

Abstract
We performed whole-genome sequencing on an individual from a family with variable psychiatric phenotypes that had a sensory processing disorder, apraxia, and autism. The proband harbored a maternally inherited balanced translocation (46,XY,t(11;14)(p12;p12)mat) that disrupted LRRC4C, a member of the highly specialized netrin G family of axon guidance molecules. The proband also inherited a paternally derived chromosomal inversion that disrupted DPP6, a potassium channel interacting protein. Copy Number (CN) analysis in 14,077 cases with neurodevelopmental disorders and 8,960 control subjects revealed that 60% of cases with exonic deletions in LRRC4C had a second clinically recognizable syndrome associated with variable clinical phenotypes, including 16p11.2, 1q44, and 2q33.1 CN syndromes, suggesting LRRC4C deletion variants may be modifiers of neurodevelopmental disorders. In vitro, functional assessments modeling patient deletions in LRRC4C suggest a negative regulatory role of these exons found in the untranslated region of LRRC4C, which has a single, terminal coding exon. These data suggest that the proband’s autism may be due to the inheritance of disruptions in both DPP6 and LRRC4C, and may highlight the importance of the netrin G family and potassium channel interacting molecules in neurodevelopmental disorders.
Keywords: Autism, sensory processing, LRRC4C, Netrin G, DPP6
INTRODUCTION
While much progress has been made on the genetics of neurodevelopmental disorders (NDDs), over 50% of cases assessed clinically on any genetic platform are considered idiopathic [Krumm et al., 2014; Sanders et al., 2015]. While more NDD cases would likely be associated with genetic variation if all cases were assessed using the full extent of state-of-the-art technology, different strategies need to be employed to further unravel the genetics of NDDs. We have previously shown that balanced chromosomal rearrangement (BCR) sequencing is a powerful strategy to discover new genes for NDDs [Talkowski et al., 2011; Talkowski et al., 2012c], and here we report on a child who inherited two BCRs, one from each parent, each of which disrupted only a single gene: leucine rich repeat containing 4C (LRRC4C [MIM 608817]) and dipeptidyl-peptidase 6 (DPP6 [MIM 126141]). Analysis of rare copy gains and losses in these genes in thousands of NDD cases suggest that that exon-disrupting CNVs in DPP6 may be a contributor to NDDs and deletions in LRRC4C may be a modifier of genetic lesions associated with variable NDD phenotypes.
METHODS
Family recruitment and assessment
We obtained blood from all carriers of a BCR identified by karyotyping in a multigenerational family (46,XY,t(11;14)(p12;p12)mat), from which the proband, and the proband’s mother, and grandmother were available for clinical and genetic testing. Participants or their guardians gave informed consent, using consent forms approved by the Douglas Institute Ethics Board. The proband was assessed with both the ADOS and ADI as part of a full clinical work-up done at the Montreal Children’s Hospital. The mother of the proband confirmed that he met criteria for autism on both measures. The mother and the grandmother of the proband were assessed with the SCID-I for Axis-I mental disorders and SCID-II for personality disorders according to DSM-IV criteria. Current level of depression was assessed by the Beck Depression Inventory and the Hamilton Depression Rating Scale (HDRS-24). Cognitive functioning for these two individuals was investigated with the Stroop Color Test and the Hayling Sentence Completion test for cognitive inhibition, the Trail Making Test (TMT) A and B for flexibility/shifting, a categorical and semantic verbal fluency test, the National Adult Reading Test (NART) for a raw estimate of verbal IQ, the working memory subscale of the WAIS-III and the Iowa Gambling Task (IGT) for decision-making. Individual cognitive performance was then compared to performance from 81 healthy female controls from the GREFEX control group and 72 healthy female controls from our own database.
Next generation sequencing of BCRs
Translocation mapping experiments were performed using customized large-insert, or “jumping library”, whole genome sequencing [Brand et al., 2014] [Brand et al., 2015] [Talkowski et al., 2012b]. Reads were aligned with BWA and analyzed using Samtools. After data filtering, BAM files were processed using BamStat, a program developed to tabulate mapping statistics and output lists of anomalous read pairs (defined as having ends that map to two different chromosomes, having an abnormal insert size, or unexpected strand orientations). Anomalous pairs were clustered by their mapped location with readPairCluster, a C++ program which performs a single-linkage clustering of paired-end reads if corresponding ends map within a specified distance (e.g., less than 10 kb) of each other.
Human gene expression analysis
Post-mortem prefrontal cortex brain tissue from Brodmann Area 46 (BA46) was obtained from the Douglas Brain Bank as described elsewhere [Klempan et al., 2009]. Tissue came from three control individuals, and RNA was extracted using the Qiagen RNAeasy kit (Qiagen, Hilden, Germany). Commercially available RNA from seven additional tissues was obtained from Clontech Laboratories (Mountain View, CA), as follows: Frontal Lobe (Cat#636563), Spinal cord (Cat#636554), Hippocampus (Cat# 636593), Liver (Cat# 636531), Lung (Cat# 636524), Kidney (Cat# 636529) and Fetal brain (Cat# 636526). Reverse transcription was performed using the M-MLV reverse transcriptase enzyme and poly-dT primers to obtain complementary DNA (cDNA). Real-time PCR reactions were performed on an Applied Biosystems (Foster City, CA) 7900 HT system, using 2X iTaq Universal SybrGreen Supermix (BioRad, Saint Laurent, Canada). We used exon boundary-spanning primers, as follows: isoforms 1 and 2 of LRRC4C (leucine rich repeat containing 4C) were quantified together (NM_020929.2 and NM_001258419.1; F: TAAGTGGGTTCCAGTTTTGC / R: CCAACAGGTATTGATCTTCCTGAG). For DPP6 (dipeptidyl-peptidase 6), we quantified the expression of isoform 3 (NM_001039350; primers F: AACGTGATGGAGCTGGTG / R: CCGCTGGTGTCAGAAGTATG).
Magnetic Resonance Imaging (MRI)
Two members of the family, II-6 and III-2, underwent a structural magnetic resonance imaging (sMRI) session. Scanning sessions were conducted at the Douglas Institute Brain Imaging Centre, on a 3T Siemens Magnetom MRI scanner. Structural scans consisted of a high resolution, whole brain T1 acquisition. T1 weighted data were acquired using a flow-compensated 3D RF-spoiled GRE sequence with TR/TE/flip angle = 18 ms/10 ms/30°, Nex 1, and a 256×256×180 matrix with 1 mm3 isotropic voxels. To avoid reading bias, scans from six females, including those from II-6 and III-2, were read by an experienced radiologist (NM) blinded to the genetic status of the subjects but aware of their age. A systematic assessment was run, which included: movement artifacts, atrophy (pons, vermian, cortical, corpus callosum), white matter hyperintensities (infratentorial, cerebellar, pons, dentate nuclei, cerebellar peduncles, mesencephalon, periventricular, deep, juxtacortical, basal ganglia, semi ovale centrum, corticospinal tract, corpus callosum), Scheltens and Koedam scores, enlargement (ventricular, olfactive sulcus), infarct (territorial, lacunar, junctional), perivascular spaces dilatation, fluid cavities, and basal ganglia signal abnormalities.
Cloning and luciferase assays
We cloned wildtype (long fragment, 776bp) and CNV deletion model (short fragment, 137 bp) constructs of the 5′UTR of LRRC4C into a luciferase vector (Invivogen), using the TOPO cloning kit, following manufacturer’s instructions (Invitrogren, Carlsbad, CA). cDNA was amplified using the following primers.
LRRC4C-F8-BsrG1-sense: actgag TGTACA agtgagaaagaaggga
LRRC4C-R1-Nco1-sense: ctagct CCATGG ctccactgggggtctcta
LRRC4C-F8-Nco1-antisense: actgag CCATGG agtgagaaagaaggga
LRRC4C-R1-BsrG1-antisense: ctagct TGTACA ctccactgggggtctcta
LRRC4C-F1-BsrG1-sense: agtttt TGTACA tggcttactttttggcgg
LRRC4C-F1-Nco1-antisense: agtttt CCATGG tggcttactttttggcgg
All fragments were cloned in the TOPO IV vector and Sanger sequenced to confirm orientation and to determine that fragments were mutation-free. To transfer inserts from the TOPO IV vector to the luciferase-containing vector, both vectors were digested with BsrG1 and Nco1 enzymes. Inserts were gel purified and ligated at 4°C overnight, where the ligation product was transformed into GT115 competent cells. Positive transformants were identified by Sanger sequencing. To measure luciferase activity, 250 ng of construct was transfected in HEK 293 cells using the Jetprime reagent with an equivalent amount of pGL3 control vector used for signal normalization. Luciferase activities were quantified using the dual luciferase reporter (Promega, Madison, WI) kit 24 h after transfection in the cell medium and cellular extracts.
Clinical CNV cohort
From Signature Genomic Laboratories (SG), we analyzed a total of 14,077 non-prenatal NDD samples submitted for clinical genetic testing using oligonucleotide-based whole-genome array comparative genomic hybridization (aCGH), either a 105K-feature platform (SignatureChip OS version 1 or 4; custom-designed by SG, manufactured by Agilent Technologies, Santa Clara, CA) or a 135K-feature platform (SignatureChip OS version 2 or 3; custom-designed by SG, manufactured by Roche NimbleGen, Laval, Canada). This cohort has been fully defined and characterized in our previous work [Talkowski et al., 2012c]. The ethnic distribution in the samples from SG was estimated from a sampling cross-section where 75% were white individuals, 7% African American individuals, and 18% individuals of other race/ethnicity. The sex distribution was 59% male and 41% female.
Control cohorts
Controls used were a combination of several datasets, for a total of 8,960 individuals. We used control datasets from SickKids Toronto (OHI n=1,234, POPGEN n=1,123[Krawczak et al., 2006], and the Ontario Population Genomics Platform (OPGP), n=416 (http://www.tcag.ca/facilities/cyto_population_control_DNA.html), all of whom (n=2,773) had CNVs called from Affymetrix 6.0 SNP arrays, and all of which were carefully screened for false positives. The POPGEN cohort is a sample of control individuals from Northern Germany, while the OHI cohort is a set of control individuals from the Ottawa Heart Institute. The OPGP cohort are control individuals from Ontario consented for use as controls in genomic studies. From a Swedish control cohort [Bergen et al., 2012; Szatkiewicz et al., 2014], all subjects were born in Sweden and identified via the Swedish Hospital Discharge Register containing all individuals hospitalized in Sweden since 1973. There were 5,917 psychiatrically screened controls that were hospitalized only for non-psychiatric reasons. DNA was extracted from blood from the Swedish control cohort using standard methods at the Karolinska Institutet. All genotyping was conducted at the Broad Institute of Harvard and MIT, and genotypes and CNVs were called using the Birdsuite algorithm using the Affymetrix (Santa Clara, CA) 6.0 platform, as previously described. CNVs present in > 0.5% of controls were removed from analyses, and only CNVs >50Kb were included. All (n=8,960) controls are Caucasian.
RESULTS
Two independent BCR in a subject with autism, a sensory processing disorder, and apraxia
Individual II-6, who was recruited at the Douglas Hospital for an unrelated study, reported that her daughter, III-2, had a 46,XX,t(11;14)(p12;p12) translocation (Fig. 1), identified after four miscarriages, and a son with autism and other developmental problems (described below). To understand better the transmission and precise breakpoints of this reported chromosomal rearrangement, we collected DNA from II-6, III-2, and IV-1. After performing an aCGH (OMNI 2.5 array) on III-2 and IV-1, which revealed no other genetic anomalies, we performed jumping library sequencing [Talkowski et al., 2011] in all three subjects. We defined the translocation breakpoint, identical in all three subjects, at (GRCh37/hg19) Chr11:40614960 and ChrUn_gl000220:140245 (Fig. 1A; Supplemental Table I). gl000220.1 is a 161.8 kb unplaced genomic contig, which has still not been placed into the newest primary reference genome assembly (hg38); however, our results show gl000220.1 localizes at least to 14p12. Acrocentric short arms contain highly repetitive sequences, with most shared with other acrocentric short arms and which contain rDNA genes [Bandyopadhyay et al., 2001a; Bandyopadhyay et al., 2001b]. Additional characterization, outside the scope of this work, would be necessary to establish whether this sequence is unique to 14p or shared among more acrocentric short arms. The Chr11 breakpoint results in the direct disruption of LRRC4C, which encodes the netrin G1 ligand important in cortical and thalamic axon guidance [Lin et al., 2003]. The disruption occurs in intron three of this seven-exon gene. In the male with autism, IV-1, we also detected a paternally inherited Chr7 inversion [inv(7)(q34q36.2)]. We localized the breakpoints (hg19) to Chr7:140416412 and Chr7:153725012 (Fig. 1B, Supplemental Table SII). The Chr7:140416412 breakpoint localizes to an intergenic region, 9,965 bp from the 3′UTR of NDUFB2 and 17,952 bp from the 3′UTR of BRAF. The Chr7:153725012 breakpoint localizes to a highly polymorphic region of intron 1 of the longest and lowest expressed isoform of DPP6 (Dipeptidyl peptidase-like protein 6), a component of Kv4 channel complexes that may be important in the neuronal A-type potassium current [Maffie et al., 2013]. Our previous work suggested revised terminology for these findings [Ordulu et al., 2014], so the next-generation cytogenetic nomenclature for this case is:
46,XY,inv(7)(q34q36.2)pat,t(11;14)(p12;p12)mat.seq[GRCh37/hg19] inv(7)(pter- >q34(140,416,412)::GGATACTGTATCTGGATCCT::q36.2q34(153,725,012- 140,416,421)::ATGTACATAGT::q36.2(153,725,220)- >qter),t(11;Un_gl000220)(Un_gl000220(+)(140,236)::TGTCTTTTGTGATA::11p12(40,614,960)- >11qter;11pter->11p12(40,614,957)::TATATAG::Un_gl000220(+)(140,245))
FIG. 1.
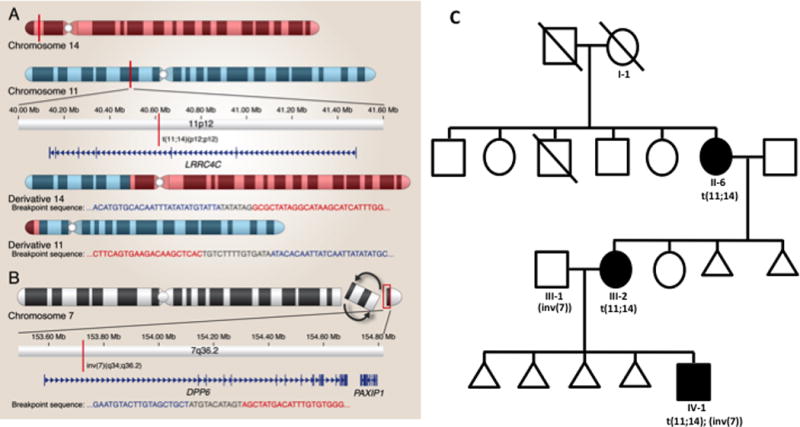
A subject with a sensory processing disorder, apraxia, and autism carries both a maternally inherited translocation affecting LRRC4C and a paternally inherited inversion on chromosome 7 affecting DPP6.
(A) Balanced translocation of chromosomes 11 and 14 that disrupts a highly repetitive region with no known genes on Chr14 and on Chr11 at Chr11:40614960, within the gene LRRC4C that encodes a netrin G1 ligand important in cortical and thalamic axon guidance. This disruption occurs in intron 3 of this seven exon gene. (B) Inversion with breakpoints mapped to Chr7:140416412, which falls in an intergenic region, and Chr7:153725012, which is in intron 1 of isoform 3 of DPP6 (dipeptidyl peptidase-like protein 6), a component of Kv4 channel complexes and thought to be important in the neuronal A-type potassium current and dendritic stability during neurodevelopment. (C) The family pedigree: I-1 died by suicide at age 54 and had multiple episodes of major depressive disorder (MDD) but is of unknown carrier status. II-6 had two serious episodes of panic disorder at age 27 and 40 years, both of which were successfully treated. III-2 had anorexia at 8 and 14 years and two episodes of MDD at 16 and 20 years. IV-1 was diagnosed with autism spectrum disorder, an oral hypersensory disorder, and apraxia, at age 3.5 years. No other members of this pedigree were assessed for translocation status or clinical phenotype.
Clinical and neurological information for members with chromosomal rearrangements
Individual IV-1 (born in: 2009) is the only son of III-2 (Fig. 1C). Pregnancy and delivery were without issue, though the family noticed lack of eye contact at an early age and delayed milestones including complete lack of speech until age 3 and walking after age 2. The proband was evaluated at a specialty clinic at the Montreal Children’s Hospital and diagnosed with autism using the ADOS and ADI. He also experiences some intellectual disability in that by age 5 he could only read some words, say the alphabet, and count as high as 30. He is able to socialize with family members on his own terms but does not like to be touched and rarely makes eye contact when speaking. He is energetic, has no concept of danger, struggles with transitions from one activity to the next, and has some trouble balancing and jumping. Physiotherapy and speech and language training after age 3 led to significant improvements in speech, implying a lack of regression.
Besides meeting criteria for autism, the mother reports other particular features specific to the proband. Specifically, that he never chews food or moves his tongue despite the ability to do so. Because of this, he was assessed for an oral sensory processing disorder which revealed extreme hypersensitivity to most textured foods in his mouth, unrelated to taste or desire for food. He also has a severe case of apraxia, two examples of which are his lack of response to painful stimuli (despite being able to sense pain), and not going to the bathroom despite acknowledging the need to do so. He has a fixation with trains that interferes with daily activities.
II-6 (born in 1950) is a 64-year-old woman and has five siblings. Her mother, I-1, suffered from major depression and committed suicide at age 54. No DNA is available for individual I-1, and the family was unsure of any history of miscarriages she may have had. II-6 reported a history of panic disorder associated with depressive symptoms for which she received successful treatment with psychological therapy and later recurrence was successfully treated with an antidepressant throughout a 9-month period. She had two miscarriages. She has completed high school and has a 2-year college diploma. The cognitive battery showed that II-6 is within the 33% lowest performance for TMT-B, categorical verbal fluency and IGT, and the 10% lowest performance for TMT-A and Stroop Interference score and errors.
Individual III-2 (born in 1980) suffered from anorexia nervosa at age 8 during a 1-year period and again at age 14, when she was followed in a specialty clinic. III-2 presented with two major depressive episodes at 16 and 20 years of age and was treated with several antidepressants. She has had four miscarriages and has a current diagnosis of ADHD. III-2 was within normal range for all psychiatric assessment and her highest level of education completed is a Bachelor’s degree. Neuropsychological profiling showed that III-2 is within the 33% lowest performance for IGT, category verbal fluency, Stroop interference score and error, and TMT-A. We conclude that the translocation does not associate with strong effect cognitive or structural deficits.
There was limited information available on III-1, the father of IV-1 who carries the inv(7)(q34q36.2), whom we did not consent to a clinical evaluation. Spousal report suggests he is a healthy individual, who is shy and works in computing.
Because the translocation disrupts a gene encoding an axon guidance-related molecule [DeNardo et al., 2012; Kim et al., 2006], which could affect brain structure, we tested II-6 and III-2 using structural MRI and cognitive performance evaluations. II-6’s MRI scan revealed moderate cortical and vermian hypoplasia, a moderate ventricular enlargement, and a moderate olfactive sulcus enlargement compared to six control individuals (Supplemental Fig. S1). There were no anomalies in III-2’s MRI scan.
Translocation and inversion breakpoints are located in genomic regions that are transcribed in human brain
To assess whether the chromosomal rearrangements, that is, inv(7) and t(11;14), were in transcriptional products, we undertook RNA analysis in healthy tissues. There are only two protein-coding transcript variants of LRRC4C. Two other transcripts produced from the 3’ end of this locus are out of frame and non-coding. Both of the gene’s protein-coding isoforms, NM_020929.2 and NM_001258419.1 [Rajasekharan and Kennedy 2009], are predicted to be disrupted by the translocation (Fig. 2A). Primers targeting both isoforms show expression in adult human brain regions as well as in fetal brain, while only mild expression was detected in spinal cord with extremely low or absent expression in the kidney, liver, or lung (Fig. 2A).
FIG. 2.
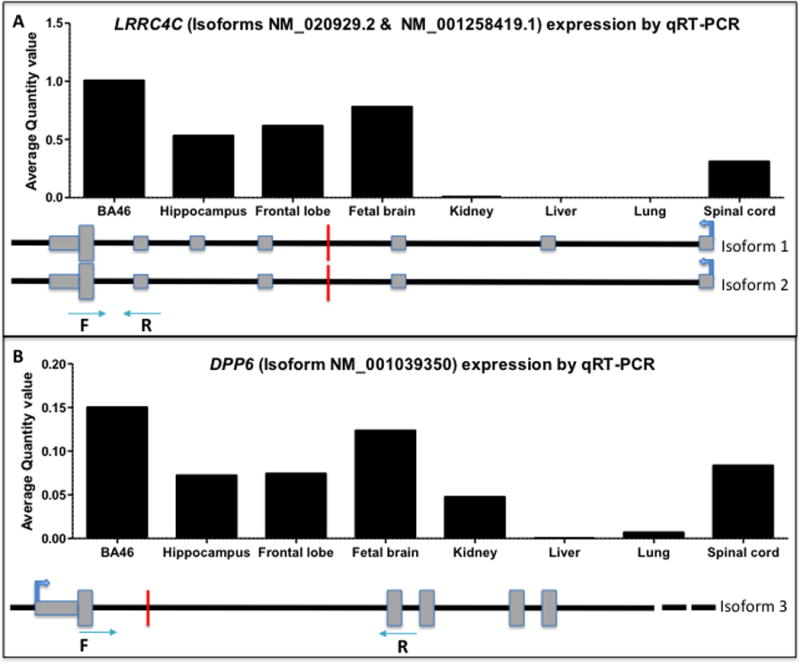
Translocation breakpoints are in genomic regions encoding for mRNAs in control human brain.
(A) mRNA expression of LRRC4C was quantified for both isoforms disrupted by the balanced translocation, NM_020929.2 and NM_001258419.1, in adult human brain regions (frontal cortex, BA46, and hippocampus) and fetal brain, as well as spinal cord, kidney, liver, and lung tissues. (B) mRNA expression of DPP6 was quantified for isoform 3 (NM_001039350) in adult human brain regions (frontal cortex, BA46, and hippocampus) and fetal brain, as well as spinal cord, kidney, liver, and lung tissues, as this isoform was the only one affected by the Chr7 inversion.
There are three predicted isoforms of DPP6, all of which have distinct transcription start sites. The inversion identified in family member IV-1 disrupts the promoter and exon 1 of isoform 3 (NM_001039350). The 5’UTR for DPP6 isoforms 1 and 2, which were the first to be identified [Wada et al., 1992], start ~171 kb and 423 kb downstream from exon 1 of isoform 3, respectively, and ~25 kb from the inversion breakpoint. Still, we observed expression of DPP6 isoform 3 in adult human brain regions as well as in fetal brain, while only mild expression was detected in spinal cord and low or absent expression in the kidney, liver, or lung (Fig. 2B).
Examination of CNVs in LRRC4C
To determine if LRRC4C variation are associated with NDDs, we took advantage of cases referred to Signature Genomics for clinical microarrays (N=14,077 NDD cases, no prenatal samples). To assess significance of LRRC4C-specific disruptions, we calculated a two-tailed Chi-square value using seven NDD cases (Fig. 3A and Table I) from a pool of 14,077 cases, and compared these with the 8,690 control cases where there was one control with an exon disrupting CNV (Χ2 = 2.23, p=0.14). We conclude that CNVs in LRRC4C have a non-significant association with NDDs.
FIG. 3.

Copy number analysis of LRRC4C and DPP6 in subjects referred for genetic diagnostic screening.
A total of 14,077 non-prenatal NDD subjects referred to Signature Genomics for clinical genetic testing were screened for intragenic exon-disrupting copy number variants (CNVs). Deletion cases (Red bars), duplication cases (Blue bars).
Table I.
Intragenic CNVs in LRRC4C and DPP6 from 14,077 neurodevelopmental disorder cases referred for clinical microarrays
| GCAD ID | Sex | Age | Indication | Inheritance | Platform | CNV type | Max coordinates (hg19) | Array ISCN (hg18 unless otherwise specified) |
|---|---|---|---|---|---|---|---|---|
| LRRC4C | ||||||||
| 60128 | Male | 2y | Mixed development disorder, Multiple congenital anomalies, Hearing loss | Unknown | Signature v2.0 12-plex | Loss | chr11:40223248-40756156 | none |
| 12480 | Female | 10y | Developmental Delay, Seizure Disorder | Unknown | Signature v1.1 2-plex | Loss | chr11:40339674-40477655 | arr cgh 1q44(243,006,404->244,452,092)x1 |
| 24623 | Male | 5y | Developmental Delay | Maternal | Signature v1.1 Rev. B 2-plex | Loss | chr11:40710283-41059922 | arr 2q33.1(203,099,433-203,183,738)x3 dn |
| 64648 | Female | 7y | ADHD, Congenital heart defect | Unknown | Signature v3.0 12-plex | Loss | chr11:40390398-40806705 | arr 16p11.2(29,564,890-30,100,123)x1 |
| 81154 | Male | 6y | Dysmorphic features, Speech delay | Unknown | Signature v4.0 4-plex – CGH | Loss | chr11:40585689-40784701 | none |
| 55397 | Male | 9y | Autism spectrum disorder | Unknown | Signature v2.0 12-plex | Gain | chr11:40059697-40292517 | none |
| 63559 | Male | 7y | Developmental delay, ADHD, Other psychological or physical stress, not elsewhere classified | Unknown | Signature v3.0 12-plex | Gain | chr11:41439036-41688684 | none |
| 56338 | Male | 1M | Clover leaf shaped head, Hypospadias, Dilated renal pelvis, Intrauterine growth restriction | Unknown | Signature v3.0 12-plex | Loss | chr11:40756156-41107103 | arr 2q11.2(96,104,157-97,384,378)x1 mat |
| DPP6 | ||||||||
| 50965 | Female | 11m | Delayed milestones | Unknown | Signature v2.0 12-plex | Loss | chr7: 154063870-154179914 | none; |
| 76774 | Male | NA | Autism spectrum disorder | Unknown | Signature v4.0 4-plex | Loss | chr7: 154113977-154142083 | none |
| 82653 | Female | 5y | Developmental delay | Unknown | Signature v4.0 4-plex – CGH | Loss | chr7: 154430846-154459512 | arr[hg19] 7q36.1(150,844,263-151,091,892)x1 |
| 2082 | Male | NA | Autism | Unknown | Signature v2.0 12-plex | Gain | chr7: 153805929-154190828 | none; |
| 72989 | Male | 0m | Fetal loss/stillbirth/POC, Premature rupture of membranes, Central nervous system malformation in fetus | Unknown | Signature v3.1 12-plex | Gain | chr7: 153522549-153653554 | none |
| GC12628 | Male | 15y | Autism | Maternal | Signature v4.0 1-plex | Gain | none | |
Examination of CNVs in DPP6
There is high CNV variation in isoform 3 exon 1 of DPP6 (hg19; Chr7: 153485627- 153682815, occurring in 26 control subjects) in the database of genomic variation, and this CNV is also within the site of the inversion breakpoint, >25kb distant from the transcription start sites and promoter of isoforms 1 and 2. This CNV was excluded from all analyses in DPP6. We identified six rare CNVs in DPP6 in NDD cases in isoforms 1 and 2 and no controls (Fig. 3B and Table I). Two-tailed Χ2-square test revealed a marginally non-significant value (Χ2 = 3.7, p=0.05). Similar to LRRC4C, we suggest that variation in DPP6 has a suggestive but non-significant association with NDDs.
DPP6 mutations leading to increased mRNA have been previously associated with familial ventricular fibrillation [Alders et al., 2009]. There was no cardiac phenotype reported in any of the NDD cases with CNVs over this locus; however, to further assess the potential association between DPP6 variation and heart anomalies, we analyzed DPP6 CNVs derived from 7,006 prenatal Signature samples (Supplemental Table SIII). Nine cases had CNV duplications, six of whom had a heart anomaly noted on the ultrasound. Two of three cases with CNV deletions affecting DPP6 had heart anomaly. There are two important caveats for this observation: (i) heart anomalies are unlikely to be rare from a population of prenatal cases sent for genomic analysis: (ii) These are structural heart deficits observed by ultrasound and so are very different the ventricular fibrillation phenotype identified by Anders et al.
In vitro functional analysis of LRRC4C 5’UTR length
The identified translocation in LRRC4C generates a predicted loss of the 5’ portion of LRRC4C, affecting the first three exons; however, only the terminal exon of LRRC4C is coding, meaning that this gene has a long, spliced 5’UTR. While this suggests a haploinsufficiency model for the proband since the promoter is also translocated, we wanted to explore this unusually long 5’UTR since several NDD cases from the Signature cohort had intragenic deletions in this region. To assess the affect of the length of the LRRC4C 5’UTR on gene expression, we cloned both a long 5’UTR and a short 5’UTR into a vector with a minimal promoter and the Luciferase gene (Fig. 4A–D). We found that a shorter length of 5’UTR led to a strong increase in activity of Luc compared to a longer form, suggesting that the 5’UTR of LRRC4C might be an important regulatory mechanism to dampen expression.
FIG. 4.
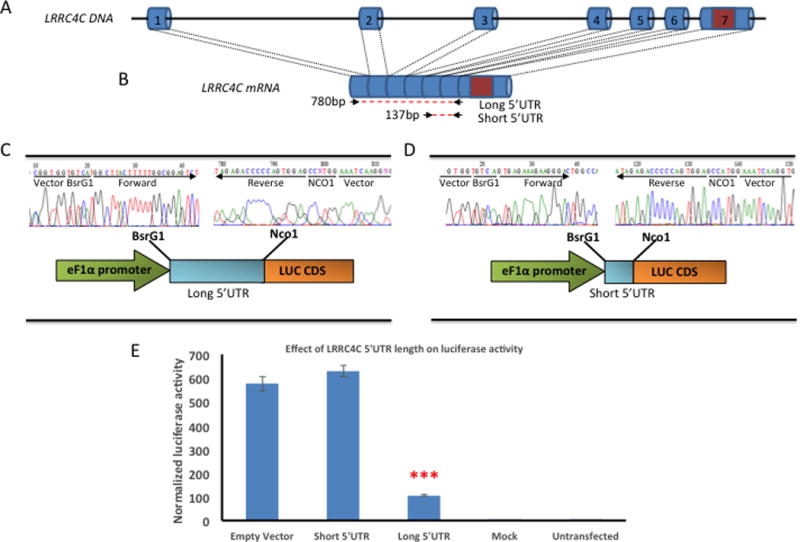
Functional impact of LRRC4C 5’UTR length on expression.
(A) Genomic locus of LRRC4C, with exons numbered. Non-coding exons are colored blue, while the orange exon represents the single protein coding exon. (B) mRNA of LRRC4C with approximate positions of long and short cloning targets shown with red-dotted line. Numbers represent the cloning product length, in basepairs. (C) Sanger sequencing of vector-amplicon junction demonstrating successful cloning of LRRC4C long fragment. Cartoon fragment shows vector with promoter (green), restriction enzyme sites, cloned amplicon (blue), and luciferase gene (green). (D) Sanger sequencing of vector-amplicon junction demonstrating successful cloning of LRRC4C short fragment. Cartoon fragment shows vector with promoter (green), restriction enzyme sites, cloned amplicon (blue), and luciferase gene (green). (E) Luciferase assay: No signal in untransfected or mock cells, no significant difference between the short 5’UTR and promoter-only constructs (p=0.237), but a very significant decrease in Luc activity with the long LRRC4C 5’UTR compared to the short LRRC4C 5′UTR (p=1.2×10−9).
LRRC4C as a potential genetic modifier of NDDs
In the Signature cohort, we found that 3/5 subjects with deletions only in LRRC4C also have a second genomic abnormality (Table I), while a single subject with a deletion in DPP6 had a CNV associated with a genomic syndrome CNV (1/6). The pathogenic CNVs associated with LRRC4C mutations are all known to be associated with syndromes of variable expressivity, including 1q44 [Thierry et al., 2012] (deletion), 2q33.1 [Docker et al., 2014] (duplication), and 16p11.2 [Hanson et al., 2010] (deletion). No LRRC4C duplication cases had co-incident secondary pathogenic CNVs, implying that if a genetic modifier effect exists it may be specific to deletions. We identified another subject from the non-NDD Signature cohort, who was too young (1-month old) to diagnose with an NDD (Table I; subject 56338), and who had a secondary anomaly consistent with variable expressivity (2q11.2 deletion [Riley et al., 2015]). Finally, we also accessed all cases from DECIPHER that have CNVs affecting LRRC4C (Supplemental Table SIV). Four of eight cases with intragenic deletions in LRRC4C had secondary lesions, three of which were on chromosome 16, with one on chromosome 8. No lesions were in genomic regions unambiguously associated with disease.
Assessment of other Netrin G family members in neurodevelopmental disorders
LRRC4C is also known as the netrin G ligand 1 (NGL-1), which is one member of a highly specialized family of neuronal guidance molecules. The netrin G family includes LRRC4 (NGL-2) and LRRC4B (NGL-3) [Woo et al., 2009b]. Receptors for netrin G ligands include NTNG1 (which binds LRRC4C), NTNG2 (which binds LRRC4), and PTPRF (aka LAR) [Takahashi and Craig 2013], which binds LRRC4B [Nishimura-Akiyoshi et al., 2007]. Netrin G ligands and receptors are expressed in non-overlapping patterns in mouse brain, specifically with differences between the thalamus and cortical regions, which are suggested to guide thalmocortical and corticothalamic projections [Nakashiba et al., 2002; Yin et al., 2002]. We confirmed this relationship in human brain using microarray gene expression data from the six available adult brains from the Allen Brain Atlas and extracted all microarray probes for netrin G ligands and receptors (Supplemental Fig. S2)
Netrin G members regulate excitatory synapse formation [Kim et al., 2006; Kwon et al., 2010; Linhoff et al., 2009; Matsukawa et al., 2014; Takahashi and Craig 2013; Woo et al., 2009a], and NTNG1 has been previously implicated in neurodevelopment and sensory processing in different investigations [Aoki-Suzuki et al., 2005; Nectoux et al., 2007; O’Roak et al., 2012]. Similarly, absent startle responses have been identified in mice with deletions in NTNG2 or LRRC4, despite normal hearing [Zhang et al., 2008], supporting a potential role for netrin G ligands and receptors in sensory processing. These data together with the particular sensory phenotype of the probands in this study, led us to reason that the netrin G family of ligands and receptors may be important for sensory processing disorders in human. In the case and control datasets used here, we examined clinically reported CNV data for LRRC4, LRRC4B, NTNG1, NTNG2, and PTPRF (Supplemental Table SV). These data suggest that LRRC4B (Two NDD duplications and one NDD deletion; zero controls) and NTNG2 (three NDD duplications and zero controls) may be important in neurodevelopmental disorders and warrant further investigation in larger cohorts. Larger datasets should provide a more complete assessment of the potential role of CNVs affecting the Netrin G family and NDDs.
DISCUSSION
We identified a boy with autism, apraxia, and a sensory processing disorder who had a maternally inherited translocation (46,XY,t(11;14)(p12;p12)mat) and a paternally inherited inversion of chromosome 7. Similar to our previous studies [Talkowski et al., 2012a], this study highlights the strength of BCR sequencing to discover new genes important in NDD, complemented by follow-up in many thousands of affected subjects.
The relationship between the phenotype and the translocation genotype within the family was ambiguous, though we could identify clinically relevant psychiatric features in all generations. There are some possible explanations as to why the affected son with autism, apraxia, and a sensory processing disorder had such a profound but dissimilar phenotype compared to his parents or grandparents (i) The combination of inv(7) and t(11;14) produced a novel phenotype not previously observed in the family (Fig. 5). (ii) The translocation breakpoint in LRRC4C shows a more deleterious phenotype in males, consistent with the female protective effect in some inherited ASD loci [Jacquemont et al., 2014]. Finally, (iii) While there were no clinically relevant CNVs in this case as assessed by clinical aCGH, the subject may harbor an unidentified mutation not detected in our assays.
FIG. 5.
Potential model for the influence of genetic variation in DPP6 and LRRC4C on behavior and disease.
We found suggestive but non-significant evidence for the involvement of LRRC4C and DPP6 mutations in NDDs. We used a cohort of 14,077 non-prenatal samples with an NDD indication and found non-significant P-values for independent association of each gene; one caveat for even a suggestive association here is that the control cohort is called with lower resolution arrays, so it is possible that the frequency of these CNVs in controls is higher than we observed.
We investigated the functionality of intragenic deletion CNVs in LRRC4C. Our investigations into this suggested that the 5’UTR of LRRC4C functions as a negative regulatory element. We do not suggest these mutations are pathological, but rather that they may be a predisposing factor to disease, dependent upon genomic background for actual disease expression. Notably, the translocation case was missing not only three exons (so had a truncated 5’UTR), but also had no promoter, meaning that the mechanism of action in the translocation case may be different than in LRRC4C CNV deletion cases. The lack of conclusive evidence for a direct effect of LRRC4C disruption on any clinical phenotype is consistent with it not being a strong-effect contributor to NDDs, yet the presence of this chromosomal rearrangement in a child with autism suggests that a contribution of the LRRC4C disruption may act to modify the penetrance or expression of NDD lesions. This modifier effect is supported by the finding that 3/5 cases with LRRC4C deletions had secondary clinically significant genetic lesions associated with reduced penetrance. Together these data argue that exonic deletions of LRRC4C may modify other genetic lesions.
The data reported here represent information from a family that carried a translocation in three generations, where the proband also inherited an inversion from his father. We sequenced the breakpoints from these karyotypic anomalies and found that they disrupted two different genes, LRRC4C and DPP6. We suggest that the co-incident inheritance of both BCRs may explain the autism and sensory deficits in the probands.
Supplementary Material
Acknowledgments
We thank the index family in this study for their time and collaboration. This work is supported by operating funds from the Canada Research Chairs program and the Scottish Rite Charitable Foundation to CE. SB was supported by a Banting and Best MSc Award from the CIHR. IK is supported by a NSERC summer undergraduate award. CCM and JFG acknowledge support from the Developmental Genome Anatomy Project.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest
References
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MW, Zeppenfeld K, Loh P, Koch KT, Demolombe S, Mannens MM, Bezzina CR, Wilde AA. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84(4):468–476. doi: 10.1016/j.ajhg.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki-Suzuki M, Yamada K, Meerabux J, Iwayama-Shigeno Y, Ohba H, Iwamoto K, Takao H, Toyota T, Suto Y, Nakatani N, Dean B, Nishimura S, Seki K, Kato T, Itohara S, Nishikawa T, Yoshikawa T. A family-based association study and gene expression analyses of netrin-G1 and -G2 genes in schizophrenia. Biol Psych. 2005;57(4):382–393. doi: 10.1016/j.biopsych.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay R, Berend SA, Page SL, Choo KH, Shaffer LG. Satellite III sequences on 14p and their relevance to Robertsonian translocation formation. Chromosome Res. 2001a;9(3):235–242. doi: 10.1023/a:1016652621226. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay R, McQuillan C, Page SL, Choo KH, Shaffer LG. Identification and characterization of satellite III subfamilies to the acrocentric chromosomes. Chromosome Res. 2001b;9(3):223–233. doi: 10.1023/a:1016648404388. [DOI] [PubMed] [Google Scholar]
- Bergen SE, O’Dushlaine CT, Ripke S, Lee PH, Ruderfer DM, Akterin S, Moran JL, Chambert KD, Handsaker RE, Backlund L, Osby U, McCarroll S, Landen M, Scolnick EM, Magnusson PK, Lichtenstein P, Hultman CM, Purcell SM, Sklar P, Sullivan PF. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol Psych. 2012;17(9):880–886. doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand H, Collins RL, Hanscom C, Rosenfeld JA, Pillalamarri V, Stone MR, Kelley F, Mason T, Margolin L, Eggert S, Mitchell E, Hodge JC, Gusella JF, Sanders SJ, Talkowski ME. Paired-Duplication Signatures Mark Cryptic Inversions and Other Complex Structural Variation. Am J Hum Genet. 2015;97(1):170–176. doi: 10.1016/j.ajhg.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand H, Pillalamarri V, Collins RL, Eggert S, O’Dushlaine C, Braaten EB, Stone MR, Chambert K, Doty ND, Hanscom C, Rosenfeld JA, Ditmars H, Blais J, Mills R, Lee C, Gusella JF, McCarroll S, Smoller JW, Talkowski ME, Doyle AE. Cryptic and complex chromosomal aberrations in early-onset neuropsychiatric disorders. Am J Hum Genet. 2014;95(4):454–461. doi: 10.1016/j.ajhg.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo LA, de Wit J, Otto-Hitt S, Ghosh A. NGL-2 regulates input-specific synapse development in CA1 pyramidal neurons. Neuron. 2012;76(4):762–775. doi: 10.1016/j.neuron.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docker D, Schubach M, Menzel M, Munz M, Spaich C, Biskup S, Bartholdi D. Further delineation of the SATB2 phenotype. European journal of human genetics. Eur J Hum Genet. 2014;22(8):1034–1039. doi: 10.1038/ejhg.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Nasir RH, Fong A, Lian A, Hundley R, Shen Y, Wu BL, Holm IA, Miller DT, p11.2 Study Group C Cognitive and behavioral characterization of 16p11.2 deletion syndrome. J Dev Behav Pediatr. 2010;31(8):649–657. doi: 10.1097/DBP.0b013e3181ea50ed. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Coe BP, Hersch M, Duyzend MH, Krumm N, Bergmann S, Beckmann JS, Rosenfeld JA, Eichler EE. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 2014;94(3):415–425. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Burette A, Chung HS, Kwon SK, Woo J, Lee HW, Kim K, Kim H, Weinberg RJ, Kim E. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat Neurosci. 2006;9(10):1294–1301. doi: 10.1038/nn1763. [DOI] [PubMed] [Google Scholar]
- Klempan TA, Rujescu D, Merette C, Himmelman C, Sequeira A, Canetti L, Fiori LM, Schneider B, Bureau A, Turecki G. Profiling brain expression of the spermidine/spermine N1-acetyltransferase 1 (SAT1) gene in suicide. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(7):934–943. doi: 10.1002/ajmg.b.30920. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9(1):55–61. doi: 10.1159/000090694. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Shendure J, Eichler EE. A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci. 2014;37(2):95–105. doi: 10.1016/j.tins.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SK, Woo J, Kim SY, Kim H, Kim E. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. The J Biol Chem. 2010;285(18):13966–13978. doi: 10.1074/jbc.M109.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JC, Ho WH, Gurney A, Rosenthal A. The netrin-G1 ligand NGL-1 promotes the outgrowth of thalamocortical axons. Nature Neurosci. 2003;6(12):1270–1276. doi: 10.1038/nn1148. [DOI] [PubMed] [Google Scholar]
- Linhoff MW, Lauren J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61(5):734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffie JK, Dvoretskova E, Bougis PE, Martin-Eauclaire MF, Rudy B. Dipeptidyl-peptidase-like-proteins confer high sensitivity to the scorpion toxin AmmTX3 to Kv4-mediated A-type K+ channels. J Physiol. 2013;591(Pt 10):2419–2427. doi: 10.1113/jphysiol.2012.248831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa H, Akiyoshi-Nishimura S, Zhang Q, Lujan R, Yamaguchi K, Goto H, Yaguchi K, Hashikawa T, Sano C, Shigemoto R, Nakashiba T, Itohara S. Netrin-G/NGL complexes encode functional synaptic diversification. J Neurosci. 2014;34(47):15779–15792. doi: 10.1523/JNEUROSCI.1141-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashiba T, Nishimura S, Ikeda T, Itohara S. Complementary expression and neurite outgrowth activity of netrin-G subfamily members. Mech Dev. 2002;111(1-2):47–60. doi: 10.1016/s0925-4773(01)00600-1. [DOI] [PubMed] [Google Scholar]
- Nectoux J, Girard B, Bahi-Buisson N, Prieur F, Afenjar A, Rosas-Vargas H, Chelly J, Bienvenu T. Netrin G1 mutations are an uncommon cause of atypical Rett syndrome with or without epilepsy. Pediatric Neuro. 2007;37(4):270–274. doi: 10.1016/j.pediatrneurol.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Nishimura-Akiyoshi S, Niimi K, Nakashiba T, Itohara S. Axonal netrin-Gs transneuronally determine lamina-specific subdendritic segments. Proc Natl Acad Sci USA. 2007;104(37):14801–14806. doi: 10.1073/pnas.0706919104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordulu Z, Wong KE, Currall BB, Ivanov AR, Pereira S, Althari S, Gusella JF, Talkowski ME, Morton CC. Describing sequencing results of structural chromosome rearrangements with a suggested next-generation cytogenetic nomenclature. Am J Hum Genet. 2014;94(5):695–709. doi: 10.1016/j.ajhg.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekharan S, Kennedy TE. The netrin protein family. Genome Biol. 2009;10(9):239. doi: 10.1186/gb-2009-10-9-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KN, Catalano LM, Bernat JA, Adams SD, Martin DM, Lalani SR, Patel A, Burnside RD, Innis JW, Rudd MK. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am J Med Genet A. 2015;167A(11):2664–2673. doi: 10.1002/ajmg.a.37269. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF, 3rd, Klei L, Mandell JD, Moreno-De-Luca D, Poultney CS, Robinson EB, Smith L, Solli-Nowlan T, Su MY, Teran NA, Walker MF, Werling DM, Beaudet AL, Cantor RM, Fombonne E, Geschwind DH, Grice DE, Lord C, Lowe JK, Mane SM, Martin DM, Morrow EM, Talkowski ME, Sutcliffe JS, Walsh CA, Yu TW, Autism Sequencing C, Ledbetter DH, Martin CL, Cook EH, Buxbaum JD, Daly MJ, Devlin B, Roeder K, State MW. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatkiewicz JP, O’Dushlaine C, Chen G, Chambert K, Moran JL, Neale BM, Fromer M, Ruderfer D, Akterin S, Bergen SE, Kahler A, Magnusson PK, Kim Y, Crowley JJ, Rees E, Kirov G, O’Donovan MC, Owen MJ, Walters J, Scolnick E, Sklar P, Purcell S, Hultman CM, McCarroll SA, Sullivan PF. Copy number variation in schizophrenia in Sweden. Mol Psych. 2014;19(7):762–773. doi: 10.1038/mp.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Craig AM. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends Neurosci. 2013;36(9):522–534. doi: 10.1016/j.tins.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, Kirby A, Liu S, Muddukrishna B, Ohsumi TK, Shen Y, Borowsky M, Daly MJ, Morton CC, Gusella JF. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011;88(4):469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Maussion G, Crapper L, Rosenfeld JA, Blumenthal I, Hanscom C, Chiang C, Lindgren A, Pereira S, Ruderfer D, Diallo AB, Lopez JP, Turecki G, Chen ES, Gigek C, Harris DJ, Lip V, An Y, Biagioli M, Macdonald ME, Lin M, Haggarty SJ, Sklar P, Purcell S, Kellis M, Schwartz S, Shaffer LG, Natowicz MR, Shen Y, Morton CC, Gusella JF, Ernst C. Disruption of a large intergenic noncoding RNA in subjects with neurodevelopmental disabilities. Am J Hum Genet. 2012a;91(6):1128–1134. doi: 10.1016/j.ajhg.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Ordulu Z, Pillalamarri V, Benson CB, Blumenthal I, Connolly S, Hanscom C, Hussain N, Pereira S, Picker J, Rosenfeld JA, Shaffer LG, Wilkins-Haug LE, Gusella JF, Morton CC. Clinical diagnosis by whole-genome sequencing of a prenatal sample. N Engl J Med. 2012b;367(23):2226–2232. doi: 10.1056/NEJMoa1208594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Rosenfeld JA, Blumenthal I, Pillalamarri V, Chiang C, Heilbut A, Ernst C, Hanscom C, Rossin E, Lindgren AM, Pereira S, Ruderfer D, Kirby A, Ripke S, Harris DJ, Lee JH, Ha K, Kim HG, Solomon BD, Gropman AL, Lucente D, Sims K, Ohsumi TK, Borowsky ML, Loranger S, Quade B, Lage K, Miles J, Wu BL, Shen Y, Neale B, Shaffer LG, Daly MJ, Morton CC, Gusella JF. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012c;149(3):525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry G, Beneteau C, Pichon O, Flori E, Isidor B, Popelard F, Delrue MA, Duboscq-Bidot L, Thuresson AC, van Bon BW, Cailley D, Rooryck C, Paubel A, Metay C, Dusser A, Pasquier L, Beri M, Bonnet C, Jaillard S, Dubourg C, Tou B, Quere MP, Soussi-Zander C, Toutain A, Lacombe D, Arveiler B, de Vries BB, Jonveaux P, David A, Le Caignec C. Molecular characterization of 1q44 microdeletion in 11 patients reveals three candidate genes for intellectual disability and seizures. Am J Med Genet A. 2012;158A(7):1633–1640. doi: 10.1002/ajmg.a.35423. [DOI] [PubMed] [Google Scholar]
- Wada K, Yokotani N, Hunter C, Doi K, Wenthold RJ, Shimasaki S. Differential expression of two distinct forms of mRNA encoding members of a dipeptidyl aminopeptidase family. Proc Natl Acad Sci USA. 1992;89(1):197–201. doi: 10.1073/pnas.89.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J, Kwon SK, Choi S, Kim S, Lee JR, Dunah AW, Sheng M, Kim E. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nature Neurosci. 2009a;12(4):428–437. doi: 10.1038/nn.2279. [DOI] [PubMed] [Google Scholar]
- Woo J, Kwon SK, Kim E. The NGL family of leucine-rich repeat-containing synaptic adhesion molecules. Molecular and cellular neurosciences. 2009b;42(1):1–10. doi: 10.1016/j.mcn.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Yin Y, Miner JH, Sanes JR. Laminets: laminin- and netrin-related genes expressed in distinct neuronal subsets. Mol Cell Neurosci. 2002;19(3):344–358. doi: 10.1006/mcne.2001.1089. [DOI] [PubMed] [Google Scholar]
- Zhang W, Rajan I, Savelieva KV, Wang CY, Vogel P, Kelly M, Xu N, Hasson B, Jarman W, Lanthorn TH. Netrin-G2 and netrin-G2 ligand are both required for normal auditory responsiveness. Genes Brain Behav. 2008;7(4):385–392. doi: 10.1111/j.1601-183X.2007.00361.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.