis G.; Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419 (6903), 135–141. 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- Vujanovic M.; Krietsch J.; Raso M. C.; Terraneo N.; Zellweger R.; Schmid J. A.; Taglialatela A.; Huang J.-W.; Holland C. L.; Zwicky K.; et al. Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol. Cell 2017, 67 (5), 882–890. e5 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagou M. E.; Zuazua-Villar P.; Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Molecular biology of the cell 2010, 21 (5), 739–752. 10.1091/mbc.E09-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.; Voss K. O.; Tobias F.; Jakob B.; Durante M.; Taucher-Scholz G. Clustered DNA damage induces pan-nuclear H2AX phosphorylation mediated by ATM and DNA-PK. Nucleic Acids Res. 2013, 41 (12), 6109–6118. 10.1093/nar/gkt304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T.; Walker S. A.; Cerosaletti K.; Goodarzi A. A.; Petermann E.; Concannon P.; O’Driscoll M.; Jeggo P. A. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25 (24), 5775–5782. 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae C. A.; Peterson R. T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discovery 2015, 14 (10), 721–731. 10.1038/nrd4627. [DOI] [PubMed] [Google Scholar]
- Yi C.; He C. DNA repair by reversal of DNA damage. Cold Spring Harbor Perspect. Biol. 2013, 5 (1), a012575. 10.1101/cshperspect.a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M. Diseases Associated with Defective Responses to DNA Damage. Cold Spring Harbor Perspect. Biol. 2012, 4 (12), a012773. 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.