

Abstract
Background
We observed the effects of nuclear factor E2-related factor 2 (Nrf2) downregulation via intrahippocampal injection of a lentiviral vector on cognition in senescence-accelerated mouse prone 8 (SAMP8) to investigate the role of the (Nrf2)/antioxidant response element (ARE) pathway in age-related changes.
Material/Methods
Control lentivirus and Nrf2-shRNA-lentivirus were separately injected into the hippocampus of 4-month-old SAMR1 and SAMP8 mice and then successfully downregulated Nrf2 expression in this brain region. Five months later, cognitive function tests, including the novel object test, the Morris water maze test, and the passive avoidance task were conducted. Glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (Iba1) immunohistochemistry was performed to observe an inflammatory response. Presynaptic synapsin (SYN) were observed by immunofluorescence. We then determined the Nrf2-regulated, heme oxygenase-1 (HO-1), P65, postsynaptic density protein (PSD), and SYN protein levels. The ultrastructure of neurons and synapses in the hippocampal CA1 region was observed by transmission electron microscopy.
Results
Aging led to a decline in cognitive function compared with SAMR1 mice and the Nrf2-shRNA-lentivirus further exacerbated the cognitive impairment in SAMP8 mice. Nrf2, HO-1, PSD, and SYN levels were significantly reduced (all P<0.05) but high levels of inflammation were detected in SAMP8 mice with low expression of Nrf2. Furthermore, neurons were vacuolated, the number of organelles decreased, and the number of synapses decreased.
Conclusions
Downregulation of Nrf2 suppressed the Nrf2/ARE pathway, activated oxidative stress and neuroinflammation, and accelerated cognitive impairment in SAMP8 mice. Downregulation of Nrf2 accelerates the aging process through neuroinflammation and synaptic plasticity.
MeSH Keywords: Aging, Inflammation, Mild Cognitive Impairment
Background
Alzheimer disease (AD) is a type of age-related dementia, and its incidence increases with age. Its pathogenesis, which involves multiple aspects of neuronal damage, is complex, including autophagy dysfunction, mitochondrial dysfunction, neurotoxicity, aggregation of amyloid-β peptides, production of reactive oxygen species, and microglial proliferation [1]. As a result, AD treatments are very limited. Neuroinflammation has a central role in the pathophysiological aspects and may be a potential therapeutic target for AD [2,3].
Nuclear factor E2-related factor 2 (Nrf2)/antioxidant response element (ARE) is the most important antioxidant stress pathway identified to date [4]. Nrf2 binds to pro-inflammatory cytokines interleukin-6 (IL-6) and interleukin1β (IL1β), and tumor necrosis factor-alpha (TNF-α) plays a crucial role in neurodegeneration. Another transcription factor, inhibiting nuclear factor-κB (NF-κB), which is blind to Nrf2 induction, exerts an anti-inflammatory response [5]. Furthermore, Nrf2 might also contribute to mitochondrial biogenesis [6]. Accumulating evidence shows that Nrf2/ARE pathway is a link that connects apoptosis, autophagy, inflammation, and mitochondrial function, all of which are implicated in the progression of neurodegeneration. Nrf2 translocates into the nucleus and binds with AREs to activate the expression of a series of protective molecules, resulting in upregulation of Nrf2-dependent heme oxygenase-1 (HO-1), an antioxidant response gene [7,8]. Decreased age-related inflammation and oxidative stress increases the resistance of cells to various stimuli. Nrf2/ARE pathway adjustment is increasingly recognized to be involved in cancer and neurodegeneration diseases.
Senescence-accelerated mouse prone 8 (SAMP8) is an aging model that is mainly characterized by aging-accelerated decline in learning and memory functions. SAMP8 mice possess distinct features typical of early-onset memory impairment in several established behavioral tasks. The age of 4–8 months in SAMP8 mice is an important period in age-related changes in cognition [9]. In SAMP8 mice, increased oxidative stress, activated microglia, and decline in memory and learning ability are found in brain tissue, especially the hippocampus [10]. SAMP8 mice may be an excellent model for studying early AD-related neurodegenerative changes and cognitive aging [11].
Recently, a new function of Nrf2 was found in regulating mitochondrial function. However, the underlying mechanisms by which Nrf2 activation/inhibition is associated with synaptic dysfunction remain unclear, and this response may eventually lead to cognitive changes in AD. The effect of Nrf2 on structural synaptic plasticity in AD-related cognitive deficits is still unknown [12]. The present study provides theoretical and experimental bases for a new AD drug target by investigating changes in cognitive function in SAMP8 mice and by exploring the mechanism of the Nrf2/ARE pathway. We also explored neuroinflammation-synapse pathways.
Material and Methods
Animals
Forty-eight clean male SAMR1 and SAMP8 mice aged 4 months and weighing 22–28 g were purchased from the Animal Experiment Center of Peking University Health Science Center in China. Mice were randomized into 4 groups: SAMR1 with control lentivirus (R1+GFP), SAMR1 with Nrf2-RNAi-lentivirus (R1+Nrf2 ShRNA), SAMP8 with control lentivirus (P8+GFP), and SAMP8 with Nrf2-RNAi-lentivirus (P8+Nrf2 ShRNA). Mice were injected in the hippocampus with control lentivirus or Nrf2-RNAi-lentivirus. All mice were allowed to recover for 1 week after injection. The mice were housed in 50–60% humidity at 22–24°C with a 12-h light/dark cycle and allowed free access to food and water.
Viral vectors
For Nrf2 RNAi, the mouse shRNA sequence (5′-CTTACTCTC CCAGTGAATA-3′) was subcloned into the GV248-RNAi lentiviral vector by Genechem (Shanghai, China); shRNA was expressed under the U6 promoter and green fluorescence protein (GFP) was expressed under the ubiquitin promoter. The lentiviral vectors expressing green fluorescence protein (GFP) only were used as the RNA interference (RNAi) control.
Total RNA was isolated from hippocampal tissues using TRIzol reagent (Invitrogen). mRNA was reverse-transcribed to cDNA using a Transcriptor first-strand cDNA synthesis kit (Roche) according to the manufacturer’s instructions, and real-time Q-PCR analysis was performed using the ABI StepOnePlus Real-Time PCR system (ABI VERITI96 PCR) with q-PCR Master Mix (Qiagen). Cycling conditions were as follows: 40 cycles of 95°C for 15 s, 60°C for 30 s, and 70°C for 30 s. Primer sequences are listed in Table 1. Target gene expression was normalized to that of GAPDH. Relative gene expression was calculated with the 2–ΔΔCt formula (Figure 1).
Table 1.
The primer sequences for quantitative real-time polymerase chain reaction.
| Name | Sequences |
|---|---|
| Nrf2 | F: 5′-CGAGATCTGAGCTCTGAACTGCAAAGCAGAGTGAAAGCAGGTGGTGAAG-3′ |
| R: 5′-CGAGATCTGAGCTCCGCGCTCTGCCTCCGCCTCCGCCGCGC-3′ | |
| GAPDH | F: 5′-TTCACCACCATGGAGAAGGC-3′ |
| R: 5′-GGCATGGACTGTGGTCATGA-3′ |
F – forward; R – reverse.
Figure 1.
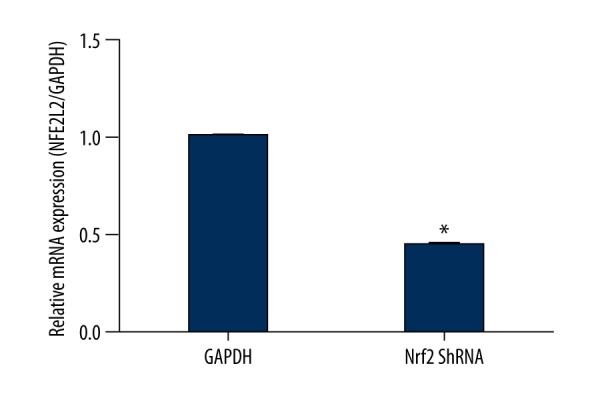
Nrf2 ShRNA in hippocampus was detected by Q-PCR (P<0.05).
Intrahippocampal injection of the lentivirus
At the age of 4 months, SAMP8 and SAMR1 mice were anesthetized with isoflurane and placed in a stereotactic frame (stereotaxic apparatus 51600, Stoelting, USA), positioned in a stereotaxic instrument and 2 μL lentivirus Nrf2-RNAi, or lenti-GFP was injected bilaterally into the hippocampus using the following coordinates: 3.2 mm medial/lateral, 2.7 mm anterior/posterior, 2.7 mm dorsal/ventral from bregma [13]. The preparation was injected at a speed of 0.5 L/min over a period of 4 min by using a syringe (Syringe pumps 51600z, Stoelting, USA) and a 27-gauge needle. The animal experiments were carried out according to the Policies on the Use of Animals and Human Neuroscience Research approved by the Society for Neuroscience in 1995 and were supervised by the Animal Administration and Ethics Committee of HeBei Medical university of Science and Technology.
Novel object recognition test (NORT)
All the behavior tasks were performed 5 months after injections. The novel object recognition test (NORT) was performed at 27±1°C, as described previously [5]. The whole experiment was carried out during a 2-day period. The complete experiment included 4 stages: open-field adaptation, familiarization period with identical objects, novel object recognition 1 h after training, and novel object recognition 24 h after training. During open-field adaptation, mice from each group were acclimatized in the bright open field for 30 min. During the familiarization period with identical objects, 2 wooden blocks of the same color and shape (A and B) were fixed on the floor of the maze 15 cm from the side wall. The mouse was gently placed in the middle of the open field with the objects, so that it could freely explore the 2 identical objects for 10 min. During novel object recognition 1 h after training, object B was replaced by object C, and the location was unchanged. The ANY-MAZE video-tracking system was used to record the contact of the mouse with the 2 objects within 5 min, including the time that the mouse’s nose or mouth touched the object or was within 2–3 cm from the object. During novel object recognition 24 h after training, the object C was replaced by object D. The time that mice explored object D was recorded. After each experiment, mice were returned to their original cages. After each test, the apparatus and all objects were individually cleaned using 75% ethanol with wet and dry cloths to eliminate olfactory cues.
After NORT, the percentage of time that a mouse spent touching a new object and the sum of the time it spent touching the objects on both sides was calculated for each group as a recognition index.
Step-through passive avoidance task
The step-through passive avoidance apparatus consisted of 2 compartments of the same size (20.3×15.9×21.3): a bright compartment and a dark compartment [14]. A lighting device was installed above the bright compartment. There was a door connecting the 2 compartments. Stainless steel bars (3.175 cm in diameter and spaced at 0.5-cm intervals) were fitted to the floors of the 2 compartments. The bars in the dark compartment were connected to a stimulator, so the mice could be given electric shocks when they entered the dark compartment. The bars in the bright compartment were not connected to the stimulator, so no intermittent electric shocks were given.
The step-through passive avoidance test contained 3 stages: adaptation, training, and memory retention. During adaptation, mice were placed into the bright compartment with their backs to the door so that they could become fully familiar with the compartment for 5 min. During training, each mouse was placed into the bright compartment. When the mouse entered the dark compartment, the door was closed immediately. Two seconds later, the mouse received 1 electric shock (10 s, 0.3 A). After that, the mouse stayed in the dark compartment for 10 s, so that the mouse formed a connection between the dark compartment and the electric shock. The mice were then returned to their original cages. At 1 h and 24 h after electric shocks, the memory retention test was conducted. The mouse was placed in the bright compartment, and the door was opened, but electric shocks were not given. We used the Super Passive Avoidance Video Recording System and Image Analysis System to record the first time that the mouse entered the dark compartment (i.e., step-through latency) and to record the number of entries into the dark compartment within 3 min (i.e., error times). If the mouse failed to enter the dark compartment within 3 min, the step-through latency was recorded as 180 s. The step-through latency was considered as an indicator of electric shock-stimulated memory. The maximum step-through latency was set as 3 min.
Morris water maze test
The procedures were modified from Morris [15]. The diameter of the water maze pool was 120 cm, and the height was 50 cm. The depth of the water was 30 cm (2 cm above the platform). The water temperature was kept at about 25°C. The platform was placed in the first quadrant. All mice were acclimatized on day 1. The position of the platform was fixed. During the training, the animals were placed into the water from 3 entry points (excluding the entry point of the fourth quadrant) with face toward the pool wall. The mice stayed on the platform for 10 s. If an animal failed to climb onto the platform within 60 s, it was manually guided onto the platform and made to stay for 10 s. In such cases, the escape latency was recorded as 60 s. In 1 session, mice were placed into the pool from 3 entry points. The mean escape latency from the 3 quadrants was considered as the final value of the navigation test. All mice from each group were tested for escape latency for 5 days, twice a day. The mean value on each day was considered as the final value on that day.
Immunocytochemistry
After fixation with 4% paraformaldehyde, sections were washed 3 times with PBS, and then permeabilized with 0.3% Triton X-100 at 37°C for 15 min. After rinsing 3 times with PBS, the sections were treated with goat serum for half an hour at 37°C. Then, they were incubated with an anti-MAP2 antibody (1: 200, Proteintech, 17490-1-AP) or an anti-SYN antibody (1: 200, Abcam, ab32127) overnight at 4°C. Sections were then incubated with fluorescent secondary antibody for 1 h at 37°C followed by counterstaining with DAPI for 15 min. Finally, labeling was visualized using a fluorescence microscope (Olympus FV1000) and analyzed using an Olympus Fluoview Ver.1.7a viewer.
Immunohistochemistry for Iba1 and GFAP
Animals were perfused with 4% paraformaldehyde and brains were post-fixed with 4% paraformaldehyde. After dehydration, clearing, and embedding, sections were cut using an RM2125 microtome (Leica Biosystems, Nussloch, Germany). Then, the sections were dewaxed by xylene and hydrated in an ethanol gradient. Antigen recovery was performed by microwaving and the sections were pretreated with 3% H2O2 for 10 min at 37°C. Subsequently, sections were incubated with rabbit anti-GFAP primary antibody (1: 200, Abcam, ab7260) or goat anti-Iba1 primary antibody (1: 200, Abcam, ab5076) at 4°C overnight. On the next day, sections were washed with PBS 3 times, and incubated with HRP-labeled goat anti-rabbit IgG polymer (PV-9001, Zhongshan Golden Bridge Biotechnology, Beijing, China) or HRP-labeled rabbit anti-goat IgG polymer (PV-9003, Zhongshan Golden Bridge Biotechnology), respectively, at room temperature for 30 min. After visualization with DAB (ZLI-9018, Zhongshan Golden Bridge Biotechnology), images were captured with an Olympus microscope (Tokyo, Japan) and analyzed by Image J software (NIH, Bethesda, MD, USA).
Western blotting
Protein was extracted using a protein extraction kit (ApexBio, K2113-50) according to standard procedures [16]. Protein concentration was quantified with a BCA Protein Quantitative Kit (TransGen, DQ101-01). Equal amounts of protein were loaded into SDS-PAGE gels for electrophoresis and then transferred onto PVDF membranes. The membranes were blocked with 5% BSA in TBST for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: rabbit anti-Nrf2 (1: 200, Abcam, ab137550), rabbit anti-HO1 (1: 500, Abcam, ab13248), rabbit anti-P65(1: 200, Abcam, ab16502), rabbit anti-PSD (1: 200, Abcam, ab76115), rabbit anti-syn (1: 200, Abcam, ab32127), and anti-beta actin (1: 200). Then, the membranes were washed 3 times with TBST and incubated with HRP-conjugated secondary antibodies at room temperature for 2 h. The bands were detected using the ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA) and analyzed with Quantity One software (Bio-Rad) normalized for beta actin density.
Tissue preparation and ultrastructure of the mouse hippocampus observed by transmission electron microscopy
The electron microscopy tissue preparation [17] for 3 mice from each group who were randomly anesthetized with 10% chloral hydrate (0.35 mg/kg) and perfused with 2% precooled glutaraldehyde and 2% precooled paraformaldehyde. Briefly, islets were fixed immediately after isolation at 4°C in 0.1 M sodium cacodylate buffer, pH 7.4, 2% paraformaldehyde, 2.5% glutaraldehyde (Electron Microscopy Sciences), and 3 μM CaCl2. Samples were then post-fixed with osmium tetraoxide (1% wt/vol in H2O) and counterstained with uranyl acetate (2% wt/vol in H2O). After embedding in Durcupan resin (Millipore Sigma), ultrathin sections (70-nm) were prepared, mounted on 300-mesh gold grids, and counterstaining with uranyl acetate (1% wt/vol in H2O) and Sato lead (1% wt/vol in H2O). Ultrathin sections were imaged at 80 keV using an electron microscope (JEOL JEM-1230) equipped with an AMT XR80 CCD camera. Three copper screens of each sample were observed, and 5 photos of each copper screen were randomly photographed. The final magnification was 5000× and 20 000×.
Statistical analysis
All data were analyzed using SPSS 21.0 software (SPSS, Chicago, IL, USA). Measurement data are expressed as the mean ± standard deviation. The difference between groups was compared using the Student’s t-test. Differences in the place navigation test in the Morris water maze were detected using repeated measures ANOVA followed by least significant difference (LSD) post hoc analysis. All other experimental data were analyzed using one-way ANOVA. A value of P<0.05 was defined as statistically significant.
Results
Lentivirus-mediated expression of Nrf2 ShRNA and microtubule-associated protein-2 (MAP2) in the hippocampus
Intrahippocampal injection of control lentivirus and Nrf2-shRNA-lentivirus were carried out in 4-month-old mice. Five months after intrahippocampal injection, to determine the stability and efficiency of lentivirus-mediated transfer, fluorescence microscopy was used to analyze GFP fluorescence in the brains of 9-month-old mice. Green fluorescence was mainly distributed in the cytoplasm, but partially in the nucleus (Figure 2).
Figure 2.

Immunofluorescence from brain sections of mice injected with lentivirus-Nrf2 shRNA with neuronal marker microtubule-associated protein-2 (Map2) (red) revealed Nrf2 shRNA (green) in the cytoplasm or nucleus (blue) of neurons present in regions of the hippocampus (n=3 per group, scale bar=50 um).
Nrf2 and Nrf2-dependent protein levels in the hippocampus
The P8 mice at 9 months had lower Nrf2 levels compared with R1 mice. The significant reduction of Nrf2 protein level, both total Nrf2 (T-Nrf2) and nuclear Nrf2 (N-Nrf2) (Figure 3A), is clear, which proves that Nrf2 was successfully downregulated in the nuclei. Similarly, Nrf2-mediated HO-1 induction has been shown in the brain. P65 was moderately increased in the P8-ShRNA group compared to the P8+GFP group, indicating the activation of inflammation response caused by downregulated Nrf2 (P<0.05). A significant reduction of total (T-Nrf2) and nuclear (N-Nrf2) Nrf2 protein levels was observed in the Nrf2 downregulated group compared with the control group (Figure 3A, 3B) (P<0.05).
Figure 3.

Expression of Nrf2 and its downstream genes in the hippocampus of SAMR1 and SAMP8 mice at 9 months (n=5 per group). (A) Protein levels of representative western blots for total-Nrf2, nuclear-Nrf2, heme oxygenase-1(HO-1), and p65. (B) Densitometry analysis of total Nrf2(T-Nrf2), nuclear Nrf2(N-Nrf2), HO-1, and p65. * P<0.05, P8+GFP group vs. R1+GFP group. # P<0.05, R1+GFP vs. R1+Nrf2 ShRNA group. ## P<0.05, P8+GFP vs. P8+Nrf2 ShRNA group.
Cognition tests in mice
NORT
In the NORT, 9-month-old SAMP8 mice were worse at distinguishing the novel object compared with the SAMR1 mice, showing the impaired situational memory at the age of 9 months in SAMP8 mice. There was no significant difference (0.32±0.12 vs. 0.41±0.14) in the recognition index (Figure 4) in the P8+Nrf2 shRNA group after 1 h, but this became worse after 24 h (P8+Nrf2 shRNA group vs. P8+GFP group: 0.36±0.15 vs. 0.53±0.24).
Figure 4.
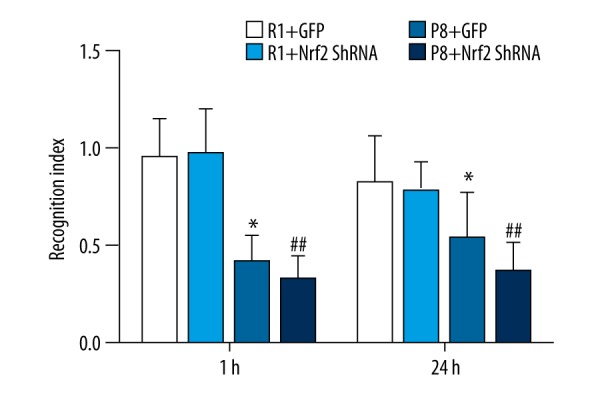
Recognition index in novel object recognition test (n=12 for R1+GFP group, n=11 for R1+Nrf2 ShRNA group; n=12 for P8+GFP group; n=11 for P8 +Nrf2 ShRNA group). * P<0.05, P8+GFP group vs. R1+GFP group. # P<0.05, R1+GFP vs. R1+Nrf2 ShRNA group. ## P<0.05, P8+GFP vs. P8+Nrf2 ShRNA group.
Morris water maze
Escape latency was not significantly different between the R1+GFP and R1+Nrf2-ShRNA groups (P>0.05). Compared with the R1 groups, escape latency was significantly prolonged in the P8 groups (P<0.05). However, Nrf2 knockdown further increased the prolongation of escape latency (P<0.05, P8+Nrf2-ShRNA group vs. P8+GFP group, Figure 5A).
Figure 5.

Results of Morris water maze test: (n=12 for R1+GFP group, n=11 for R1+Nrf2 ShRNA group, n=12 for P8+GFP group, n=11 for P8 +Nrf2 ShRNA group). (A) Escape latency (P<0.05, P8+Nrf2-ShRNA group vs. P8+GFP group). (B) Platform crossings (P<0.05, P8+Nrf2-ShRNA group vs. P8+GFP group). * P<0.05, P8+GFP group vs. R1+GFP group. # P<0.05, R1+GFP vs. R1+Nrf2 ShRNA group. ## P<0.05, P8+GFP vs. P8+Nrf2 ShRNA group.
In the spatial probe test, the platform was removed. The number of times test animals crossed the platform was not significantly different between the R1+GFP (6.2±1.04) and R1+Nrf2-ShRNA groups (6.6±0.88) (P>0.05). Compared with R1 groups, the number of times mice crossed the platform was significantly less in the P8 groups (P<0.05, Figure 5B). The number of times mice crossed the platform was further reduced in the P8+Nrf2-ShRNA group (P<0.05, P8+Nrf2-ShRNA group 1.6±0.72 vs. P8+GFP group 3.8±0.64, Figure 5B). These results indicated that aging in SAMP8 mice resulted in decreased cognition, whereas Nrf2 knockdown further aggravated cognitive impairment in SAMP8 mice, but Nrf2 knockdown did not have an obvious effect on cognitive function in SAMR1 mice.
Step-through passive avoidance task
The passive avoidance task further suggested emotional changes in SAMP8 mice. The times of the electric shock in all 9-month-old SAMP8 mice deteriorated modestly compared with the SAMR1 group, and the passive avoidance latency of P8 group mice also decrease (Figure 6B). Compared with P8+GFP group, the times of the electric shock increased significantly in the P8+Nrf2-ShRNA group (3.2±1.2 vs. 2.7±0.99, Figure 6B). However, passive avoidance latency in the P8+Nrf2-ShRNA group vs. the P8+GFP group (168±62 s vs. 198±36s) showed a modest decrease (P=0.02 <0.05, Figure 6A).
Figure 6.

The differences between P8 and R1 groups in the passive avoidance test, the passive avoidance latency (A), and the times of the electric shock (B) (n=12 for R1+GFP group, n=11 for R1+Nrf2 ShRNA group, n=12 for P8+GFP group, n=11 P8 +Nrf2 for ShRNA group). * P<0.05, P8+GFP group vs. R1+GFP group. # P<0.05, R1+GFP vs. R1+Nrf2 ShRNA group. ## P<0.05, P8+GFP vs. P8+Nrf2 ShRNA group.
Increased Iba1 and GFAP expression as measured by immunohistochemistry
As neuroimmunological response parameters, microglial processes at the site of Iba1 cell aggregation were numerous and thick in the P8 group (Figure 7B). Iba1-positive cells were more numerous in a unit area of the hippocampus of 9-month-old mice in the P8+Nrf2-ShRNA group compared with the P8+GFP group (4.322±0.168 vs. 2.726±0.176 P=0.000, Figure 7B, 7D). Similarly, GFAP immunoreactivity was increased in the CA1 region of 9-month-old mice in the P8+Nrf2-ShRNA group compared with the P8+GFP group (5.178±0.613 vs. 3.684±0.562, P<0.05, Figure 7A, 7C).
Figure 7.

Immunohistochemistry staining of GFAP (A) and Iba1 (B) in CA1 region. The results of immunohistochemistry staining of GFAP (C) and Iba1(D); (n=4 per group, scale bar=50 um).
PSD-95 and SYN expression in the hippocampal CA1 region
PSD-95 and SYN are markers for synaptic plasticity. Compared with R1 groups, synaptophysin and PSD95 proteins in P8 mice were significantly decreased (Figure 8A). Similarly, western blot assays demonstrated that PSD and SYN expression was reduced in the P8 groups and was significantly decreased in the P8+Nrf2 ShRNA group (P<0.05, Figure 8B), indicating that Nrf2 downregulation significantly affects the synapse. To further investigate the effects of Nrf2 depletion on the hippocampus, SYN immunofluorescence staining was monitored using confocal microscopy (Figure 9A). The area fraction of SYN also was dramatically diminished in the P8+Nrf2 ShRNA group (P8+GFP group vs. P8+Nrf2 ShRNA group: 30.268±2.233 vs. 22.495±1.387, P<0.05, Figure 9B).
Figure 8.

Protein expression level (PSD and SYN) were determined (B) and representative western blot (A) for PSD and SYN (n=5 per group).
Figure 9.

Results of immunofluorescence staining. (A) Expression results of SYN (scale bar=25 μM). (B)% area fraction of SYN (n=3 per group).
Ultrastructural differences in hippocampal CA1 region neurons in each group observed by transmission electron microscopy
Morphological changes of neurons
In the R1+GFP group, neuronal cell membranes were clearly visible. The cytoplasmic matrix and ribosomes were uniformly distributed. Mitochondrial membranes and mitochondrial cristae were distinct and complete (Figure 10A). In the R1+Nrf2 ShRNA group, hippocampal neurons did not have obvious changes compared with the R1+GFP group; lipid droplets and lipofuscin were occasionally visible within the cell matrix (Figure 10B). In the R1+Nrf2 ShRNA group, the cell membranes of hippocampal neurons were still intact. However, high levels of lipofuscin were observed in the cell matrix, some of which were surrounded by a bilayer membrane structure. Some Golgi complex membranes were fuzzy. The nucleus and nuclear membrane were intact. Pyknosis was observed (Figure 10C). In the P8+Nrf2 ShRNA group, hippocampal neuron membranes were blurred and there was cytoplasmic vacuolization, and a pale and uneven matrix was observed. High levels of lipofuscin and autophagosomes were seen in the cell matrix. The number of mitochondria and the amount of endoplasmic reticulum and Golgi complex were reduced (Figure 10D).
Figure 10.

Ultrastructural alterations in neurons in CA1 region. (A) R1+GFP group: cell membrane of normal neurons. (B) R1+ Nrf2 ShRNA group: the rough endoplasmic reticulum. (C) P8+GFP group: a large amount of lipofuscin in the cell matrix. (D) P8+Nrf2 ShRNA group: height edema in cytoplasm and nucleoplasm of neurons (magnification: 5000× and 20 000×).
Synaptic changes
The main morphological changes were in the synaptic cleft, the number of synapses, and the width of the postsynaptic density. In the R1 group, there were many synapses. The synaptic glomerulus, presynaptic membrane, postsynaptic membrane, and synaptic cleft were distinct (Figure 11A, 11B). Links between synapses were reduced (Figure 11C, 11D). In the P8+Nrf2 ShRNA group, postsynaptic density thickness was remarkably diminished. The postsynaptic membrane was thickened, the synaptic cleft disappeared, and there were few synaptic vesicles (Figure 11D).
Figure 11.

Synaptic structure by transmission electron microscopy. (A) R1+GFP group. (B) R1+Nrf2 ShRNA group. (C) P8+GFP group. (D) P8+Nrf2 ShRNA group (magnification: 20 000×).
Discussion
Learning and memory in this study were evaluated by 3 behavioral tests that involve different aspects of cognition: the novel object recognition test (NORT) is used to judge situational and recognition memory; the Morris water maze test identifies higher-level memory, such as spatial memory; and the passive avoidance test leads to aversive emotional memory, especially fear memory [18,19].
In our study, the lentivirus injected into the hippocampus of SAMP8 mice successfully reduced the expression of Nrf2 in the hippocampus by 55%. Five months after injection, Nrf2 protein levels were obviously decreased. Compared with R1 mice, both total and nuclear Nrf2 levels were lower in P8 mice. Reduced expression of Nrf2 has also been reported in AD brains [20].
Previous studies have shown that Nrf2 mediation by lentiviral expression improves spatial memory. In another AD model mice, Nrf2-knockout mice exhibited reduced expression of an autophagy gene involved in learning and memory processes [21,22]. However, few studies have assessed cognitive domain changes in SAMP8 mice through the Nrf2/ARE pathway. SAMP8 is a valuable animal model, with young SAMP8 mice in particular showing impairment in emotional behavior with regard to fear and anxiety [23]. The varying tendencies of cognitive parameters is consistent with oxidative stress disorder in SAMP8 mice [24–26]. Our results showed that downregulation of Nrf2 not only affected spatial learning, but also impaired fear memory in SAMP8 mice. Other studies have reported that Nrf2-knockout in mice impairs autophagy such that it is insufficient to maintain proteostasis [27]. Correspondingly, enhancing the hippocampal Nrf2/ARE signaling pathways improves aging-induced cognitive dysfunction. In conclusion, deficiency of Nrf2 is associated with adverse outcomes or reduced neuroprotection.
We observed that partial downregulation of Nrf2 had no effect on learning and memory in SAMR1 mice. Compared with SAMR1 mice, the expression of Nrf2 and downstream HO-1 in P8 mice was significantly reduced. Lower Nrf2 protein level was associated with worse memory in the P8+Nrf2 ShRNA group. A previous study [28] showed that low levels of Nrf2 are associated with high levels of oxidative stress in the pathogenesis of neurodegenerative diseases, including AD. Increasing Nrf2 levels or decreasing some Nrf2-negative regulators, such as GSK-3β or keap1, reduced stress, and improved cognition in 12-month-old SAMP8 mice [29].
In this study, we observed a significant increase in the inflammatory response in the P8+ShRNA group. In APP/PS1 mice, a decrease in Nrf2 also increased many inflammatory reactions, including activation of microglia [30]. Nrf2 is involved in regulation of anti-inflammatory cytokines via increasing p65 [31]. As a key regulator of inflammation, P65 were measured in all groups. P65 in the P8+ShRNA group was clearly increased. P65 has been shown to function as a negative regulator of Nrf2 activation [32]. It is interesting that downregulation of Nrf2 failed to vary the p65 protein level between R1 groups. The lack of Nrf2 in APP/PS1 mice drives p65 into more insoluble aggregates. Increased hippocampal levels of GFAP were also observed in our study, as in earlier reports, clearly indicating the presence of reactive astrocytes, which accompany an inflammatory process.
GFAP and Iba1 label astrocytes and microglia, respectively. As markers to detect inflammatory reactions, GFAP and Iba1 directly reflect the function of the brain and are significantly associated with cognitive changes [33]. Microglia interact with astrocytes to activate and drive inflammation in the brain. In the brain of AD patients, levels of GFAP and Iba1 are increased [12,34]. In the hippocampus of Nrf2 knockdown mice, GFAP and Iba1 expression is increased and microglial proliferation is activated, which accelerates the impairment of neurological function. The Nrf2/ARE pathway is also called the cholinergic anti-inflammatory pathway [35,36]. The importance of Nrf2 had been shown in the anti-inflammatory response, as demonstrated in Nrf2 knockout mice. Nrf2-mediated glutathione is released from astrocytes and provides neuroprotection [37,38].
Spatial learning and memory impairment are early clinical features of AD caused by synaptic dysfunction rather than neuronal loss. We also measured PSD95 and SYN proteins. PSD95 and downstream molecular signals play an important role in synaptic development and neural information transfer. Previous studies in our lab showed that decreased SYN and PSD95 expression and cognitive impairment caused chronic stress in the SAMP8 mice [39]. Our study demonstrates that lower PSD95 and SYN expression in P8 mice was associated with synapse density and the degree of cognitive impairment. The correlation between synapse density and the degree of cognitive impairment is well established in patients with AD. Maintaining the expression of Nrf2/PSD95 may contribute to normal behavior in mice. The confocal microscopy results showing the reduction in the area expressing SYN indicate that Nrf2 depletion also disrupted mitochondrial function and synaptic integrity. Mitochondrial function, regulation of stress, and inflammation response all contribute to the loss of synaptic integrity, which is probably the cause of cognitive impairment in mice. Enhanced synaptic markers (SYN, PSD95, and Drebrin) modulate the synaptic structure and function of the hippocampus, all of which result in improved cognitive function [40].
Some studies have confirmed that Nrf2- and ARE-dependent signaling pathways mediate detoxification and antioxidant genes, providing a new perspective for age-related diseases. Some factors that promote Nrf2, such as pyrrolidine, lycopene, and sulfuretin, improve cognition [7,41]. H2S alleviates cognitive impairment in APP/PS1 transgenic mice by upregulating Nrf2 [42]. As a result, some pharmacological Nrf2 activators are applied in clinical trials [43]. The mechanism of Nrf2-mediated neuroprotection will be further studied in future research.
Emerging evidence suggests that overexpression of Nrf2 is important in improving mitochondrial integrity during oxidative damage and inflammatory response and alleviating the attack of Aβ peptides on neurons [44,45]. The activity of Nrf2 protects cells and makes the cell resistant to endothelial dysfunction. Furthermore, elevated Nrf2 activity promotes cell survival and proliferation. Nrf2 activators could exert positive effect for AD, which will alleviate brain aging and improve cognitive functions. Despite some limitations, our study still provides some learning and emotional changes in SAMP8 mice that were rarely reported before, further elucidating the possible mechanism.
Conclusions
We found a significant decrease in Nrf2 expression in the hippocampus in SAMP8 mice, as well as activated oxidative stress and disrupted synaptic integrity, ultimately worsening the cognitive function of SAMP8 mice. Therefore, the Nrf2/ARE pathway may be a new therapeutic target for age-related disease. In particular, activators of Nrf2 or reducing downstream products of Nrf2 may be new therapeutic strategies.
Footnotes
Conflict of interest
None.
Source of support: This work was supported by Provincial Major Medical Programs (ZD2013077, ZD2013079) and the Hebei Health and Family Planning Commission Directive Program (ZL20140184)
References
- 1.Hopperton KE, Mohammad D, Trepanier MO, et al. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol Psych. 2017;23(2):177–198. doi: 10.1038/mp.2017.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakrabarty P, Li A, Ceballos-Diaz C, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85:519–33. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pan W, Han S, Kang L, et al. Effects of dihydrotestosterone on synaptic plasticity of the hippocampus in mild cognitive impairment male SAMP8 mice. Exp Ther Med. 2016;12:1455–63. doi: 10.3892/etm.2016.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pajares M, Jimenez-Moreno N, Garcia-Yague AJ, et al. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016;12:1902–16. doi: 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kandimalla R, Manczak M, Fry D, et al. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet. 2016;25:4881–97. doi: 10.1093/hmg/ddw312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rojo AI, Pajares M, Rada P, et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017;13:444–51. doi: 10.1016/j.redox.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwon SH, Ma SX, Hwang JY, et al. Involvement of the Nrf2/HO-1 signaling pathway in sulfuretin-induced protection against amyloid beta25–35 neurotoxicity. Neuroscience. 2015;304:14–28. doi: 10.1016/j.neuroscience.2015.07.030. [DOI] [PubMed] [Google Scholar]
- 8.Zhao X, Bian Y, Sun Y, et al. Effects of moderate exercise over different phases on age-related physiological dysfunction in testes of SAMP8 mice. Exp Gerontol. 2013;48:869–80. doi: 10.1016/j.exger.2013.05.063. [DOI] [PubMed] [Google Scholar]
- 9.Mottahedin A, Ardalan M, Chumak T, et al. Effect of neuroinflammation on synaptic organization and function in the developing brain: implications for neurodevelopmental and neurodegenerative disorders. Front Cell Neurosci. 2017;11:190. doi: 10.3389/fncel.2017.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baltanas A, Solesio ME, Zalba G, et al. The senescence-accelerated mouse prone-8 (SAM-P8) oxidative stress is associated with upregulation of renal NADPH oxidase system. J Physiol Biochem. 2013;69:927–35. doi: 10.1007/s13105-013-0271-6. [DOI] [PubMed] [Google Scholar]
- 11.Akiguchi I, Pallas M, Budka H, et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda’s legacy and future directions. Neuropathology. 2017;37:293–305. doi: 10.1111/neup.12373. [DOI] [PubMed] [Google Scholar]
- 12.Branca C, Ferreira E, Nguyen TV, et al. Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2017;26:4823–35. doi: 10.1093/hmg/ddx361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanninen K, Heikkinen R, Malm T, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Pro Natl Acad Sci USA. 2009;106:16505–10. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brodziak A, Kolat E, Rozyk-Myrta A. In search of memory tests equivalent for experiments on animals and humans. Med Sci Monit. 2014;20:2733–39. doi: 10.12659/MSM.891056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vorhees CV, Williams MT. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–58. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J, Yuan J, Pang J, et al. Effects of chronic stress on cognition in male SAMP8 mice. Cell Physiol Biochem. 2016;39:1078–86. doi: 10.1159/000447816. [DOI] [PubMed] [Google Scholar]
- 17.Heidari MH, Amini A, Bahrami Z, et al. Effect of chronic morphine consumption on synaptic plasticity of rat’s hippocampus: A transmission electron microscopy study. Neurol Res Int. 2013;2013:290414. doi: 10.1155/2013/290414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.d’Isa R, Brambilla R, Fasano S. Behavioral methods for the study of the Ras-ERK pathway in memory formation and consolidation: Passive avoidance and novel object recognition tests. Methods Mol Biol. 2014;1120:131–56. doi: 10.1007/978-1-62703-791-4_9. [DOI] [PubMed] [Google Scholar]
- 19.Tabassum S, Ahmad S, Madiha S, et al. Impact of oral supplementation of Glutamate and GABA on memory performance and neurochemical profile in hippocampus of rats. Pak J Pharm Sci. 2017;30:1013–21. [PubMed] [Google Scholar]
- 20.Cao H, Wang L, Chen B, et al. DNA Demethylation upregulated Nrf2 expression in Alzheimer’s disease cellular model. Front Aging Neurosci. 2015;7:244. doi: 10.3389/fnagi.2015.00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao M, Lewis Wang FS, Hu X, et al. Acrylamide-induced neurotoxicity in primary astrocytes and microglia: Roles of the Nrf2-ARE and NF-kappaB pathways. Food Chem Toxicol. 2017;106:25–35. doi: 10.1016/j.fct.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 22.Sardari M, Rezayof A, Khodagholi F. Hippocampal signaling pathways are involved in stress-induced impairment of memory formation in rats. Brain Res. 2015;1625:54–63. doi: 10.1016/j.brainres.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 23.Flood JF, Morley JE. Learning and memory in the SAMP8 mouse. Neurosci Biobehav Rev. 1998;22:1–20. doi: 10.1016/s0149-7634(96)00063-2. [DOI] [PubMed] [Google Scholar]
- 24.Grinan-Ferre C, Palomera-Avalos V, Puigoriol-Illamola D, et al. Behaviour and cognitive changes correlated with hippocampal neuroinflammaging and neuronal markers in female SAMP8, a model of accelerated senescence. Exp Gerontol. 2016;80:57–69. doi: 10.1016/j.exger.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 25.Yanai S, Endo S. Early onset of behavioral alterations in senescence-accelerated mouse prone 8 (SAMP8) Behav Brain Res. 2016;308:187–95. doi: 10.1016/j.bbr.2016.04.026. [DOI] [PubMed] [Google Scholar]
- 26.Qiang M, Xiao R, Su T, et al. A novel mechanism for endogenous formaldehyde elevation in SAMP8 mouse. J Alzheimers Dis. 2014;40:1039–53. doi: 10.3233/JAD-131595. [DOI] [PubMed] [Google Scholar]
- 27.Wong YC, Holzbaur EL. Autophagosome dynamics in neurodegeneration at a glance. J Cell Sci. 2015;128:1259–67. doi: 10.1242/jcs.161216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshmukh P, Unni S, Krishnappa G, Padmanabhan B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys Rev. 2017;9:41–56. doi: 10.1007/s12551-016-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farr SA, Ripley JL, Sultana R, et al. Antisense oligonucleotide against GSK-3beta in brain of SAMP8 mice improves learning and memory and decreases oxidative stress: Involvement of transcription factor Nrf2 and implications for Alzheimer disease. Free Radic Biol Med. 2014;67:387–95. doi: 10.1016/j.freeradbiomed.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer’s disease-like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36:664–79. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye F, Zhao T, Liu X, et al. Long-term Autophagy and Nrf2 signaling in the hippocampi of developing mice after carbon ion exposure. Sci Rep. 2015;5:18636. doi: 10.1038/srep18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dinkova-Kostova AT, Kostov RV, Kazantsev AG. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018 doi: 10.1111/febs.14379. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rice RA, Spangenberg EE, Yamate-Morgan H, et al. Elimination of microglia improves functional outcomes following extensive neuronal loss in the hippocampus. J Neurosci. 2015;35:9977–89. doi: 10.1523/JNEUROSCI.0336-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buendia I, Michalska P, Navarro E, et al. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther. 2016;157:84–104. doi: 10.1016/j.pharmthera.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: A critical review. Auton Neurosci. 2014;182:65–69. doi: 10.1016/j.autneu.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gan L, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci. 2012;32:17775–87. doi: 10.1523/JNEUROSCI.3049-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 39.Grinan-Ferre C, Puigoriol-Illamola D, Palomera-Avalos V, et al. Environmental enrichment modified epigenetic mechanisms in SAMP8 mouse hippocampus by reducing oxidative stress and inflammaging and achieving neuroprotection. Front Aging Neurosci. 2016;8:241. doi: 10.3389/fnagi.2016.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou C, Shi Y, Ohli J, et al. Neuroinflammation impairs adaptive structural plasticity of dendritic spines in a preclinical model of Alzheimer’s disease. Acta Neuropathol. 2016;131:235–46. doi: 10.1007/s00401-015-1527-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liddell JR, Lehtonen S, Duncan C, et al. Pyrrolidine dithiocarbamate activates the Nrf2 pathway in astrocytes. J Neuroinflammation. 2016;13:49. doi: 10.1186/s12974-016-0515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y, Deng Y, Liu H, et al. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol Biochem Behav. 2016;150–51:207–16. doi: 10.1016/j.pbb.2016.11.002. [DOI] [PubMed] [Google Scholar]
- 43.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–92. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 44.Karkkainen V, Pomeshchik Y, Savchenko E, et al. Nrf2 regulates neurogenesis and protects neural progenitor cells against Abeta toxicity. Stem Cells. 2014;32:1904–16. doi: 10.1002/stem.1666. [DOI] [PubMed] [Google Scholar]
- 45.Wu G, Liu Z. Nuclear factor erythroid 2-related factor 2 (Nrf2) mediates neuroprotection in traumatic brain injury at least in part by inactivating microglia. Med Sci Monit. 2016;22:2161–66. doi: 10.12659/MSM.896568. [DOI] [PMC free article] [PubMed] [Google Scholar]