

Abstract
HMG20A (also known as iBRAF) is a chromatin factor involved in neuronal differentiation and maturation. Recently small nucleotide polymorphisms (SNPs) in the HMG20A gene have been linked to type 2 diabetes mellitus (T2DM) yet neither expression nor function of this T2DM candidate gene in islets is known. Herein we demonstrate that HMG20A is expressed in both human and mouse islets and that levels are decreased in islets of T2DM donors as compared to islets from non-diabetic donors. In vitro studies in mouse and human islets demonstrated that glucose transiently increased HMG20A transcript levels, a result also observed in islets of gestating mice. In contrast, HMG20A expression was not altered in islets from diet-induced obese and pre-diabetic mice. The T2DM-associated rs7119 SNP, located in the 3′ UTR of the HMG20A transcript reduced the luciferase activity of a reporter construct in the human beta 1.1E7 cell line. Depletion of Hmg20a in the rat INS-1E cell line resulted in decreased expression levels of its neuronal target gene NeuroD whereas Rest and Pax4 were increased. Chromatin immunoprecipitation confirmed the interaction of HMG20A with the Pax4 gene promoter. Expression levels of Mafa, Glucokinase, and Insulin were also inhibited. Furthermore, glucose-induced insulin secretion was blunted in HMG20A-depleted islets. In summary, our data demonstrate that HMG20A expression in islet is essential for metabolism-insulin secretion coupling via the coordinated regulation of key islet-enriched genes such as NeuroD and Mafa and that depletion induces expression of genes such as Pax4 and Rest implicated in beta cell de-differentiation. More importantly we assign to the T2DM-linked rs7119 SNP the functional consequence of reducing HMG20A expression likely translating to impaired beta cell mature function.
Introduction
Type 2 Diabetes Mellitus (T2DM) is a metabolic disease characterized by impaired insulin secretion and/or action in target organs that leads to elevations in blood glucose levels. Environmental factors as well as strong genetic components contribute to the pathogenesis of T2DM. Thus far, 100–120 susceptibility loci have been associated to T2DM by Genome Wide Association Studies (GWAS)1–3. Although functional defects remain to be assigned, many of these loci point to primary defects in beta cell function rather than to insulin resistance4. In this context, several SNPs within the HGM20A gene (also known as iBRAF) have been associated to T2DM5–8. Of particular interest is the rs7119 SNP located within the 3′ UTR of the HMG20A transcript. In silico analysis revealed that rs7119 modifies a functional microRNA (miRNA) cis-regulatory element9. However, the molecular consequences of this SNP on the expression of HMG20A and/or its regulation and impact on islet physiology are still unknown.
HMG20A is a member of the high mobility group (HMG) box-containing genes that binds to chromatin and exerts global genomic changes through establishing active or silent chromatin10. HMG20A is highly expressed in mature neurons and plays a key role in promoting neuronal differentiation during development11. In this context, HMG20A relieves the transcriptional repression imposed by the complex LSD1–CoREST histone demethylase12, which function is to silence neuronal genes in non-neuronal tissues through its interaction with the transcription factor REST. In analogy, epigenetic repression of the Rest gene in pancreatic precursors was shown to coincide with the activation of the core beta cell program13. Mature pancreatic beta cells do not express REST while forced expression results in inhibition of neuronal proteins of the insulin exocytotic machinery such as SNAP25 and SYNAPTOTAGMIN VII (SYT7) leading to impaired glucose-induced insulin secretion14. Interestingly, REST was shown to repress expression of key beta cell development genes, such as NEUROD and PAX415,16. NEUROD is a bona fide target gene of HMG20A in neuronal cells11 and mutations in this gene predispose individuals to maturity onset diabetes of the young 6 (MODY6)17.
HMG20A is also implicated in epithelial-to-mesenchymal transition (EMT) through interaction with specific key regulators of this process such as SNAIL18. EMT is an example of cell plasticity and a key process during embryonic development, and together with the reverse transformation, the mesenchymal-to-epithelial transition (MET), are required for the formation of organs in the final destinations of embryonic migratory cells19,20. Both processes occur during pancreatic and islet development and require extensive reorganization of the epigenetic information of the cells. Remarkably, not only in development but also in response to different physiological demands, beta cells may de-differentiate in order to acquire plasticity capabilities and increase survival21,22, two processes that may implicate PAX423,24.
Based on these findings, we hypothesized that HMG20A may contribute to the regulation of key genes such as NeuroD and Pax4 which are essential for beta cell functional maturity as well as survival. Towards this goal, herein we investigate the expression profile and the gene regulatory function of HMG20A in pancreatic islets, as well as determine whether the SNP rs7119 associated with T2DM impacts HMG20A expression. We report that T2DM-associated SNP rs7119 leads to altered HMG20A expression, and that HMG20A regulates metabolism-secretion coupling genes as well as functional maturity of beta cells.
Results
HMG20A is expressed in pancreatic islets and transcript levels are decreased in islets from T2DM donors
As a first step to assign a potential role of the HMG20A gene in pancreatic islet physiology, we determined its transcript levels in islets as compared to other tissues. Mouse pancreatic islets displayed comparable expression levels of Hmg20a to other organs such as adipose tissue (white and brown), brain and muscle whereas the liver displayed highest levels (Fig. 1a). HMG20A co-stained with INSULIN (beta cells), GLUCAGON (alpha cells) and SOMATOSTATIN (delta cells) whereas its expression was rarely detected in exocrine pancreas (Fig. 1b). A similar endocrine cell expression pattern was detected in human islets (Fig. 1c). We next assessed whether expression levels of HMG20A were altered in islets isolated from T2DM donors. The rational was to establish a correlation between HMG20A levels and the hyperglycemic environment that may be altered by T2DM-linked SNPs. HMG20A transcript levels were decreased by ~60 % in islets from T2DM patients as compared to islets purified from normoglycemic control donors (Fig. 1d).
Fig. 1. HMG20A is expressed in both mouse and human islets and is decreased in islets from T2DM donors.

a HMG20A transcript levels were assessed by qPCR in islets, brain, liver, muscle, white adipose tissue (WAT), and brown adipose tissue (BAT) from mice (n = 6). Representative images of b mouse and c human islets co-stained for HMG20A (green) along with INSULIN (INS), GLUCAGON (GLUC) or SOMATOSTATIN (SMT) (red). Nuclei are stained using DAPI (blue). Magnification 40×. White boxes define areas enlarged in panels bellow. d HMG20A mRNA levels were measured by qPCR in human islets isolated from normoglycemic (control) or type 2 diabetic (T2DM) organ donors (n = 7). Data are depicted as dot plots with means ± S.E.M. p values were determined using unpaired two-tailed Student's t-test. **p < 0.01
Metabolic stressors modulate HMG20A expression in islets
As HMG20A transcript levels were decreased in T2DM islets, we reasoned that lipids and/or glucose might alter its expression. Towards addressing this premise, we used our mouse model of 45% high fat diet (HFD)-induced obesity and pre-diabetes to assess the contribution of lipids. These mice exhibit a typical 20% increase in body weight, develop glucose intolerance with the concomitant increases in insulin and leptin levels and decreased adiponectin levels25. Although these mice developed insulin resistance, glycaemia is normal due to increased insulin levels (Fig. 2a, b). Under these conditions, HMG20A protein levels were not significantly changed in islets as compared to levels in chow-fed control mice (Fig. 2c). In contrast, short-term exposure of INS-1E cells to palmitate resulted in a transient reduction in HMG20A transcript levels (Fig. 2d).
Fig. 2. HMG20A protein expression is not altered in islets of high fat diet (HFD)-induced obesity and pre-diabetic mice.
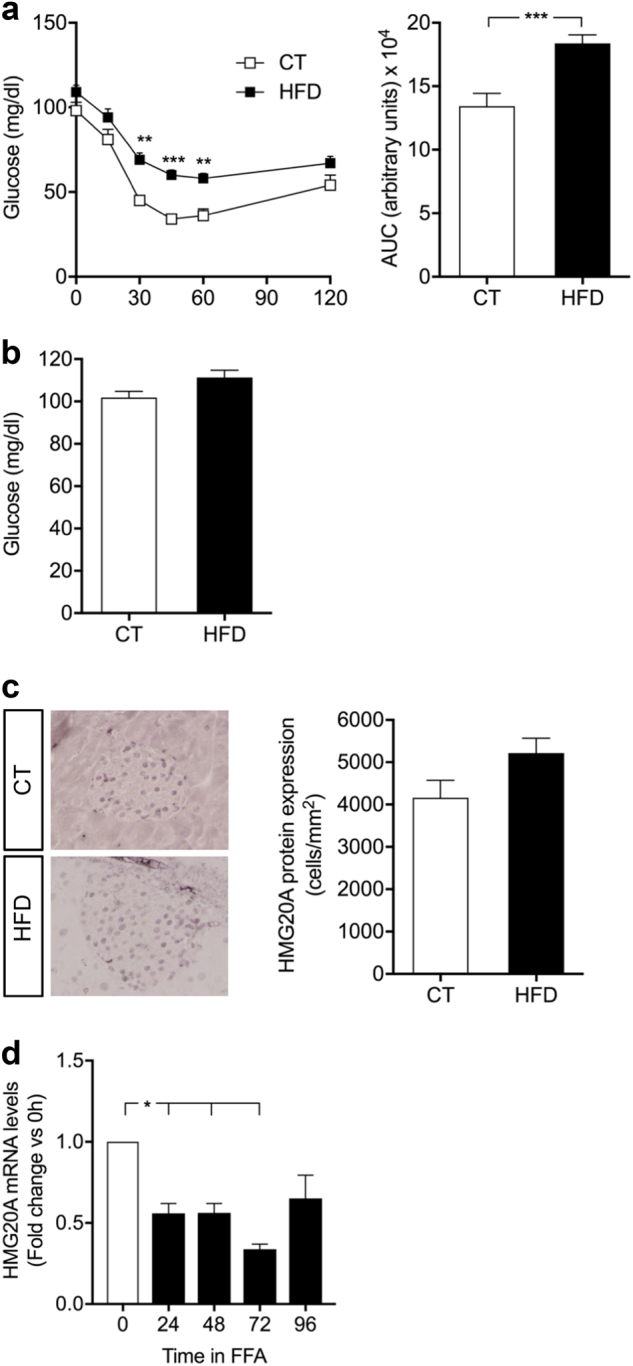
a Insulin tolerance test (ITT) and area under the curve (AUC) analysis in both control (CT) and HFD mice (CT, n = 10; HFD, n = 15). b Fasting blood glucose levels in both control and HFD mice (CT, n = 10; HFD, n = 15). c Representative images of pancreatic sections from HFD and CT mice stained and quantified for HMG20A (n = 4). d Hmg20a mRNA levels were assessed by qPCR in INS-1E cells cultured with 0.05 mM palmitate for 24, 48, 72 or 96 h (n = 3 independent experiments performed in triplicates). Data are represented as means + S.E.M. p values were determined by ordinary one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.0001
We next assessed the direct action of high glucose on HMG20A levels in isolated islets. In human islets, HMG20A expression levels were transiently increased by approximately twofold at 72 h returning to basal levels by 96 h (Fig. 3a). A similar transient induction pattern, albeit attaining maximum levels at 48 h, was also observed in mouse islets and in INS-1E beta cells (Fig. 3b,c). Interestingly, a significant decrease was detected INS-1E cells at 72 h of high glucose exposure (Fig. 3c). It is noteworthy that expression levels of NEUROD, a bona fide target of HMG20A, mirrored those of HMG20A in human islets as well as in INS-1E (Fig. 3d,f), following the same tendency in mouse islets (Fig. 3e). In order to satisfy metabolic demand by fetus and mother, glucose metabolism is altered during pregnancy inducing metabolic stress that, if ill adapted, will lead to gestational diabetes. We therefore pondered whether Hmg20a expression levels were modulated during pregnancy. Consistent with this premise, transcript and protein levels of the Hmg20a gene were increased reaching a peak of maximum signal at 12.5 and 14.5 days post coitum, respectively, and thereafter declining (Fig. 4a–c). Pregnant females displayed mild glucose intolerance at 14.5 correlating with the peak in HMG20A protein levels (Fig. 4d). Taken together, these data suggest that increased glucose levels both in vitro and in vivo stimulate HMG20A expression.
Fig. 3. HMG20A is regulated by glucose.

a–c HMG20A and d–f NEUROD mRNA levels were assessed by qPCR in human (n = 3 donors performed in triplicates) and mouse islets (three independent preparations of pooled islets from three different isolation) as well as INS-1E cells cultured in 24 mM glucose for 24, 48, 72, or 96 h (n = 3 independent experiments of all time points executed in triplicates). Data are represented as means + S.E.M. p values were determined by ordinary one-way ANOVA. *p < 0.05, compared to the control 0 time point
Fig. 4. HMG20A is transiently increased in mouse islets during pregnancy.

a Representative images of pancreatic sections from pregnant mice co-stained for HMG20A (green) and INSULIN (INS) (red). Nuclei were stained using DAPI (blue). Magnification 40×. White boxes define areas enlarged in panels below. p.c. post coitum. b Quantification of integrated fluorescence intensity for HMG20A in beta cells of islets from pregnant mice (n = 3 mice per time point with 35-71 islets counted per time point). Data are depicted as dot plots with means ± S.E.M. c HMG20A mRNA levels in islets from pregnant mice (n = 5 mice) at 12.5 and 14.5 days of pregnancy. d Oral glucose tolerance test (OGTT) and area under the curve (AUC) analysis of non-pregnant (NP) and pregnant (day 14.5 of pregnancy; D14.5) mice (NP, n = 8; D14.5, n = 6). Data are represented as means ± S.E.M. p values were determined by the unpaired two-tailed Student´s t-test (d for AUC and c) or ordinary one-way ANOVA with Dunnet’s multiple comparison test (b). *p < 0.05, **p < 0.01, ****p < 0.0001, compared to the control 0 time point
The rs7119 SNP variant within the 3′ untranslated region (UTR) of human HMG20A reduces activity of a luciferase reporter gene
Although GWAS have highlighted a panoply of SNPs correlating with T2DM, few have been assigned a functional consequence to the associated gene. Towards addressing this question, we assessed whether the human HMG20A SNPs associated to T2DM may impact HMG20A levels. Of particular interest is the SNP rs7119 located in the 3′ UTR of the HMG20A transcript that is encoded in part by exon 10 and the entirety of exon 11 (Fig. 5c). The SNP rs7119 causes a “C” (reference allele “wt”) to “T” (“mut” allele) substitution. In silico analysis of the HMG20A 3′UTR revealed the binding site of a single miRNA (miR-571) to the wt allele whereas the “mut” allele results in the ablation of this miRNA binding site (Table 1). Interestingly expression of miR-571 was not detected in mouse islets as compared to the highly expressed miR375 (data not shown). Notwithstanding, the “mut” allele generates binding sites for six alternative miRNAs (Table 1) of which four, miR-143 (3p and 5p), miR-490-3p, miR-1246, and miR-1261, were expressed in the human 1.1E7 pancreatic cell line (Fig. 5a). In contrast mouse islets only expressed miR-143 and very low levels of miR-1246 (Fig. 5b). The latter findings suggest that the “mut” variant may alter expression of the HMG20A transcript due to the action of those miRNAs. To assess this premise, we generated two Gaussia luciferase (Gluc) reporter constructs harboring either the “wt” or “mut” 3′ UTR of the human HMG20A transcript. Expression of these constructs was under the transcriptional regulation of the SV40 basal promoter (Fig. 5c). Transient transfections were then performed in the human pancreatic beta 1.1E7 and retinal pigmented epithelial RPE1 cell lines. The rational of using human cell lines was to maintain species-specific effects targeting the human 3′ UTR of the HMG20A transcript, which is distinct than that of the mouse Hmg20a transcript. We initially confirmed that glucose also stimulated HMG20A expression in 1.1E7 beta cells (Fig. 5d). The “mut” variant displayed significantly less Gluc activity as compared to the construct bearing the “wt” variant 48 h post transfection in either cell lines (Fig. 5e, f). These results suggest that the diabetes-linked SNP rs7119 alters expression of the human HMG20A.
Fig. 5. The rs7119 SNP within the HMG20A 3′ UTR decreases protein levels.

Expression profile of putative miRNAs binding to the “mut” allele of the HMG20A transcript in a human 1.1E7 cells and b mouse islets. Data are presented respect to miR375 expression levels. c Schematic representation of the HMG20A gene depicting the promoter region (HMG20A promoter), transcription starting site (tss) and 11 exons. Gaussia luciferase (GLuc) reporter constructs harboring the 3′ UTR corresponding to either wild type (wt) or rs7119 mutant (mut) allele under pSV40 promoter are shown below. d HMG20A mRNA levels assessed by qPCR in the human pancreatic beta cell line 1.1E7 cultured in 24 mM glucose for 24, 48, 72, or 96 h (n = 5 independent experiments performed in triplicates). e Human pancreatic beta 1.1E7 and f RPE1 cell lines were transfected with the reporter construct bearing either wt or rs7119 mut variant of the HMG20A 3′ UTR. Both the Gaussia Luciferase activity and SEAP activity were determined 24 h post transfection (n = 3 independent experiments). Data are represented as means + S.E.M. p values were determined by ordinary one-way ANOVA (d) or unpaired Student's t-test (e, f). *p < 0.05
TABLE 1.
In silico identification of miRNAs binding sites within the rs7119 SNP located in the 3′UTR of HMG20A
| “C” reference alelle | “T” risk allele |
|---|---|
| (wt) | (mut) |
| hsa-miR-571 | hsa-miR-1261 |
| hsa-miR-1257 | |
| hsa-miR-1246 | |
| hsa-miR-586 | |
| hsa-miR-490 (5p, 3p) | |
| hsa-miR-143 (5p, 3p) |
HMG20A regulates the expression of beta cell genes involved in metabolism-secretion coupling and beta cell maturity
To determine the specific contribution of HMG20A to islet beta cell function and the impact of its reduced levels in T2DM islets, we silenced HMG20A expression by RNA interference and then assessed transcript levels of key factors involved in beta cell identity and function. A 60% depletion of Hmg20a in INS-1E cells (Fig. 6a, b) resulted in a concomitant 60% inhibition of NeuroD transcript levels (Fig. 6c). Mafa, which is essential to maintain the beta cell phenotype, was decreased by 50% whereas Pdx1 was unaltered. Interestingly, Pax4 as well as Rest were increased by 1.8- and 1.5-fold, respectively, subsequent to Hmg20a silencing (Fig. 6c). We previously demonstrated that chronic overexpression of Pax4 leads to loss of beta cell identity characterized by decreased Mafa and Insulin expression23. We thus, pondered whether HMG20A could directly regulate Pax4 thereby maintaining its expression within a range that would not interfere with beta cell function. To validate this premise, chromatin immunoprecipitation (ChIP) experiments were performed using the islet specific Pax4 gene promoter24. As expected H3K4me2, a marker for active promoters and enhancers specifically occupied this region (Fig. 6d)26. More importantly, HMG20A also occupied the Pax4 gene promoter region as well as exon 1 while control anti-IgG exhibited no binding (Fig. 6e). Since NEUROD and MAFA are involved in metabolism-secretion coupling in beta cell, genes implicated in this process were assessed after Hmg20a silencing. Glucokinase (Gck) and Insulin transcript levels were significantly repressed by 30% (Fig. 6f). In contrast, Pi3k and Glut2 levels were increased 1.8 and 2.6-fold, respectively (Fig. 6f). Transcript levels of Syt7 and Snap25, two factors involved in insulin granule membrane docking and fusion were not altered in Hmg20a-repressed INS-1E cells (Fig. 6f). The modulation of key HMG20A target genes was validated using a second independent siHMG20A (Fig. 6g). Taken together our results indicate that HMG20A coordinates expression of genes that will establish the mature beta cell phenotype.
Fig. 6. HMG20A regulates metabolism-secretion coupling genes in INS-1E cells.

a Hmg20a was silenced by specific siRNA in INS-1E cells (n = 6 independent experiments, each performed in triplicates). b Representative images of INS-1E cells immuno-stained for HMG20A (green) and DAPI (blue) confirming decreased protein level after treatment with siHMG20A. Magnification 40×. c Beta cell development and maturity genes, as Neurod, Pax4, Mafa, Pdx1, and Rest transcripts levels after silencing of Hmg20a in INS-1E cells (n = 4 independent experiments performed in triplicates). d H3K4m2 and e HMG20A occupancy of promoter, exon 1 and ATG regions of the Pax4 gene after chromatin immunoprecipitation using anti-H3K4m2 or anti-HMG20A antibodies, with IgG as a control for non-specific interactions (n = 4 independent experiments performed in triplicates). f Beta cell metabolism-secretion coupling genes transcripts levels after silencing of Hmg20a in INS-1E cells (n = 4 independent experiments, performed in triplicates). g Alteration in the expression of key genes was confirmed in INS-1E cells using a second siHMG20A (n = 4 independent experiments performed in triplicates). Data are represented as means + S.E.M. p values were determined by one-way ANOVA with Tukey’s multiple comparisons test (a, c, f, g). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Glucose-stimulated insulin secretion is impaired by Hmg20a depletion
We next assess the cellular and functional consequences of Hmg20a depletion in beta cells. Neither cell death nor proliferation was altered by HMG20A depletion in INS-1E cells (Fig. 7a, b). In contrast, glucose-stimulated insulin secretion (GSIS) was blunted in these cells (Fig. 7c). Similarly, subsequent to a 40% depletion of Hmg20a in mouse islets (Fig. 7d), cell proliferation was unaltered while GSIS was drastically decreased as compared to control islets (Fig. 7e, f). Taken together, Hmg20a expression is essential for insulin secretion.
Fig. 7. Hmg20a silencing impairs insulin secretion in INS-E1 cells as well as in mouse islets without affecting cell death or proliferation.

a Cell death (n = 3), b proliferation (n = 3), and c glucose-induced insulin secretion (GSIS, n = 4) were assessed subsequent to siRNA-mediated HMG20A depletion in INS-1E cells. d Effect of siRNA-mediated HMG20A depletion on e cell proliferation (n = 6 islet preparations) and f GSIS in mouse islets (n = 6 islet preparations). Data are represented as mean + S.E.M. p values were determined by unpaired two-tailed Student's t-test. *p < 0.05, **p < 0.01
Discussion
The overall aim of the current study was to assign a functional role of the putative diabetes-linked HMG20A gene to islet physiology, and determine the pathophysiological consequence of the rs7119 SNP associated with this disease. We find that: (1) HMG20A is expressed in pancreatic islet beta cells, (2) HMG20A levels are decreased in T2DM islets, (3) high glucose stimulates HMG20A expression, (4) the T2DM-associated SNP within the 3′ UTR of the HMG20A transcript decreases luciferase activity, (5) HMG20A regulates key genes in beta cell function and maturity, and (6) HMG20A depletion impairs GSIS. Therefore, we provide first evidence that HMG20A is a key regulator of mature beta cell function.
Our results indicate that variations in glucose levels are a main rheostat of islet HMG20A expression, which in turn appears to modulate islet adaptive measures by increasing insulin biosynthesis. The latter premise is supported by our findings that NEUROD expression levels mirrored those of HMG20A, being stimulated by high glucose and inhibited under siRNA-mediated Hmg20a downregulation. Mutations in the NEUROD gene predispose individuals to develop maturity onset diabetes of the young (MODY6)17 while adult mouse islets lacking Neurod respond poorly to glucose27. We also found reduced expression levels of Mafa as a consequence of Hmg20a depletion. This transcription factor along with NEUROD activates expression of the Gck and Insulin gene27–29, consistent with reduced levels of both transcripts in Hmg20A-silenced cells. Intriguingly, Glut2 was increased following Hmg20a repression perhaps as a compensatory mechanism to increase cellular glucose influx due to decreased GCK-mediated phosphorylation and glycolysis30. Thus, the coordinated regulation of NeuroD and Mafa by HMG20A may be a key molecular hub essential for beta cell function and adaptation to physiological stress such as during pregnancy in which glucose metabolism is altered. Interestingly, although HMG20A was repressed by palmitate in vitro, expression levels were not significantly altered in islets of a mouse model of high fat diet-induced obesity and pre-diabetes. Thus, it is tempting to speculate that in vivo, glucose and lipids may have opposite effect on HMG20A expression nullifying any significant increased in expression, which may in long-term hinder islet adaptation, precipitating beta cell dysfunction and T2DM. The latter hypothesis may rationalize the observed association of HMG20A risk variants with T2DM in obese cases8.
Our study also identifies the Pax4 gene as a direct target repressed by HMG20A. Although the Pax4 gene promoter was shown to interact with REST15, the absence of this “disallowed” gene in beta cells suggest that HMG20A acts independent of the LSD1–coREST complex that requires REST to be recruited onto DNA14. Rather, our results are in agreement with recent findings that HMG20A in cohort with LSD1 is sufficient to repress Snail1 and induce TGF beta-triggered EMT, demonstrating a mechanism independent of REST as well as the capacity of HMG20A for binding directly to DNA18. Notwithstanding, Hmg20a silencing also evoked REST re-expression indicating that in cohort with PAX4, these two factors may promote de-differentiation of beta cells conveying increased survival under unfavorable physiological conditions such as chronic hyperglycemia.
To date, regulation of the endogenous Pax4 gene in mature islets conveying protection and adaptation in response to physiological or pathophysiological conditions has remained obscure. Our data suggest that HMG20A may be an important epigenetic regulator of Pax4 gene expression that will dictate the faith of beta cells under stress conditions such as in T2DM. In accord with this premise, we previously demonstrated that PAX4 expression was increased in islets derived from T2DM patients31. We now demonstrate that HMG20A levels are repressed in T2DM islets. These combined human data are in line with increased expression of Pax4 subsequent to Hmg20a silencing in either islets or INS-1E cells without altering apoptosis and proliferation. Accumulating evidence suggest that the main characteristic event in T2DM is not massive beta cell death but rather beta cell de-differentiation32. We previously demonstrated that chronic expression of Pax4 is implicated in this process through downregulation of Mafa and Insulin expression33. Our current data substantiate these findings as we detected a decrease in Mafa expression after Hmg20a silencing. Thus, the role of HMG20A may be to supress Pax4 transcription thereby permitting expression of Mafa as well as Insulin. Such cross talk between the two factors is highlighted also during pregnancy. Indeed, we previously found a peak of Pax4 expression and beta-cell proliferation at 10.5 days and 12.5 days of pregnancy respectively24. Herein, we now find that HMG20A expression in islets is increased at 14.5 days of pregnancy, just after the peak of proliferation and correlating with decreased Pax4 and increased Mafa expression levels. These findings are consistent with the premise that beta cells after proliferation have to re-differentiate to reach their metabolic mature phenotype and adequately secrete insulin, a process requiring MAFA34. A similar regulatory cross talk between HMG20A and PAX4 may also be operative in early stages of hyperglycemia, which is characterized by active beta cell adaptation. Nonetheless, long-term exposure may favor de-differentiation and survival (without altering proliferation) to the detriment of function, as we observed in vitro in which HMG20A expression was transiently increased but then inhibited by glucose. Although further studies will be needed to specify the mechanism by which HMG20A regulates those islet-enriched genes, our work provides the basis for the involvement of a non-mutually exclusive cross regulation of HGM20A and PAX4 in islet/beta cell physiology.
A significant finding of our study is the potential decreased in HMG20A levels associated with the diabetes-linked rs7119 SNP within the 3′ UTR of the transcript. This SNP opens the putative binding site of several new miRNAs as compared to the normal wild type allele. Although a previous study has linked T2DM-associated genes to islet-expressed miRNA35, our study, to the best of our knowledge, provides first evidence that a diabetes-linked SNP alters the putative binding site for miRNAs resulting in the deregulate repression of the target transcript. Further indication for a dysfunctional role of the rs7119 variant in aberrant HMG20A repression was highlighted by the expression of SNP-associated miRNAs, the most abundant being miR-143 and 1246, in human 1.1E7 cells and mouse islets. In contrast, miR-571 associated with the wild type allele miRNA binding site was not express suggesting the potential absence of post-transcriptional regulation conveyed by the normal allele. Although scarce details are available on the role of miR-143 and 1246 in islet function, miR-143 was shown to be among the ten most abundant miRNAs expressed in human islets beta cells whereas miR-1246 was predominantly expressed in other islet cell types35. Elsewhere, miR-143 expression was found to be essential for human pre-adipocyte differentiation partly through repression of its target gene ERK5 involved in cell growth and proliferation36. Although it remains to be validated, the potential serendipitous binding of miR-143 to the HMG20A rs7119 gene variant may induce aversely de-differentiation through activation of Pax4 and Rest.
The overall data presented in this work clearly describes a functional link between HMG20A and islet physiology. HMG20A regulates metabolism-secretion coupling genes in beta cells and could be also involved in beta cell plasticity through PAX4 and REST, regulating phenotypic changes needed for beta cells in order to respond to different physiological situations as pregnancy and obesity. Failure on those HMG20A-mediated processes could lead to the development and establishment of T2DM. As consequence, our study opens a venue to consider targeting of HMG20A expression and/or regulation as potential therapies for T2DM.
Materials and methods
Animals
The experimental mouse procedures were approved by the Institutional Animal Care Committee of the Andalusian Center of Molecular Biology and Regenerative Medicine (CABIMER) and by the ethic committee of the University of Malaga, Biomedical Research Institute of Málaga (IBIMA) and performed according to the Spanish law on animal use RD 53/2013. Animal studies were performed in compliance with the ARRIVE guidelines37. Mice were housed in ventilated plastic cages and maintained on a 12-h light–dark cycle with ad libitum access to pelleted chow and water. C57BL/6J of both genders were used to obtain pancreatic islets and other tissues (liver, adipose tissue, skeletal muscle, and brain) and females for pregnancy studies. For diet-induced obesity studies, 8-weeks-old male C57BL/6J mice (Janvier Labs, Saint-Berthevin Cedex, France) were housed in individual cages under a 12 h light/dark cycle (8:00 pm light off) in a room with controlled temperature (21 ± 2 °C) and humidity (50 ± 10%). For induction of obesity, groups of ten mice were fed a high fat diet (D12451 Research Diets Inc., New Brunswick, NJ, USA), containing 45% of Kcal from saturated fat, for 15 weeks. In parallel, groups of ten age-matched mice were fed a control diet containing 10% of Kcal from fat (D12450 Research Diets Inc.). Circulating glucose levels were measured from tail vein blood samples using an Optium Xceed glucometer (Abbott Scientifica SA, Barcelona, Spain). Insulin tolerance tests (ITT) and glucose tolerance test (OGTT) were performed as previously described38. At 15 weeks, mice were sacrificed by cervical dislocation and pancreas extracted for histological processing.
Pancreatic islet and cell line cultures
Mouse pancreatic islets were isolated from 2 to 4-months-old C57BL/6J mice by intraductal collagenase as previously described24,39 . After isolation, mouse islets were cultured in 5.5 mM glucose/RPMI 1640 medium (Life Technologies, Madrid, Spain) supplemented with 10 % Fetal bovine serum (FBS, Sigma-Aldrich, Madrid, Spain), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich), 2 mM l-glutamine (Life Technologies), 1 mM sodium pyruvate (Sigma-Aldrich), 50 μM β-mercaptoethanol (Life Technologies), and 10 mM HEPES (Life Technologies) prior to different experimental treatments. Islets from non-diabetic or T2DM organ donors were obtained in Pisa or purchased from Tebu-Bio (Barcelona, Spain). After reception, human islets were cultured in CMRL-1066 (Gibco) medium containing 5.5 mM glucose and supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 100 μg/ml gentamycin. To assess glucose influence in HMG20A expression, both human and mice islets were cultured in normal (5.5 mM) or high (24.4 mM) glucose concentrations for 24, 48, 72, and 96 h, and lately harvested for RNA extraction.
The rat insulinoma INS-1E beta cell line (kindly provided by Dr. P Maechler, Geneva, CH) was cultured between passages 50–90 and maintained in 11.1 mM glucose/RPMI 1640 medium (Life Technologies) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, and 10 mM HEPES. Following overnight glucose deprivation (3 mM), cells were cultured in 11.1 mM or 24.4 mM glucose RPMI 1640 medium for 24, 48, 72, and 96 h. Alternatively INS-1E cells were also exposed to 0.5 mM palmitate for up to 96 h. Cells were harvested at each time point for RNA extraction.
The human retinal pigmented epithelial RPE1 (ATCC, CRL-4000) and the islet beta 1.1E7 cell lines (The European Collection of Authenticated Cell Cultures) were cultured in F-12 Ham and RPMI media, respectively, prior to luciferase reporter assays. The 1.1E7 cell line was also cultured in the presence of either 11.1 or 24.4 mM glucose for up to 96 h. RNA was extracted at 24, 48, 72, and 96 h post treatment.
RNA interference
Mouse pancreatic islets and INS-1E cells were cultured as previously described40 and transfected with either 50 μmol of two independent HMG20A small interfering (si)RNAs (5′-AGGCAAAUCUCAUAGGCAA-3′ and 5′-UCACAAGGAUGUUGGGCAA-3′) or scramble siRNA (Sigma-Aldrich) using Oligofectamine (Life Technologies). Samples were processed for RNA, immunofluorescence, GSIS, cell proliferation and death assessment 72 h after transfection.
RNA extraction and quantitative-PCR expression analysis
Qiagen RNeasy Micro and Mini kits (Qiagen, Madrid, Spain) were used for the extraction of total RNA from the different tissues, islets and cell line samples. After quantification of the RNA concentration using a NanoDrop 1000 Spectrophotometer (Wilmington, DE, USA), single-stranded cDNA was synthesized with the Superscript II First-Strand cDNA synthesis kit (Invitrogen, Carlsbad, CA, USA) and Anchored OligodT as primers (Sigma-Aldrich). Quantitative PCR was performed on a 7500 Real-Time PCR System (Applied Biosystems). Gene-specific primers (HMG20A, NEUROD1, PDX1, PAX4, REST, GLUKOKINASE, INSULIN, PI3K, MAFA, GLUT1, GLUT2, SNAP25, and SYT7) were designed using Power Primer3 (Supplementary Table 1, 2 and 3). Expression levels of the housekeeping genes β-ACTIN or CYCLOPHILIN were used for normalization. The relative gene expression was calculated using the 2−ΔΔCt method24. For miRNA expression profiling, microRNAs were extracted from the 1.1E7 beta cell line and mouse pancreatic islets using the miRNAeasy kit (Qiagen) and cDNA synthesis was conducted using the Universal cDNA Synthesis Kit (Exiqon) as previously described41. ExiLENT SYBR Green Master Mix Kit and primers for each assay to perform quantitative-PCR (qPCR) were obtained from Exiqon.
Immunofluorescence and immunohistochemistry studies
Dissected pancreases were fixed overnight in 4% paraformaldehyde at 4 °C and processed for embedding in paraffin at the Histology Core Facility, CABIMER. Pancreatic sections from non-pregnant/pregnant mice sacrificed at 10.5, 12.5, 14.5, and 17.5 days post coitum were analyzed by immunohistochemistry using specific antibodies against HMG20A, insulin, glucagon, and somatostatin (Supplemental Table 4). Counterstaining was performed with 5 μg/ml DAPI (Life Technologies) to stain the nuclei and slides were mounted using fluorescent mounting medium (DAKO). Images were acquired using either the epifluorescence (Leica AF6000, Leica, England) or confocal (Leica TCS SP5) microscopes. Expression of HMG20A in beta cells was quantified using Metamorph Analysis Software (Molecular Devices). Single frames were acquired using a confocal TCS SP5 microscope (Leica). Same illumination settings were applied for all the samples (image parameters including pinhole size, photomultiplier offset and gain, and laser intensity were first set for non-pregnant control samples, and then, the same settings were used for all conditions). For Image analysis/quantification, the integrated intensity values (∑ pixel intensity at region of interest) were measured as previously described42–44. Integrated intensity of nuclear HMG20A in islet cells co-expressing insulin were measured, and the average of intensity per islet was calculated. Three independent animals and an average of 50 islets were used for each time point. Pancreatic sections from control and HFD mice were performed and analyzed as previously described25.
Luciferase 3′ UTR reporter assays
The effect of the 3′ UTR SNP rs7119, previously linked to T2DM was evaluated by luciferase reporter assays. 3′ UTR constructs containing the wt allele (reference allele) “C” or the DMT2-linked allele (“mut” allele) “T” cloned downstream of the luciferase gene were obtained from GeneCopoeia. Luciferase contained in these constructs is a secreted one; therefore luciferase activity can be measured in the medium. These constructs also have a secreted embryonic alkaline phosphatase (SEAP) gene driven by a constitutive promoter, used as internal control. The Secrete-Pair TM Dual Luminescence Assay Kit (GeneCopoeia) was used as luciferase reporter system according to manufacturer’s instructions. RPE1 and 1.1E7 cell lines were chosen for transfection studies using those constructs. Both are cell lines of human origin and from different tissues. RPE1 are cells from retina pigmentosum ephitelia45 meanwhile 1.1E746 is a human beta cell line. The RPE1 and 1.1E7 cell lines were transfected and luciferase activity was measured 48 h after in the collected culture media.
Glucose-stimulated insulin secretion assays
Insulin secretion in response to 2.8 mM or 22 mM glucose was measured in static incubations as previously described47,48 in both mouse pancreatic islets and INS-1E cells. Following the incubations, supernatants were collected and islets and cells were lysated with HCl-ethanol to obtain the insulin content. Insulin from supernatants and cell contents was measured by ELISA (Mercodia, Uppsala, Sweden). All experiments were run in triplicate.
Cell death and proliferation
Cell death (apoptosis) and proliferation was measured using either the Cell Death Detection ELISA kit or the 5-Bromo-2′-deoxy-uridine labeling and detection kit (Roche Diagnostics, Madrid, Spain) as described by the manufacturer.
ChIP assays
ChIP assays were performed as previously described12,18. INS-1E cells were treated with 1% formaldehyde for 15 min at 37 °C for crosslinking. Chromatin was sonicated to an average fragment size of 400 to 500 bp using the Diagenode Bioruptor. Immunoprecipitations were performed using the following reagents: Dynabeads Protein A (Invitrogen), rabbit polyclonal anti-HMG20A (Sigma-Aldrich) and rabbit monoclonal to Histone H3 (di methyl K4) (Abcam). Rabbit IgG (Sigma) was used as a control for non-specific interactions. Input was prepared with 10% of the chromatin used for immunoprecipitation. Quantification of immunoprecipitated DNA was performed by real-time PCR with the Applied Biosystems 7500 FAST real-time PCR system, using Applied Biosystems Power SYBR green master mix. Each sample was quantified in triplicate. Provided data are the average of at least three independent experiments.
Statistical analysis
The Ruth Lenth's power of analysis was applied to the different animal models to ensure that adequate numbers of animals had been studied to detect significant changes. Results are expressed as mean ± SEM (line plots as a function of time) or as mean + SEM (bar graphs). Statistical analyses were performed using the GraphPad Prism software version 7 (GraphPad Software, La Jolla, USA).
Electronic supplementary material
Acknowledgements
This work was supported by the Consejería de Salud, Fundación Pública Andaluza Progreso y Salud, Junta de Andalucía (PI-0727-2010 to B.R.G., PI-0085-2013 to P.I.L., PI-0006-2016 to E.F.-M. and PI-0574-2012 to S.Y.R.-Z.), the Consejería de Economía, Innovación y Ciencia (P10.CTS.6359 to B.R.G. and P09-CTS-5445 to A.C.-C.), the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III co-funded by Fondos FEDER (PI10/00871 and PI13/00593 to B.R.G., and BFU2014-5343-P to J.C.R.) and the Red TerCel program (RD12/0019/0028 to B.S. and K.H.). E.F.M. is recipient of a Juan de la Cierva Incorporación Fellowship from the Ministerio de Economía y Competitividad (IJCI-2015-26238). S.Y.R.Z is a recipient of a postdoctoral fellowship from Consejería de Salud, Junta de Andalucía (RH-0070-2013). F.J.B.S. is recipient of a “Nicolás Monardes” research contract from Consejería de Salud Junta de Andalucía, (C-0070-2012). A.M.M. is supported by CP14/00105 and PI15/00134 from the Instituto de Salud Carlos III co-funded by Fondes FEDER. CIBERDEM is an initiative of the Instituto de Salud Carlos III. We thank Noelia García-Rodriguez, Lourdes Sanchez-Salido, and Leopoldo Perez-Rosendo for their excellent technical assistance. B.R.G. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
These authors contributed equally: Jose M. Mellado-Gil, Esther Fuente-Martín.
Edited by M. Agostini
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41419-018-0272-z).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Frayling TM, Hattersley AT. Physiology helps GWAS take a step closer to mechanism. Diabetes. 2014;63:1836–1837. doi: 10.2337/db14-0130. [DOI] [PubMed] [Google Scholar]
- 2.Prasad RB, Groop L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel) 2015;6:87–123. doi: 10.3390/genes6010087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mohlke KL, Boehnke M. Recent advances in understanding the genetic architecture of type 2 diabetes. Hum. Mol. Genet. 2015;24:R85–R92. doi: 10.1093/hmg/ddv264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry JR, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr. Opin. Clin. Nutr. Metab. Care. 2008;11:371–377. doi: 10.1097/MCO.0b013e32830349a1. [DOI] [PubMed] [Google Scholar]
- 5.Kooner JS, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011;43:984–989. doi: 10.1038/ng.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sim X, et al. Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLoS Genet. 2011;7:e1001363. doi: 10.1371/journal.pgen.1001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukuda H, et al. A single nucleotide polymorphism within DUSP9 is associated with susceptibility to type 2 diabetes in a Japanese population. PLoS ONE. 2012;7:e46263. doi: 10.1371/journal.pone.0046263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perry JR, et al. Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet. 2012;8:e1002741. doi: 10.1371/journal.pgen.1002741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold M, Ellwanger DC, Hartsperger ML, Pfeufer A, Stumpflen V. Cis-acting polymorphisms affect complex traits through modifications of microRNA regulation pathways. PLoS ONE. 2012;7:e36694. doi: 10.1371/journal.pone.0036694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianchi ME, Agresti A. HMG proteins: dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 2005;15:496–506. doi: 10.1016/j.gde.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 11.Wynder C, Hakimi MA, Epstein JA, Shilatifard A, Shiekhattar R. Recruitment of MLL by HMG-domain protein iBRAF promotes neural differentiation. Nat. Cell Biol. 2005;7:1113–1117. doi: 10.1038/ncb1312. [DOI] [PubMed] [Google Scholar]
- 12.Ceballos-Chavez M, et al. Control of neuronal differentiation by sumoylation of BRAF35, a subunit of the LSD1-CoREST histone demethylase complex. Proc. Natl Acad. Sci. USA. 2012;109:8085–8090. doi: 10.1073/pnas.1121522109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Arensbergen J, et al. Derepression of polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010;20:722–732. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin D, Grapin-Botton A. The importance of REST for development and function of beta cells. Front. Cell Dev. Biol. 2017;5:12. doi: 10.3389/fcell.2017.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemp DM, Lin JC, Habener JF. Regulation of Pax4 paired homeodomain gene by neuron-restrictive silencer factor. J. Biol. Chem. 2003;278:35057–35062. doi: 10.1074/jbc.M305891200. [DOI] [PubMed] [Google Scholar]
- 16.Martin D, et al. REST represses a subset of the pancreatic endocrine differentiation program. Dev. Biol. 2015;405:316–327. doi: 10.1016/j.ydbio.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malecki MT, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat. Genet. 1999;23:323–328. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 18.Rivero S, Ceballos-Chavez M, Bhattacharya SS, Reyes JC. HMG20A is required for SNAI1-mediated epithelial to mesenchymal transition. Oncogene. 2015;34:5264–5276. doi: 10.1038/onc.2014.446. [DOI] [PubMed] [Google Scholar]
- 19.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342:1234850. doi: 10.1126/science.1234850. [DOI] [PubMed] [Google Scholar]
- 21.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19:872–882. doi: 10.1016/j.cmet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu HeKH, et al. In vivo conditional Pax4 overexpression in mature islet beta-cells prevents stress-induced hyperglycemia in mice. Diabetes. 2011;60:1705–1715. doi: 10.2337/db11-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenzo PI, et al. PAX4 defines an expandable beta-cell subpopulation in the adult pancreatic islet. Sci. Rep. 2015;5:15672. doi: 10.1038/srep15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romero-Zerbo SY, et al. The cannabinoid ligand LH-21 reduces anxiety and improves glucose handling in diet-induced obese pre-diabetic mice. Sci. Rep. 2017;7:3946. doi: 10.1038/s41598-017-03292-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu C, et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell. Metab. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moates JM, Nanda S, Cissell MA, Tsai MJ, Stein R. BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta-cells and gut endocrine cells. Diabetes. 2003;52:403–408. doi: 10.2337/diabetes.52.2.403. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Brun T, Kataoka K, Sharma AJ, Wollheim CB. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–358. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Remedi MS, Nichols CG. Hyperinsulinism and diabetes: genetic dissection of beta cell metabolism-excitation coupling in mice. Cell. Metab. 2009;10:442–453. doi: 10.1016/j.cmet.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brun T, et al. The diabetes-linked transcription factor Pax4 is expressed in human pancreatic islets and is activated by mitogens and GLP-1. Hum. Mol. Genet. 2008;17:478–489. doi: 10.1093/hmg/ddm325. [DOI] [PubMed] [Google Scholar]
- 32.Hunter CS, Stein RW. Evidence for loss in identity, de-differentiation, and trans-differentiation of islet beta-cells in type 2 diabetes. Front. Genet. 2017;8:35. doi: 10.3389/fgene.2017.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lorenzo P. I., Juarez-Vicente F., Cobo-Vuilleumier N., Garcia-Dominguez M., Gauthier B. R. The diabetes-linked transcription factor PAX4: from gene to functional consequences. Genes(Basel)8, E101 (2017). [DOI] [PMC free article] [PubMed]
- 34.Kaneto H, et al. Role of MafA in pancreatic beta-cells. Adv. Drug Deliv. Rev. 2009;61:489–496. doi: 10.1016/j.addr.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 35.van de Bunt M, et al. The miRNA profile of human pancreatic islets and beta-cells and relationship to type 2 diabetes pathogenesis. PLoS ONE. 2013;8:e55272. doi: 10.1371/journal.pone.0055272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Esau C, et al. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 37.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez-Noriega L, et al. Levothyroxine enhances glucose clearance and blunts the onset of experimental type 1 diabetes mellitus in mice. Br. J. Pharmacol. 2017;174:3795–3810. doi: 10.1111/bph.13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellado-Gil JM, et al. PAX4 preserves endoplasmic reticulum integrity preventing beta cell degeneration in a mouse model of type 1 diabetes mellitus. Diabetologia. 2016;59:755–765. doi: 10.1007/s00125-016-3864-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brun T, Duhamel DL, Hu He KH, Wollheim CB, Gauthier BR. The transcription factor PAX4 acts as a survival gene in INS-1E insulinoma cells. Oncogene. 2007;26:4261–4271. doi: 10.1038/sj.onc.1210205. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Comas J, et al. Stress-induced microRNA-708 impairs beta-cell function and growth. Diabetes. 2017;66:3029–3040. doi: 10.2337/db16-1569. [DOI] [PubMed] [Google Scholar]
- 42.Mahoney E, et al. ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood. 2012;120:1262–1273. doi: 10.1182/blood-2011-12-400184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monteiro P, et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J. Cell Biol. 2013;203:1063–1079. doi: 10.1083/jcb.201306162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wheelock MS, Wynne DJ, Tseng BS, Funabiki H. Dual recognition of chromatin and microtubules by INCENP is important for mitotic progression. J. Cell Biol. 2017;216:925–941. doi: 10.1083/jcb.201609061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rambhatla L, Chiu CP, Glickman RD, Rowe-Rendleman C. In vitro differentiation capacity of telomerase immortalized human RPE cells. Invest. Ophthalmol. Vis. Sci. 2002;43:1622–1630. [PubMed] [Google Scholar]
- 46.McCluskey JT, et al. Development and functional characterization of insulin-releasing human pancreatic beta cell lines produced by electrofusion. J. Biol. Chem. 2011;286:21982–21992. doi: 10.1074/jbc.M111.226795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauthier BR, et al. PDX1 deficiency causes mitochondrial dysfunction and defective insulin secretion through TFAM suppression. Cell. Metab. 2009;10:110–118. doi: 10.1016/j.cmet.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jimenez-Moreno CM, et al. A simple high efficiency intra-islet transduction protocol using lentiviral vectors. Curr. Gene Ther. 2015;15:436–446. doi: 10.2174/1566523215666150630121557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.