

Abstract
Among metabolic rearrangements occurring in cancer cells, lipid metabolism alteration has become a hallmark, aimed at sustaining accelerated proliferation. In particular, fatty acids (FAs) are dramatically required by cancer cells as signalling molecules and membrane building blocks, beyond bioenergetics. Along with de novo biosynthesis, free FAs derive from dietary sources or from intracellular lipid droplets, which represent the storage of triacylglycerols (TAGs). Adipose triglyceride lipase (ATGL) is the rate-limiting enzyme of lipolysis, catalysing the first step of intracellular TAGs hydrolysis in several tissues. However, the roles of ATGL in cancer are still neglected though a putative tumour suppressor function of ATGL has been envisaged, as its expression is frequently reduced in different human cancers (e.g., lung, muscle, and pancreas). In this review, we will introduce lipid metabolism focusing on ATGL functions and regulation in normal cell physiology providing also speculative perspectives on potential non-energetic functions of ATGL in cancer. In particular, we will discuss how ATGL is implicated, mainly through the peroxisome proliferator-activated receptor-α (PPAR-α) signalling, in inflammation, redox homoeostasis and autophagy, which are well-known processes deregulated during cancer formation and/or progression.
Facts
ATGL is the rate-limiting enzyme of lipolysis, mainly expressed in adipose tissues but virtually functioning in all other organs.
ATGL has been found downregulated in several human cancers including lung and pancreas.
In most immune cells ATGL activity contributes to pro-inflammatory responses mainly providing precursors for eicosanoids. In non-immune cells (e.g., myocytes, hepatocytes and adipocytes) the ATGL-PPAR-α axis maintains an anti-inflammatory phenotype.
ATGL favours redox homoeostasis in many cell types mainly through a PPAR-α-mediated control of antioxidant enzymes.
ATGL triggers autophagy through SIRT1 in hepatocytes and its involvement in lipophagy facilitates lipid droplets mobilisation.
Open questions
Identification of upstream signalling pathways that account for ATGL downregulation in cancer
Analysis of the ATGL-PPAR-α axis in cancer development and/or progression
Identification of downstream molecular targets linking ATGL downregulation to tumorigenesis
Introduction
Lipids, including triacylglycerols (TAGs) and cholesterol, essentially derive from dietary sources and are transported in the blood mainly packaged in lipoprotein complexes. Free fatty acids (FAs) are obtained from TAGs-rich lipoprotein through the action of lipoprotein lipases, located at the capillary walls of most tissues, or by lysosomal acidic hydrolysis following lipoprotein endocytosis. Moreover, FAs also derive from de novo biosynthesis and those released by the adipose tissue are substrates for mitochondrial β-oxidation in most cell types. Beyond energetic purposes, FAs are essential for membranes biosynthesis and also serve as signalling molecules1.
Intracellular fat depots consist of lipid droplets (LDs), which store neutral lipids such as TAGs and cholesteryl esters to avoid lipotoxicity. LDs are dynamic organelles with complex biogenesis and multiple functions, present in many cell types beyond adipocytes2–4. Consistently, the enzymatic machinery for LDs remodelling/utilisation to release FAs is ubiquitously expressed and equipped with specific hydrolases designated lipases. The adipose triglyceride lipase (ATGL) selectively catalyses the first and rate-limiting step of intracellular TAGs hydrolysis to generate diacylglycerol (DAG) and FAs5,6. DAGs are then sequentially hydrolysed by the hormone-sensitive lipase (HSL) and the monoacylglycerol lipase (MAGL), achieving the release of FAs and glycerol. Overall, this process is known as lipolysis (Fig. 1)7,8. The molecular mechanisms involved in lipolysis are well-characterised in adipocytes. Indeed, fasting and hormone stimulation, including epinephrine and glucagon, trigger the activation of cAMP-dependent protein kinase A (PKA). The phosphorylation of HSL by PKA is required for translocation to LDs and consequent activation9. On the contrary, ATGL hydrolytic activity on LDs occurs also in a hormone-independent fashion and is further enhanced upon fasting, depending on AMPK-mediated phosphorylation10, and following β-adrenergic stimulation via PKA-mediated phosphorylation (Fig. 1)11.
Fig. 1. Canonical lipolytic function of ATGL in adipocytes.

Schematic representation of lipid droplet (LD) components and the lipolytic machinery. LDs are organelles storing triacylglycerols (TAGs) and cholesteryl esters (CEs) and are composed of a phospholipid monolayer anchoring different proteins, including the perilipins (PLINs). In the unstimulated condition, ATGL is inhibited by the association with PLIN5 and G0S2. However, a basal TAG hydrolysis by ATGL could be achieved through AMPK-mediated phosphorylation and/or interaction with CGI-58, when the latter is not associated with PLIN1. In the fasted state, the hormones epinephrine or glucagon bind to their cognate receptors stimulating the adenylyl cyclase (AC) activity. Increased cAMP levels activate cAMP-dependent protein kinase (PKA), which phosphorylates ATGL, PLIN1 and hormone-sensitive lipase (HSL). Following PLIN1 phosphorylation, CGI-58 is released and thus it can further activate ATGL. PKA-mediated phosphorylation of HSL favours its association with LDs and its activity, leading to diacylglycerol (DAG) conversion into monoacylglycerol (MAG). The lipolytic cascade culminates with the MAG lipase (MAGL) activity, which induces the release of free fatty acid (FFA) and glycerol. FFAs are then delivered to mitochondria to produce energy through β-oxidation or used as signalling molecules for nuclear receptors, such as peroxisome proliferator-activated receptors (PPARs)
ATGL is a member of the patatin-like phospholipase domain containing (PNPLA) family, specifically codified by the PNPLA2 gene and highly expressed in adipose tissue, whereas moderate to low levels are detectable in all other tissues, including liver, heart and skeletal muscle5. The relevance of ATGL for whole-body energetics is confirmed by TAGs systemic accumulation in ATGL knockout (KO) mice, which face premature death from cardiac dysfunction due to massive fat accumulation12. Organ-specific features of ATGL KO mice include hepatic steatosis13 and altered insulin signalling pathway in adipose tissue, liver and skeletal muscle14,15. In humans, PNPLA2 gene bi-allelic loss-of-function mutations also cause extensive LDs accumulation in several organs leading to a disorder named “neutral lipid storage disease with myopathy” (NLSDM), characterised by progressive myopathy, cardiomyopathy and hepatomegaly16,17.
Most aspects of ATGL functions in lipolysis have been characterised in adipocytes. ATGL association with LDs in basal conditions is primarily mediated by a highly conserved C-terminal hydrophobic sequence5,18. Moreover, an intricate crosstalk with different protein partners controls ATGL localisation and activity. Indeed, the interaction with comparative gene identification-58 (CGI-58) is known to stimulate ATGL activity19 while the small basic protein G0/G1 switch gene 2 (G0S2) was identified as a selective inhibitor of ATGL impeding substrate accessibility19,20. Furthermore, the LD-associated scaffold proteins perilipins (PLINs) are implicated in the regulation of ATGL function. PLIN1 is highly expressed in adipose tissue and, in basal conditions, sequesters CGI-58, which is released by hormone stimulation with consequent activation of ATGL21,22. PLIN5 is instead expressed in muscle, liver and brown adipose tissue and, although promoting ATGL association with LDs, impairs its hydrolase activity23.
ATGL-mediated lipolysis releases FAs that are used for β-oxidation as well as intracellular signalling. Indeed, FAs are cognate ligands for nuclear receptors/transcription factors, among which the peroxisome proliferator-activated receptor (PPAR) family is the most characterised24. In particular, ATGL activity is associated with increased levels of PPAR-α downstream targets, which are mainly involved in increased uptake and oxidation of lipids25. Consistently, ATGL-released free FAs favour energy expenditure through the upregulation of mitochondrial biogenesis and β-oxidation genes in combination with peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α) in cardiac muscle, liver and adipose tissue25–27. An alternative mechanism describes ATGL-mediated activation of Sirtuin 1 (SIRT1) as the linking event that triggers PGC-1α/PPAR-α signalling28. Mitochondrial functionality in brown adipocytes and pancreatic β cells is instead regulated by ATGL-mediated activation of PPAR-δ27,29. Along with the described ATGL/PPAR-α signalling impinging on mitochondrial metabolism, this axis impacts on additional processes that will be deeply examined throughout this review.
Metabolic rewiring and ATGL deregulation in cancer
Metabolic reprogramming is a key feature of cancer cells to sustain fast proliferation rate, which is at the basis of tumour progression30,31. In order to cope with the required energetic demand, most cancer cells exploit a faster ATP production by enhancing the glycolytic rate rather than the oxidative phosphorylation. This metabolic adaption is defined as Warburg effect, or aerobic glycolysis32. Importantly, the metabolic requirement of cancer is not uniquely limited to energetics as it also accounts for the generation of new cells in the tumour mass by microenvironment rearrangement and augmented macromolecule synthesis31. In this regard, glutamine is largely engaged as nitrogen donor for nucleotide and amino-acid biosynthesis while sustaining mitochondrial metabolism33. Interestingly, also increased de novo lipid synthesis has been established as key metabolic footprint of nearly all cancers and is achieved through the upregulation of lipogenic enzymes. Unlike most adult tissues except adipose tissue and liver, cancer cells reactivate FAs synthesis for the generation of membranes building blocks and for the sustenance of oncogenic lipid signalling and protein post-translational modification34. Remarkably, also lipid catabolic pathways are re-adapted in transformed cells. For instance, non-glycolytic tumours, like prostate cancer and diffuse large B-cell lymphoma, are highly dependent on mitochondrial β-oxidation35,36. Moreover, elevated activity of lipases, including phospholipase A2 (producing lysophospholipids and FAs from glycerophospholipids) and phospholipase D (producing phosphatidic acid and free choline from phosphatidylcholine), has been documented37,38.
Although the fundamental role of ATGL in LDs catabolism has been extensively investigated in the last decade, only recent evidence has highlighted a deregulation of ATGL in cancer specimens. However, the few data available on the mechanisms by which ATGL might impinge on cancer formation and progression are still elusive and controversial.
Most of in vitro studies have proposed pro-neoplastic features of ATGL. Consistently, reduced proliferation and invasiveness were observed upon ATGL depletion in colorectal cancer cells39 and non-small-cell lung carcinoma cell lines40. The role of ATGL in prostate cancer cells is instead ambiguous as opposite evidence has been reported up to now41,42. Interestingly, ATGL upregulation in breast cancer was associated with a tumour microenvironment enriched in adipocytes, contributing to aggressiveness of high-grade tumours43. Similarly, pancreatic ductal adenocarcinoma with elevated levels of ATGL were characterised by higher adiposity and stromal proliferation (desmoplasia)44.
This scenario is further complicated by the contribution of ATGL interactors to cancer. In this regard, an antitumor property for the ATGL co-activator CGI-58 was proposed. Indeed, deregulation of CGI-58 in prostate and colorectal cancer cells did not mimic the effects of ATGL on cell proliferation and invasion, suggesting that CGI-58 regulates such processes independently of ATGL39,41,45. On the contrary, the tumour suppressor function of G0S2 actually occurs by inhibition of ATGL, attenuating cell growth and motility in cancer cells40. Nevertheless, ATGL is not required for oncogene-mediated transformation of G0S2 null fibroblasts45.
Strikingly, recent in vivo insights unambiguously shed light on anti-neoplastic effects of ATGL in mouse models and human cancer. Indeed, mice lacking ATGL displayed spontaneous development of pulmonary neoplasia46, whereas adipose-specific ablation of both ATGL and HSL-induced liposarcoma in brown adipose tissue between 11 and 14 months of age47. Consistently, the expression of ATGL was found extremely reduced in human specimens of lung adenocarcinoma and lung squamous cell carcinoma with respect to normal epithelium46. Analogously, loss of ATGL was also depicted in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia, being progressive and stage-dependent46. Reduction of ATGL levels were also disclosed in malignant smooth muscle tumour (leiomyosarcoma), with respect to normal counterpart46. Accordingly, unpublished data from our laboratory highlighted reduced transcript levels of ATGL in both human biopsies and in a murine model of induced hepatocellular carcinoma. Although no causative molecular mechanisms for ATGL downregulation has been revealed yet, it is noteworthy to mention that the 11p15.5 chromosome arm harbouring the human PNPLA2 gene results frequently deleted in cancer and considered a hot-spot of cytogenetic alterations in human malignancies48,49. Indeed, PNPLA2 is deleted in the 38% of lung cancer and in other cancer types, including ovarian serous cystadenocarcinoma, glioblastoma, oesophageal and stomach carcinomas and paraganglioma46.
While the role of ATGL in cancer is still debated due to incongruence between in vitro and in vivo evidence, the oncogenic role of MAGL, the third lipolytic enzyme, in cancer is well established50–52. Indeed, MAGL is highly expressed in many aggressive human cancers where it orchestrates lipid signalling-mediated tumorigenesis, migration and invasion, acting as a critical regulator of metastatization52–54. Given this evidence, it is enticing to hypothesise that each lipase exerts functions beyond metabolism (i.e., lipolysis) that account for their contribution to cancer. In this context, it is noteworthy that ATGL affects many cellular processes including inflammation, oxidative stress and autophagy, which are highly perturbed in malignant cells. These ATGL effects highly rely on the activation of PPAR-α signalling, are largely conserved among tissues and may correlate with its contribution to tumour biology, as reported in the following sections.
ATGL and inflammation
Inflammation is critically involved in cancer initiation and progression. In fact, sites of infection or chronic injury can originate solid tumours due to local persistent inflammation, which induces genome instability, DNA damage and activation of cancer-promoting genes55,56. Alternatively, inflammatory cells can be channelled later in tumour sites, where they build up a signalling network that promotes disease progression. Consistently, inflammatory tumour microenvironment fosters survival and migration promoting the formation of blood and lymphatic vessels that feed and disseminate cancer cells57,58. Tumour microenvironment is directly orchestrated by cancer cells that have equipped their secretome with signalling inflammatory molecules, such as cytokines and chemokines, enabling the recruitment of immune and endothelial cells in order to further amplify inflammatory and angiogenetic processes59.
A possible anti-neoplastic feature of ATGL can be envisaged in its ability to restrain inflammatory response in non-immune cells. In fact, heart, adipose tissue and skeletal muscle from ATGL KO mice exhibited increased basal mRNA levels of pro-inflammatory genes including tumour necrosis factor (TNF)α, interleukin (IL)-6, IL-1β and monocyte chemotactic protein 1 (MCP-1) (Fig. 2)60–62. Livers from ATGL KO mice also displayed augmented inflammatory response, in terms of high transcript levels of pro-inflammatory markers, after the induction of hepatic inflammation dependent on steatohepatitis or upon endotoxin challenge63.
Fig. 2. ATGL effects in inflammation.
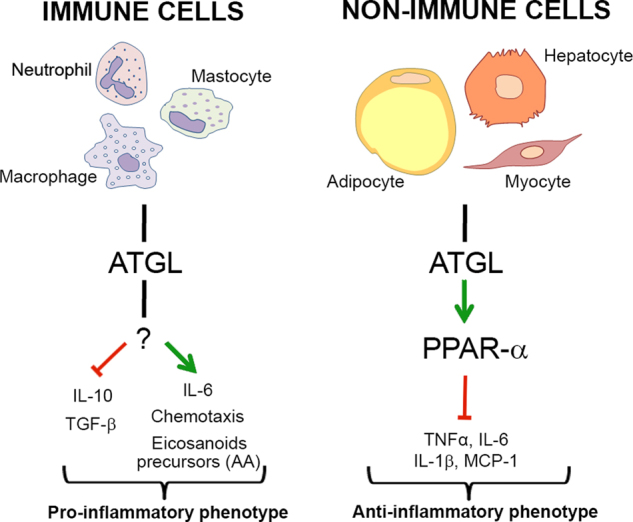
In most immune cells ATGL activity supports pro-inflammatory responses and chemotaxis mainly contributing to the production of the pro-inflammatory cytokines IL-6, and of arachidonic acid (AA), the precursor of eicosanoids. The protein effectors downstream ATGL activity are still poorly characterised in immune cells. In some non-immune cells, namely adipocytes, myocytes and hepatocytes, the ATGL-PPAR-α axis hinders the production of pro-inflammatory cytokines including IL-6, TNFα, IL-1β and MCP-1
Attenuation of inflammatory response mediated by ATGL in non-immune cells has been largely associated with the activation of PPAR-α signalling (Fig. 2). Consistently, myocytes and adipocytes depleted for ATGL showed decreased levels of PPAR-α-target cytokine genes61,64. Moreover, treatment of ATGL KO mice with PPAR-α agonists ameliorated the massive inflammatory response occurring after the induction of hepatic inflammation63. Therefore, the ATGL-mediated FAs release may partially contribute to the already established anti-inflammatory role of PPAR-α65, at least in non-immune cells.
It has to be mentioned that ATGL also participates to proper inflammatory response of immune cells. The first indirect evidence of this action derived from the prominent accumulation of LDs in leucocytes from subjects affected by NLSDM, a hallmark of the disease known as Jordan’s anomaly17. Later studies on animal models corroborated this evidence. Elevated content of LDs was observed in peripheral blood cells from ATGL KO mice, particularly neutrophils66, which exhibited an even more evident increase of LDs as well as of chemotaxis when challenged with pro-inflammatory stimuli. Notably, LDs can be a source of arachidonic acid, a precursor of the inflammatory signalling molecules eicosanoids, the biosynthesis of which is compromised by ATGL deletion/inhibition in neutrophils and mastocytes (Fig. 2)66,67.
Broad aspects of macrophage functionality are also affected by ATGL depletion. In particular, macrophages from ATGL KO mice show an anti-inflammatory phenotype characterised by inefficient phagocytic activity, impaired migration and decreased release of the pro-inflammatory cytokine IL-6, associated with an increase of the anti-inflammatory molecules IL-10 and TGF-β (Fig. 2)68–70. Interestingly, the ATGL-mediated regulation of all these processes in macrophages can underpin the attenuated formation of atherosclerotic lesions in a mouse model of atherosclerosis transplanted with bone marrow from ATGL KO mice69.
ATGL and oxidative stress
Intracellular redox environment is fundamental for several biological functions, the most straightforward of which are the redox-dependent transduction pathways that control cell cycle progression, growth and death71. Intracellular redox status results from the balance between pro-oxidant molecules, such as reactive oxygen species (ROS) and antioxidants, namely enzymatic (e.g., catalase, superoxide dismutase, glutathione peroxidase) and non-enzymatic (e.g., vitamin E, glutathione) ones72,73. Oxidative stress is the detrimental condition occurring upon excessive ROS production or impairment of antioxidant response74,75. Notably, cancer cells possess inherent higher levels of ROS, which have been implicated in several aspects of tumour progression. Tumour-associated oxidative stress can be responsible for genetic and epigenetic mutations that affect expression of oncogenes and tumour suppressors76,77.
Mitochondrial activity is the major source of endogenous ROS with a well-recognised impact on proliferative and survival signalling pathways78,79. In addition, impingement of ROS on proliferation, angiogenesis or metastatization has been also ascribed to increased activity of NADPH oxidases80–82, a family of enzyme whose primary function is to catalyse the transfer of electrons from NADPH to oxygen generating superoxide anion radical (O2.-) and H2O2.
FAs mitochondrial oxidation generates massive ATP production through oxidative phosphorylation, but how lipids influence mitochondrial redox status has not been fully elucidated yet. It can be envisaged that enhanced FAs oxidation causes sustained mitochondrial ROS emission and ATGL may have a leading role especially in cells that store large amounts of LDs. Nevertheless, this hypothesis is not supported by literature as reports mostly argue for an antioxidant role of ATGL. Heart-specific ATGL KO was associated with increased oxidative stress caused by NADPH oxidase activity62. Indeed, mRNA and protein levels of NOX2 and NOX4, two catalytic subunits of NADPH oxidase complexes, were raised in ATGL-deficient cardiomyocytes (Fig. 3)62. Similarly, increased levels of ROS were observed in ATGL-depleted macrophages as a consequence of higher expression of the NOX1 subunit (Fig. 3)83. Additional data highlighted the suppression of oxidative stress by the ATGL-PPAR-α pathway through the upregulation of antioxidant enzymes. ATGL KO mice displayed an increased amount of carbonylated proteins and reduced glutathione content in skeletal muscle61. This phenotype was due to impaired ATGL-PPAR-α signalling that caused decreased expression of PGC-1α and of nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) transcription factor, whose target genes include superoxide dismutase 2 (SOD2) and γ-glutamylcysteine ligase (GCLC)61 (Fig. 3). In another study, intestines from ATGL KO mice showed reduced mRNA levels of PPAR-α target genes, among which are the antioxidant enzymes glutathione-S-transferases (Fig. 3)84. Remarkably, PPAR-α expression itself was recently shown to be reduced in ATGL KO mice kidney, which also displayed increased ROS levels and tubulointerstitial damage, with resulting renal dysfunction85.
Fig. 3. ATGL effects in redox homoeostasis.
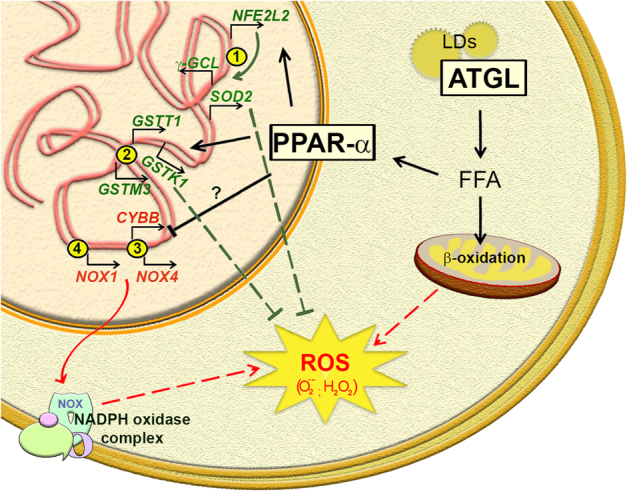
ATGL-mediated release of free fatty acids (FFAs) from lipid droplets (LDs) is a source of mitochondrial reactive oxygen species (ROS). The ATGL-PPAR-α axis mitigates oxidative stress favouring the expression of antioxidant enzymes in (1) skeletal muscle (i.e., SOD2 and γ-GCL, two downstream targets of NFE2L2) and in (2) intestines (i.e., GSTT1, GSTK1 and GSTM3). ATGL activity impairs the expression of catalytic subunits of the NADPH oxidase complex in (3) cardiac muscle (i.e., CYBB, codifying for NOX2 protein, and NOX4) and in (4) macrophages (i.e., NOX1). The influence of PPAR-α in these latter events has been not investigated, yet
Overall, the antioxidant function of ATGL represents a broad feature of different tissues and is mainly achieved through the activation of the PPAR-α-dependent transcriptional programmes. Antioxidant properties of PPAR-α have been already shown in different disease models. Indeed, oxidative stress following ischaemia/reperfusion injury in rat hippocampus was mitigated by PPAR-α agonist WY1464386, which also attenuated ROS-mediated hepatic fibrosis via catalase expression87. In this scenario, it is intriguing to speculate that disabling the antioxidant functions of ATGL/PPAR-α could increase ROS-dependent cancer proliferative signalling.
ATGL and autophagy
Autophagy is a highly complex set of regulated events carried out by autophagy-related proteins (ATGs), aimed at degrading in-excess/misfolded proteins and damaged organelles in order to maintain cellular homoeostasis or to recycle macromolecules upon stress stimuli88. Due to its high dynamism, autophagy has a complex role in cancer, largely dependent on the stage of tumour development. Indeed, a tumour suppressor function of autophagy is linked to the clearance of damaged organelles such as mitochondria, a source of ROS leakage and DNA mutations. Conversely, by providing metabolic substrates, autophagy might have a pro-survival role in tumours facing nutrient shortage89.
Autophagy is also a key regulator of lipid homoeostasis: in particular, the autophagy-mediated degradation of lipids is referred to as lipophagy90. The first evidence of autophagy as a critical player in lipid metabolism goes back to the evidence that TAGs were increased upon autophagy impairment by ATG5 knockdown91. Interestingly, LDs, which can be targets of autophagy, are surrounded by the autophagic protein microtubule-associated protein 1A/1B-light chain 3 (LC3)92,91. However, the role of LC3 on LDs surface is still debated because, even though it is fundamental for the autophagic/lipophagic process, LC3 is also responsible for LDs formation93.
Notably, ATGL per se seems to play an active role in lipophagy regulation. In fact, ATGL protein sequence harbours the LC3-interacting region (LIR), a motif mediating the association between autophagy receptors and LC3-coated autophagosomes94. ATGL/LC3 interaction contributes to the recruitment of ATGL on LDs for a more efficient LDs degradation achieved by the synergistic effect of lipolysis and lipophagy. Moreover, a recent report has demonstrated that ATGL stimulates autophagy and lipophagy via SIRT1 signalling in liver95. In this context, it has to be mentioned that lipid homoeostasis in liver largely relies on lipophagy and that its impairment favours hepatic steatosis, one of the leading risk factors for hepatocellular carcinoma development91,96. Remarkably, ATGL KO mouse models develop progressive hepatic steatosis13. Moreover, ATGL repression mediated by the steroid receptor RNA activator (SRA) represents the mechanism by which also this long non-coding RNA mediates hepatic steatosis97. These observations together with our unpublished evidence of ATGL reduction in liver cancer suggest that deregulation of ATGL lipolytic and lipophagic activity is detrimental for liver homoeostasis with potential tumorigenic consequences. Further efforts are necessary to validate this hypothesis and to extend it to other tissues that face excessive lipid accumulation, such as pancreas.
Concluding remarks
Research work over the past decade has undoubtedly established the pivotal role of ATGL in lipid metabolism in organs other than adipose tissue. Lipid accumulation due to ATGL dysfunction affects liver and skeletal muscle causing steatohepatitis and myopathies, respectively. Moreover, diverse types of neoplasia exhibit decreased levels of ATGL or deregulated expression of its protein partners, particularly the activator CGI-58 and the inhibitor G0S239,40. Most of the current in vivo evidence supports an anti-neoplastic role for ATGL, although the underlying up- and down-stream mechanisms have not been elucidated yet. On the contrary, other lipases, including MAGL and phospholipase D, are mainly upregulated in cancer triggering a lipid signalling network associated with enhanced tumour growth, invasion and metastasis. This inconsistency needs further clarification but the different non-energetic functions of ATGL illustrated in the present review may provide an explanation.
We offered insights into several aspects of ATGL biology that are not directly dependent on energetic purposes (i.e., FAs β-oxidation) and we revised all the available information about ATGL deregulation in cancer. We hypothesise that ATGL may have a broad influence on processes linked to cancer (Fig. 4), such as redox homoeostasis, inflammation and autophagy, through PPAR-α signalling. This is suggested by the evidence that the ATGL-PPAR-α axis has similar outcomes on inflammatory and antioxidant responses in several tissues, whereas tissue-specificity effects of PPAR-α target genes involved in fatty acid uptake and oxidation have been observed25,98. Absence of ATGL in non-immune cells is associated with oxidative stress and enhanced production of pro-inflammatory cytokines in basal and/or stimulated conditions. Considering this, the downregulation of ATGL in cancer cells might prime the set-up of an inflammatory microenvironment necessary for fibroblasts, endothelial cells and leucocytes recruitment and the induction of redox-based proliferative signalling, two events that favour cancer formation and progression99,100.
Fig. 4. Multifaceted role of ATGL in cancer.

Changes of ATGL levels can have different impact on several aspects of tumour biology. In fact, beyond providing lipids for membrane building blocks and for energy production through oxidative metabolism, ATGL could also manage tumour-associated inflammation, oxidative stress response and autophagic/lipophagic process. ECM extracellular matrix, PL phospholipids
Although a connection between ATGL and autophagy exists, no definitive molecular mechanism has been established yet. A tentative speculation on the pro-lipophagic role of ATGL in the repression of hepatosteatosis and of liver carcinogenesis has been provided. However, no evidence on ATGL KO mice and on the actual regulation of autophagic/lipophagic flux in pathological conditions has been provided. Further investigation may be interestingly addressed to understand whether ATGL contributes to the pro-autophagic action of PPAR-α signalling. Indeed, it has been demonstrated that treatment with PPAR-α agonists increases LC3 protein levels in fed mice101 as well as transcript levels of autophagic genes in a murine model of acute liver failure, leading to an active autophagic flux102.
Overall, some tumours might take advantage from ATGL deregulation to suppress the non-energetic functions herewith described, which would otherwise hinder tumour promotion/progression. At the same time, ATGL downregulation might contribute to the switch from mitochondrial metabolism to a glycolytic phenotype typical of many cancers. Furthermore, dampening ATGL activity may be necessary for those tumours that rely on high amount of intracellular LDs. In fact, LDs accumulation in cancer has been associated with resistance to therapeutic treatments103 and to endoplasmic reticulum stress conditions104.
Although the molecular mechanisms underlying ATGL additional roles in cancer biology are still elusive, its anti-neoplastic features could be useful for targeting cancer cells. Particularly, it could be intriguing to use approaches aimed at increasing ATGL expression/activity or mimicking its functions. In this regard, the alkaloid berberine and the steroid hormone dehydroepiandrosterone (DHEA) have been documented to increase ATGL expression in adipose tissue in vitro and in vivo, respectively, and to ameliorate lipid mobilisation105,106. Furthermore, considering the role of PPAR-α in mediating ATGL signalling, the use of PPAR-α agonists, such as fenofibrate, could be an enticing approach to counteract cancer progression. Indeed, fenofibrate was reported to disturb glioma cell growth and melanoma metastatic activity both in vitro and in vivo107–109. Overall, future efforts are needed to deeply characterise ATGL molecular mechanisms in cancer biology and to develop selective therapeutic strategies.
Acknowledgements
This work was supported by a Grant from Italian Association for Cancer Research (AIRC-IG 15403).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Rolando Vegliante, Luca Di Leo contributed equally to this work.
Edited by A. Stephanou
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Young SG, Zechner R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013;27:459–484. doi: 10.1101/gad.209296.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cermelli S, Guo Y, Gross SP, Welte MA. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol.: CB. 2006;16:1783–1795. doi: 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- 3.Barba G, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl Acad. Sci. USA. 1997;94:1200–1205. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walther TC, Farese RV., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zimmermann R, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 6.Reid BN, et al. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J. Biol. Chem. 2008;283:13087–13099. doi: 10.1074/jbc.M800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 8.Holm C, et al. Hormone-sensitive lipase: sequence, expression, and chromosomal localization to 19 cent-q13.3. Science. 1988;241:1503–1506. doi: 10.1126/science.3420405. [DOI] [PubMed] [Google Scholar]
- 9.Sztalryd C, et al. Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J. Cell Biol. 2003;161:1093–1103. doi: 10.1083/jcb.200210169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SJ, et al. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell. Biol. 2016;36:1961–1976. doi: 10.1128/MCB.00244-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagnon J, et al. Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology. 2012;153:4278–4289. doi: 10.1210/en.2012-1127. [DOI] [PubMed] [Google Scholar]
- 12.Haemmerle G, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 13.Wu JW, et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54:122–132. doi: 10.1002/hep.24338. [DOI] [PubMed] [Google Scholar]
- 14.Kienesberger PC, et al. Adipose triglyceride lipase deficiency causes tissue-specific changes in insulin signaling. J. Biol. Chem. 2009;284:30218–30229. doi: 10.1074/jbc.M109.047787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ong KT, Mashek MT, Bu SY, Mashek DG. Hepatic ATGL knockdown uncouples glucose intolerance from liver TAG accumulation. FASEB J. 2013;27:313–321. doi: 10.1096/fj.12-213454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer J, et al. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007;39:28–30. doi: 10.1038/ng1951. [DOI] [PubMed] [Google Scholar]
- 17.Schweiger M, Lass A, Zimmermann R, Eichmann TO, Zechner R. Neutral lipid storage disease: genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 2009;297:E289–E296. doi: 10.1152/ajpendo.00099.2009. [DOI] [PubMed] [Google Scholar]
- 18.Murugesan S, Goldberg EB, Dou E, Brown WJ. Identification of diverse lipid droplet targeting motifs in the PNPLA family of triglyceride lipases. PLoS ONE. 2013;8:e64950. doi: 10.1371/journal.pone.0064950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lass A, et al. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006;3:309–319. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Lu X, Liu J. Identification of a novel splicing isoform of murine CGI-58. FEBS Lett. 2010;584:903–910. doi: 10.1016/j.febslet.2009.12.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian V, et al. Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J. Biol. Chem. 2004;279:42062–42071. doi: 10.1074/jbc.M407462200. [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi T, Omatsu N, Matsushita S, Osumi T. CGI-58 interacts with perilipin and is localized to lipid droplets. Possible involvement of CGI-58 mislocalization in Chanarin-Dorfman syndrome. J. Biol. Chem. 2004;279:30490–30497. doi: 10.1074/jbc.M403920200. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011;286:15707–15715. doi: 10.1074/jbc.M110.207779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michalik L, Wahli W. PPARs mediate lipid signaling in inflammation and cancer. PPAR Res. 2008;2008:134059. doi: 10.1155/2008/134059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haemmerle G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong KT, Mashek MT, Davidson NO, Mashek DG. Hepatic ATGL mediates PPAR-alpha signaling and fatty acid channeling through an L-FABP independent mechanism. J. Lipid Res. 2014;55:808–815. doi: 10.1194/jlr.M039867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mottillo EP, Bloch AE, Leff T, Granneman JG. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) alpha and delta in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 2012;287:25038–25048. doi: 10.1074/jbc.M112.374041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan SA, et al. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1alpha/PPAR-alpha signaling. Diabetes. 2015;64:418–426. doi: 10.2337/db14-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang T, et al. Desnutrin/ATGL activates PPARdelta to promote mitochondrial function for insulin secretion in islet beta cells. Cell Metab. 2013;18:883–895. doi: 10.1016/j.cmet.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017;284:3132–3144. doi: 10.1111/febs.14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic. Biol. Med. 2015;79:253–263. doi: 10.1016/j.freeradbiomed.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer. 2017;3:169–180. doi: 10.1016/j.trecan.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5:e189. doi: 10.1038/oncsis.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Zuckier LS, Ghesani NV. Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach. Anticancer Res. 2010;30:369–374. [PubMed] [Google Scholar]
- 36.Caro P, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–560. doi: 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai Q, et al. Elevated and secreted phospholipase A(2) activities as new potential therapeutic targets in human epithelial ovarian cancer. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2012;26:3306–3320. doi: 10.1096/fj.12-207597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene. 2013;32:5551–5562. doi: 10.1038/onc.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ou J, et al. Loss of abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep. 2014;9:1798–1811. doi: 10.1016/j.celrep.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zagani R, El-Assaad W, Gamache I, Teodoro JG. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget. 2015;6:28282–28295. doi: 10.18632/oncotarget.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen G, et al. Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci. Rep. 2017;7:13021. doi: 10.1038/s41598-017-13398-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mitra R, Le TT, Gorjala P, Goodman OB., Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer. 2017;17:631. doi: 10.1186/s12885-017-3589-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang YY, et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight. 2017;2:e87489. doi: 10.1172/jci.insight.87489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grace SA, et al. Adipose triglyceride lipase (ATGL) expression is associated with adiposity and tumor stromal proliferation in patients with pancreatic ductal adenocarcinoma. Anticancer Res. 2017;37:699–703. doi: 10.21873/anticanres.11366. [DOI] [PubMed] [Google Scholar]
- 45.Yim CY, et al. G0S2 suppresses oncogenic transformation by repressing a MYC-regulated transcriptional program. Cancer Res. 2016;76:1204–1213. doi: 10.1158/0008-5472.CAN-15-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Zoughbi W, et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget. 2016;7:33832–33840. doi: 10.18632/oncotarget.9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu JW, et al. Epistatic interaction between the lipase-encoding genes Pnpla2 and Lipe causes liposarcoma in mice. PLoS Genet. 2017;13:e1006716. doi: 10.1371/journal.pgen.1006716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fearon ER, Feinberg AP, Hamilton SH, Vogelstein B. Loss of genes on the short arm of chromosome 11 in bladder cancer. Nature. 1985;318:377–380. doi: 10.1038/318377a0. [DOI] [PubMed] [Google Scholar]
- 49.Karnik P, et al. Two distinct tumor suppressor loci within chromosome 11p15 implicated in breast cancer progression and metastasis. Hum. Mol. Genet. 1998;7:895–903. doi: 10.1093/hmg/7.5.895. [DOI] [PubMed] [Google Scholar]
- 50.Zhu W, et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016;9:127. doi: 10.1186/s13045-016-0361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye L, et al. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. 2011;307:6–17. doi: 10.1016/j.canlet.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 52.Nomura DK, et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu WR, et al. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2014;7:3704–3713. [PMC free article] [PubMed] [Google Scholar]
- 54.Nomura DK, et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 2011;18:846–856. doi: 10.1016/j.chembiol.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury- and death-induced inflammation. Immunity. 2011;35:467–477. doi: 10.1016/j.immuni.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib. Microbiol. 2006;13:118–137. doi: 10.1159/000092969. [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Song X, Mohri Y, Qiao L. Role of inflammation and tumor microenvironment in the development of gastrointestinal cancers: what induced pluripotent stem cells can do? Curr. Stem Cell Res. Ther. 2015;10:245–250. doi: 10.2174/1574888X09666140812112305. [DOI] [PubMed] [Google Scholar]
- 59.Capece D., et al. Cancer secretome and inflammation: The bright and the dark sides of NF-kappaB. Semin. Cell Dev. Biol. 2017, 10.1016/j.semcdb.2017.08.004. [DOI] [PubMed]
- 60.Lettieri Barbato D, et al. Proline oxidase-adipose triglyceride lipase pathway restrains adipose cell death and tissue inflammation. Cell Death Differ. 2014;21:113–123. doi: 10.1038/cdd.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aquilano K, et al. Adipose triglyceride lipase decrement affects skeletal muscle homeostasis during aging through FAs-PPARalpha-PGC-1alpha antioxidant response. Oncotarget. 2016;7:23019–23032. doi: 10.18632/oncotarget.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schrammel A, et al. Cardiac oxidative stress in a mouse model of neutral lipid storage disease. Biochim. Et. Biophys. Acta. 2013;1831:1600–1608. doi: 10.1016/j.bbalip.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jha P, et al. Role of adipose triglyceride lipase (PNPLA2) in protection from hepatic inflammation in mouse models of steatohepatitis and endotoxemia. Hepatology. 2014;59:858–869. doi: 10.1002/hep.26732. [DOI] [PubMed] [Google Scholar]
- 64.Lettieri Barbato D, Tatulli G, Aquilano K, Ciriolo MR. Inhibition of age-related cytokines production by ATGL: a mechanism linked to the anti-inflammatory effect of resveratrol. Mediat. Inflamm. 2014;2014:917698. doi: 10.1155/2014/917698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 66.Schlager S, et al. Adipose triglyceride lipase acts on neutrophil lipid droplets to regulate substrate availability for lipid mediator synthesis. J. Leukoc. Biol. 2015;98:837–850. doi: 10.1189/jlb.3A0515-206R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dichlberger A, Schlager S, Maaninka K, Schneider WJ, Kovanen PT. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. J. Lipid Res. 2014;55:2471–2478. doi: 10.1194/jlr.M048553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chandak PG, et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J. Biol. Chem. 2010;285:20192–20201. doi: 10.1074/jbc.M110.107854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lammers B, et al. Macrophage adipose triglyceride lipase deficiency attenuates atherosclerotic lesion development in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2011;31:67–73. doi: 10.1161/ATVBAHA.110.215814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aflaki E, et al. Impaired Rho GTPase activation abrogates cell polarization and migration in macrophages with defective lipolysis. Cell. Mol. life Sci.: CMLS. 2011;68:3933–3947. doi: 10.1007/s00018-011-0688-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr. Biol.: CB. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rahal A, et al. Oxidative stress, prooxidants, and antioxidants: the interplay. BioMed. Res. Int. 2014;2014:761264. doi: 10.1155/2014/761264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Filomeni G, Desideri E, Cardaci S, Rotilio G, Ciriolo MR. Under the ROS…thiol network is the principal suspect for autophagy commitment. Autophagy. 2010;6:999–1005. doi: 10.4161/auto.6.7.12754. [DOI] [PubMed] [Google Scholar]
- 74.Valko M, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 75.Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012;5:9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hussain SP, Aguilar F, Amstad P, Cerutti P. Oxy-radical induced mutagenesis of hotspot codons 248 and 249 of the human p53 gene. Oncogene. 1994;9:2277–2281. [PubMed] [Google Scholar]
- 77.Kang KA, Zhang R, Kim GY, Bae SC, Hyun JW. Epigenetic changes induced by oxidative stress in colorectal cancer cells: methylation of tumor suppressor RUNX3. Tumour Biol. 2012;33:403–412. doi: 10.1007/s13277-012-0322-6. [DOI] [PubMed] [Google Scholar]
- 78.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl Acad. Sci. USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bell EL, Emerling BM, Ricoult SJ, Guarente L. SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene. 2011;30:2986–2996. doi: 10.1038/onc.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arbiser JL, et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl Acad. Sci. USA. 2002;99:715–720. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xia C, et al. Reactive oxygen species regulate angiogenesis and tumor growth through vascular endothelial growth factor. Cancer Res. 2007;67:10823–10830. doi: 10.1158/0008-5472.CAN-07-0783. [DOI] [PubMed] [Google Scholar]
- 82.Kim JS, Huang TY, Bokoch GM. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell. 2009;20:2650–2660. doi: 10.1091/mbc.E09-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aflaki E, et al. Triacylglycerol accumulation activates the mitochondrial apoptosis pathway in macrophages. J. Biol. Chem. 2011;286:7418–7428. doi: 10.1074/jbc.M110.175703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Obrowsky S, et al. Adipose triglyceride lipase is a TG hydrolase of the small intestine and regulates intestinal PPARalpha signaling. J. Lipid Res. 2013;54:425–435. doi: 10.1194/jlr.M031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen W, et al. Atgl gene deletion predisposes to proximal tubule damage by impairing the fatty acid metabolism. Biochem. Biophys. Res. Commun. 2017;487:160–166. doi: 10.1016/j.bbrc.2017.03.170. [DOI] [PubMed] [Google Scholar]
- 86.Collino M, et al. Oxidative stress and inflammatory response evoked by transient cerebral ischemia/reperfusion: effects of the PPAR-alpha agonist WY14643. Free Radic. Biol. Med. 2006;41:579–589. doi: 10.1016/j.freeradbiomed.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 87.Toyama T, et al. PPARalpha ligands activate antioxidant enzymes and suppress hepatic fibrosis in rats. Biochem. Biophys. Res. Commun. 2004;324:697–704. doi: 10.1016/j.bbrc.2004.09.110. [DOI] [PubMed] [Google Scholar]
- 88.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi KS. Autophagy and cancer. Exp. Mol. Med. 2012;44:109–120. doi: 10.3858/emm.2012.44.2.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu K, Czaja MJ. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013;20:3–11. doi: 10.1038/cdd.2012.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zamani M, Taher J, Adeli K. Complex role of autophagy in regulation of hepatic lipid and lipoprotein metabolism. J. Biomed. Res. 2016;30:377–385. doi: 10.7555/JBR.30.20150137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shibata M, et al. LC3, a microtubule-associated protein1A/B lightchain3, is involved in cytoplasmic lipid droplet formation. Biochem. Biophys. Res. Commun. 2010;393:274–279. doi: 10.1016/j.bbrc.2010.01.121. [DOI] [PubMed] [Google Scholar]
- 94.Martinez-Lopez N, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 2017;19:1–9. doi: 10.1016/j.celrep.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singh R, Cuervo AM. Lipophagy: connecting autophagy and lipid metabolism. Int J. Cell Biol. 2012;2012:282041. doi: 10.1155/2012/282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen G, et al. LncRNA SRA promotes hepatic steatosis through repressing the expression of adipose triglyceride lipase (ATGL) Sci. Rep. 2016;6:35531. doi: 10.1038/srep35531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ong KT, Mashek MT, Bu SY, Greenberg AS, Mashek DG. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53:116–126. doi: 10.1002/hep.24006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 100.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee JM, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiao M, et al. Peroxisome proliferator-activated receptor alpha activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 2014;5:e1397. doi: 10.1038/cddis.2014.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pan X, et al. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metab.: Off. J. Metab. Soc. 2013;9:722–729. doi: 10.1007/s11306-012-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Qiu B, et al. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015;5:652–667. doi: 10.1158/2159-8290.CD-14-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karbowska J, Kochan Z. Fat-reducing effects of dehydroepiandrosterone involve upregulation of ATGL and HSL expression, and stimulation of lipolysis in adipose tissue. Steroids. 2012;77:1359–1365. doi: 10.1016/j.steroids.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 106.Jiang D, et al. Berberine increases adipose triglyceride lipase in 3T3-L1 adipocytes through the AMPK pathway. Lipids Health Dis. 2016;15:214. doi: 10.1186/s12944-016-0383-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Drukala J, et al. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARalpha-mediated inhibition of glioma cell motility in vitro. Mol. Cancer. 2010;9:159. doi: 10.1186/1476-4598-9-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006;12:3028–3036. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 109.Grabacka M, et al. Inhibition of melanoma metastases by fenofibrate. Arch. Dermatol. Res. 2004;296:54–58. doi: 10.1007/s00403-004-0479-y. [DOI] [PubMed] [Google Scholar]