

Abstract
Background
Abdominal aortic aneurysm (AAA) is a deadly cardiovascular disease characterised by the gradual, irreversible dilation of the abdominal aorta. AAA is a complex genetic disease but little is known about the role of epigenetics. Our objective was to determine if global DNA methylation and CpG-specific methylation at known AAA risk loci is associated with AAA, and the functional effects of methylation changes.
Results
We assessed global methylation in peripheral blood mononuclear cell DNA from 92 individuals with AAA and 93 controls using enzyme-linked immunosorbent assays, identifying hyper-methylation in those with large AAA and a positive linear association with AAA diameter (P < 0.0001, R2 = 0.3175).
We then determined CpG methylation status of regulatory regions in genes located at AAA risk loci identified in genome-wide association studies, using bisulphite next-generation sequencing (NGS) in vascular smooth muscle cells (VSMCs) taken from aortic tissues of 44 individuals (24 AAAs and 20 controls). In IL6R, 2 CpGs were hyper-methylated (P = 0.0145); in ERG, 13 CpGs were hyper-methylated (P = 0.0005); in SERPINB9, 6 CpGs were hypo-methylated (P = 0.0037) and 1 CpG was hyper-methylated (P = 0.0098); and in SMYD2, 4 CpGs were hypo-methylated (P = 0.0012).
RT-qPCR was performed for each differentially methylated gene on mRNA from the same VSMCs and compared with methylation. This analysis revealed downregulation of SMYD2 and SERPINB9 in AAA, and a direct linear relationship between SMYD2 promoter methylation and SMYD2 expression (P = 0.038). Furthermore, downregulation of SMYD2 at the site of aneurysm in the aortic wall was further corroborated in 6 of the same samples used for methylation and gene expression analysis with immunohistochemistry.
Conclusions
This study is the first to assess DNA methylation in VSMCs from individuals with AAA using NGS, and provides further evidence there is an epigenetic basis to AAA. Our study shows that methylation status of the SMYD2 promoter may be linked with decreased SMYD2 expression in disease pathobiology. In support of our work, downregulated SMYD2 has previously been associated with adverse cardiovascular physiology and inflammation, which are both hallmarks of AAA. The identification of such adverse epigenetic modifications could potentially contribute towards the development of epigenetic treatment strategies in the future.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0460-9) contains supplementary material, which is available to authorized users.
Keywords: Epigenetics, Vascular disease, Aneurysm, Inflammation
Background
Abdominal aortic aneurysm (AAA) is a degenerative cardiovascular disease and a global health concern. AAA is responsible for between 2 and 4% of deaths in white males over the age of 65 [1–3]. Progressive aneurysm growth may lead to rupture, characterised by internal aortic haemorrhage, which results in death in approximately 80% of cases. Surgical repair is the only proven therapeutic option in order to prevent or treat AAA rupture. The exact etiological basis of AAA is still not fully understood, making it difficult to develop a successful pharmaco-therapeutic strategy [4]. However, it is well known that AAA is multifactorial with known risk factors that contribute towards aneurysm development (smoking, male sex, increased age, white European ancestry, atherosclerosis and hyperlipidaemia) [5, 6]. AAA also demonstrates strong heritability [7, 8] and is polygenic. To date, 10 genomic risk loci have been identified through the conduct of genome-wide association studies (GWASs) [9–16].
Epigenetics refers to modifications of the genome that are not exclusively a result of change in the primary DNA sequence and includes DNA methylation, histone modifications and non-coding RNAs [6, 17]. These mechanisms are essential for cell, tissue and organ development and directly interact with DNA sequence/structure to manipulate gene expression at the transcriptional and post-transcriptional levels. However, aberrant epigenetic modifications have been implicated in many complex diseases [18–20]. DNA methylation is the most extensively studied epigenetic modification to DNA and involves the addition of a methyl group to a cytosine base 5′ to a guanine (CpG dinucleotide) by DNA methyltransferase enzymes, which can inhibit gene transcription; however, increased methylation has also been associated with increased expression, since methylation can be necessary for transcriptional binding [21–23]. DNA methylation patterns are long-term inherited signatures that are passed through generations and can be affected by both genetic and environmental factors [24, 25].
There is currently very limited investigation surrounding the role of DNA methylation in AAA, and the only published study to date was conducted by Ryer et al. [26], who performed genome-wide methylation analysis in peripheral blood DNA from 20 AAA patients (11 smokers and 9 non-smokers) and 21 control samples (10 smokers and 11 non-smokers) using Illumina 450k micro-arrays. They identified differentially methylated regions in ADCY10P1, CNN2, KLHL35 and SERPINB9, and differential expression in SERPINB9 and CNN2.
Variations in DNA sequence at polymorphic loci can result in variable patterns of DNA methylation, known as methylation quantitative trait loci (meQTL) [27–29]. Disease variants can alter transcription factor levels and methylation of their binding sites [30]. The direct mechanisms of the variants associated with AAA are not fully known, but many are intronic and it is likely that their effects are regulatory in nature as opposed to being directly functional. It is therefore feasible that disease-specific meQTLs exist for AAA, and considering many meQTL act in cis [28], their likely effects on methylation are in the genes surrounding the risk loci identified in previous GWASs.
In this study, targeted bisulphite next-generation sequencing (NGS) was performed in vascular smooth muscle cell (VSMC) DNA, isolated from whole aortic tissues of AAA and controls, to investigate the methylation status of CpG islands in regulatory regions of genes located near AAA genomic risk loci. This approach offers a more refined way to assess the methylation status of genes in proximity to AAA risk loci than other methodologies such as the 450k microarray. The identification of adverse epigenetic modifications directly linked to patients with AAA could offer a more comprehensive understanding of AAA pathobiology, and an alternative research avenue in the search for a future treatment strategy [31, 32].
Results
Global DNA methylation and homocysteine analysis in AAA patients and controls
Global genomic DNA methylation was assessed in peripheral blood DNA of 185 individuals, and circulating homocysteine (HCY) was assessed in blood plasma from 137 of the same individuals using ELISAs. Global DNA methylation was significantly higher in men with large AAA (> 55 mm, n = 48, global DNA methylation 1.86% (± 0.6%)) compared to men with small AAA (30–55 mm, n = 45, global DNA methylation 0.93% (± 0.52)) and controls (< 25 mm, n = 92, global DNA methylation 0.79% (± 0.43%)) (Fig. 1a). There was a linear relationship between AAA size and global DNA methylation which was adjusted for patient age (Fig. 1b) (Pearson coefficient R2 value = 0.3175, P < 0.0001). Circulating blood plasma HCY levels were slightly higher in men with AAA compared to controls (9.6 μmol/L ± 0.62, n = 70, vs 7.94 μmol/L ± 0.52, n = 67, P = 0.0433) (Additional file 1: Figure S1a). However, there was no association between global methylation and circulating HCY (P = 0.095) (Additional file 1: Figure S1b).
Fig. 1.
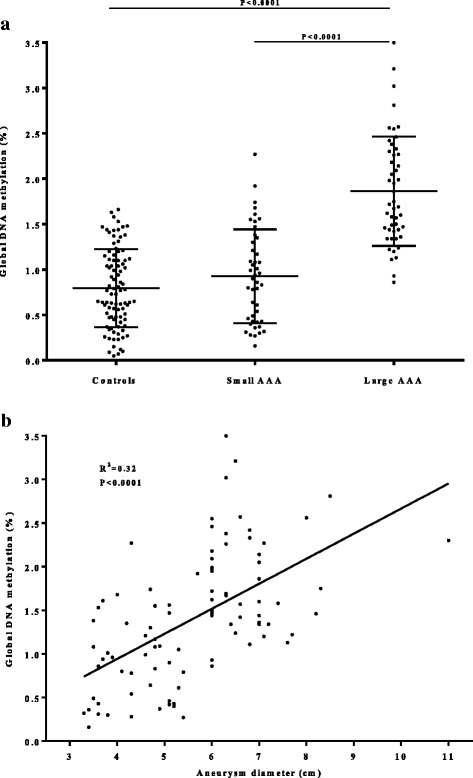
a Global DNA methylation levels of peripheral blood DNA in controls (aortic diameter < 25 mm, n = 92), small aneurysms (aneurysm diameter 30–55 mm, n = 45) and large aneurysms (aneurysm diameter > 55 mm, n = 48). b The linear relationship between AAA size and DNA methylation (n = 93)
Bisulphite NGS of vascular smooth muscle cell DNA in AAA patients and controls
We isolated CpG islands within regulatory regions of genes associated with AAA (LRP1, ERG, MMP9, LDLR, IL6R, SORT1, SERPINB9, SMYD2 and DAB2IP—Table 1) in DNA extracted from the VSMCs of 20 controls and 24 large AAAs (Additional file 1: Table S2). Using targeted bisulphite NGS, we determined the methylation status of these regions and subsequently compared AAA to controls. We identified significant differences in methylation between AAA and controls in four genes: ERG, IL6R, SERPINB9 and SMYD2 (Fig. 2 and Table 2). There were no significant differences in methylation status between AAA and controls in DAB2IP, LDLR, LRP1, MMP9, SORT1 and an alternative promoter in ERG (Additional file 1: Figure S2). A consistently high mapped sequencing depth was observed for each gene, and there were no significant differences in sequencing coverage between AAA and controls in any gene. Mean mapped sequencing coverage for AAA and controls for each gene is shown in Additional file 1: Figure S3.
Table 1.
Candidate genes and amplicons for VSMC bisulphite NGS: Genomic locations, primer sequences, annealing temperatures and CpG coverage are displayed for each amplicon targeting candidate genes
| Gene symbol | Chromosome and RefSeq ID | RefSeq ID coordinates | Primer sequence 5′-3’ | Annealing temp (°C) | CpG coverage |
|---|---|---|---|---|---|
| LRP1 | 3875–4240 | F: TAGAAGGGGGTAGTGATTAAAAGTA | 54 | 13 | |
| R: AAAACAAACCCTAATTTAAAAAAAA | |||||
| Chr: 12 | 4274–4640 | F: GGGTATTAAGGTGGGTTTTATTTT | 57 | 25 | |
| NG_016444.1 | R: TACTCTAAAATTTCAAACTCCCTCC | ||||
| 4680–5036 | F: GTATTAGGGAGGAGGGTTTAGTTAG | 54 | 24 | ||
| R: TCCTCAATACATAAACCTAAAACTC | |||||
| ERG | 282787–283319 | F:TTTTATTAGGTAGTGGTTAGATTTAGTTTT | 54 | 22 | |
| Chr: 21 | R: TACCCCCAATTAATAAATTCCAATATA | ||||
| NG_029732.1 | 283331–283680 | F: TTGGAATTTATTAATTGGGGGTATATAT | 57 | 6 | |
| R: ACACTATCTTTTACAAAATCAATCCAC | |||||
| 5162–5540 | F: AAAATTTTTGGAAGGGGTTTAGTT | 56 | 37 | ||
| R: ATAATATTTTTCCAACCTCATTAAAAAC | |||||
| MMP-9 | 3980–4579 | F: TTGGGTTTAAGTAATTTTTTTATTT | 52 | 7 | |
| Chr: 20 | R: TAACCCATCCTTAACCTTTTACAAC | ||||
| NG_011468.1 | 4640–5600 | F: GATGGGGGATTTTTTTAGTTTTATT | 57 | 8 | |
| R: TACCCATTTCTAACCATCACTACTC | |||||
| LDLR | 4376–4725 | F: TTTTTTAAGGGGAGAAATTAATATTTA | 54 | 5 | |
| R: CACAAAAAAATAACAACAACCTTTC | |||||
| 4776–5355 | F: GAAAGGTTGTTGTTATTTTTTTGTG | 57 | 28 | ||
| Chr: 19 | R: AAACTCCCTCTCAACCTATTCTAAC | ||||
| NG_009060.1 | 5464–5972 | F: TTTAAGTTTTTATAGGGTGAGGGAT | 56 | 31 | |
| R: ACACCCAACTCAAAATAACAATAAC | |||||
| 6054–6785 | F: AATTTTATTGGGTGTAGTTTAATAGGTTAT | 54 | 49 | ||
| R: CTCAAAATCATACACTAACCAACCTC | |||||
| IL6R | 2781–3061 | F: TTTTTGTTTAGGTTGGAGTGTAGTG | 52 | 8 | |
| Chr: 1 | R: CAAAATTTTAATATTATAATTCACATAAAA | ||||
| NG_012087.1 | 3095–3729 | F: GTTTGTTTTGGTGTAGAGATAGGTG | 58 | 7 | |
| R: ATAAAACTCCCAAATAAACAAAACC | |||||
| SORT1 | Chr: 1 | 4363–4804 | F: AGATTATTTTTTAGGTTTGTAGGAGTTA | 55 | 24 |
| NG_028280.1 | R: AACAAAAACTACTAAAATCAACCCC | ||||
| SMYD2 | 214280141–214280630 | F: TTTTATTTTGAAGTAGTGGTTTTTGTTAA | 55 | 13 | |
| R: TTTTCCAAAATTAAAAATTTTTAAACCT | |||||
| Chr: 1 | 214280631–214281330 | F: AAGGTTTAAAAATTTTTAATTTTGGAAA | 58 | 82 | |
| NC_000001.11 | R: AAATCCCCCACCTAAAAAAACTAC | ||||
| 214281331–214281733 | F: TTTTTTTAGGTGGGGGATTT | 55 | 46 | ||
| R: CCCACATTTAAAAACAAAAACCTAC | |||||
| SERPINB9 | 2891544–2892079 | F: GGTGTTGAAAATATTTTTGGAGGTA | 57 | 26 | |
| R: AAAATCTACCATTCATCAAACTAACAAA | |||||
| 2890564–2891263 | F: TTAGAGGGTGGGATTAGAGGTAGTT | 54 | 9 | ||
| Chr: 6 | R: TTTCCACCTAAAAAACCAAAATTAA | ||||
| NC_000006.12 | 2889934–2890423 | F: GTATTTGGGAATTGTTGATGTTTTT | 55 | 11 | |
| R: ACAACAAAAAATCATTATAATCTATTTTCT | |||||
| 2889425–2889933 | F: AGAATTTTATATGTAATTTATTTTGG | 54 | 31 | ||
| R: TATAATCCCAACACTTTAAAAAACC | |||||
| DAB2IP | 121689899–121690502 | F: TTGGAGTAGTTTGTTTGTTGTGTT | 57 | 10 | |
| R: TAAAAAATTAAATACCCAAAACCTC | |||||
| 121690573–121691062 | F: TTGTTATATTTTAAGTTGAGATTTTGGG | 57 | 6 | ||
| Chr: 9 | R: TAACCACATAAAAACATTCCAAACA | ||||
| NC_000009.12 | 121767430–121767872 | F: GGAGAGGAGATAGGAAAGTTTTTAG | 57 | 6 | |
| R: CTAACCTTATCCCTTACAACCACTAC | |||||
| 121768273–121768692 | F: AATGGAGTGGGAGTTTGTTATAGTG | 57 | 11 | ||
| R: TAATAACAACTTTTAACCCCACCTC |
Fig. 2.
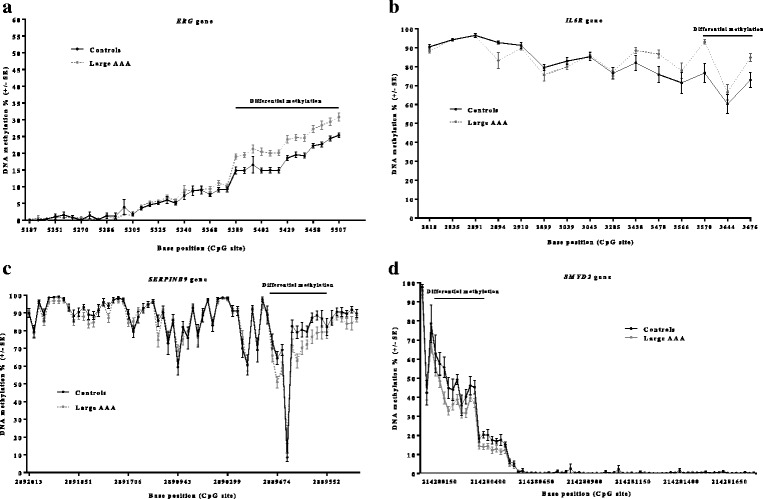
Vascular smooth muscle cell DNA methylation status of bisulphite-sequenced CpG islands where significant differential methylation was observed in 20 controls vs 24 AAAs (ERG (a), IL6R (b), SERPINB9 (c) and SMYD2 (d)). See Tables 2 and 3 for descriptive statistics of differentially methylated CpGs
Table 2.
Mean observed CpG site methylation in vascular smooth muscle cell DNA after bisulphite sequencing of candidate genes (total n = 24 AAAs and 20 controls—Additional file 1: Table S2)
| Gene, chromosome and RefSeq ID | RefSeq ID coordinate | Overall mean meth % | Overall mean meth % | Overall meth difference | P value | Q value | Age-adjusted |
|---|---|---|---|---|---|---|---|
| (CpG site) | ± SD (AAA n = 24) | ± SD (controls n = 20) | (% ± SE) | odds ratio | |||
| ERG | 5389 | 19.00 (3.48) | 14.83 (4.42) | 4.17 (1.23) | 0.0019 | 0.0037 | NS |
| Chr: 21 | 5393 | 19.43 (3.72) | 14.83 (3.618) | 4.60 (1.16) | 0.0004 | 0.0010 | NS |
| NG_029732.1 | 5400 | 21.26 (5.76) | 16.50 (10.57) | 4.76 (2.58) | 0.0002 | 0.0010 | 6.70 |
| 5402 | 20.43 (5.45) | 14.83 (3.50) | 5.60 (1.48) | 0.0001 | 0.0009 | NS | |
| 5405 | 19.96 (4.15) | 14.89 (3.43) | 5.07 (1.21) | 0.0001 | 0.0008 | 6.18 | |
| 5408 | 20.13 (4.20) | 14.89 (3.51) | 5.24 (1.23) | 0.0001 | 0.0008 | 6.19 | |
| 5429 | 24.13 (5.26) | 18.56 ((3.22) | 5.58 (1.41) | 0.0003 | 0.0010 | NS | |
| 5440 | 24.65 (4.49) | 19.50 (3.40) | 5.15 (1.28) | 0.0001 | 0.0008 | NS | |
| 5442 | 24.57 (5.12) | 19.28 (3.25) | 5.29 (1.38) | 0.0002 | 0.0009 | NS | |
| 5458 | 27.22 (4.88) | 22.22 (3.15) | 5.00 (1.33) | 0.0003 | 0.0010 | NS | |
| 5468 | 28.35 (6.01) | 22.67 (3.16) | 5.68 (1.56) | 0.0015 | 0.0032 | NS | |
| 5474 | 29.39 (5.55) | 24.39 (3.48) | 5.00 (1.50) | 0.0012 | 0.0027 | NS | |
| 5507 | 30.83 (5.57) | 25.33 (3.09) | 5.49 (1.46) | 0.0004 | 0.0010 | NS | |
| IL6R Chr: 1 | 3570 | 93.17 (5.62) | 76.70 (23.02) | 16.47 (4.85) | 0.0113 | 0.0743 | 3.11 |
| NG_012087.1 | 3676 | 84.92 (10.89) | 73.00 (17.49) | 11.92 (4.31) | 0.0176 | 0.0773 | NS |
| SERPINB9 | 2889674 | 51.00 (16.19) | 64.47 (16.19) | −13.47 (4.97) | 0.0063 | 0.0375 | NS |
| Chr: 6 | 2889644 | 20.46 (20.29) | 8.579 (10.64) | 11.88 (5.14) | 0.0098 | 0.0478 | 3.29 |
| NC_000006.12 | 2889627 | 71.67 (18.37) | 82.53 (14.94) | −10.86 (5.20) | 0.0058 | 0.0375 | NS |
| 2889620 | 62.88 (19.54) | 79.11 (15.15) | −16.23 (5.47) | 0.0017 | 0.0273 | NS | |
| 2889612 | 70.33 (19.08) | 80.37 (17.96) | −10.04 (5.71) | 0.0064 | 0.0375 | NS | |
| 2889568 | 76.58 (19.21) | 87.21 (11.26) | −10.63 (4.97) | 0.0012 | 0.0273 | 0.33 | |
| 2889564 | 78.67 16.34) | 88.58 (10.65) | −9.91 (4.35) | 0.0010 | 0.0273 | NS | |
| SMYD2 | 214280412 | 39.50 (14.74) | 55.67 (16.24) | −16.17 (6.00) | 0.0004 | 0.0200 | NS |
| Chr: 1 | 214280441 | 32.91 (10.53) | 44.80 (17.29) | −11.89 (4.87) | 0.0014 | 0.0299 | 0.09 |
| NC_000001.11 | 214280507 | 38.78 (12.67) | 49.22 (7.93) | −10.44 (4.56) | 0.0016 | 0.0299 | 0.06 |
| 214280600 | 37.04 (11.02) | 45.30 (11.43) | −8.26 (4.22) | 0.0014 | 0.0299 | 0.03 |
NS not significant
The greatest differences in methylation were seen in the ERG gene, where 13 CpGs were consecutively hyper-methylated (Fig. 2a) in AAAs compared to controls. The mean methylation of these sites was 18.67% ± 3.92 in controls versus 23.8% ± 4.11 in cases with a mean increase in methylation of 5.13% ± 0.43 (P = 0.0005, Q = 0.0014). The 13 CpGs in ERG span a region of 118 bp (NG_029732.1:g.5389_5507|gom) which is located within an intronic region directly downstream from the first ERG exon.
We observed a more conservative site of potential differential methylation in the IL6R gene promoter upstream of the transcriptional start site, where we identified significant hyper-methylation of 2 CpGs (NG_012087.1:g.3570_3676|gom) in AAA (89% ± 5.83) vs controls (74.85% ± 2.62) (Fig. 2b). The mean increase in methylation was 14.2% ± 3.22 (P = 0.0145, Q = 0.076).
In the SERPINB9 gene, seven CpGs located in the 3′ untranslated region downstream of exon 7 were found differentially methylated in AAA compared to controls. Overall, hypo-methylation of the region was observed (NC_000006.12:g.2889674_2889564|lom) where six CpGs were hypo-methylated (P = 0.0037, Q = 0.032) and one CpG was hyper-methylated (P = 0.0098, Q = 0.0478) (NC_000006.12:g.2889644|gom) (Fig. 2c). This is in contrast to the overall AAA hyper-methylation reported by Ryer et al., [26], which was conducted on peripheral blood DNA.
Finally in the SMYD2 gene, we identified four significantly hypo-methylated CpG sites in AAA compared to controls (Fig. 2d) in the gene promoter upstream of the transcriptional start site (NC_000001.11:g 214280412_214280600|lom). Mean control methylation was 48.75% ± 5.02, mean methylation in the cases was 37.06% ± 2.95, and the mean overall decrease in methylation in AAA compared to controls was 11.69% ± 8.2 (P = 0.0012, Q = 0.027). Figure 3a displays a more detailed, CpG-specific illustration of the differentially methylated sites in the SMYD2 gene promoter.
Fig. 3.
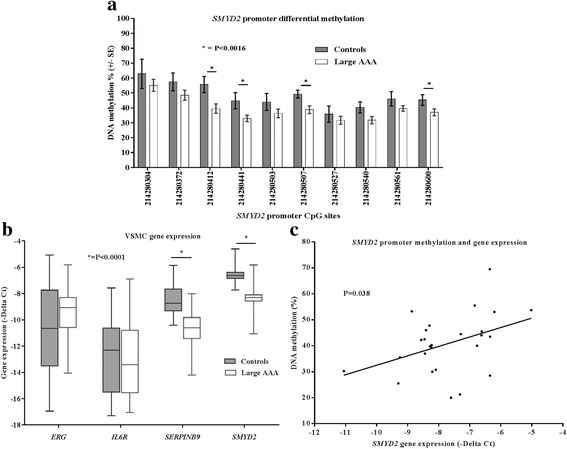
a Differentially methylated CpGs in the SMYD2 gene promoter identified after bisulphite sequencing of DNA from 24 AAAs and 20 controls. b Relative gene expression levels of differentially methylated candidate genes (SMYD2, SERPINB9, IL6R and ERG) in vascular smooth muscle cells from 20 controls and 24 AAAs. c Linear relationship between gene expression and mean DNA methylation status of the differentially methylated CpGs (NC_000001.11: 214280412, 214280441, 214280507 and 214280600) in SMYD2 (n = 26 where sufficient sequencing coverage and expression data was acquired)
Logistic regression was conducted on statistically significant differentially methylated CpGs in each gene to assess the potential effects of age (controls (56 years ±35) and AAA (68 years ±25)) on disease status. Some sites lost significance when applying age as an additional variable for differential methylation; however, there still remained CpGs in each differentially methylated gene that were significant independently of age. In addition, the effects of smoking and sex were assessed in our data by excluding all non-smokers and females respectively (Table 3).
Table 3.
Mean observed CpG site methylation in vascular smooth muscle cell DNA after bisulphite sequencing of candidate genes after consideration of sex and smoking status as co-variates of methylation (total n = 24 AAAs, 16 male controls and 16 smoking controls—Additional file 1: Table S2)
| Gene, chromosome and RefSeq ID | RefSeq ID coordinate | Meth % ± SD—all male smokers | Smokers only meth % ± SD | Smokers only meth difference (% ± SE) | Men only meth % ± SD | Men only meth difference (% ± SE) | Smokers only P value | Men only P value |
|---|---|---|---|---|---|---|---|---|
| (CpG site) | (AAA—n = 24) | (Controls—n = 16) | n = 16 | (Controls—n = 16) | n = 16 | (24 AAAs vs 16 controls) | (24 AAAs vs 16 controls) | |
| ERG | 5389 | 19.00 (3.48) | 14.43 (3.99) | 4.57 (1.25) | 15.07 (4.65) | 3.93 (1.32) | 0.0011 | 0.0059 |
| Chr: 21 | 5393 | 19.43 (3.72) | 14.86 (3.65) | 4.58 (1.25) | 14.80 (3.82) | 4.63 (1.25) | 0.0013 | 0.0009 |
| NG_029732.1 | 5400 | 21.26 (5.76) | 17.36 (11.73) | 3.90 (2.88) | 16.87 (11.58) | 4.39 (2.82) | 0.0011 | 0.0007 |
| 5402 | 20.43 (5.45) | 15.14 (3.55) | 5.29 (1.64) | 14.80 (3.73) | 5.63 (1.61) | 0.0009 | 0.0004 | |
| 5405 | 19.96 (4.15) | 15.50 (3.54) | 4.46 (1.33) | 14.93 (3.65) | 5.02 (1.32) | 0.0014 | 0.0003 | |
| 5408 | 20.13 (4.20) | 15.36 (3.69) | 4.77 (1.36) | 14.87 (11.57) | 5.26 (1.33) | 0.0007 | 0.0002 | |
| 5429 | 24.13 (5.26) | 19.21 (2.83) | 4.92 (1.53) | 18.73 (3.26) | 5.40 (1.52) | 0.0028 | 0.0010 | |
| 5440 | 24.65 (4.49) | 20.07 (2.92) | 4.58 (1.35) | 19.60 (3.48) | 5.05 (1.37) | 0.0010 | 0.0003 | |
| 5442 | 24.57 (5.12) | 19.86 (2.51) | 4.71 (1.47) | 19.40 (3.36) | 5.17 (1.50) | 0.0009 | 0.0006 | |
| 5458 | 27.22 (4.88) | 22.93 (2.49) | 4.29 (1.41) | 22.40 (3.31) | 4.82 (1.44) | 0.0030 | 0.0010 | |
| 5468 | 28.35 (6.01) | 23.50 (2.34) | 4.85 (1.68) | 22.80 (3.39) | 5.55 (1.71) | 0.0127 | 0.0045 | |
| 5474 | 29.39 (5.55) | 25.36 (2.56) | 4.03 (1.58) | 24.53 (3.70) | 4.86 (1.63) | 0.0125 | 0.0037 | |
| 5507 | 30.83 (5.57) | 26.21 (1.85) | 4.61 (1.54) | 25.20 (3.36) | 5.63 (1.60) | 0.0040 | 0.0008 | |
| IL6R Chr: 1 | 3570 | 93.17 (5.62) | 78.31 (22.41) | 14.85 (4.76) | 75.37 (23.77) | 17.79 (5.02) | NS | 0.0037 |
| NG_012087.1 | 3676 | 84.92 (10.89) | 75.56 (16.55) | 9.35 (4.33) | 73.63 (16.97) | 11.29 (4.39) | NS | NS |
| SERPINB9 | 2889674 | 51.00 (16.19) | 66.27 (15.98) | −15.27 (5.30) | 64.07 (16.84) | −13.07 (5.41) | 0.0037 | 0.0120 |
| Chr: 6 | 2889644 | 20.46 (20.29) | 8.13 (9.93) | 12.33 (5.63) | 9.27 (11.59) | 11.19 (5.76) | 0.0139 | 0.0230 |
| NC_000006.12 | 2889627 | 71.67 (18.37) | 84.93 (12.93) | −13.27 (5.44) | 82.13 (15.91) | −10.47 (5.76) | 0.0015 | 0.0100 |
| 2889620 | 62.88 (19.54) | 80.00 (15.17) | −17.13 (5.95) | 81.00 (14.06) | −18.13 (5.84) | 0.0018 | 0.0007 | |
| 2889612 | 70.33 (19.08) | 80.27 (20.23) | −9.933 (6.43) | 78.93 (19.58) | −8.60 (6.34) | 0.0081 | 0.0168 | |
| 2889568 | 76.58 (19.21) | 88.33 (12.19) | −11.75 (5.56) | 87.67 (9.31) | −11.08 (5.33) | 0.0003 | 0.0018 | |
| 2889564 | 78.67 16.34) | 87.80 (11.82) | −9.13 (4.88) | 90.47 (8.57) | −11.80 (4.59) | 0.0039 | 0.0002 | |
| SMYD2 | 214280412 | 39.50 (14.74) | 59.00 (17.12) | −19.50 (6.64) | 55.38 (17.33) | −15.88 (6.37) | 0.0002 | 0.0015 |
| Chr: 1 | 214280441 | 32.91 (10.53) | 48.57 (17.80) | −15.66 (5.37) | 41.56 (14.78) | −8.64 (4.64) | 0.0004 | NS |
| NC_000001.11 | 214280507 | 38.78 (12.67) | 50.14 (8.61) | −11.36 (5.14) | 50.13 (7.97) | −11.34 (4.81) | 0.0012 | 0.0009 |
| 214280600 | 37.04 (11.02) | 47.57 (10.63) | −10.53 (4.72) | 45.78 (12.02) | −8.73 (4.44) | 0.0001 | 0.0020 |
NS not significant
Functional corroboration of differential methylation in AAA patients and controls
Gene expression analysis of differentially methylated genes
In genes where differentially methylated CpGs were identified between AAA and controls (ERG, IL6R, SERPINB9 and SMYD2), we performed mRNA expression analysis to assess the relationship between DNA methylation and gene expression. We identified a significant overall reduction in the expression of SMYD2 (P < 0.0001) and SERPINB9 mRNA (P < 0.0001) in people with AAA compared to controls. However, there were no differences in expression in the IL6R and ERG mRNA (Fig. 3b). Subsequently, the methylation values of significantly associated CpG regions were correlated with expression levels in their respective genes using linear regression. In the SERPINB9 gene, where significant expression and methylation differences were observed, neither the hypo- nor hyper-methylated CpG sites were associated with SERPINB9 expression levels (P = 0.243 and P = 0.31 respectively). Finally, the mean DNA methylation percentage of differentially methylated CpGs (214280412, 214280441, 214280507 and 214280600) in the SMYD2 gene promoter was significantly associated with SMYD2 gene expression (P = 0.0383, R2 = 0.17, (Fig. 3c). However SMYD2 gene expression did not correlate with aortic diameter.
Immunohistochemistry to assess Smyd2 in aortic tissues
To further corroborate the SMYD2 methylation/expression relationship and to ensure that the differential gene activity seen between AAA and controls was applicable to the site of aneurysm in the aortic wall, histological staining of the Smyd2 protein was conducted in whole aortic tissues from six of the same samples used in methylation and gene expression analysis (three AAAs and three controls).
For each of the six samples, three individual stains were conducted (total = 18 slides) on frozen tissue cuts from the same aortic sections. The first was for Smyd2 staining, the second was a negative Smyd2 control with no primary antibody, and the last was for smooth muscle actin (SMA) staining. The controls were checked for the presence of Smyd2, and all were absent of brown colouring, which represents the presence of primary antibody. The SMA slides were then visualised, and where the densest section of smooth muscle fibres was seen, the Smyd2 slides were visualised in the same regions, where photographs were taken (Fig. 4).
Fig. 4.

Immuno-histochemical staining: staining of aneurysmal (n = 3) and non-aneurysmal (n = 3) abdominal aortic frozen tissue sections for smooth muscle actin (SMA) and Smyd2. Brown = presence of primary specific antibody
The histological technique used for this assay is hard to quantify and is not as objective as other methodologies. However, Fig. 4 subjectively shows a reduced abundance of the Smyd2 protein (brown colouring) in the tissues of those with AAA compared to controls. Whilst a statistical analysis is not available, it appears that the same pattern of downregulated Smyd2 activity (from the mRNA gene expression analysis) is evident in AAA compared to controls. This demonstrates that transcript expression differences are also reflected from a translational perspective at the site of aneurysm, which is in turn associated with SMYD2 promoter methylation.
Discussion
Peripheral blood global DNA hyper-methylation is commonly a hallmark of chronic inflammation and has been observed as a potential pathological marker of disease [19, 33–37], which supports our findings. In turn, inflammation is a pathological hallmark of AAA, yet up until now, no work has investigated global methylation in AAA. Our study has identified global DNA hyper-methylation in patients with a large AAA, and a clear linear relationship with AAA diameter in peripheral blood DNA. These results were adjusted for age, and both groups (cases and controls) consisted exclusively of white males with at least a 10-year smoking history. Age, smoking and inflammation, as stated, can have considerable impact upon global and Cp-specific DNA methylation patterns, consisting of global hypo-methylation (ageing), hyper- and hypo-methylation (smoking) and global hyper-methylation (inflammation). Each of these factors is also significant a risk factor for AAA [6].
In addition, circulating HCY has previously been associated with global methylation [38], is a proposed biomarker for cardiovascular disease [39] and has also been suggested to have a role in AAA [40]. However, the differences in HCY between AAA and controls in this study were not clinically different (hyper-homocysteinemia is described above 15 μmol/L) and support the idea that HCY is not an appropriate biomarker for AAA, which is also suggested by Lindqvist et al., [41]. In addition, there was no significant relationship between HCY and global DNA methylation, suggesting that global DNA hyper-methylation is associated with AAA independently of circulating blood HCY. Similarly to other inflammatory diseases, increased global DNA methylation may be a factor in the pathobiology of AAA and provides a solid rationale for further study in to the role of DNA methylation in AAA.
This study is the first to adopt bisulphite NGS, a gold standard technique in studies of DNA methylation [42], to identify differentially methylated CpG sites associated with AAA in VSMCs. VSMCs are the main constituents of the tunica media, where cellular apoptosis occurs as a hallmark of AAA during aneurysm development and growth [43]. Ryer et al. [26] previously conducted a methylation study on AAA in peripheral blood DNA using Illumina 450k micro-arrays and found differential methylation and gene expression in CNN2 and SERPINB9. In VSMCs, we also identified differential methylation in SERPINB9, but the relationship was converse to that seen in DNA isolated from peripheral blood. In our study, after assessing the association between differential methylation and expression in SERPINB9 with linear regression analysis, it was determined that there was not a direct relationship between the two. SERPINB9 could however still be important in AAA, as reduced SERPINB9 expression is associated with atherosclerotic disease progression and is inversely related to the extent of apoptosis within the intima [44].
Most importantly in this study, we identified significant DNA hypo-methylation in the SYMD2 gene promoter in those with AAA (NC_000001.11:g214280412_214280600|lom), and the mean methylation percentage of the differentially methylated CpGs were significantly correlated with gene expression. SMYD2 gene expression however was not correlated with aortic diameter, indicating that downregulated SMYD2 expression, a potential result of SMYD2 promoter methylation, may be associated with the development, and not progression, of AAA. Our analysis is limited by the fact that all VSMC samples in this study were from individuals with AAA greater than 55 mm. The lack of data from those with smaller AAA limits our ability to make conclusions on whether the level of SMYD2 expression is linked to AAA size. Future studies should therefore address this. Further to this analysis, the decrease in expression of SMYD2 in AAA was corroborated in aortic tissues using immunohistochemistry. SMYD2 codes for an important lysine methyltransferase and is well known for its role in transcriptional regulation [45]. Our results are particularly relevant when considering the role of Smyd2 in inflammation and cardio-physiology, as previous studies have identified a role in cardio-protection and suppression of inflammation. In one study, protein levels of Smyd2 were decreased in cardiomyocytes after cellular apoptosis and after myocardial infarction. In addition, SMYD2 deletion in cardiomyocytes in vivo promoted apoptotic cell death upon myocardial infarction [46]. It has also been reported that Smyd2 is a negative regulator for macrophage activation. Elevated SMYD2 expression suppresses the production of pro-inflammatory cytokines, including IL-6 and TNF. In addition, macrophages with high SMYD2 expression promote regulatory T cell differentiation as a result of increased TGF-β production and decreased IL-6 secretion [47]. In summary, previous work has demonstrated that down-regulation of SMYD2 is linked with adverse cardio-physiology and an increase in inflammation, both of which are key hallmarks of AAA pathobiology.
Looking more closely at the relationship between SMYD2 expression and DNA methylation in our study, it is clear that the observed association is opposite to the traditional notion of promoter hyper-methylation inhibiting gene transcription. However, it is becoming better known that this is not definitively correct, as multiple pieces of evidence go against this paradigm [48, 49]. In some cases, methylation is required for activation of transcription and is therefore positively correlated with gene expression [49], which could be appropriate to our findings. Due to this, it must be stated that the exact mechanisms by which methylation controls gene expression is still not definitively understood and is not always the same.
The results presented in this article are preliminary. Future work will concentrate on proving up the SMYD2 relationship with large-scale replication, whilst also establishing whether there is a causal genetic basis to differential methylation patterns in AAA (meQTL analysis). There is some knowledge of meQTLs, but this is still very scarce and currently there is no comprehensive database highlighting phenotype-specific associations between genotype and methylation. It is therefore likely that existing disease-specific meQTLs are unreported, and in our case, genes that are known AAA risk loci represent potentially insightful targets for further genetic and methylation analysis. Finally, after replication, it is suggested that mechanistic analysis to assess the causal role of SMYD2 promoter methylation on gene function would be insightful.
There were limitations to this work, including limited sample size which was hindered due to the difficulty of obtaining aortic tissues from individuals. This problem in turn had an effect on sample group demographics, and we could not control for gender, smoking and age as effectively as we would have liked—as we did in the peripheral blood global methylation assay.
Conclusion
This pilot study is the first to assess DNA methylation in VSMCs of people with AAA, and the first to assess methylation in AAA using bisulphite NGS. The results presented in this article demonstrate further evidence that there is an epigenetic basis to AAA and is proof of concept for further large-scale analysis. Global peripheral blood DNA hyper-methylation is associated with large AAA, increasing AAA size, and this relationship appeared independently of circulating HCY. Gene-specific changes in DNA methylation were also identified in VSMC DNA (IL6R, SMYD2, SERPINB9 and ERG). In particular, there was a significant association between the methylation status of CpGs in the SMYD2 promoter and SMYD2 gene expression, which was further corroborated in whole aortic tissues using immunohistochemistry. In summary, this article demonstrates that global and gene-specific DNA methylation changes exist in AAA and are potentially involved in the pathobiology of the disease and could be useful in the future development of epigenetic therapies.
Methods
Population
Patients and controls were recruited from two sources: The UK aneurysm growth study (UKAGS) (http://www2.le.ac.uk/projects/ukags) and our local AAA research programme based at the NIHR Leicester Cardiovascular Biomedical Research Centre. The UKAGS collects peripheral blood samples from men with AAA (aortic diameter 30 mm or greater) and healthy controls (all screened for AAA) recruited from the English NHS AAA Screening Programme. Furthermore, AAA biopsies (aortic wall) are taken from patients undergoing open AAA repair in our regional vascular unit, where we have also established an aortic tissue collection from cadaveric organ donors. Ethical approval by an NHS Research Ethics Committee was obtained for both studies. Risk factors associated with AAA can be confounders of DNA methylation and include age, smoking, gender and ethnicity. Where possible, to help prevent any confounding effects of these factors, white males over the age of 65 with at least a 10-year history of smoking were selected for inclusion.
Vascular smooth muscle cell culture from aortic biopsies
Aortic tissues were collected by vascular surgeons within the vascular surgery group at the University of Leicester. There were no signs of AAA or atherosclerosis in the abdominal aortas obtained from organ donors, which were therefore used as controls. For each aortic tissue sample collected from those undergoing open AAA surgery (n = 24) and from cadaveric organ donors (n = 20), explant culture was performed to isolate and grow VSMCs in vitro. It was important to isolate individual cells given that independent cell lines have different epigenetic profiles. VSMCs are a good surrogate for such analysis considering aneurysmal formation is characterised by inflammation and VSMC apoptosis in the tunica media of the aortic wall.
Thin sections of the tunica media were carved from the whole aortic tissues no longer than 48 h after surgery (AAA samples), or death (cadaveric organ donor samples), and placed in T80 cell culture flasks containing 10 ml smooth muscle cell Medium 231 (ThermoFisher Scientific - M2315005) with added Smooth Muscle Growth Supplement (ThermoFisher Scientific - S00725). A 25-ml bottle of growth serum was added to each 500 ml 231 media prior to use. The flasks containing the media, growth supplement and aortic tissues were left to incubate at 37 °C until visible primary cell growth was observed (~ 2 weeks). After primary growth of VSMCs, confluent cells were detached from the flask with the addition of 2× trypsin-EDTA solution and incubated at 37 °C for 3 min. Five-millilitre sterile phosphate buffered saline (PBS) and fetal bovine serum solution (20:1 ratio) was added to neutralise the trypsin. Solutions containing detached cells were aspirated from the flasks into sterile universal tubes and centrifuged at 500×g for 6 min at 20 °C. Supernatants were discarded and cell pellets were re-suspended in 3 ml media. One millilitre of the 3 ml suspensions were added to three new T80 cell culture flasks, each containing 10 ml of media. This process was repeated until sufficient amounts of isolated VSMCs (two clearly visible cell pellets) were available for DNA and RNA extraction, however, never beyond passage 3 to attenuate any potential changes in cellular gene expression or DNA methylation patterns because of the artificial media/culture environment. Isolated cultured cells stored by cryogenic freezing at − 80 °C until needed for experimental analysis. In the earliest stages of establishing our explant culture work flows, α-smooth muscle immunofluorescent cell type staining (internal work not published) was conducted to assess for fibroblast contamination in our standard protocol, and this protocol was used in this study.
DNA extraction
DNA was extracted from 185 peripheral blood mononuclear cell (PBMC) samples from the UKAGS for global methylation analysis (Additional file 1: Table S1), and 44 VSMC samples (Additional file 1: Table S2) isolated from whole aortic tissues for bisulphite NGS analysis using the DNeasy Blood & Tissue Kit (Qiagen) and according to the manufacturer’s standard protocol. DNA concentrations were measured using a NanoDrop Spectrophotometer. Each sample was diluted to 25 ng/μl using sterilised distilled water in a 100 μl final volume working solution.
Global methylation analysis in peripheral blood DNA
Genome-wide global DNA methylation was assessed in DNA derived from the 185 PBMC samples from our UKAGS resource (Additional file 1: Table S1) using a colorimetric enzyme linked immunosorbent-assay (ELISA) following the manufacturers’ protocol (Epigentek - Methyl flash DNA quantification (colorimetric)). All reactions were performed in duplicate, and for each new 96-well plate, new standardisation controls were conducted. Mixed plate repeats were also performed to assess for potential batch effects. Absorbance (optical density (OD)) was measured with a micro-plate colorimetric spectrophotometer (Bio-Tek ELX808IU Ultra Microplate Reader), and ODs were converted to genome methylation percentage with the use of positive (5 ng of 50% methylated DNA) and negative control (20 ng of un-methylated DNA) reactions, which were also conducted in duplicate.
Homocysteine analysis in circulating blood plasma
A total of 137 of the blood plasma samples from the global methylation assay were available (67 controls and 70 AAAs) for homocysteine (HCY) analysis (Additional file 1: Table S3). Colorimetric ELISAs were performed following the manufacturers’ protocol (Cell Biolabs Inc. – Homocysteine ELISA kit – STA-670). All reactions were conducted in duplicate with new controls and mixed plate repeats for each new 96-well ELISA plate. ODs were measured with a micro-plate colorimetric spectrophotometer and HCY levels were determined using the standard curve method.
Bisulphite conversion of DNA and targeted Illumina NGS
Each of the 44 VSMC DNA samples isolated from aortic biopsies was bisulphite-converted using the MOD50 kit (Sigma–Aldrich) according to the manufacturer’s standard protocol. Eluted bisulphite-treated DNA was concentrated at 25 ng/μl in sterilised distilled water.
Genes for this study were chosen based on the results from the recent AAA GWAS meta-analysis conducted by Jones et al., [15], and included the following: low-density lipoprotein receptor-related protein 1 (LRP1), ETS-related gene (ERG), matrix metallopeptidase 9 (MMP9), low-density lipoprotein receptor (LDLR), interleukin 6 receptor (IL6R), sortilin 1, (SORT1), SET and MYND domain containing 2 (SMYD2) and DAB2-interacting protein (DAB2IP). In addition, SERPINB9 was included, which was differentially methylated in PBMCs from patients with AAA in the Ryer et al. study previously described. We also tried to target the differentially methylated CpG Island in the CNN2 gene from the Ryer et al. study but bisulphite-specific PCR primers could not be designed due to the repetitive nature of the target sequence.
Within all candidate genes (with the exception of the SERPINB9 CpG Island which is located in the gene body), promoters and transcriptional start sites were identified using the transcriptional regulatory element promoter database (http://rulai.cshl.edu/TRED) and the NCBI gene database (https://www.ncbi.nlm.nih.gov/gene). All candidate gene sequences (GRCh38) were compiled in a single file for use as an in-house reference sequence for bioinformatics analysis.
Bisulphite-specific PCR primers were designed to isolate genomic regions of interest within candidate genes using methprimer [50] (Table 1). Each 20 μl bisulphite-specific PCR reaction consisted of 8 μl sterilised distilled water, 10 μl 2× jumpstart red-taq polymerase ready mix (SIGMA), 1 μl mixed forward and reverse primers (5 μM, SIGMA) and 1 μl bisulphite converted DNA (25 ng/μl). Each bisulphite-specific primer pair was optimised via PCR:
94 °C for 2 min—1 cycle
94 °C for 15 s—40 cycles
52 °C - 58 °C for 35 s—40 cycles
72 °C for 35 s—40 cycles
72 °C for 5 min—1 cycle
Amplicons were checked by fractionation using agarose gel electrophoresis (10 μl per well, 2% agarose gel).
All bisulphite-specific PCR reactions (total n = 1146) were cleaned with ExoSAP-IT PCR Clean-up (Affymetrix). On ice, 5 μl of the PCR reactions were mixed with 2 μl ExoSAP-IT solution and incubated at 37 °C for 15 min, then at 80 °C for 15 min. The cleaned products were pooled together (2 μl of each reaction) to create a single pooling of each separate DNA sample (44 individual samples containing 26 amplified genomic regions spanning 9 genes).
The NEBNext Ultra™ DNA Library Prep Kit for Illumina (E7370) and NEBNext Mutiplex Oligos for Illumina dual index kit (E7600) were used for end repair and adaptor ligation and quantification (bar coding for multiplex sequencing) using the standard manufacturer protocols. Clean-up of adaptor-ligated DNA was performed using Agencourt AMPure XP - PCR Purification (A63880) following the manufacturer’s standard protocol.
Each of the adaptor ligated, bar-coded DNA samples was ran on an Agilent 2100 Bioanalyzer Instrument (expert High-Sensitivity DNA chip) to assess sample DNA concentration and the presence and distribution of correct fragment sizes [51]. These readings were used to pool the individual samples prior to sequencing at 4 nM. Indexed libraries were pooled, and paired-end multiplexed sequencing (2 × 310 bp) was performed on an Illumina Miseq platform using MiSeq Reagent Kit v3. Sequencing was conducted in the NUCLEUS Genomics Centre, University of Leicester.
Bioinformatics workflow and data analysis
A provisional data quality filter (Phred quality score (Q) = 15) was applied to fastq files with Trimmomatic [52] to remove all low-quality raw sequencing reads, remove contaminating Illumina adaptor sequences, and to clip and discard partially poor-quality sequencing reads four bases at a time using the sliding window function.
BWA-meth (https://github.com/brentp/bwa-meth) was used to align the provisionally filtered sequencing reads to the in house reference sequence. BWA-meth is specifically designed for targeted bisulphite NGS analysis.
PCR duplicates for all BAM files were marked and excluded with the Picard utility MarkDuplicates (http://broadinstitute.github.io/picard/>), and the final processed files were sorted and indexed with Samtools [53].
Finally, for each sample, the methylation values (normalised as methylation %) of each sequenced CpG in each individual were extracted to a bedGraph file at a threshold of Q50 (read alignment filter) and Q20 (base quality filter) with a minimum read depth of 5 using PileOMeth (https://github.com/dpryan79/PileOMeth). Sequencing data that did not meet these criteria were excluded from the study.
The data extracted using PileOMeth was taken forward for statistical analysis using IBM SPSS Statistics 24 and GraphPad prism 7. To correct for non-normal distribution, each methylation value at CpG sites in each individual were ranked with rank-based inverse normal transformations (Blom normal scores in SPSS). Unpaired multiple t tests were conducted on the ranked normal scores at each sequenced CpG site between cases vs controls for each gene separately. For multiple comparison testing, the false discovery rate approach was adopted. Discovery was determined using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 10%. CpG sites with significant P values and Q values were then manually checked to ensure only sites with a noticeably visual difference in methylation between cases, and controls were included to investigate further. The effects of smoking and sex were assessed in our original data by excluding all non-smokers and females respectively, where we then reported P values. Logistic regression was performed using SPSS at each significant CpG site to adjust for age differences between cases vs controls. Age-adjusted odds ratios were reported for each CpG unless significance was lost.
RNA extraction, genomic DNA removal and cDNA synthesis
VSMCs from the same samples as used in the NGS assay (n = 44) were used for gene expression analysis. RNA isolation was performed following the manufacturer’s protocol (QIAGEN RNeasy Mini Kit). Total RNA was eluted in 30 μl RNase-free water and taken forward for contaminating genomic DNA digestion using the manufacturer’s standard protocol (DNase I - RNase-Free: New England BioLabs M0303). CDNA was then synthesised from the DNase digested RNA using the ThermoFisher High-Capacity cDNA Reverse Transcription Kit (4368814) according to the manufacturer’s protocol. CDNA was eluted in a final volume of 40 μl and stored at − 20 °C until further use.
TaqMan gene expression analysis
For each gene where differential methylation was observed after VSMC bisulphite sequencing (SMYD2, IL6R, SERPINB9 and ERG), pre-designed TaqMan gene expression assays were purchased from Fisher Scientific UK. GAPDH was used as the normalisation reference gene:
Hs01554629_m1 (ERG gene FAM-MGB dye)
Hs00220210_m1 (SYMD2 gene: FAM-MGB dye)
Hs01075666_m1 (IL6R gene: FAM-MGB dye)
Hs00394497_m1 (SERPINB9 gene: FAM-MGB dye)
Hs02758991_g1 (GAPDH gene: FAM-MGB dye)
The TaqMan qPCR assays were all performed on an Applied Biosystems Step One Plus qPCR system in duplicate and the qPCR constituents were as follows in a 20 μl total volume: 10 μl 2× TaqMan Gene Expression Master Mix (ThermoFisher), 1 μl 20× TaqMan Gene Expression primers/probes (ThermoFisher), 8 μl sterilised distilled water and 1 μl cDNA (25 ng/μl).
The TaqMan qPCR cycling conditions were as follows:
50 °C for 2 min – 1 cycle
95 °C for 20 s – 1 cycle
95 °C for 1 s – 40 cycles
60 °C for 20 s – 40 cycles
Mean Ct values of the target gene were subtracted from the mean Ct values of the housekeeper gene to acquire inverse delta Ct values (higher value represents higher expression).
Immunohistochemistry to assess protein expression in aortic tissues
Histological staining of the Smyd2 protein was conducted on frozen sections from aortic tissues to corroborate differential SMYD2 gene expression and promoter methylation in three AAAs and three controls. This work was conducted in the Histology Facility, Core Biotechnology Services, University of Leicester.
Three assays were conducted for each individual aortic tissue sample, meaning a total of 18 slides were prepared for microscopy. Six slides (three controls and three AAAs) were prepared with a Smyd2 polyclonal rabbit antibody at 50: 1 (ThermoFisher - PA5-51339), six control slides (three controls and three AAAs) were prepared with no primary antibody, and six slides (three controls and three AAAs) were prepared with a smooth muscle actin (SMA) polyclonal rabbit antibody at 100: 1 (ThermoFisher - PA5-19465). Visualisation of Smyd2 staining was concentrated on the regions where the highest density of smooth muscle fibres were seen from the SMA. The primary antibodies were detected using the NOVOLINK polymer detection system (Novocastra RE7140-K) using the standard protocol, and visualisation of the slides was performed on the Hamamatsu Nanozoomer 2.0HT Slide Scanner with the use of NDP VIEW2 software (U12388-01).
Summary of statistical analysis
Statistical analyses were conducted using GraphPad Prism 7 (GraphPad Software, Inc., CA, USA) and IBM SPSS Statistics 24 (IBM, NY, USA). Continuous parametric data are presented as mean value ± standard error (SE) and non-parametric data are presented as median value and range; categorical data are presented as absolute value or percentage. The chi-square test was used to compare categorical data. Student’s t test was used to compare continuous parametric data. Pearson’s correlation coefficient was calculated to assess linear dependence between two variables. A P value of < 0.05 was considered statistically significant. Where applicable, a Q value has been reported (Q < 0.1), adjusted for false discovery rate (FDR).
Additional file
Supplemental material. (DOCX 592 kb)
Acknowledgements
Not applicable. List of UKAGS collaborators: Mr. Rajiv Pathak: rajiv.pathak@dgh.nhs.uk, Mr. Marcus J Brooks: marcus.brooks@uhbristol.nhs.uk, Mr. Paul Hayes: paul.hayes@addenbrookes.nhs.uk, Prof Chris H Imray: Christopher.Imray@uhcw.nhs.uk, Mr. John Quarmby: John.Quarmby@derbyhospitals.nhs.uk, Mr. Sohail A Choksy: sohailchoksy@nhs.net, Mr. Jonothon J Earnshaw: Jonothan.Earnshaw@glos.nhs.uk, Prof Cliff P Shearman: C.P.Shearman@soton.ac.uk, Mr. Eric Grocott: eric.grocott@googlemail.com, Mr. Thomas Rix: thomasrix@nhs.net, Prof Ian C Chetter: Ian.chetter@hey.nhs.uk, Mr. William Tennant: william.tennant@nuh.nhs.uk, Mr. Gabor Libertiny: gabor.libertiny@ngh.nhs.uk, Mr. Tim Sykes: Tim.Sykes@sath.nhs.uk, Mr. Mark Dayer: mark.dayer@tst.nhs.uk, Ms. Lynda Pike: lynda.pike@nhs.net, Mr. Arun Pherwani: Arun.Pherwani@uhns.nhs.uk, Mr. Colin Nice: colin.nice@ghnt.nhs.uk, Mr. Neil Browning: neil.browning@asph.nhs.uk, Prof Charles N McCollum: charles.mccollum@manchester.ac.uk, Mr. Syed W Yusuf: Syed.Yusuf@bsuh.nhs.uk, Mr. Mark Gannon: mark.gannon@heartofengland.nhs.uk, Mr. Jamie Barwell: Jamie.Barwell@nhs.net, Mrs. Sara Baker: Sara.Baker@rbch.nhs.uk, Mr. Srinivasa R Vallabhaneni: fempop@liverpool.ac.uk, Mr. JV Smyth: JV.Smyth@cmft.nhs.uk, Prof Alun H Davies: a.h.davies@imperial.ac.uk, Mr. Tim Lees: Tim.Lees@nuth.nhs.uk, Mr. Louis Fligelstone: Louis.Fligelstone@wales.nhs.uk, Prof Rob Sayers: rs152@leicester.ac.uk, Prof Nilesh J Samani: njs@leicester.ac.uk, Dr. Mike J Sweeting: mjs212@medschl.cam.ac.uk, Prof John Thompson: trj@le.ac.uk.
Funding
Sample collection for this study was funded by the British Heart Foundation (UK Aneurysm Growth Study, CS/14/2/30841) and the National Institute for Health Research (NIHR Leicester Cardiovascular Biomedical Research Unit Biomedical Research Informatics Centre for Cardiovascular Science, IS_BRU_0211_20033). BJT held a PhD fellowship (2014–2017) awarded by the University Of Leicester, Department Of Cardiovascular Sciences, which funded this study.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AAA
Abdominal aortic aneurysm
- ELISA
Enzyme-linked immunosorbent assay
- HCY
Homocysteine
- MeQTL
Methylation quantitative trait loci
- NGS
Next-generation sequencing
- PBMC
Peripheral blood mononuclear cell
- UKAGS
UK aneurysm growth study
- VSMC
Vascular smooth muscle cell
Authors’ contributions
BJT conceived the study, designed the experiments, carried out the experiments, conducted the bioinformatics and statistical analysis, and wrote the manuscript. MJB conceived the study, helped design the experiments, supervised the study and critically reviewed the manuscript. AS conceived the study, helped supervise the study and critically reviewed the manuscript. PJF and NS assisted with next generation sequencing preparations and reactions, and critically reviewed the manuscript. All authors approved the final manuscript for submission.
Ethics approval and consent to participate
Patients and controls were recruited from two sources: The UK aneurysm growth study (UKAGS) and the local AAA research programme based at the NIHR Leicester Biomedical Research Centre. Ethical approval by an NHS Research Ethics Committee was obtained for both studies. Consent was obtained for each individual sample used in this research.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0460-9) contains supplementary material, which is available to authorized users.
Contributor Information
Bradley J. Toghill, Email: bt96@le.ac.uk
UKAGS collaborators:
Rajiv Pathak, Marcus J. Brooks, Paul Hayes, Chris H. Imray, John Quarmby, Sohail A. Choksy, Jonothon J. Earnshaw, Cliff P. Shearman, Eric Grocott, Thomas Rix, Ian C. Chetter, William Tennant, Gabor Libertiny, Tim Sykes, Mark Dayer, Lynda Pike, Arun Pherwani, Colin Nice, Neil Browning, Charles N. McCollum, Syed W. Yusuf, Mark Gannon, Jamie Barwell, Sara Baker, Srinivasa R. Vallabhaneni, J. V. Smyth, Alun H. Davies, Tim Lees, Louis Fligelstone, Rob Sayers, Nilesh J. Samani, Mike J. Sweeting, and John Thompson
References
- 1.Sidloff D, Stather P, Dattani N, Bown M, Thompson J, Sayers R, Choke E. Aneurysm global epidemiology study: public health measures can further reduce abdominal aortic aneurysm mortality. Circulation. 2014;129:747–753. doi: 10.1161/CIRCULATIONAHA.113.005457. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Earnshaw JJ. Triumphs and tribulations in a new national screening programme for abdominal aortic aneurysm. Acta Chir Belg. 2012;112:108–110. doi: 10.1080/00015458.2012.11680806. [DOI] [PubMed] [Google Scholar]
- 4.Meijer CA, Stijnen T, Wasser MN, Hamming JF, van Bockel JH, Lindeman JH. Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann Intern Med. 2013;159:815–823. doi: 10.7326/0003-4819-159-12-201312170-00007. [DOI] [PubMed] [Google Scholar]
- 5.Toghill BJ, Saratzis A, Bown MJ. Abdominal aortic aneurysm—an independent disease to atherosclerosis? Cardiovasc Pathol. 2017;27:71–75. doi: 10.1016/j.carpath.2017.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Toghill BJ, Saratzis A, Harrison SC, Verissimo AR, Mallon EB, Bown MJ. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis. 2015;241:121–129. doi: 10.1016/j.atherosclerosis.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Wahlgren CM, Larsson E, Magnusson PK, Hultgren R, Swedenborg J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg. 2010;51:3–7. doi: 10.1016/j.jvs.2009.08.036. [DOI] [PubMed] [Google Scholar]
- 8.Joergensen TM, Christensen K, Lindholt JS, Larsen LA, Green A, Houlind K. High heritability of liability to abdominal aortic aneurysms: a population based twin study. Eur J Vasc Endovasc Surg. 2016;52:41–46. doi: 10.1016/j.ejvs.2016.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Bown MJ, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Badger SA, Bradley DT, Burnand K, et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am J Hum Genet. 2011;89:619–627. doi: 10.1016/j.ajhg.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bradley DT, Hughes AE, Badger SA, Jones GT, Harrison SC, Wright BJ, Bumpstead S, Baas AF, Gretarsdottir S, Burnand K, et al. A variant in LDLR is associated with abdominal aortic aneurysm. Circ Cardiovasc Genet. 2013;6:498–504. doi: 10.1161/CIRCGENETICS.113.000165. [DOI] [PubMed] [Google Scholar]
- 11.Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, Kranendonk SE, Zeebregts CJ, van Sterkenburg SM, Geelkerken RH, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–697. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison SC, Smith AJ, Jones GT, Swerdlow DI, Rampuri R, Bown MJ, Folkersen L, Baas AF, de Borst GJ, Blankensteijn JD, et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm. Eur Heart J. 2013;34:3707–3716. doi: 10.1093/eurheartj/ehs354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, Jones GT, Rinkel GJ, Blankensteijn JD, Ronkainen A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 14.Jones GT, Bown MJ, Gretarsdottir S, Romaine SP, Helgadottir A, Yu G, Tromp G, Norman PE, Jin C, Baas AF, et al. A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum Mol Genet. 2013;22:2941–2947. doi: 10.1093/hmg/ddt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones GT, Tromp G, Kuivaniemi H, Gretarsdottir S, Baas AF, Giusti B, Strauss E, van’t Hof FN, Webb T, Erdman R, et al. Meta-analysis of genome-wide association studies for abdominal aortic aneurysm identifies four new disease-specific risk loci. Circ Res. 2016;120: 341–53. [DOI] [PMC free article] [PubMed]
- 16.Toghill BJ, Saratzis A, Liyanage L. S, Sidloff D, Bown MJ. Genetics of Aortic Aneurysmal Disease. 2016. eLS. 1–10. 10.1002/9780470015902.a0026851.
- 17.Verma M, Banerjee HN. Epigenetic inhibitors. Methods Mol Biol. 2015;1238:469–485. doi: 10.1007/978-1-4939-1804-1_24. [DOI] [PubMed] [Google Scholar]
- 18.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 19.Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Goncalves I, Moran S, Esteller M. A DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014;7(5):692–700. [DOI] [PubMed]
- 20.Wongtrakoongate P. Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries. World J Stem Cells. 2015;7:137–148. doi: 10.4252/wjsc.v7.i1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aslibekyan S, Claas SA, Arnett DK. Clinical applications of epigenetics in cardiovascular disease: the long road ahead. Transl Res. 2014;165(1):143–53. [DOI] [PMC free article] [PubMed]
- 25.Cornuz J, Sidoti Pinto C, Tevaearai H, Egger M. Risk factors for asymptomatic abdominal aortic aneurysm: systematic review and meta-analysis of population-based screening studies. Eur J Pub Health. 2004;14:343–349. doi: 10.1093/eurpub/14.4.343. [DOI] [PubMed] [Google Scholar]
- 26.Ryer EJ, Ronning KE, Erdman R, Schworer CM, Elmore JR, Peeler TC, Nevius CD, Lillvis JH, Garvin RP, Franklin DP, et al. The potential role of DNA methylation in abdominal aortic aneurysms. Int J Mol Sci. 2015;16:11259–11275. doi: 10.3390/ijms160511259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drong AW, Nicholson G, Hedman AK, Meduri E, Grundberg E, Small KS, Shin SY, Bell JT, Karpe F, Soranzo N, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS One. 2013;8:e55923. doi: 10.1371/journal.pone.0055923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richmond RC, Sharp GC, Ward ME, Fraser A, Lyttleton O, McArdle WL, Ring SM, Gaunt TR, Lawlor DA, Davey Smith G, Relton CL. DNA methylation and BMI: investigating identified methylation sites at HIF3A in a causal framework. Diabetes. 2016;65:1231–1244. doi: 10.2337/db15-0996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhi D, Aslibekyan S, Irvin MR, Claas SA, Borecki IB, Ordovas JM, Absher DM, Arnett DK. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics. 2013;8:802–806. doi: 10.4161/epi.25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonder MJ, Luijk R, Zhernakova DV, Moed M, Deelen P. Disease variants alter transcription factor levels and methylation of their binding sites. Nature Genetics. 2017;49:131–138. [DOI] [PubMed]
- 31.Valdespino V, Valdespino PM. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag Res. 2015;7:241–251. doi: 10.2147/CMAR.S70358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Napoli C, Grimaldi V, De Pascale MR, Sommese L, Infante T, Soricelli A. Novel epigenetic-based therapies useful in cardiovascular medicine. World J Cardiol. 2016;8:211–219. doi: 10.4330/wjc.v8.i2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuggle NR, Howe FA, Allen RL, Sofat N. New insights into the impact of neuro-inflammation in rheumatoid arthritis. Front Neurosci. 2014;8:357. doi: 10.3389/fnins.2014.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenvinkel P, Karimi M, Johansson S, Axelsson J, Suliman M, Lindholm B, Heimburger O, Barany P, Alvestrand A, Nordfors L, et al. Impact of inflammation on epigenetic DNA methylation—a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–499. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S, Zhong B, Chen M, Yang L, Yang G, Li Y, Wang H, Wang G, Li W, Cui J, et al. Epigenetic reprogramming reverses the malignant epigenotype of the MMP/TIMP axis genes in tumor cells. Int J Cancer. 2014;134:1583–1594. doi: 10.1002/ijc.28487. [DOI] [PubMed] [Google Scholar]
- 36.Liyanage VR, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR. DNA modifications: function and applications in normal and disease states. Biology (Basel) 2014;3:670–723. doi: 10.3390/biology3040670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Long TI, Arakawa K, Wang R, Yu MC, Laird PW. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One. 2010;5:e9692. doi: 10.1371/journal.pone.0009692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pushpakumar S, Kundu S, Narayanan N, Sen U. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney. FASEB J. 2015;29:4713–4725. doi: 10.1096/fj.15-272443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antoniades C, Antonopoulos AS, Tousoulis D, Marinou K, Stefanadis C. Homocysteine and coronary atherosclerosis: from folate fortification to the recent clinical trials. Eur Heart J. 2009;30:6–15. doi: 10.1093/eurheartj/ehn515. [DOI] [PubMed] [Google Scholar]
- 40.Krishna SM, Dear A, Craig JM, Norman PE, Golledge J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis. 2013;228:295–305. doi: 10.1016/j.atherosclerosis.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 41.Lindqvist M, Hellstrom A, Henriksson AE. Abdominal aortic aneurysm and the association with serum levels of homocysteine, vitamins B6, B12 and folate. Am J Cardiovasc Dis. 2012;2:318–322. [PMC free article] [PubMed] [Google Scholar]
- 42.Patterson K, Molloy L, Qu W, Clark S. DNA methylation: bisulphite modification and analysis. J Vis Exp. 2011;56:e3170. [DOI] [PMC free article] [PubMed]
- 43.Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8:623–631. doi: 10.1097/00019501-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 44.Hendel A, Cooper D, Abraham T, Zhao H, Allard MF, Granville DJ. Proteinase inhibitor 9 is reduced in human atherosclerotic lesion development. Cardiovasc Pathol. 2012;21:28–38. doi: 10.1016/j.carpath.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 46.Sajjad A, Novoyatleva T, Vergarajauregui S, Troidl C, Schermuly RT, Tucker HO, Engel FB. Lysine methyltransferase Smyd2 suppresses p53-dependent cardiomyocyte apoptosis. Biochim Biophys Acta. 1843;2014:2556–2562. doi: 10.1016/j.bbamcr.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu G, Liu G, Xiong S, Liu H, Chen X, Zheng B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J Biol Chem. 2015;290:5414–5423. doi: 10.1074/jbc.M114.610345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, Kawaji H, Lassmann T, Harbers M, Forrest AR, Bajic VB. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014;15:119. doi: 10.1186/1471-2164-15-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rishi V, Bhattacharya P, Chatterjee R, Rozenberg J, Zhao J, Glass K, Fitzgerald P, Vinson C. CpG methylation of half-CRE sequences creates C/EBPalpha binding sites that activate some tissue-specific genes. Proc Natl Acad Sci U S A. 2010;107:20311–20316. doi: 10.1073/pnas.1008688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 51.Mueller O, Hahnenberger K, Dittmann M, Yee H, Dubrow R, Nagle R, Ilsley D. A microfluidic system for high-speed reproducible DNA sizing and quantitation. Electrophoresis. 2000;21:128–134. doi: 10.1002/(SICI)1522-2683(20000101)21:1<128::AID-ELPS128>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 52.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. (DOCX 592 kb)
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.