

Abstract
Background
The contribution of airway remodeling in chronic obstructive pulmonary disease (COPD) has been well documented, with airway smooth muscle cell proliferation and migration playing a role in the remodeling process. Here, we aimed to verify the effects of fine particulate matter (PM2.5) on human bronchial smooth muscle cell (HBSMC) migration and to explore the underlying signaling pathways.
Methods
HBSMC apoptosis, proliferation and migration were measured using flow cytometry, cell counting and transwell migration assays, respectively. The role of the hedgehog pathway in cell migration was assessed by western blotting to measure the expression of Sonic hedgehog (Shh), Gli1 and Snail. Furthermore, siRNA was used to knock down Gli1 or Snail expression.
Results
PM2.5 induced HBSMC apoptosis in a dose-dependent manner, although certain concentrations of PM2.5 did not induce HBSMC proliferation or apoptosis. Interestingly, cell migration was stimulated by PM2.5 doses far below those that induced apoptosis. Additional experiments revealed that these PM2.5 doses enhanced the expression of Shh, Gli1 and Snail in HBSMCs. Furthermore, PM2.5-induced cell migration and protein expression were enhanced by recombinant Shh and attenuated by cyclopamine. Similar results were obtained by knocking down Gli1 or Snail.
Conclusions
These findings suggest that PM2.5, which may exert its effects through the Shh signaling pathway, is necessary for the migration of HBSMCs. These data define a novel role for PM2.5 in airway remodeling in COPD.
Electronic supplementary material
The online version of this article (10.1186/s12931-017-0702-y) contains supplementary material, which is available to authorized users.
Keywords: COPD, PM2.5, Human bronchial smooth muscle cell, Migration, Sonic hedgehog signaling pathway
Background
Chronic obstructive pulmonary disease (COPD) has become an increasingly serious threat to human health, but its pathogenesis remains poorly understood. Airway remodeling is widely recognized as the core hallmark of chronic bronchiolitis leading to COPD [1]. Many studies have shown that COPD is associated with airway wall thickening [2–4]. Moreover, Dournes and colleagues have shown that the progression of airflow obstruction in COPD is strongly associated with thickening of the smooth muscle layer [5]. The abnormal migration of airway smooth muscle cells (ASMCs) is thought to play a role in airway remodeling [6]. Biopsy sections from COPD and asthma patients have shown that the smooth muscle mass moves closer to the epithelium [7, 8]. The causes of these abnormalities are complex.
Air pollution is an increasingly serious problem, with environmental particulate matter (PM) posing a significant hazard to human health, especially in developing countries. Furthermore, our previous study established an association between exposure to ambient PM and COPD [9]. PM with an aerodynamic diameter smaller than 2.5 μm (PM2.5) can deposit in the respiratory tract and enter alveolar cells, inducing or worsening pulmonary diseases. Road traffic is known to be a major source of PM2.5 in urban environments, while residential biomass (e.g., wood, straw and animal feces) combustion for cooking or heating contributes considerably to indoor PM2.5 exposure in many rural environments. Investigating the effects of traffic ambient PM2.5 (TAPM2.5) and wood smoke PM2.5 (WSPM2.5) on human bronchial smooth muscle cells (HBSMCs) is therefore imperative for providing experimental evidence for the effects of different types of PM2.5 on airway remodeling.
Increasing evidence suggests that the Sonic hedgehog (Shh) pathway is involved in adult lung diseases such as pulmonary fibrosis, asthma, COPD and lung cancer [10]. The hedgehog (Hh) family of extracellular signaling proteins includes Shh, Indian hedgehog (Ihh) and Desert hedgehog (Dhh). Shh is the most broadly expressed and the most potent member of this family [11]. The presence of Shh releases Smoothened (SMO) from the inhibition exerted by the Patched (Ptch) receptor. SMO is a G protein associated with a receptor that transduces the Shh signal inside the cell. Activation of the Shh pathway results in nuclear translocation of the transcription factor Gli, which regulates the expression of genes involved in cell differentiation, survival and growth, such as cyclins and Snail, as well as Hh pathway genes including Gli and Ptch [12]. Three Gli genes (Gli1, Gli2 and Gli3) have been identified in mammalian tissues, of which Gli1 is considered the only specific marker of Shh pathway activity [13]. Several recent studies have shown that the Shh pathway is associated with cancer cell migration in pancreatic, ovarian and gastric carcinomas [14–16]. However, to date, the role of the Shh pathway in HBSMC migration has not been elucidated. Furthermore, the exact regulatory mechanisms by which the Shh pathway affects HBSMC migration remain unclear. A recent study has shown that Snail genes act primarily as survival factors and as inducers of cell migration, rather than as inducers of epithelial-mesenchymal transition (EMT) or cell fate [17]. Thus, further investigation of the mechanisms of HBSMC migration is required.
In this study, we tested the hypothesis that PM2.5 induces HBSMC proliferation, apoptosis and migration. We found that PM2.5 induces HBSMC migration and Shh pathway activation at concentrations far lower than those required for inducing apoptosis. We further confirmed that the Shh signaling pathway was required for the PM2.5-dependent migration of these cells.
Methods
Cell culture
HBSMCs and human bronchial fibroblasts (HBFs) were purchased from ScienCell (Carlsbad, CA, USA). These cell lines were cultured in the recommended medium supplemented with 2% fetal bovine serum and 1% growth supplement, and maintained at 37 °C in a humidified atmosphere with 5% CO2. The medium was replaced every 2 days. Cells from passages 3–8 were used for experiments.
Particle collection and preparation
TAPM2.5 was collected from a busy road beside Dongfeng West Road, Guangdong City, Guangzhou Province (traffic jam hours, 8:00–21:00 during March–April 2015). Each filter membrane was collected after about 13 h and the machine was placed 3 m away from the road edge and 2 m away from the ground. WSPM2.5 was collected from the burning of China fir in a conventional Chinese wood stove (April 23–May 6, 2015), as previously described [18]. Each filter membrane was collected after about 1 h at full combustion and the machine was placed 2 m away from the oven. Both PMs were collected with a high-volume sampling machine (TE-6070, Tisch, USA) equipped with a PM2.5 selective-inlet head (1.13 m3/min). TAPM2.5 and WSPM2.5 were collected on glass fiber membrane filters with a 1.6-μm pore size and a 406-cm2 sampling area. The amount PM2.5 was defined as the weight increase of each filter. The filters were soaked in dimethyl sulfoxide (DMSO), and the solutions were then filtered through a 5-μm needle filter. The supernatant was collected, and the particles recovered from different filters were pooled to ensure a homogenous batch of particles. The 15 mg/ml TAPM2.5 and 40 mg/ml WSPM2.5 stock solutions were stored at − 20 °C until use. In the control group, filters without particles were prepared in the same conditions containing 0.1% DMSO. The final concentration of DMSO in the well did not exceed 0.5%. The presence of DMSO did not affect cell viability.
Flow cytometry
HBSMCs were treated with TAPM2.5 (0, 15, 30, 60, 120, 240 and 480 μg/ml) or WSPM2.5 (0, 3, 6, 12, 24, 48 and 96 μg/ml) for 72 h. Cells were then harvested, washed and resuspended in phosphate-buffered saline (PBS). Apoptotic cells were identified using an annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) cell apoptosis kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Briefly, the cells were washed and subsequently incubated with 100 μl of 1× annexin binding buffer containing 5 μl of annexin V-FITC and 1 μl of PI for 15 min in the dark. Apoptosis was then analyzed by flow cytometry (BD Biosciences).
Cell proliferation assays
Cell proliferation was analyzed by cell counting and EdU immunostaining. For cell counting, 7.5 × 104 HBSMCs were plated in 12-well plates in 1 ml of smooth muscle growth medium (SMGM). The following day, the cells were washed and transferred to 0.2% FBS-containing DMEM-F12 for 24 h, and subsequently treated with TAPM2.5 (15 μg/ml) or WSPM2.5 (3 μg/ml) for an additional 48 h. At the end of each experimental period, the cells were harvested with trypsin-EDTA and counted using a Zeiss Coulter Counter. At least three wells were analyzed per condition per experiment. For the analysis of cell proliferation by EdU immunostaining, a Cell-Light™ EdU staining kit was used to label the nuclei of dividing cells. HBSMCs were seeded in triplicate into 96-well plates at a density of 5000 cells per well and cultured in SMGM overnight. The cells were then treated with TAPM2.5 (15 μg/ml) or WSPM2.5 (3 μg/ml) for 24 h, and then subjected to EdU staining. EdU-incorporation assays were performed according to the manufacturer’s protocol [19, 20]. Briefly, 100 μL of 50 μM EdU were added to each well for 4 h. The cells were fixed in 4% paraformaldehyde and incubated with 2 mg/ml aminoacetic acid for 5 min with constant shaking. The cells were then incubated with 100 μL of penetrant per well for 10 min with constant shaking, followed by 100 μL of 1× EdU solution for 30 min. Hoechst 33,342 was used to stain cell nuclei. Images were acquired using a fluorescence microscope equipped with a × 20 objective. In each experiment, the proportion of EdU-positive cells was determined by analyzing cells from three random fields.
Cell migration assays
Cell migration was assessed using the transwell chamber method. Assays were performed using Corning cell culture inserts (6.5 mm diameter, 8-μm pore size). TAPM2.5 (15 μg/ml) or WSPM2.5 (3 μg/ml) was prepared in DMEM-F12 supplemented with 10% FBS and added to the lower chamber. HBSMCs or HBFs (1 × 105) in serum-free DMEM-F12 were added to the upper chamber. After a 6-h incubation at 37 °C, cells on both sides of the membrane were fixed and stained with 0.1% crystal violet solution. Cells on the upper side of the membrane were then removed with a cotton swab. Cell migration was evaluated by counting the number of cells that migrated to the lower side of the membrane in five random fields using a microscope equipped with a × 20 objective. In some experiments, 100 ng/ml recombinant Shh protein (r-Shh; R&D Systems, Minneapolis, MN, USA) was added to the lower chamber. In experiments where the SMO inhibitor, cyclopamine (Sigma-Aldrich, St. Louis, MO, USA), was used, cells were pretreated with the drug for 30 min prior to PM2.5 or r-Shh exposure.
Western blotting
HBSMCs were harvested in RIPA cell lysis buffer supplemented with protease inhibitors (Thermo, Rockford, IL, USA), and protein concentrations were determined using the BCA protein assay. Protein extracts (40 μg) were separated by SDS-PAGE using 10% polyacrylamide gels, and then transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with PBS/0.5% Tween 20 containing 5% skim milk, incubated overnight at 4 °C with primary antibodies against Shh, Gli1 and Snail (Abcam, Cambridge, MA, USA), and then incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (Proteintech) for 1 h at room temperature. Immunoreactivity was detected using an enhanced chemiluminescence kit according to the manufacturer’s instructions. Protein expression levels were normalized against GAPDH expression.
RNA interference
Gli1, Snail and negative control (NC) siRNAs were purchased from Life Technologies. HBSMCs were grown in six-well plates to 60% confluence, and then transfected with 50 nM siRNA using 7.5 μL Lipofectamine™ RNAiMAX according to the manufacturer’s instructions. Gli1 or Snail knockdown efficiency was evaluated 72 h post-transfection by western blotting.
Statistical analysis
Data are expressed as the mean ± SD of at least three independent experimental repeats. Comparisons between groups were performed using one-way ANOVA and Student’s t-test. A value of P < 0.05 was considered statistically significant.
Results
Effects of PM2.5 on the proliferation and apoptosis of HBSMCs
Cell apoptosis assays were performed to evaluate the effects of PM2.5 on cell survival. HBSMCs were treated with varying concentrations of TAPM2.5 or WSPM 2.5 for 72 h before evaluation of apoptosis by flow cytometry. PM2.5 treatment increased apoptosis in HBSMCs (Fig. 1a and b). The proportion of apoptotic cells observed following treatment of HBSMCs with 120 μg/ml and 240 μg/ml TAPM2.5 was 29.24 ± 3.47% and 34.64 ± 7.51%, respectively. Following treatment with 48 μg/ml and 96 μg/ml WSPM2.5, the proportion of apoptotic cells was 23.57 ± 6.05% and 23.09 ± 12.37%, respectively. These values differed significantly from that of control HBSMCs, where 14.00 ± 2.92% of cells were apoptotic. However, treatment of HBSMCs with TAPM2.5 concentrations above 240 μg/ml resulted in a decrease in apoptosis to a level similar to that observed in the control population. Far lower doses of TAPM2.5 (15 μg/ml) or WSPM2.5 (3 μg/ml) had no significant effect on HBSMC apoptosis (Fig. 1c and d).
Fig. 1.
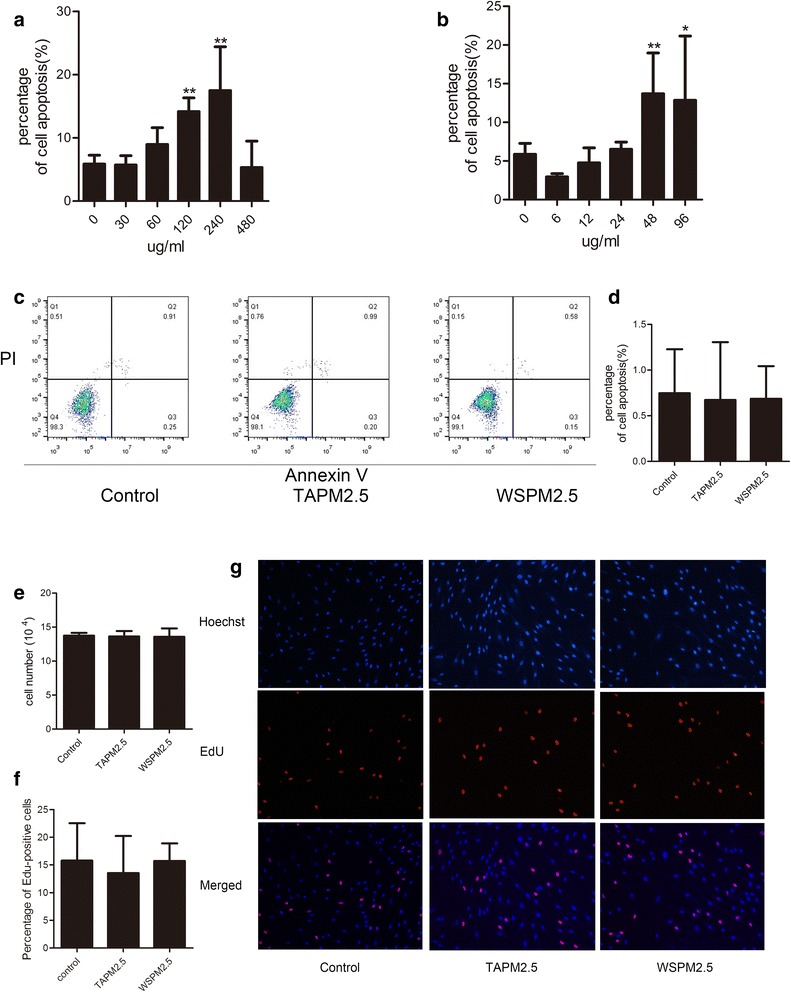
Effects of PM2.5 on HBSMCs. a Effects of various TAPM2.5 concentrations on HBSMC apoptosis. b Effects of various WSPM2.5 concentrations on HBSMC apoptosis. c and d Effects of PM2.5 (15 μg/ml TAPM2.5 or 3 μg/ml WSPM2.5) on HBSMC apoptosis. e Effects of PM2.5 on HBSMC number. f and g Evaluation of cell proliferation by EdU incorporation. *, P < 0.05; **, P < 0.001 compared with the control
Because little is known about the effects of PM2.5 on the growth of HBSMCs, we next investigated the effects of PM2.5 on cell proliferation. As shown in Fig. 1e–g, exposure to 15 μg/ml TAPM2.5 or 3 μg/ml WSPM2.5 had no significant effect on the proliferation of HBSMCs. Taken together, our results clearly established that low concentrations of TAPM2.5 or WSPM2.5 do not induce HBSMC proliferation and apoptosis, but high PM2.5 concentrations induce apoptosis.
Effects of PM2.5 on the migration of HBSMCs and HBFs
We next characterized the effects of PM2.5 on the migration of HBSMCs and HBFs. As shown in Fig. 2a–d, a 6.6-fold and 9.6-fold increase in HBSMC migration was observed in response to 15 μg/ml TAPM2.5 (Fig. 2b) and 3 μg/ml WSPM2.5 (Fig. 2c), respectively. However, PM2.5 had no significant effect on the migration of HBFs (Additional file 1: Figure S1A–D, Fig. 2e–h). These results provide the first evidence that PM2.5 promotes HBSMC migration.
Fig. 2.
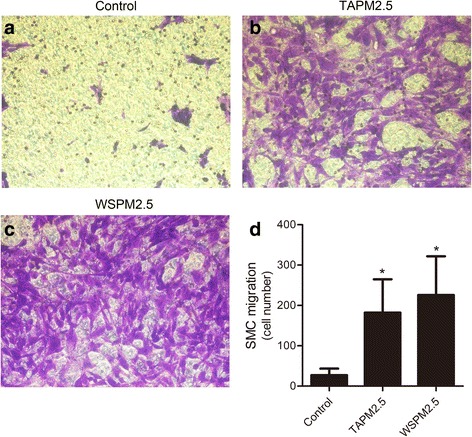
Effects of PM2.5 on HBSMC migration. a HBSMC migration in the control group. b Effects of TAPM2.5 on HBSMC migration. c Effects of WSPM2.5 on HBSMC migration. d Quantitative analysis of HBSMC migration. *, P < 0.05 compared with the control
Effects of PM2.5 on the Shh pathway in HBSMCs
To elucidate the potential mechanisms by which PM2.5 augments HBSMC migration, we examined the Shh pathway following PM2.5 stimulation. We first checked whether PM2.5 exposure activates the Shh pathway. Interestingly, we observed a dramatic, time-dependent increase in Shh protein expression in TAPM2.5-treated HBSMCs (Fig. 3a). Shh expression increased significantly from 0.5–12 h, and then decreased from 24 to 48 h, following HBSMC treatment with TAPM2.5. Similar changes in Shh expression were observed in WSPM2.5-treated HBSMCs (Fig. 3b). Furthermore, we found that PM2.5-induced Shh activation was accompanied by a significant increase in Gli1 (Fig. 3a and b) and Snail (Fig. 3c and d) protein expression. We further investigated the possibility that Shh directly upregulates Gli1 expression following PM2.5 exposure. The SMO antagonist, cyclopamine, binds to the SMO heptahelical bundle and inhibits its activity, thereby suppressing the Shh pathway. Cyclopamine significantly attenuated Gli1 (Fig. 3e and f) and Snail (Fig. 3g and h) expression. Knocking down Gli1 with siRNA decreased Snail expression (Fig. 3k and l). Taken together, these results suggest that the Shh pathway plays a key role in the response to PM2.5 exposure.
Fig. 3.
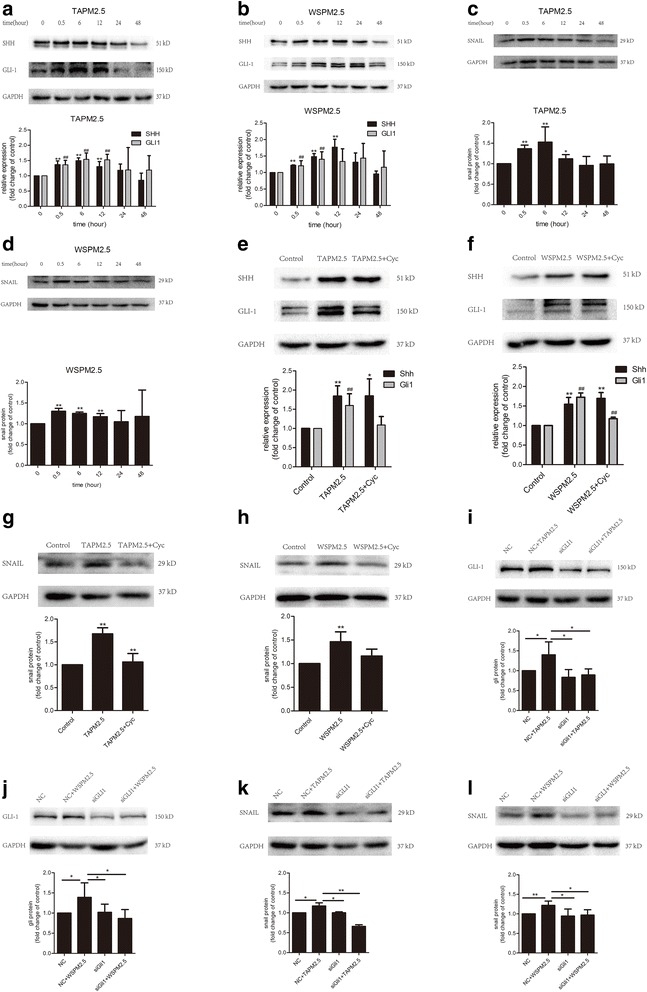
Effects of PM2.5 on the Shh pathway in HBSMCs. a TAPM2.5 and (b) WSPM2.5 increased Shh and Gli1 expression in HBSMCs in a time-dependent manner. c TAPM2.5 and (d) WSPM2.5 increased Snail expression in HBSMCs in a time-dependent manner. e and f Cyclopamine significantly attenuated the effects of TAPM2.5 and WSPM2.5 on Gli1 expression, but had no effect on Shh expression. g and h Cyclopamine significantly attenuated the effects of TAPM2.5 and WSPM2.5 on Snail expression. i and j siRNA knockdown of Gli1 significantly attenuated the effects of TAPM2.5 and WSPM2.5 on Gli1 expression. k and l siRNA knockdown of Gli1 significantly attenuated the effects of TAPM2.5 and WSPM2.5 on Snail expression. *, P < 0.05; **, P < 0.001; #, P < 0.05; ##, P < 0.001 compared with the control group
Effects of the Shh pathway on HBSMC migration
To examine the role of the Shh pathway in the PM2.5-dependent migration of HBSMCs, we performed experiments using r-Shh protein in combination with cyclopamine, or Gli1 and Snail siRNAs. Treatment with r-Shh enhanced HBSMC migration in a dose-dependent manner (Fig. 4a), while cyclopamine significantly inhibited this response (Fig. 4b). Moreover, pretreatment of HBSMCs with cyclopamine (40 μM) attenuated the PM2.5-induced migration (Fig. 4c). Knocking down Gli1 or Snail significantly inhibited the PM2.5-dependent HBSMC migration (Fig. 4g). Taken together, these data suggest that the Shh pathway is necessary for PM2.5-induced HBSMC migration.
Fig. 4.
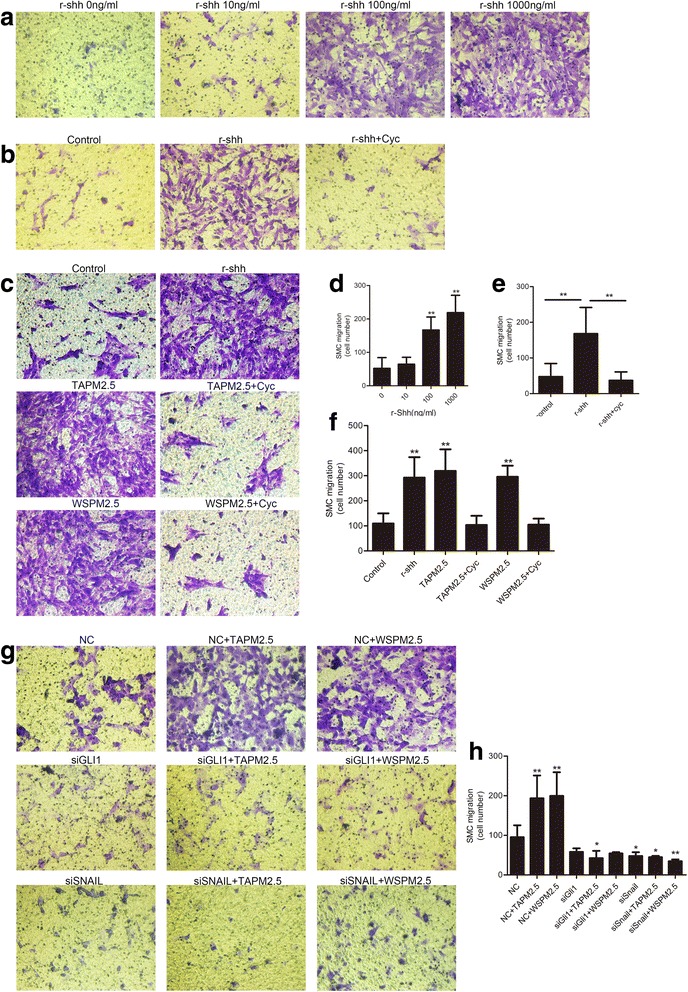
Effects of the Shh pathway on HBSMC migration. a and d HBSMC migration in response to increasing concentrations of Shh. b and e Cyclopamine significantly inhibited r-Shh-induced HBSMC migration. c and f Cyclopamine significantly inhibited TAPM2.5- and WSPM2.5-induced HBSMC migration. g and h siRNA knockdown of Gli1 or Snail significantly inhibited TAPM2.5- and WSPM2.5-induced HBSMC migration. *, P < 0.05; **, P < 0.001 compared with the control group
Discussion
Numerous studies have shown that exposure to environmental PM is associated with adverse health effects, including the development of pulmonary disease, cardiovascular disease and cancer, and leads to increased morbidity and mortality. Moreover, our previous studies have demonstrated that exposure to elevated PM concentrations is strongly associated with increased COPD prevalence and reduced respiratory function [9]. Recently, a study by our group has demonstrated that PM2.5 exposure leads to the thickening of the smooth muscle layer in a rat model [21]. To examine the effect of PM2.5 on airway remodeling and to elucidate the underlying mechanisms, we assessed the effect of PM2.5 on several aspects of HBSMC behavior, namely, proliferation, apoptosis and migration.
In this study, we investigated the pro-apoptotic and proliferative effects of PM2.5 on HBSMCs. Our results indicated that a low dose of TAPM2.5 (15 μg/ml) or WSPM2.5 (3 μg/ml) did not induce significant proliferation and that only high concentrations of PM2.5 induced HBSMC apoptosis in vitro. Conversely, a marked increase in cell migration occurred following PM2.5 exposure, and at doses far lower than those required to induce apoptosis. These results demonstrated that HBSMCs were more sensitive to the migratory effects of PM2.5 at low concentrations. According to Gualtieri et al. and Mercer et al., who estimated the bronchial surface area and the deposition of PM2.5 [22, 23], a dose of 15 μg/ml of TAPM2.5 is equivalent to approximately 15 days of atmospheric exposure (assuming a daily exposure of 6 h), and a dose of 3 μg/ml of WSPM2.5 is equivalent to 22 min of rural kitchen wood smoke exposure. Thus, the concentrations that caused HBSMC migration in our experiments can be achieved in practice. The dose of WSPM2.5 required for promoting HBSMC migration was five times lower than that of TAPM2.5, indicating that the migration-inducing potential of WSPM2.5 is greater than that of TAPM2.5. The PM2.5 doses used in our study were non-lethal doses, designed to investigate the effect of PM2.5 on the biological function of normal ASMCs. We showed that cell proliferation and apoptosis did not occur in ASMCs at the low PM2.5 doses, but cell migration occurred significantly. Thus, low doses of PM2.5 stimulated airway remodeling by promoting ASMC migration. Cells that undergo migration change from muscle type to secretory type also have the ability to proliferate and further promote airway remodeling. However, longer time and/or higher PM2.5 doses may be needed, hence further experiments are required. The results of our study are inconsistent with the increase in PM2.5 doses and the severity of COPD clinical manifestations, but only with different emphases. In addition, we believe that the pathogenesis of COPD is caused by low-dose and long-term exposure to PM2.5.
In our study low doses of PM2.5 promoted HBSMC migration, but failed to stimulate HBF migration, suggesting that PM2.5 plays a major role in the migration of HBSMCs at low doses, which is a unique phenomenon in the airway structure. However, HBFs are important airway structural cells, whose proliferation and secretion function contribute to airway fibrosis leading to airway remodeling. HBFs also have the ability to migrate. It has been reported that HBFs differentiate into HBSMCs under some external stimulation, thereby promoting muscle layer thickening. However, under our exposed experimental conditions, HBFs were less sensitive than HBSMCs. The effect of PM2.5 as an important pathogenic factor on HBFs needs to be further studied, and our group has already carried out some related studies.
Our experimental results can be explained by the effects of Snail genes, which have been linked to cell migration. Analysis of Snail expression and function in triploblasts (animals with three germ layers) of the Lophotrochozoan, Ecdysozoan and Deuterostome families supports a role for Snail in regulating cell migration. For instance, it has been shown that Snail homologues were not expressed in the mesoderm, but rather in ectodermal tissues that undergo changes in cell shape or morphogenetic movements [24, 25]. Similarly, Snail is expressed in the developing skin of mice when skin cells invaginate to form hair follicle buds [26]. In conclusion, these data suggest that Snail can regulate cell migration. Although they play a crucial role in embryonic development, Snail genes can have pathological effects in the adult. In embryos, the Snail-mediated induction of cell migration facilitates the generation of different tissues and organs that are located far from the site of origin of their precursors [17]. In cancer, however, Snail genes facilitate the delamination of cells from the primary tumor and to metastasize in other parts of the body. Snail is upregulated in non-small cell lung cancer (NSCLC), which is associated with poor prognosis, and promotes tumor progression in vivo. Moreover, it has been reported that overexpression of Snail leads to upregulation of secreted proteins, acidic and rich in cysteine (SPARC) in models of premalignancy and established disease, as well as in lung carcinoma tissues in situ [27]. Snail expression was generally high in all subjects throughout the airway wall with marked cytoplasmic to nuclear shift in COPD. Furthermore, Snail expression was associated with airflow obstruction and with expression of a canonical EMT biomarker (S100A4) [28]. The Snail pathway plays critical roles in pulmonary fibrosis animal models [29, 30]. In COPD, Snail genes facilitate ASMC migration into the airway intima, where they can secrete a series of factors leading to airway remodeling.
We next investigated the molecular mechanism by which PM2.5 promotes HBSMC migration. We examined the effects of PM2.5 on the expression of proteins associated with the Shh pathway. Obstructive airway diseases such as COPD and asthma are also associated with altered Hh signaling [10]. Growing evidence has highlighted the importance of the Shh pathway in cell migration [31–33]. The canonical Shh pathway, which depends on SMO and the transcription factor Gli, is also implicated in cell migration via gene regulation. In addition, when Gli1 expression is induced in epithelial cells, Snail is rapidly and dramatically upregulated [34]. In pancreatic cancer cells, knocking out Gli1 reduces Snail expression [35]. Inhibiting the Hh pathway with cyclopamine, a steroid that blocks SMO, decreases Gli1 and Gli2 expression, as well as Snail expression [36]. The Shh pathway has also been found to be reactivated and associated with cell migration in lung cancer [37, 38]. In summary, previous studies have revealed the involvement of the Shh pathway in cancer cell migration. However, little is known about the role of the Shh pathway in HBSMC migration. Thus, there is an urgent need to identify the mechanisms of HBSMC migration.
Our data show that PM2.5 caused a dramatic, time-dependent increase in Shh protein expression. Similar effects were observed for Gli1 and Snail expression. Intriguingly, Shh pathway activity increased significantly from 0.5–12 h, and then decreased from 24 to 48 h. The reason for this pattern remains unclear, but it is possible that Shh signaling activates other pathways, resulting in a complex network of signaling events following PM2.5 exposure. These interesting possibilities require further investigation. Our results demonstrated the importance of Shh in the response to PM2.5 exposure, given that inhibition of Shh signaling by cyclopamine was able to downregulate Gli1 and Snail expression, thereby inhibiting the effects of PM2.5 on HBSMC migration. Taken together, these results indicate that PM2.5 exposure activates the Shh signaling pathway.
Furthermore, our results demonstrated that PM2.5-induced migration was accompanied by activation of the Shh pathway. r-Shh induced HBSMC migration in a dose-dependent manner, and cyclopamine significantly reduced this effect. Moreover, cyclopamine attenuated the PM2.5-induced HBSMC migration. Knocking down Gli1 or Snail expression also significantly inhibited HBSMC migration (Fig. 4). Based on existing evidence and our current data, we propose that the Shh pathway plays a vital role in PM2.5-induced HBSMC migration (Fig. 5).
Fig. 5.
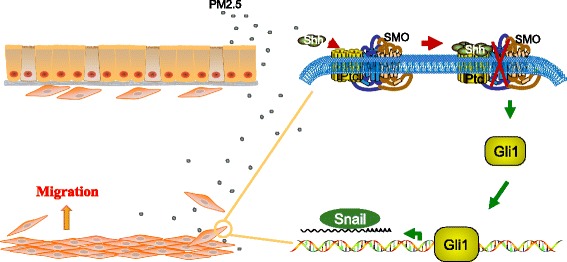
Schematic depicting the migration signaling pathway induced by PM2.5 in HBSMCs. Here, we showed that PM2.5 regulated HBSMC migration by activating the Shh signaling pathway. PM2.5 increases the expression of Shh. Shh binds to Ptch and inhibits the repression of SMO, thereby activating Gli1. Gli1 translocates to the nucleus and activates Snail transcription, triggering HBSMC migration to the epithelium
Conclusion
To the best of our knowledge, this is the first study to establish that the Shh signaling pathway is a promising therapeutic target for preventing PM2.5-induced HBSMC migration. Further studies and a greater understanding of the changes that occur in the airway are required to elucidate how airway remodeling and ASMC migration are involved in the pathology of COPD, and whether early therapeutic interventions will prevent the progression of airway remodeling caused by PM2.5 exposure.
Additional files
Figure S1A–D. Effects of PM2.5 on the migration of HBFs. (A) HBF migration in the control group. (B) Effects of TAPM2.5 on HBF migration. (C) Effects of WSPM2.5 on HBF migration. (D) Quantitative analysis of HBF migration. *, P < 0.05 compared with the control. (TIFF 9170 kb)
Acknowledgements
The authors thank Fang Peng and Baoling Liao for valuable suggestions and technical assistance. We thank James Monypenny, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work was supported by National Natural Science Foundation of China (81470233, 81670040, 81570035, 81570045), National key basic research and development program (973 Program 20l5CB553403), Science and Technology program of Guangzhou (201504010018), Scientific Research Grant of Guangzhou City (201510010226), National Natural Science Foundation of Guangdong (S2011020002789, 2014A030311040, 2017A030313683, 2017A030310419) and Grant of State Key Laboratory of Respiratory Disease (SKLRD2016ZJ013).
Availability of data and materials
Please contact author for data requests.
Abbreviations
- ASMCs
Airway smooth muscle cells
- COPD
Chronic obstructive pulmonary disease
- HBFs
Human bronchial fibroblasts
- HBSMC
Human bronchial smooth muscle cell
- PM2.5
Fine particulate matter
- r-Shh
Recombinant Shh protein
- Shh
Sonic hedgehog
- TAPM2.5
Traffic ambient PM2.5
- WSPM2.5
Wood smoke PM2.5
Authors’ contributions
XY, WH, MZ, CL, EY and JP performed the experiments. XY, WH, BL and PR designed the study. WH, YZ, BH and LH analyzed the data. XY, WH, GP, ZZ, CL and BL wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
For over a decade, ScienCell Research Laboratories has been a leading provider of human and animal primary cells, cell-derived DNA, RNA and protein, as well as other biological materials. For our business practices, we strictly comply with the following policies:
We are committed to operating under the highest ethical and legal standards in the acquisition of human and animal tissues used for the preparation of primary cells and other biological materials.
We warrant that human tissues have been obtained in compliance with local, state, and federal laws and regulations.
We strictly adhere to the guidelines for human tissue collection and distribution according to established protocols.
Human tissue used for the isolation of primary cells is derived from donors who have signed informed consent by the donor themselves or an authorized agent acting on the donor’s behalf.
We protect the privacy and autonomy of all donors.
We believe that human actions have social, economic, and environmental impact, and that they should promote good and avoid harm.
We believe in the integrity of our activities in the field of biotechnology and that they should be conducted honestly, truthfully, lawfully, impartially, competently, and with considered regard for transparency of process.
We recognize that individuals and institutions are inherently responsible for their actions and the justification, purpose, and consequences of any action should be taken into account when it is determined. We rely on the protocols, policies, and procedures noted here to provide guidance for the application of ethical behavior in our work as members of the life science society.
True to our mission, ScienCell provides our products to serve the needs of the life science research community around the world in their pursuit of curing disease and improving life.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12931-017-0702-y) contains supplementary material, which is available to authorized users.
Contributor Information
Xiuqin Ye, Email: 36494437@qq.com.
Wei Hong, Email: hongwei@gzhmu.edu.cn.
Binwei Hao, Email: haobinwei@126.com.
Gongyong Peng, Email: gongyong19761@163.com.
Lingmei Huang, Email: huanglingmei2013@163.com.
Zhuxiang Zhao, Email: zhaozhuxiang@126.com.
Yumin Zhou, Email: zhouyumin410@126.com.
Mengning Zheng, Email: mengningzheng@126.com.
Chenglong Li, Email: 278209094@qq.com.
Chunxiao Liang, Email: LiangChunxiao310@163.com.
Erkang Yi, Email: 15915774986@163.com.
Jinding Pu, Email: try20005@126.com.
Bing Li, Email: libing@gzhmu.edu.cn.
Pixin Ran, Email: pxran@gzhmu.edu.cn.
References
- 1.Tam A, Sin DD. Pathobiologic mechanisms of chronic obstructive pulmonary disease. Med Clin North Am. 2012;96:681–698. doi: 10.1016/j.mcna.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 2.Kuwano K, Bosken CH, Pare PD, Bai TR, Wiggs BR, Hogg JC. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1993;148:1220–1225. doi: 10.1164/ajrccm/148.5.1220. [DOI] [PubMed] [Google Scholar]
- 3.Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164:S28–S38. doi: 10.1164/ajrccm.164.supplement_2.2106061. [DOI] [PubMed] [Google Scholar]
- 4.Rutgers SR, Timens W, Kauffman HF, Postma DS. Markers of active airway inflammation and remodelling in chronic obstructive pulmonary disease. Clin Exp Allergy. 2001;31:193–205. doi: 10.1046/j.1365-2222.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 5.Dournes G, Laurent F. Airway Remodelling in asthma and COPD: findings, similarities, and differences using quantitative CT. Pulm Med. 2012;2012:670414. doi: 10.1155/2012/670414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.An SS, Fredberg JJ. Biophysical basis for airway hyperresponsiveness. Can J Physiol Pharmacol. 2007;85:700–714. doi: 10.1139/Y07-059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamid Q, Cosio M, Lim S. Inflammation and remodeling in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2004;114:1479–1481. doi: 10.1016/j.jaci.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Joubert P, Hamid Q. Role of airway smooth muscle in airway remodeling. J Allergy Clin Immunol. 2005;116:713–716. doi: 10.1016/j.jaci.2005.05.042. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Zhou Y, Liu S, Chen X, Zou W, Zhao D, Li X, Pu J, Huang L, Chen J, Li B, Liu S, Ran P. Association between exposure to ambient particulate matter and chronic obstructive pulmonary disease: results from a cross-sectional study in China. Thorax. 2016; 10.1136/thoraxjnl-2016-208910. [DOI] [PMC free article] [PubMed]
- 10.Kugler MC, Joyner AL, Loomis CA, Munger JS. Sonic hedgehog signaling in the lung. From development to disease. Am J Respir Cell Mol Biol. 2015;52:1–13. doi: 10.1165/rcmb.2014-0132TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 12.Stecca B, Ruiz IAA. Context-dependent regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG signals. J Mol Cell Biol. 2010;2:84–95. doi: 10.1093/jmcb/mjp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsushita S, Onishi H, Nakano K, Nagamatsu I, Imaizumi A, Hattori M, Oda Y, Tanaka M, Katano M. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014;105:272–280. doi: 10.1111/cas.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steg AD, Burke MR, Amm HM, Katre AA, Dobbin ZC, Jeong DH, Landen CN. Proteasome inhibition reverses hedgehog inhibitor and taxane resistance in ovarian cancer. Oncotarget. 2014;5:7065–7080. doi: 10.18632/oncotarget.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samadani AA, Akhavan-Niaki H. Interaction of sonic hedgehog (SHH) pathway with cancer stem cell genes in gastric cancer. Med Oncol. 2015;32:48. doi: 10.1007/s12032-015-0492-3. [DOI] [PubMed] [Google Scholar]
- 16.Miyazaki Y, Matsubara S, Ding Q, Tsukasa K, Yoshimitsu M, Kosai K, Takao S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI inhibitor GANT61 in combination with mTOR inhibition. Mol Cancer. 2016;15:49. doi: 10.1186/s12943-016-0534-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrallo-Gimeno A, Nieto MA. The snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151–3161. doi: 10.1242/dev.01907. [DOI] [PubMed] [Google Scholar]
- 18.Huang L, Pu J, He F, Liao B, Hao B, Hong W, Ye X, Chen J, Zhao J, Liu S, Xu J, Li B, Ran P. Positive feedback of the amphiregulin-EGFR-ERK pathway mediates PM2.5 from wood smoke-induced MUC5AC expression in epithelial cells. Sci Rep. 2017;7:11084. doi: 10.1038/s41598-017-11541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chehrehasa F, Meedeniya AC, Dwyer P, Abrahamsen G, Mackay-Sim A. EdU, a new thymidine analogue for labelling proliferating cells in the nervous system. J Neurosci Methods. 2009;177:122–130. doi: 10.1016/j.jneumeth.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 21.He F, Liao B, Pu J, Li C, Zheng M, Huang L, Zhou Y, Zhao D, Li B, Ran P. Exposure to ambient particulate matter induced COPD in a rat model and a description of the underlying mechanism. Sci Rep. 2017;7:45666. doi: 10.1038/srep45666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gualtieri M, Mantecca P, Corvaja V, Longhin E, Perrone MG, Bolzacchini E, Camatini M. Winter fine particulate matter from Milan induces morphological and functional alterations in human pulmonary epithelial cells (A549) Toxicol Lett. 2009;188:52–62. doi: 10.1016/j.toxlet.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Mercer RR, Russell ML, Roggli VL, Crapo JD. Cell number and distribution in human and rat airways. Am J Respir Cell Mol Biol. 1994;10:613–624. doi: 10.1165/ajrcmb.10.6.8003339. [DOI] [PubMed] [Google Scholar]
- 24.Lespinet O, Nederbragt AJ, Cassan M, Dictus WJ, van Loon AE, Adoutte A. Characterisation of two snail genes in the gastropod mollusc Patella Vulgata. Implications for understanding the ancestral function of the snail-related genes in Bilateria. Dev Genes Evol. 2002;212:186–195. doi: 10.1007/s00427-002-0228-1. [DOI] [PubMed] [Google Scholar]
- 25.Yamazaki K, Akiyama-Oda Y, Oda H. Expression patterns of a twist-related gene in embryos of the spider Achaearanea Tepidariorum reveal divergent aspects of mesoderm development in the fly and spider. Zool Sci. 2005;22:177–185. doi: 10.2108/zsj.22.177. [DOI] [PubMed] [Google Scholar]
- 26.Jamora C, Lee P, Kocieniewski P, Azhar M, Hosokawa R, Chai Y, Fuchs E. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLoS Biol. 2005;3:e11. doi: 10.1371/journal.pbio.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant JL, Fishbein MC, Hong LS, Krysan K, Minna JD, Shay JW, Walser TC, Dubinett SM. A novel molecular pathway for snail-dependent, SPARC-mediated invasion in non-small cell lung cancer pathogenesis. Cancer Prev Res (Phila) 2014;7:150–160. doi: 10.1158/1940-6207.CAPR-13-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahmood MQ, Walters EH, Shukla SD, Weston S, Muller HK, Ward C. Sohal SS: beta-catenin, twist and snail: transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci Rep. 2017;7:10832. doi: 10.1038/s41598-017-11375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyoshi K, Yanagi S, Kawahara K, Nishio M, Tsubouchi H, Imazu Y, Koshida R, Matsumoto N, Taguchi A, Yamashita S, Suzuki A, Nakazato M. Epithelial Pten controls acute lung injury and fibrosis by regulating alveolar epithelial cell integrity. Am J Respir Crit Care Med. 2013;187:262–275. doi: 10.1164/rccm.201205-0851OC. [DOI] [PubMed] [Google Scholar]
- 30.Yan W, Wu Q, Yao W, Li Y, Liu Y, Yuan J, Han R, Yang J, Ji X, Ni C. MiR-503 modulates epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by targeting PI3K p85 and is sponged by lncRNA MALAT1. Sci Rep. 2017;7:11313. doi: 10.1038/s41598-017-11904-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagase T, Nagase M, Machida M, Fujita T. Hedgehog signalling in vascular development. Angiogenesis. 2008;11:71–77. doi: 10.1007/s10456-008-9105-5. [DOI] [PubMed] [Google Scholar]
- 32.Renault MA, Roncalli J, Tongers J, Thorne T, Klyachko E, Misener S, Volpert OV, Mehta S, Burg A, Luedemann C, Qin G, Kishore R, Losordo DW. Sonic hedgehog induces angiogenesis via rho kinase-dependent signaling in endothelial cells. J Mol Cell Cardiol. 2010;49:490–498. doi: 10.1016/j.yjmcc.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roncalli J, Renault MA, Tongers J, Misener S, Thorne T, Kamide C, Jujo K, Tanaka T, Ii M, Klyachko E, Losordo DW. Sonic hedgehog-induced functional recovery after myocardial infarction is enhanced by AMD3100-mediated progenitor-cell mobilization. J Am Coll Cardiol. 2011;57:2444–2452. doi: 10.1016/j.jacc.2010.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Deng W, Nail CD, Bailey SK, Kraus MH, Ruppert JM, Lobo-Ruppert SM. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene. 2006;25:609–621. doi: 10.1038/sj.onc.1209077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joost S, Almada LL, Rohnalter V, Holz PS, Vrabel AM, Fernandez-Barrena MG, McWilliams RR, Krause M, Fernandez-Zapico ME, Lauth M. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 2012;72:88–99. doi: 10.1158/0008-5472.CAN-10-4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohta H, Aoyagi K, Fukaya M, Danjoh I, Ohta A, Isohata N, Saeki N, Taniguchi H, Sakamoto H, Shimoda T, Tani T, Yoshida T, Sasaki H. Cross talk between hedgehog and epithelial-mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer. 2009;100:389–398. doi: 10.1038/sj.bjc.6604846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castellone MD, Laukkanen MO, Teramoto H, Bellelli R, Ali G, Fontanini G, Santoro M, Gutkind JS. Cross talk between the bombesin neuropeptide receptor and sonic hedgehog pathways in small cell lung carcinoma. Oncogene. 2015;34:1679–1687. doi: 10.1038/onc.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giroux Leprieur E, Vieira T, Antoine M, Rozensztajn N, Rabbe N, Ruppert AM, Lavole A, Cadranel J, Wislez M. Sonic hedgehog pathway activation is associated with resistance to platinum-based chemotherapy in advanced non-small-cell lung carcinoma. Clin Lung Cancer. 2016;17:301–308. doi: 10.1016/j.cllc.2015.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1A–D. Effects of PM2.5 on the migration of HBFs. (A) HBF migration in the control group. (B) Effects of TAPM2.5 on HBF migration. (C) Effects of WSPM2.5 on HBF migration. (D) Quantitative analysis of HBF migration. *, P < 0.05 compared with the control. (TIFF 9170 kb)
Data Availability Statement
Please contact author for data requests.