

Abstract
Background
The median age of diagnosis of primary sclerosing cholangitis (PSC) is ∼30–40 years.
Objective
We aimed to analyse disease progression and liver-dependent survival in patients diagnosed with PSC after 50 years of age.
Methods
Patients with PSC were analysed with regard to their age at diagnosis. Patients with a first diagnosis of PSC after the age of 50 years were considered as the late-onset group.
Results
A total of 32/215 (14.9%) patients were diagnosed with PSC after 50 years of age. The proportion of females was significantly higher among patients with late-onset PSC (48.4 vs. 27.3%; p = 0.02). Patients with later diagnosis required dilatation therapy more often due to dominant stenosis (84.2 vs. 53.1%; p = 0.01) and suffered from recurrent cholangitis more often (48.3 vs. 21.0%; p = 0.003). Patients with late-onset PSC had reduced transplantation-free survival (10.5 ± 0.6 years vs. 20.8 ± 1.7 years, p < 0.0001), with progredient liver failure and cholangiocarcinoma as the leading causes of death.
Conclusions
Patients with later age at diagnosis of PSC displayed a different clinical phenotype with a different sex ratio, immune status and an increased risk for progressive liver failure and biliary malignancies.
Keywords: Primary sclerosing cholangitis, immunosenescence, dominant stenosis, cholangiocarcinoma, inflammatory bowel disease, liver transplantation
Key summary
Patients with later age at diagnosis of PSC display a different clinical phenotype with a significantly higher proportion of females.
Patients with later diagnosis require biliary dilatation therapy more often due to dominant stenosis and re-stenosis and suffer from recurrent cholangitis more often.
Patients with late-onset PSC had reduced transplantation-free survival with progredient liver failure and cholangiocarcinoma as the leading causes of death.
Introduction
Primary sclerosing cholangitis (PSC) is a chronic cholestatic liver disease characterised by inflammation and stricture formation in the biliary system, progressing to liver cirrhosis in the majority of cases.1-3 The aetiology is still not resolved, but many studies suggest an immunological context.4,5 Besides the frequent association with inflammatory bowel disease (IBD) and other immune-mediated diseases,6–8 there is a clear male predominance. The median age at onset of the disease varies between different ethnic groups, but is generally between 30 and 40 years.9–11 In many cases, time of onset of disease until clinical obvious symptoms is unknown. A recent study found a high prevalence of subclinical PSC in patients with IBD, detected by magnetic resonance cholangiography.12
In Japan, a second peak in age distribution was reported, with an onset occurring over 50 years of age. PSC occurring after 50 years of age shows distinct clinical features, such as a weaker association with ulcerative colitis (UC) and lower IgM serum levels.13 In many immune-mediated diseases, late onset is associated with a different clinical phenotype.14 In myasthenia gravis, early onset shows female preponderance, while late-onset patients have a male predominance, and both groups develop different alterations in the thymus tissue.15 Furthermore, in systemic lupus erythematosus, the sex ratio varies between early- and late-onset cohorts, with a higher mortality in patients with disease onset in childhood.16 In UC, late-onset disease is associated with higher rates of proctitis and left-sided colitis and an increased risk for cytomegalovirus and herpes virus infections.17 Changes in immune cell function may explain characteristics of patients with late onset of different immune-mediated diseases.
In this study, we analysed the prevalence of patients with late-onset PSC, defined as a first diagnosis after the age of 50 years, and evaluated their clinical characteristics and immunological activity markers in serum and bile fluids and its impact on liver-dependent survival in a well-characterised cohort of 215 patients that has been prospectively followed for up to 25 years.
Patients and methods
Patients/study design
The prospective cohort for evaluation the effects of first diagnosis of PSC after the age of 50 has already been described.18 Selection criteria included typical endoscopic retrograde cholangiographic findings, serum alkaline phosphatase activity at least twofold the reference range, negative antimitochondrial antibody results, and liver biopsy findings compatible with the diagnosis of PSC. Criteria for exclusion were cirrhosis at first presentation, patients in whom liver transplantation was foreseen, and patients with a history of neoplastic disease and/or coexisting hepatic disease. The end points were liver transplantation or death. The study covered the period from May 1987 to September 2012. The outcomes of all patients in the study were followed until May 2013. A total of 66 of 281 consecutive patients who presented at our institutions with PSC were not included according to our exclusion criteria. Eight patients refused to participate in the study; in 22 patients, liver transplantation was foreseen because of the advanced stage of PSC at first visit; 10 had cholangiocarcinoma (CC) diagnosed within 3 months; and one had hepatocellular carcinoma and two had colon carcinoma at first visit. Fifteen patients were lost to follow-up treatment and were not considered in the final evaluation. In eight patients, the diagnosis of PSC was not confirmed. Ten of the excluded patients were 50 years or older at time of first diagnosis. One patient with diagnosis of PSC after the age of 50 refused to participate in the study. Of the 22 patients with end-stage disease at first presentation, three had the diagnosis of PSC after the age of 50. Of the 10 patients excluded due to development of CC, six were older than 50 years. All patients were treated with ursodeoxycholic acid (UDCA) and endoscopic retrograde cholangiography (ERC), as previously reported.18 Bile samples were routinely collected during endoscopic procedures. Routine laboratory values were determined using standard methods. A dominant stenosis (DS) was defined as a stenosis with a diameter 1.5 mm smaller than that of the common duct or 1.0 mm smaller than that of a hepatic duct (within 2 cm of the bifurcation).19 Patients with first diagnosis of PSC after the age of 50 years were categorised as late-onset, according to Hirano et al.,13 and were compared with patients with earlier onset of disease, meaning before or at the age of 50. In total, 215 patients were included in the final evaluation and followed up until July 2012 (Table 1).
Table 1.
Clinical characteristics of patients with earlier- and later-onset primary sclerosing cholangitis.
| Early onset | Late onset | p-value | |
|---|---|---|---|
| Patients | 183 | 32 | – |
| Age at diagnosis [years] | 30.1 ± 0.7 | 57.3 ± 1.3 | – |
| Sex [male %] | 133 (72.7) | 17 (53.1) | 0.02 |
| IBD [yes/no] | 122 (66.7) | 19 (59.4) | 0.4 |
| Overlap with autoimmune hepatitis | 11 (6.0) | 0 (0.0) | 0.2 |
| Dominant stenosis | 112 (61.2) | 19 (59.4) | 0.6 |
| Dominant stenosis at first diagnosis | 60 (32.8) | 11 (34.4) | 0.9 |
| Laboratory parameters at baseline | |||
| Bilirubin [mg/dL] | 0.9 ± 0.3 | 0.8 ± 0.4 | 0.9 |
| ALT [IU/L] | 117.9 ± 11.3 | 62.3 ± 36.7 | 0.03 |
| AST [IU/L] | 75.0 ± 6.9 | 44.1 ± 21.0 | 0.05 |
| AP [IU/L] | 298.5 ± 20.9 | 341.0 ± 52.7 | 0.2 |
| GGT [IU/L] | 320.0 ± 33.2 | 405.0 ± 61.3 | 0.6 |
| Albumin [g/dL] | 44.0 ± 0.7 | 41.5 ± 1.2 | 0.4 |
| Mayo risk score | 0.46 ± 0.1 | 0.69 ± 0.13 | 0.2 |
| Development of CCA [%] | 6 (3.3) | 4 (12.5) | 0.02 |
| Development of colon cancer | 11 (6.0) | 2 (6.3) | 0.4 |
| Death [%] | 7 (3.8) | 7 (21.9) | <0.0001 |
| OLT [%] | 45 (24.6) | 6 (18.8) | 0.5 |
| Death/OLT [%] | 52 (28.4) | 13 (40.6) | 0.2 |
| Survival free of liver transplantation [years] | 20.8± 1.7 | 10.5 ± 0.6 | <0.0001 |
Data are presented as n (%) or as means ± standard deviations.
IBD: inflammatory bowel disease; ALT: alanine aminotransferase; AST: aspartate aminotransferase; AP: alkaline phosphatase; GGT: gamma-glutamyl transferase; CCA: cholangiocarcinoma; OLT: orthotopic liver transplantation
Measurement of serum sCD30 and cytokines in bile fluid
Serum sCD30 at time-point of first diagnosis was quantitated in 50 patients (early-onset = 40, late-onset = 10) by Human sCD30 Instant ELISA (Bender MedSystems, Vienna, Austria) according to the manufacturer’s instructions.
In 27 patients (early-onset = 19, late-onset = 8) bile samples from first ERC were available for cytokine analysis. Cytokine levels were measured by mouse cytometric bead array (CBA) Kit (BD Biosciences). Briefly, 50 μl of bile fluid or known concentrations of standard samples (0–5000 pg/ml) were added to a mixture of 50 μl each of capture antibody bead reagent and phycoerythrin (PE)-conjugated detection antibody. The mixture was incubated at room temperature in the dark for 2 h and then washed to remove unbound detection antibody. Data were acquired using a FACS AriaII flow cytometer and analysed using CBA software 1.1 (BD Biosciences).
Statistical analysis
Calculations were carried out using PASW Statistics 20. Frequencies were compared using a chi-squared test or the Fisher’s exact test where appropriate. Continuous data were compared using the nonparametric Wilcoxon rank-sum test. Actuarial transplantation-free survival was estimated using a Kaplan–Meier product limit estimator. Differences between the actuarial estimates were tested using the log rank test. Factors that independently affected the risk of reduced transplantation-free survival were determined using Cox proportional hazard ratio models with simultaneous adjustment for Mayo risk score (MRS), sex, late-onset disease, presence of IBD, and presence of DS. Statistical significance was set at p < 0.05.
Consent
Written, informed consent was obtained from each patient included in the study. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a prior approval by the institution’s human research committee. The study was approved by the local ethics committee of Heidelberg University (S-043/2011; approval granted 19.04.2011).
Results
Clinical and laboratory characteristics and first diagnosis of PSC
A cohort of 215 patients with PSC was analysed in this study. Of these, 150 (69.8%) patients were male and 65 (30.2%) were female; 145 (67.4%) patients had concomitant IBD, of which 129 (60.0%) were UC, 13 (6.0%) were Crohn’s disease, and three (1.4%) were Colitis indeterminata. The median age at first diagnosis was 34.4 years. The clinical and laboratory baseline characteristics are depicted in Table 1. The age at PSC diagnosis was analysed and is shown in Figure 1 (a–c). Patients with first diagnosis of PSC above the age of 50 were considered as the late-onset group. The late-onset group comprised 32 patients with a median onset of PSC of 58.3 (range: 50–68) years, compared with 30.0 (range: 13–49) years in the early-onset group.
Figure 1.

Distribution of age at first diagnosis of PSC.
Distribution of age at first diagnosis of PSC differentiate into decades for the whole study cohort (N = 215) The number of patients is plotted on the y-axis. The sex ratio in patients with diagnosis after the age of 50 is significantly different compared with patients with earlier onset.
Clinical characterisation of patients with diagnosis of PSC after the age of 50
The two groups of PSC patients with earlier or later diagnosis of PSC were compared with regard to their clinical and laboratory parameters at entry into the study. We found marginally lower alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in the late-onset group (p = 0.03 and p = 0.05, respectively), whereas the other cholestatic and liver parameters, including INR and serum albumin, showed no statistically significant difference. The prevalence of DS did not differ significantly between the two groups (112/183 (61.2%) vs. 19/32 (59.4%)), nor did we find a statistically significant difference in prevalence of DS at first diagnosis (60/183 (32.8%) vs. 11/32 (34.4%)). However, patients from the late-onset group more often developed re-stenosis, requiring repeated endoscopic dilatation (56/112 (50.0%) vs. 15/19 (78.9%); p = 0.02). In 52/215 patients, episodes of recurrent cholangitis requiring hospitalisation and/or antibiotic treatment were reported. Fourteen of 32 (43.8%) belonged to the late-onset group and 38/183 (20.8%) to the earlier-onset group (p = 0.002). Numbers of collected bile samples were comparable between both groups. Furthermore, the proportion of females was significantly higher in the late-onset group compared with the earlier-onset group (50/183 (27.3%) vs. 15/32 (46.9%); p = 0.02).
Immunological serum markers
The amount of total serum immunglobulin was not significantly different between both groups. Analysis of serum immunoglobulin subclasses revealed a marginal increase of IgA in the late-onset group (4.1 ± 0.6 years vs. 2.6 ± 0.2 years; p = 0.45), whereas IgG and IgM levels did not significantly differ between the groups. Furthermore, we analysed serum levels of soluble CD30 and found decreased levels in the late-onset group (34.0 ± 13.9 years vs. 58 ± 20.7 years; p = 0.044). Levels of serum bile acids showed no significant difference between the groups (Table 2).
Table 2.
Immunological serum markers in patients with younger and late-onset primary sclerosing cholangitis.
| means ± SD |
|||
|---|---|---|---|
| Early onset | Late onset | p-value | |
| Serum bile acid [µmol/l] | 9.0 ± 10.5 | 37.0 ± 43.5 | 0.07 |
| Immunoglobulin [g/l] | 19.5 ± 0.7 | 20.9 ± 2.1 | 0.2 |
| IgG [g/l] | 17.6 ± 0.7 | 21.2 ± 2.6 | 0.8 |
| IgA [g/l] | 2.6 ± 0.2 | 4.1 ± 0.6 | 0.045 |
| IgM [g/l] | 1.7 ± 0.2 | 1.6 ± 0.2 | 0.6 |
| sCD30 [U/ml] | 58.0 ± 20.7 | 34.0 ± 13.9 | 0.044 |
Cytokines secreted into bile fluid
Cytokines secreted into bile fluid were analysed using a CBA Kit. Levels of interleukin (IL)-2 (0.78 ± 1.0 years vs. 0.03 ± 0.09) years, IL-10 (0.9 ± 0.2 years vs. 0.04 ± 0.1 years), interferon gamma (6.7 ± 11.5 years vs. 0.0 ± 0.0 years), and IL-17 (14.1 ± 14.9 years vs. 1.7 ± 4.8 years) were markedly reduced in bile fluid of patients with late-onset disease, whereas IL-4 and tumour necrosis factor showed marginal differences. Levels of IL-6 were almost similar between both groups (Table 3). We found no correlation between biliary cytokine levels with AP level or MRS at first diagnosis.
Table 3.
Cytokine profile secreted into bile fluid.
| mean ± SD |
|||
|---|---|---|---|
| Early onset | Late onset | p-value | |
| IL-2 | 0.78 ± 1.0 | 0.03 ± 0.09 | 0.045 |
| IL-4 | 0.52 ± 0.8 | 0.01 ± 0.02 | 0.06 |
| IL-6 | 28.2 ± 94.7 | 25.5 ± 68.8 | 0.3 |
| IL-10 | 0.9 ± 0.2 | 0.04 ± 0.1 | 0.009 |
| TNF | 1.1 ± 1.5 | 0.0 ± 0.0 | 0.06 |
| IFN gamma | 6.7 ± 11.5 | 0.0 ± 0.0 | 0.034 |
| IL-17 | 14.1 ± 14.9 | 1.7 ± 4.8 | 0.022 |
IL, interleukin; TNF, tumour necrosis factor; IFN, interferon
Disease progression and survival analysis
During the observation period, 51 patients (six of whom were in the late-onset group) underwent liver transplantation. Seven of 32 (21.9%) patients in the late-onset group died, compared with 7/183 (3.8%) patients in the earlier-onset group. Causes of death among the patients with younger onset were CC (3/183 (1.6%)), progressive liver failure (1/183 (0.5%)), and colon carcinoma (2/183 (1.1%)). One patient died of non-hepatic disease. Causes of death for the patients in the late-onset group were CC (4/32 (12.5%)) and progredient liver failure (3/32 (9.4%)) (Table 4). Six of 183 (3.3%) patients with earlier onset of the disease developed CC as compared with 4/32 (12.5%) patients in the late-onset group (p = 0.02). Development of colon carcinoma was almost equal between the two groups (11/183 (6.0%) vs. 2/32 (6.3%)). After a follow-up period of 24 years, the actuarial survival free of liver transplantation was 20.8 years in patients with earlier onset and 10.5 years in the late-onset group (p < 0.0001) (Figure 2). The median age at death or liver transplantation was 41.6 ± 0.8 years in the young-onset group and 64.4 ± 1.5 years in the late-onset group.
Table 4.
Clinical end points among patients with early- and late-onset primary sclerosing cholangitis.
| Early onset | Late onset | |
|---|---|---|
| Orthotopic liver transplantation | 45 (24.6) | 6 (18.8) |
| Cause of death | ||
| Progredient liver failure | 1 (0.5) | 4 (12.5) |
| Cholangiocarcinoma | 3 (1.6) | 3 (9.4) |
| Colon carcinoma | 2 (1.1) | 0 (0.0) |
| Others | 1 (0.5) | 0 (0.0) |
Figure 2.
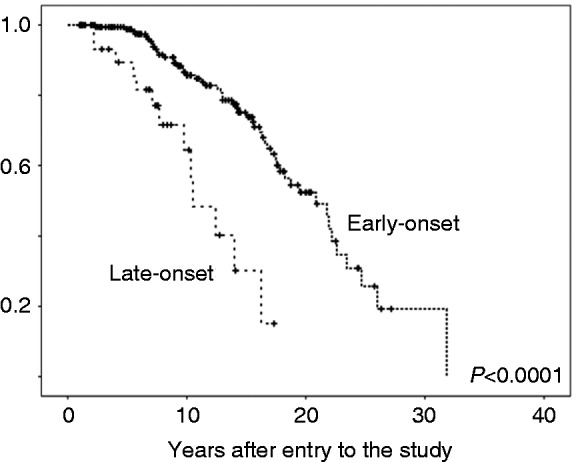
Transplantation-free survival in patients with early- and late-onset PSC.
Actuarial transplantation-free survival (Kaplan–Meier estimate) of patients with early- or late-onset of PSC. Time is given in years from the time of first diagnosis (x-axis). The percentage of patients surviving without liver transplantation is plotted on the y-axis. Patients with late-onset PSC showed significantly lower transplantation-free survival.
Prognosis factors in late-onset PSC
We performed further stratification analyses to analyse the influence of gender, presence of DS, and IBD among the late-onset cohort. In both male and female patients, late onset of the disease was associated with reduced transplantation-free survival (18.7 ± 2.1 years vs. 10.3 ± 1.6 years, p < 0.001; and 21.8 ± 1.6 years vs. 14.0 ± 1.2 years, p = 0.001, respectively). In patients with and without DS, late-onset disease was associated with reduced transplantation-free survival (17.8 ± 1.6 years vs. 10.5 ± 2.2 years, p < 0.001; and 23.5 ± 1.3 years vs. 11.3 ± 1.2 years; p < 0.001, respectively). In patients without IBD, late-onset disease was not associated with reduced transplantation-free survival (19.5 ± 1.6 years vs. 14.0 ± 1.4 years; p = 0.4) compared with a significantly reduced survival in patients with concomitant IBD (21.8 ± 2.2 years vs. 9.8 ± 2.7 years: p < 0.001). We performed single and multiple Cox proportional hazard regression models with adjustment for potential confounders, including gender, presence of DS, presence of IBD, MRS at baseline, and late-onset disease. Multivariate analysis revealed MRS at baseline and late-onset of disease as independent risk factors for reduced transplantation-free survival (Table 5). To analyse the influence of disease duration we further run a multiple regression analysis including disease duration from first diagnosis (HR 0.8 (HR 95%CI 0.8–0.9); p < 0.0001)), MRS at first diagnosis (HR 1.3 (HR 95%CI 1.0–1.8); p = 0.014)) and age at first diagnosis (HR 1.0 (HR 95%CI 1.0–1.1); p = 0.041)) showing all risk factors independently associated with reduced transplantation-free survival.
Table 5.
Cox regression analysis.
| Univariate |
Multivariate |
|||||
|---|---|---|---|---|---|---|
| Risk factor | HR | HR (95% CI) | p-value | HR | HR (95% CI) | p-value |
| Sex | 0.9 | 0.5–1.5 | 0.6 | 1.2 | 0.6–2.2 | 0.6 |
| Presence of IBD | 0.9 | 0.5–1.5 | 0.6 | 0.7 | 0.3–1.3 | 0.2 |
| Presence of DS | 0.4 | 0.2–0.8 | 0.005 | 0.5 | 0.3–1.1 | 0.09 |
| MRS | 1.6 | 1.3–2.0 | <0.001 | 1.7 | 1.3–2.2 | <0.001 |
| Late onset | 0.2 | 0.1–0.4 | <0.001 | 0.3 | 0.1–0.5 | <0.001 |
HR: hazard ratio; CI: confidence interval; IBD: inflammatory bowel disease; DS: dominant stenosis; MRS: Mayo risk score
Data show prospective factors for longer survival until liver transplantation or death. In univariate analysis presence of dominant stenosis, MRS and late-onset disease were associated with reduced actuarial survival. In multivariate analysis only MRS and late-onset disease reached significance (p < 0.05).
Discussion
In this large cohort study, we describe the clinical features of patients first diagnosed with PSC after the age of 50. We found these patients to be at increased risk of developing cholangiocarcinoma and suffer from progredient liver failure. Although these patients are reaching clinical end points at older age, compared with patients with earlier diagnosis of PSC, transplantation-free survival from first diagnosis is markedly reduced. Interestingly, the sex ratio was significantly different in the late-onset group, with a higher rate of female patients. Frequency and manifestation of IBD were not different between groups. Frequency of DS was equal between earlier and late-onset patients. However, late-onset patients developed re-stenosis requiring repeated endoscopic therapy more often, and suffered more often from recurrent episodes of cholangitis.
Worse prognosis in the late-onset cohort may be in part explained by an increased frequency of infectious complications that could aggravate disease progression.20 This may be explained by mechanisms of immunosenescence and a subsequently impaired immune response.21 Although an immunological basis of PSC is obvious22 and, in many immunological diseases, a late onset is associated with slower disease progression, in late-onset PSC, the course of the disease may be driven more rapidly by repeated or chronic biliary infections. The increased frequency of episodes of cholangitis in the late-onset group may support this hypothesis. Notably, the number of bile samples were not different between patients with earlier or later diagnosis, as routinely bile samples were collected during endoscopic intervention. One possible explanation for the increased frequency of infectious complications in the late-onset group might be a reduced immune competence. To test this hypothesis, we analysed serum levels of immunoglobulins, showing a marginal elevation of serum IgA in the late-onset group. Elevated IgA has been linked to increased risk for infections in patients with liver cirrhosis, which may reflect a sustained exposure to bacterial pathogens derived from the gut.23 Furthermore, elevated IgA seems to be associated with progressive liver fibrosis24 as well as immunological diseases like coeliac disease.25 In addition, we analysed serum levels of soluble CD30, a well-established marker of T-cell immunity, in our cohort. Increased serum levels of soluble CD30 are associated with disease activity in IBD.26 Furthermore, soluble CD30 participates in the immune response against viral infectious diseases27 and is a reliable marker for immune monitoring after solid organ transplantation.28 CD30 knockout mice display increased susceptibility to bacterial infections due to an impaired innate function of memory phenotype CD44+, CD4+, and T cells.29 An age-dependent decrease of soluble CD30 has already been demonstrated in paediatric patients.30 In line with this, we found a marked decrease in soluble CD30 in the late-onset cohort. Furthermore, we found an altered biliary cytokine composition in the late-onset cohort. However, as differences in biliary cytokines were only marginal in a small subset of patients these findings need confirmation by larger studies.
The increased frequency of CC among patients with later diagnosis of PSC accounts for a higher percentage of deaths. It is worth noting that most CC developed within the first 5 years after diagnosis of PSC in these patients. We have to emphasise that patients developing malignancies within the first 3 months after the first diagnosis were excluded according to the study protocol. Of the 10 patients excluded due to development of CC, six were older than 50 years. PSC had not been previously diagnosed in any of these patients, further supporting that late-onset disease in PSC is not an innocent diagnosis. A large multicentre cohort study also found significantly higher incidence of CC in PSC patients with advanced age at first diagnosis.31
Poorer outcome in late-onset patients may be explained by the later detection of a lengthy, pre-existing disease with a subclinical course.12 The short timeframe between the first diagnosis of PSC followed by CC may point towards this explanation. To rule out this possibility, we performed extensive comparisons of laboratory and clinical parameters at first diagnosis. There were no significant differences in baseline laboratory parameters. Liver function, expressed by INR and serum albumin level, were comparable between both groups at the time of first diagnosis.
The reliability of the date of first diagnosis is a limitation of this study. A subclinical course of the disease without significant symptoms may differ between patients and hamper interpretation of the results. However, our well-characterised cohort of PSC patients with a standardised diagnostic, screening and therapy protocol, including UDCA and endoscopic treatment, should prevent this concern. Comparable baseline characteristics between both groups at entry into the study argue against a systematically missed onset of the disease.32 Importantly, there were no patients with biliary cirrhosis at first diagnosis, arguing against delayed diagnosed end-stage patients with later PSC diagnosis. Finally, age at first diagnosis is a continuous variable and the age discriminator at the age of 50 seems arbitrary. Nevertheless, we decided for this age because it has been used in other immunological diseases, and previously in a PSC cohort from Japan. Furthermore, we found the age of 50 to be the best discriminator between both groups, although the basic results (transplantation-free survival, frequency of CC) remains equal if the discrimination is varied between the ages of 45 to 55 years.
In conclusion, we found an altered sex ratio and an increased frequency of biliary infectious complications in line with an altered immune status in late-onset PSC patients, subsequently leading to reduced transplantation-free survival with CC and progressive liver failure as leading causes of death. Patients with late-onset PSC display a different clinical phenotype, requiring adapted screening and surveillance strategies. As treatment options are often limited in elderly patients due to comorbidities and increased risk for general anaesthesia, PSC patients have to be evaluated carefully and well in advance for liver transplantation.
Acknowledgements
The authors would like to thank Petra Klöters-Plachky and Yvonne Schäfer for technical assistance.
Conflict of interest
The authors declare that they have no competing interests.
Financial support
DNG and PSa were supported by “Stiftung Elementarteilchen”, Hamburg, Germany. CR was supported by Deutsche Forschungsgemeinschaft (DFG).
Ethics approval
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a prior approval by the institution s human research committee. The study was approved by the local ethics committee of Heidelberg University (S-043/2011; approval granted 19.04.2011).
Informed consent
Written, informed consent was obtained from each patient included in the study.
References
- 1.Chapman RW, Arborgh BA, Rhodes JM, et al. Primary sclerosing cholangitis: A review of its clinical features, cholangiography, and hepatic histology. Gut 1980; 21: 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiesner RH, LaRusso NF. Clinicopathologic features of the syndrome of primary sclerosing cholangitis. Gastroenterology 1980; 79: 200–206. [PubMed] [Google Scholar]
- 3.Lee YM, Kaplan MM. Primary sclerosing cholangitis. N Engl J Med 1995; 332: 924–933. [DOI] [PubMed] [Google Scholar]
- 4.Worthington J, Cullen S, Chapman R. Immunopathogenesis of primary sclerosing cholangitis. Clin Rev Allergy Immunol 2005; 28: 93–103. [DOI] [PubMed] [Google Scholar]
- 5.Weismuller TJ, Wedemeyer J, Kubicka S, et al. The challenges in primary sclerosing cholangitis – aetiopathogenesis, autoimmunity, management and malignancy. J Hepatol 2008; 48(Suppl 1): S38–S57. [DOI] [PubMed] [Google Scholar]
- 6.Saarinen S, Olerup O, Broome U. Increased frequency of autoimmune diseases in patients with primary sclerosing cholangitis. Am J Gastroenterol 2000; 95: 3195–3199. [DOI] [PubMed] [Google Scholar]
- 7.Lamberts LE, Janse M, Haagsma EB, et al. Immune-mediated diseases in primary sclerosing cholangitis. Dig Liver Dis 2011; 43: 802–806. [DOI] [PubMed] [Google Scholar]
- 8.Ellinghaus D, Folseraas T, Holm K, et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology 2013; 58: 1074–1083. [DOI] [PubMed] [Google Scholar]
- 9.Broome U, Olsson R, Loof L, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996; 38: 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toy E, Balasubramanian S, Selmi C, et al. The prevalence, incidence and natural history of primary sclerosing cholangitis in an ethnically diverse population. BMC Gastroenterol 2011; 11: 83–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boonstra K, Weersma RK, van Erpecum KJ, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013; 58: 2045–2055. [DOI] [PubMed] [Google Scholar]
- 12.Lunder AK, Hov JR, Borthne A, et al. Prevalence of sclerosing cholangitis detected by magnetic resonance cholangiography in patients with long-term inflammatory bowel disease. Gastroenterology 2016; 151: 660–669 e4. [DOI] [PubMed] [Google Scholar]
- 13.Hirano K, Tada M, Isayama H, et al. Clinical features of primary sclerosing cholangitis with onset age above 50 years. J Gastroenterol 2008; 43: 729–733. [DOI] [PubMed] [Google Scholar]
- 14.Amador-Patarroyo MJ, Rodriguez-Rodriguez A, Montoya-Ortiz G. How does age at onset influence the outcome of autoimmune diseases? Autoimmune Dis 2012; 2012: 251730–251730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: Emerging clinical and biological heterogeneity. Lancet Neurol 2009; 8: 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tucker LB, Uribe AG, Fernandez M, et al. Adolescent onset of lupus results in more aggressive disease and worse outcomes: Results of a nested matched case-control study within LUMINA, a multiethnic US cohort (LUMINA LVII). Lupus 2008; 17: 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ha CY, Newberry RD, Stone CD, et al. Patients with late-adult-onset ulcerative colitis have better outcomes than those with early onset disease. Clin Gastroenterol Hepatol 2010; 8: 682–687 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudolph G, Gotthardt D, Kloters-Plachky P, et al. Influence of dominant bile duct stenoses and biliary infections on outcome in primary sclerosing cholangitis. J Hepatol 2009; 51: 149–155. [DOI] [PubMed] [Google Scholar]
- 19.Gotthardt DN, Rudolph G, Kloters-Plachky P, et al. Endoscopic dilation of dominant stenoses in primary sclerosing cholangitis: Outcome after long-term treatment. Gastrointest Endosc 2010; 71: 527–534. [DOI] [PubMed] [Google Scholar]
- 20.Rupp C, Bode KA, Chahoud F, et al. Risk factors and outcome in patients with primary sclerosing cholangitis with persistent biliary candidiasis. BMC Infect Dis 2014; 14: 562–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamir A, Eisenbraun MD, Garcia GG, et al. Age-dependent alterations in the assembly of signal transduction complexes at the site of T cell/APC interaction. J Immunol 2000; 165: 1243–1251. [DOI] [PubMed] [Google Scholar]
- 22.Sebode M, Peiseler M, Franke B, et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol 2014; 60: 1010–1016. [DOI] [PubMed] [Google Scholar]
- 23.Papp M, Sipeki N, Vitalis Z, et al. High prevalence of IgA class anti-neutrophil cytoplasmic antibodies (ANCA) is associated with increased risk of bacterial infection in patients with cirrhosis. J Hepatol 2013; 59: 457–466. [DOI] [PubMed] [Google Scholar]
- 24.Tomita K, Teratani T, Yokoyama H, et al. Serum immunoglobulin a concentration is an independent predictor of liver fibrosis in nonalcoholic steatohepatitis before the cirrhotic stage. Dig Dis Sci 2011; 56: 3648–3654. [DOI] [PubMed] [Google Scholar]
- 25.Salmi TT, Collin P, Jarvinen O, et al. Immunoglobulin A autoantibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming coeliac disease. Aliment Pharmacol Ther 2006; 24: 541–552. [DOI] [PubMed] [Google Scholar]
- 26.Sun X, Somada S, Shibata K, et al. A critical role of CD30 ligand/CD30 in controlling inflammatory bowel diseases in mice. Gastroenterology 2008; 134: 447–458. [DOI] [PubMed] [Google Scholar]
- 27.Fattovich G, Vinante F, Giustina G, et al. Serum levels of soluble CD30 in chronic hepatitis B virus infection. Clin Exp Immunol 1996; 103: 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Susal C, Dohler B, Sadeghi M, et al. Posttransplant sCD30 as a predictor of kidney graft outcome. Transplantation 2011; 91: 1364–1369. [DOI] [PubMed] [Google Scholar]
- 29.Umeda K, Sun X, Guo Y, et al. Innate memory phenotype CD4+ T cells play a role in early protection against infection by Listeria monocytogenes in a CD30L-dependent manner. Microbiol Immunol 2011; 55: 645–656. [DOI] [PubMed] [Google Scholar]
- 30.Chrul S, Polakowska E. Age-dependent changes of serum soluble CD30 concentration in children. Pediatr Transplant 2011; 15: 515–518. [DOI] [PubMed] [Google Scholar]
- 31.Weismuller TJ, Trivedi PJ, Bergquist A, et al. Patient age, sex, and inflammatory bowel disease phenotype associate with course of primary sclerosing cholangitis. Gastroenterology 2017; 152: 1975–1984.e8–1975–1984.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rupp C, Rossler A, Halibasic E, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther 2014; 40: 1292–1301. [DOI] [PubMed] [Google Scholar]