

Abstract
Status epilepticus (SE) is a medical emergency characterized by uncontrolled, prolonged seizures with rapid and widespread neuronal damage. Patients that suffer from longer episodes of SE are more likely to have poorer clinical outcomes and a higher cost of healthcare. Understanding novel molecular mechanisms that regulate inhibitory and excitatory neurotransmission that initiate SE and the necessary medical infrastructure to stop SE could help identify targets for early intervention. Intranasal administration of benzodiazepines may shorten the time between initiation and cessation of seizures when compared to other routes of administration. Current pharmaceutical administration guidelines are appropriate for sporadic incidences of SE, but exploring other approaches is necessary to prepare for situations involving multiple patients outside of a hospital, such as a massive chemical weapons attack. Intranasal drug delivery helps to circumvent the blood–brain barrier and offers a noninvasive way to quickly administer drugs in settings that require an immediate response, such as nerve agent exposure. In addition, examining the intranasal delivery of new drugs, such as nanotherapeutics, may lead to more effective, noninvasive, scalable, and portable methods of treating SE.
Key Points:
| Status epilepticus (SE) requires immediate therapeutic intervention. |
| Intranasal delivery methods are under-utilized as a potential therapy. |
| Current work is being done to facilitate treatment of SE with intranasal therapeutics. |
Introduction
Status epilepticus (SE) is a medical emergency in which a patient has an abnormally prolonged seizure (greater than 30 min) or multiple prolonged seizures without an intervening restoration of consciousness or physiological neurological function. The Commission on Classification and Terminology and the Commission on Epidemiology of the International League Against Epilepsy (ILAE) have recently published a definition of SE as follows [1]:
“SE is a condition resulting either from the failure of the mechanisms responsible for seizure termination or from the initiation of mechanisms which lead to abnormally prolonged seizures. It is a condition, which can have long-term consequences, including neuronal death, neuronal injury, and alteration of neuronal networks, depending on the type and duration of seizures.”
A longer time frame between the onset of SE and initial medical treatment is associated with a higher rate of mortality and a poorer neurological functional outcome as measured by the Modified Rankin Scale, which is a measure of independence and disability [2]. It has been shown that the length of the seizure is directly correlated to the degree of neuronal damage, morbidity, and mortality in rats [3]. Therefore, administering fast and effective treatments that will help end seizures is critical for first responders, emergency physicians, and neurologists.
Mass-casualty incidents involving nerve agents, such as the 1995 terrorist attack in Tokyo using sarin gas or the recent use of chemical weapons in Syria, raise many concerns about the quick and effective treatment of SE, as these agents can easily overwhelm healthcare facilities with patients suffering from SE. Many nerve agents that could conceivably cause widespread toxicity through chemical warfare or industrial/occupational exposure can cause seizures, and plausibly SE, by either increasing neuronal excitation or inhibiting neuronal inhibition [4]. For example, nerve agents typically used in chemical warfare, such as sarin and VX, cause excitotoxicity by inhibiting acetylcholinesterase, while industrial insecticides, such as lindane, block the action of inhibitory γ-aminobutyric acid (GABA) receptors [4].
Many of these seizure-inducing neurotoxins, such as dichlorodiphenyltrichloroethane (DDT), have no antidote [4]. Even if antidotes to particular toxins are available, it is not necessarily possible to confirm what substance is involved in mass-casualty exposures in the moments immediately afterwards, when treatment is crucial. This makes early treatment with anticonvulsant drugs vital in acute exposure-induced seizures. Incidents of chemical warfare, which often involve exposing a high number of people to neurotoxic agents, highlight the need for innovations in the urgent treatment of neurotoxicity and subsequent seizures. Getting patients from ground zero to medical personnel or vice versa would be hindered by the panic and chaos that invariably accompanies these events. Developing a way to administer drugs to abort seizure activity in situ would provide a way to vastly reduce the death and suffering that results from widespread neurotoxin exposure.
The problems associated with incidents involving widespread neurotoxicity are highlighted by the April 4th, 2017 chemical attack in Khan Sheikhoun, Syria [5]. The attack killed at least 85 men, women, and children and undoubtedly left many more with irreparable neurological damage from exposure to the organophosphate weapon known as sarin [5, 6]. This is not the first attack of this kind to occur over the course of the Syrian civil war; a similar gas attack took place in Damascus in 2013, and doctors without borders estimated that up to 3600 people presented with symptoms of neurotoxicity, several hundred of which perished from the attack [7].
Here, we discuss current treatments of SE and the underpinnings and implications of ongoing advances in the pursuit of improving the outcomes of early intervention in patients suffering from SE in pre-hospital settings. Current guidelines address episodes of SE involving one patient with access to medical infrastructure, but it is necessary to explore options for situations involving many patients without access to such infrastructure, such as in biochemical terrorism.
Status Epilepticus (SE) Damages the Brain, Body, and Society
Seizures are categorized as being either generalized or focal and either convulsive or non-convulsive. Any of these forms of seizures can lead to SE and can become refractory or super-refractory; focal and generalized SE are treated much the same, while non-convulsive SE treatment is dependent on the further classification and etiology of the SE [8, 9]. The ILAE recently published guidelines for classifying SE according to four axes: semiology, etiology, electroencephalography (EEG), and age [10]. The most clinically relevant of these is semiology, which classifies SE according to signs that are readily observable by healthcare providers, such as mental status and degree of convulsiveness [10]. There are several types of seizures, and they may either be primary, as a consequence of an underlying epilepsy disorder, or secondary, as in seizures resulting from some form of cerebral insult. Seizures that evolve into SE can develop from a broad range of brain injuries, including, but not limited to, stroke, brain infections, neurotoxins, and poorly managed treatment of pre-existing epilepsy. While these all constitute medical emergencies, the prognosis of SE is dependent not only on the duration of the seizure, but also on the pathophysiological mechanisms specific to each case of SE [11].
Using experimental models of epilepsy, SE is positively correlated with extensive hippocampal damage and epileptogenesis. In addition, in some cases, frequent seizure activity is associated with the loss of GABAergic interneurons and pyramidal cells in the hippocampus, suggesting its association with temporal lobe epilepsy [12, 13]. SE may trigger systemic complications that can contribute to mortality and morbidity. The most common complications are acidosis and a hypoxic state due to an increased metabolic rate and a concomitant decrease in alveolar ventilation from spasms of the diaphragm and fatigue of the ventilator muscles [14]. Other complications may arise due to sympathetic overdrive, such as hyperpyrexia, cardiac arrhythmias, and cardiomyopathies. Rhabdomyolysis, destruction of muscle tissue, is also a concern in prolonged convulsive seizures, and can lead to myoglobinuria and acute kidney failure [14]. The systemic complications of SE can prove to be lethal in vulnerable patients, such as those with pre-existing medical conditions or pregnant women. Taken together, SE and its potential complications can negatively impact society at large by increasing the cost of medical treatment and by decreasing patients’ quality of life and functional independence.
The epidemiology and prognosis of SE varies between geographical regions and patient demographics, but the two most common causes of SE tend to be cerebrovascular disease and low levels of anti-epileptic drugs (AEDs) in patients with pre-existing epilepsy [11]. SE caused by low levels of AEDs is cause for concern, as socioeconomic factors can lead to the sudden cessation of AED compliance. Patients who have poor educational instruction, social support, and financial stability may be especially at risk for this form of SE, which can drastically exacerbate existing biopsychosocial problems. SE of any etiology is a large economic burden on individual patients as well as on society at large, both in terms of resources spent and lost productivity, even when not accounting for indirect costs of treatment for patients and their families. A study of inpatients who were treated for SE at the Virginia Commonwealth University Medical Center in 1994 found that the direct cost of treating SE in inpatients admitted with SE averaged US$8417, and the average length of stay was 12.9 days [15]. The direct cost of treatment of hospital admission with a diagnosis of SE was estimated to be 1.81 times more than that of myocardial infarctions and 1.94 times more than that of congestive heart failure [15]. It is not immediately clear how federal legislation in the United States will alter the costs of healthcare for vulnerable populations. However, it is clear that SE will continue to be a major source of neurological disability and financial burden for underserved patients.
Mechanism of SE and Brief Rationale for Therapeutic Targets
Failure of Inhibitory Drive
Endogenous GABA receptor-mediated neuronal inhibition is the mechanism that terminates seizures. Activation of GABAA receptors, ligand-gated chloride channels, causes hyperpolarization of neurons, which results in less frequent firing of action potentials. Increased activation of GABA receptors and subsequent neuronal inhibition leads to seizure cessation. For this reason, the initial treatment of SE involves delivery of benzodiazepines (BZDs), which are GABA receptor agonists. Some episodes of SE can become insensitive to BZDs, which are GABA receptor agonists, only a few minutes after the onset of seizures [16]. The effectiveness of seizure termination by BZDs rapidly declines after 10 min of seizure activity in mice, and successfully stopping SE after 40 min of seizure activity requires approximately ten times the dose of BZDs required after only 10 min of seizure activity [16]. After SE becomes BZD-insensitive, it is more likely that the SE will progress to refractory or super-refractory SE, which then results in a higher risk of morbidity and mortality. This narrow window of time after the onset of SE in which BZDs can effectively halt seizures demands an urgent initiation of medical care, which is especially problematic for episodes of SE that occur outside of an inpatient setting. The mechanism of the failure of inhibitory drive that prevents spontaneous seizure termination and leads to refractory SE is the sustained activity-dependent internalization of post-synaptic GABA receptors [17] (Table 1).
Table 1.
Details the evolution of the clinical and pathophysiological characteristics of SE over time

Specifically, cycling of the synaptic isoforms of the GABA receptor between the cytosol and the synaptic membrane is governed by the state of phosphorylation of the receptor [18] (Fig. 1). On the 408/9 serine residues of the intracellular domain of the β3 subunit of type A GABA receptors, a basic patch binding motif interacts with clathrin adapter protein 2 (AP2), which promotes internalization by clathrin-mediated endocytosis. Phosphorylation of the S408/9 hinders the interaction of the GABA receptor and AP2, which blocks internalization. Conversely, dephosphorylation of this site promotes intracellular accumulation of the synaptic GABA receptors [18]. GABA receptor phosphorylation is associated with activity of protein kinase C (PKC) as well as protein kinase A (PKA), while dephosphorylation mediates the activity of protein phosphatase 1, 2A, and 2B (PP1, PP2A, and calcineurin) [19, 20]. Pilocarpine-induced SE in mice results in decreased synaptic expression of α1–4, β3, and γ2 subunit-containing GABA receptors, and that SE causes a reduction in β3 subunit phosphorylation and a decrease in PKC activity and an increase in PPA2 activity [21]. The activity of phosphatases, such as PPA2, may make them useful as potential pharmacological targets for future treatments.
Fig. 1.
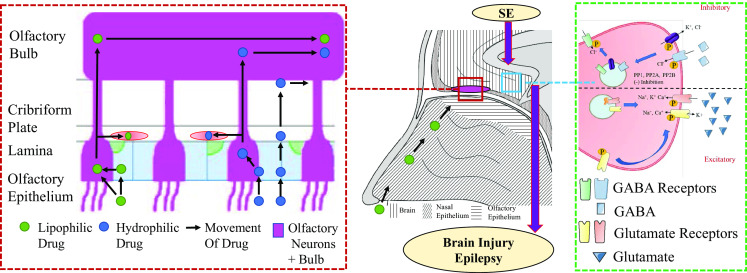
Left path of drugs through the olfactory structures; middle coronal diagram of the nasal cavity and brain; right changes in synaptic channel expression due to SE. GABA γ-aminobutyric acid, KCC2 potassium chloride cotransporter 2, PP protein phosphatase, SE status epilepticus. Representative sagittal section of the nasal cavity and forebrain showing dynamic passage of different type of drugs through the olfactory cavity(Red dash box) and its potential modulation of main receptor activation in status epilepticus (Green dash box)
On the other hand, it is thought that activity-dependent GABA receptor internalization is mediated by N-methyl-d-aspartate (NMDA) receptor activity, and is likely due, in part, to an NMDA-mediated influx of calcium ions which leads to the activation of calcineurin [22] (Fig. 1). Calcineurin is a calcium-dependent phosphatase, and the increase in the intracellular concentration of calcium due to NMDA and GluA2-lacking α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor activation is sufficient to stimulate detectable dephosphorylation and subsequent internalization of synaptic GABA receptors [23]. This suggests that SE leads to a shift in the equilibrium of GABA receptor phosphorylation, leading to a higher likelihood of GABA receptor dephosphorylation and subsequent internalization.
Interestingly, membrane expression of GABA receptors containing δ or α5 subunits is upregulated in the mouse hippocampus by SE [21]. These δ- or α5-containing receptors are extrasynaptic receptors and are responsible for tonic neuronal inhibition [24]. They are sensitive to anesthetics, and may therefore be important therapeutic targets for BZD-insensitive SE. While the mechanism underlying the regulation of their expression has not been fully elucidated, administration of allopregnanolone, a neurosteroid known to act on these receptors, induces the upregulation of δ-containing GABA receptors in the molecular layer of the hippocampal dentate gyrus through a potentiation of tonic inhibitory activity in dentate gyrus granule cells in an experimental model of SE [24].
In addition to a decrease in synaptic GABA receptors, the phosphatase-mediated decrease in postsynaptic inhibition may be exacerbated by a decrease in the activity of potassium chloride cotransporter 2 (KCC2), which is an outward chloride ion pump that is essential for rapidly integrating inhibitory GABA signals [25]. Phosphorylation of KCC2 at serine 940 enhances its activity, and PP1 dephosphorylates and inactivates KCC2.
It has previously been suggested that the process of reducing the inhibitory capability of GABA by receptor internalization marks a milestone in the progression of isolated seizure activity to established SE [26]. SE caused by exposure to neurotoxic agents has been shown to progress to refractory SE [4, 27]. Several mechanisms have been proposed to describe how neurotoxin exposure leads to SE. McDonough and Shih suggest that organophosphate exposure initiates seizures via a cholinergic mechanism, which then transitions to other non-cholinergic, atropine-resistant neurotransmitter disturbances [28]. It is not necessarily known whether organophosphate-induced SE becomes refractory to BZDs by the same mechanisms as SE resulting from other etiologies. Focal SE is also associated with a decrease in GABA receptor binding in epileptic foci in humans [29], although the extent of the similarities to the mechanisms of refractoriness in generalized SE is unclear. Primate alumina-gel models of focal SE have demonstrated a decrease in nerve terminals containing glutamic acid decarboxylase, an enzyme used in GABA synthesis, within epileptic foci [30, 31]. This suggests a decline in the number of GABAergic synapses. A similar finding has been reported in cobalt-induced focal SE in rats [32].
Increase of Excitatory Drive
While down-regulation of GABA receptors is thought to be responsible for the absence of seizure remission, release of endogenous glutamate is thought to be the mechanism of seizure-induced brain damage and neurotoxicity [33]. In experimental models of SE, brain damage is a consequence of prolonged seizures, mediated by NMDA-receptor activity [3]. In contrast to the SE-induced loss of inhibitory postsynaptic GABA receptors, SE can induce a gain of excitatory ionotropic NMDA and AMPA glutamate receptors on the postsynaptic membrane. Niquet et al. showed that NMDA receptors migrate to the synaptic membrane from intracellular stores as well as from the perisynaptic plasma membrane in mice after 1 h of experimental SE [26] (Fig. 1). Specifically, 1 h of SE in rodents increases the mean number of functional synaptic NMDA receptors by 38% in experimental models of SE [34]. In addition to NMDA receptors, it is also evident that SE increases the expression of AMPA receptors on the post-synaptic cell’s surface [35]. However, expression of the GluA2 subunits of the AMPA receptors decreases in the hippocampi during refractory SE, and leads to the characteristic influx of calcium ions that has been observed in SE due to the relatively high calcium permeability of GluA2-lacking AMPA receptors [36].
Current Treatments
There are several medical protocols that have been proposed for the pharmacological treatment of SE, all of which share common themes (Table 2). Briefly, the current preferred treatment at the onset of SE is intravenous (IV) BZDs, such as lorazepam, diazepam, or midazolam, which are GABA receptor agonists. If a patient does not respond to BZDs, AEDs are given, such as phenytoin or valproate. The third line of treatment is an anesthetic dose of barbiturates, propofol, or inhalation anesthetics [37]. The practicality of these models is limited to sporadic, isolated episodes of SE in which the patient has access to medical infrastructure. Scenarios involving many casualties in chaotic or isolated pre-hospital settings, such as biochemical warfare or terrorism, require alternative treatment guidelines.
Table 2.
| Stage of SE | Drug | Route | Dose | Mechanism/class | Additional notes |
|---|---|---|---|---|---|
| Early (< 10 min) | Lorazepam | IV | 4 mg over 2 min | Benzodiazepine | Repeat ×1 if necessary after 5 min; Preferred drug if IV is available |
| Early (< 10 min) | Diazepam | rectal | 20 mg (IV solution) | Benzodiazepine | If no IV access |
| Early (< 10 min) | Midazolam | IN/IM | 10 mg (IV solution) | Benzodiazepine | If no IV access |
| Established (10–30 min) | Valproate | IV | 40 mg/kg over 10 min | Anti-epileptic drug (AED), Modulator of GABA and cerebral metabolism [38] | Additional 20 mg/kg if needed |
| Established (10–30 min) | Fosphenytoin | IV | 20 mg PE/kg, up to 150 mg PE/min | AED, Voltage-gated cation channel modulator [39] | Additional 5 mg PE/kg if needed |
| Established (10–30 min) | Fosphenytoin | IM | 20 mg PE/kg | AED, Voltage-gated cation channel modulator [39] | Only if IV access is not achievable; Additional 5 mg PE/kg if needed |
| Established (10–30 min) | Levetiracetam | IV | 2500–4000 mg over 5–10 min | AED, Pre-synaptic calcium channel blocker [39] | Additional 1500–3000 mg if needed |
| Established (10–30 min) | Lacosamide | IV | 400 mg over several minutes | AED, Slow inactivator of sodium channels [40] | Additional 200 mg over 10 min if needed |
| Established (10–30 min) | Midazolam | IV |
0.2–0.4 mg/kg every 5 min until resolution (max 2 mg/kg), 0.1–2.9 mg/kg/h maintenance |
benzodiazepine | Only if intubated; use in conjunction with AED |
| Established (10–30 min) | Propofol | IV | 1–2 mg/kg every 2–3 min until resolution (max 10 mg/kg), 1.02–15 mg/kg/h maintenance | Induction anesthetic | Only if intubated; use in conjunction with AED |
| Refractory (> 30 min) | Pentobarbital | IV | Load 5 mg/kg at 50 mg/min, 1–5 mg/kg/h maintenance | barbiturate | Only if intubated |
| Refractory (>30 min) | Thiopental | IV | Load 1–2 mg/kg, 1–5 mg/kg/h maintenance | Barbiturate | Only if intubated |
The current standard for inpatient treatment of sustained seizures is IV BZDs, such as lorazepam. However, starting IV administration is slower and requires more training than other routes of administration, especially in convulsing patients. In many pre-hospital settings, people rendering aid to seizure victims may not have the proper training or equipment to deliver drugs by IV. These problems are complicated in rural or underserved areas. Intramuscular (IM) delivery of BZDs is popular among emergency medical services (EMS) personnel, because of its easier and faster administration compared to IV. A study of pre-hospital treatment of SE by first responders found that initiation of IM delivery of midazolam was faster than initiation of IV delivery of lorazepam (1.2 vs 4.8 min, respectively) [41]. It was also shown that IM midazolam was superior to IV lorazepam in stopping seizures before arrival in the emergency department (73.4 vs 63.4%, respectively), and patients who received IM midazolam were less likely to be admitted to the intensive care unit (ICU) (28.6 vs 36.2%, respectively) [41].
Devising and implementing effective ways of administering BZDs in a portable and scalable manner is vital in the pursuit of minimizing the damage done by SE outside of an inpatient setting, where IV administration is suboptimal. IM administration is an effective alternative to IV administration in many pre-hospital settings, such as medical transport. Many militaries, including the US Armed Forces, use autoinjectors for rapid treatment in the field [42]. However, in potential mass-casualty scenarios involving SE resulting from intoxication that occur in impoverished locales or in population centers without a massive pre-existing military or humanitarian apparatus may benefit from alternative systems, such as drone-delivered therapeutics. Delivery of drugs in emergencies by drones is already being explored, and is aimed to be deployed in some Swiss cities by 2018 [43]. Intranasal (IN) therapies may be preferable in such a system because of the ease of IN administration.
There are pros and cons to both IM and IN treatment. IM administration of many drugs is expected to have a higher bioavailability and peak plasma concentration than IN administration, and IN administration can be hindered by loss of the drug back through the nares or through oral ingestion. However, IN therapies may be easier for laypeople to administer, because of the lack of training required, low invasiveness, and general familiarity with nasal sprays. IN administration also does not carry the same cross-contamination risk that IM therapies might, particularly in pre-hospital settings; needles may introduce more of the toxin into the patient, especially if introduced through clothing. A pre-packaged nasal atomizer may be quick and easy to distribute and administer in a broader range of situations than injectable therapies. Further study is required to determine if putting this into practice would be worthwhile.
Non-IV methods have their own distinct disadvantages, however. One disadvantage of non-IV routes is the slower rate of absorption and the lower peak plasma concentration when compared to IV administration. However, a small, retrospective study comparing IV and IN administration of BZDs in SE patients showed that median time from presentation with SE to seizure cessation in stroke patients is 9.5 min when given IV diazepam compared to just 3 min when given IN diazepam. The time from drug administration to seizure termination was much lower for IV diazepam than IN (0.5 and 3 min, respectively), but IN is still quicker from presentation to seizure termination because of quicker administration [44]. Midazolam is also an option for IN administration rather than diazepam. Intranasally, midazolam is absorbed faster than diazepam, but has a lower bioavailability [45]. IN drug delivery is not always the best option, but it is likely to be the best choice in a pre-hospital setting, or when IV administration is not feasible.
Rationale for Intranasal (IN) Administration: From Bench to Bedside
Compared to other routes of non-IV administration, IN medication has several advantages. Nasal sprays are noninvasive and quick to administer, and the nasal cavity is well-vascularized with a high surface area and provides quick access to the olfactory epithelium. Drugs can bypass the blood–brain barrier by diffusing through the olfactory and trigeminal nerves via the nasal epithelium into the cerebrospinal fluid and the brain itself, and compounds that are absorbed into the blood by the nasal vasculature can reach the brain without modification by the liver [46]. The olfactory epithelium covers about 10% of the nasal cavity in the uppermost portion, just inferior to the cribriform plate [47]. This olfactory epithelium is made up of three types of cells: sustentacular, basal, and olfactory neural cells. The neural cells are directly exposed to the lumen of the nasal cavity, and synapse within the olfactory bulb, which then goes on to the orbitofrontal cortex via the primary olfactory cortex [48, 49].
The trigeminal nerve has also been shown to play a role in allowing therapeutics to reach the brain directly through the nasal cavity epithelium. The trigeminal nerve innervates the respiratory epithelium as well as the olfactory epithelium, and therefore has larger exposure to drugs delivered intranasally than the olfactory nerves. The particle size of the administered solution has been shown to influence the pattern of deposition on the walls of the nasal cavity. A pattern of deposition that hinders absorption by the olfactory epithelium can still access the trigeminal nerve fibers. The trigeminal nerve also has the advantage of having two entry points into the neurocranium, namely the cribriform plate and the anterior lacerated foramen. While the bulk of the trigeminal nerve enters the brain at the pons, the trigeminal nerve also shares connections with the olfactory system, both peripherally and centrally [47, 50]. Drug delivery via the trigeminal nerve has been demonstrated by administering lidocaine intranasally, which resulted in a concentration 20 times higher in orofacial structures that are innervated by the trigeminal nerve compared to other tissues and plasma [51]. It has also been shown that large polypeptides can reach the brainstem after IN administration via retrograde transport within the trigeminal nerve [52, 53].
There are several mechanisms that are responsible for transporting drugs from the nasal mucosa to the brain through nerves (Fig. 1). Lipophilic drugs can passively diffuse into the sustentacular cells or the olfactory neurons across the plasma membrane, which can then be transported to the olfactory bulb. The olfactory bulb directly projects axons to many structures, including the anterior olfactory nucleus, the piriform cortex, and the amygdala, and these projections serve as conduits for compounds to be transported to the brain [54]. Hydrophilic drugs can passively diffuse into the brain parenchyma and cerebrospinal fluid through the olfactory epithelium either via aquaporins located in the clefts between the sustentacular cells and the olfactory neurons or via paracellular pathways through the perineurium and perivascular spaces. The rate of passive diffusion of lipophilic drugs is greater than that of hydrophilic drugs for a given molecular weight, and is dictated by the degree of lipophilicity, with more lipophilic drugs having a higher rate of diffusion. The rate of passive diffusion of hydrophilic drugs is governed by the drug’s molecular weight, with good efficiency being achieved by compounds weighing up to 1 kDa [47]. Large polypeptides have also been shown to reach the brain through the olfactory epithelium following IN administration via several novel approaches involving both saturable and unsaturable pathways [55].
Drug absorption rates can be altered by factors like mucus production and altered nasal blood flow. A relatively small amount of liquid must be used to give drugs intranasally (200 uL–1 mL), so drugs must be either concentrated or potent, which poses problems for lipophilic drugs as they are not readily concentrated in aqueous solutions [56]. Another challenge is the interference in absorption from the enzymes present in the nasal epithelium that serve as protection against infections. These enzymes can hydrolyze drugs and reduce their efficacy [57]. IN drug delivery can also be limited by the layer of mucus that covers the respiratory epithelium. This layer of mucus increases the distance that molecules must diffuse through to reach the epithelium, and can limit epithelial exposure to compounds of high molecular weight [47]. Because the secretion of mucus from the nasal epithelium is controlled by muscarinic receptor activity, exposure to acetylcholinesterase inhibitors such as organophosphates may increase nasal mucus production thereby reducing the efficacy of IN drug delivery. However, IN administration of atropine is still effective in improving organophosphate exposure mortality in mice, even when administered after the exposure [58]. Despite the potential decrease in efficacy due to increased nasal secretions, IN therapy following organophosphate exposure may still be effective in preventing or terminating SE in settings where first responders lack the training or resources to implement large-scale IM or IV therapy.
Experimental Approaches to IN Delivery
Typical IN treatments of SE consist of standard IV solutions of BZDs delivered into the nasal cavity via syringe or atomizer, which have been shown to be effective when administered early in the absence of established IV access. BZDs in this form, while effective, are not optimized for IN delivery. Aqueous midazolam, for example, is usually 5 mg/mL or less, and therefore only 10 mg can be delivered effectively [59]. Patients that require larger doses would benefit from drug preparations that have been designed specifically for IN administration. The IN treatment of SE provides an avenue for substantial improvements to be made to patient care, and there is potential to improve the outcomes of patients who suffer from SE by improving the initial treatments in early SE.
There are several drugs and formulations under development that hope to offer real benefits over traditional BZD nasal sprays. Lipophilic molecules, such as diazepam, will be absorbed into the nasal neurovasculature more rapidly than hydrophilic drugs, but are more difficult to concentrate in aqueous solutions. Adapting the solvents and preparations of drugs to nasal administration can potentially improve the efficacy of BZDs for treating SE. A 2015 phase I clinical trial from Upsher-Smith Laboratories shows favorable results for a formulation of midazolam, referred to as USL261, that was designed specifically for IN administration. They found that their formulation had a 134% relative bioavailability compared to IV midazolam solutions delivered nasally [60]. However, they recently terminated a phase III clinical trial because of slow enrollment [61].
Work is also being done with modified drugs and drug combinations which may provide other avenues to improve urgent treatment of seizures. An approach to circumventing the problems with drug solubility and permeability without the need for special solvents is to use aqueous solutions of water-soluble prodrugs co-administered with converting enzymes. Siegel et al. explored IN delivery of diazepam and midazolam prodrugs, both of which are converted to their respective drugs by Aspergillus oryzae protease [62]. However, since this enzyme comes from a fungus, it could be allergenic. They speculate that human converting enzymes must exist, as these prodrugs can be converted in human muscle tissue, but the enzyme had not been identified as of 2015. They found that this combination can create supersaturated solutions of diazepam and midazolam, and that this supersaturated solution exhibited a several-fold increase in permeation through Madin-Darby canine kidney II-wild type (MDCKII-wt) monolayers compared to saturated aqueous diazepam. They also created a chirally pure prodrug of midazolam, which, when converted into a supersaturated solution of midazolam, permeated the monolayers 25 times faster than saturated aqueous midazolam [62].
In addition to optimizing relatively traditional treatment options to IN delivery, current efforts also seek to design new ways of delivering and targeting neurotherapeutics to the brain via IN administration. One such avenue of exploration is the use of nanoparticles to target the central nervous system through the nasal epithelium in the form of classical spheroid nanoparticles, nanogels, nanotubes, etc. [63]. Typically, the electrical, chemical, and physical properties of a bulk material do not depend on the size and shape of the material. However, these properties are dictated by the size and shape of particles of certain materials when the particle is sufficiently small [64]. This property makes nanoparticles interesting vehicles for drug delivery, as molecules can be adsorbed onto the surface of these particles and then carried into tissues. Chen et al. have demonstrated a method of efficiently targeting poly(lactic-co-glycolic acid) (PLGA) nanoparticles to the central nervous system after IN administration via endocytosis by conjugating the nanoparticles with Solanum tuberosum lectin (STL) [65]. The concentration of STL-conjugated nanoparticles is higher in the olfactory bulb, cerebrum, and cerebellum compared to unconjugated PLGA nanoparticles, while the plasma concentration is lower compared to the unconjugated nanoparticles [65]. This suggests that the STL specifically targeted the nanoparticles to the olfactory neurons. While this is not therapeutic, it may be a promising way to target difficult-to-deliver drugs to the central nervous system.
IN administration is also being explored as a means of delivering therapeutic antibodies to the brain, which has the potential to open many doors in the pursuit of immunoglobulin-based therapies that have previously been unachievable. In general, large molecules do not effectively reach the brain when administered intranasally without modification or co-administration, but there are exceptions. One exception that is particularly interesting is an oligomeric amyloid-β antibody (NU4) that was administered to 5XFAD mice, which serve as a model of Alzheimer’s disease [66]. Researchers show that this antibody reaches most of the brain parenchyma within 12 h of administration, and its path is traced from the olfactory bulb to the hippocampus (CA1) and cerebrum as well as from areas around the fourth ventricle to the brainstem, implying olfactory and trigeminal nerve traversal [66]. Impressively, the antibody finds its way into the brain in concentrations sufficient to be therapeutic as evidenced by a decrease in cerebral amyloid concentrations and by improved spatial acquisition learning in 5XFAD mice [66].
It may be possible to modify antibodies to make them more amenable to IN delivery, and several approaches have been explored to increase the efficacy of neuroimmunological treatments. One of the most elegant methods is to use small antibody fragments rather than full-fledged antibodies. A tumor necrosis factor (TNF)-α inhibitory single-chain antibody fragment (ESBA105) has been shown to effectively reach the brain via the olfactory and trigeminal nerves when administered intranasally, even compared to IV administration [67]. Maximum concentrations in the cerebellum and brainstem were recorded 1 h after administration, while maximum concentrations in the olfactory bulb and cerebrum were seen 2 h after administration [67]. When adjusted for dose, significantly higher concentrations are achieved by IN delivery than IV delivery in all brain regions [67]. Despite administering ten times more antibody intranasally as compared to intravenously, the plasma concentrations were 33 times higher after IV administration than after IN administration, implying that IN administration avoided systemic circulation [67].
Discussion
Devising novel drugs, routes of administration, and methods of distribution are necessary to provide immediate care in SE. Quick distribution of drugs directly following the 2017 chemical attack in Syria by “medical drones” may have saved lives and protected many victims’ quality of life. This is especially true when the victims are children; it has been shown that SE, but not self-limiting seizures, is associated with a worse global outcome and quality of life in children [68].
Recent advances in our understanding of the pathophysiology of SE have highlighted the need for rapid treatments that can terminate seizures before they progress to established SE. Access to effective alternative therapeutic modalities may improve the outcomes of victims of SE, particularly in pre-hospital contexts. The methods of improving the initial treatment of acute or prolonged seizures discussed here may solve some of the problems with IN anti-seizure drug delivery and may provide the keys to much more effective initial treatment of SE. There are many exciting and innovative ways of improving patient outcomes that remain to be explored. Improvements in terminating seizures and protecting the brain after seizure termination will undoubtedly improve the lives of these patients.
Western nations, even during peacetime, are not immune to possible chemical weapons attacks that could induce SE as a consequence of neurotoxicity, and the wealthiest and most protected of nations would struggle to provide adequate care to victims of such an event. Implementing methods of IN drug delivery that can be quickly distributed and administered in a wide variety of situations, such as ground zero of a chemical weapons attack, would help to lessen the burden of the resulting crisis on healthcare infrastructure. IN neurotherapeutics could also be kept aboard medical transport vehicles and in places that serve vulnerable populations, such as clinics and schools, where they could be used by untrained personnel.
Compliance with Ethical Standards
Conflict of Interest
John Benfield has no conflicts of interest to declare. Alberto Musto has no conflicts of interest to declare.
Funding
No funds were used in the writing of this manuscript.
References
- 1.Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH. A definition and classification of status epilepticus—report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia. 2015;56(10):1515–1523. doi: 10.1111/epi.13121. [DOI] [PubMed] [Google Scholar]
- 2.Cheng Jocelyn Y. Latency to treatment of SE is associated with mortality and functional status. J Neurol Sci. 2016;370:290–295. doi: 10.1016/j.jns.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Denson G. Fujikawa. The temporal evolution of neuronal damage from pilocarpine-induced stats epilepticus. Brain Res. 1996;725(1):11–22. doi: 10.1016/0006-8993(96)00203-x. [DOI] [PubMed] [Google Scholar]
- 4.Jett DA. Chemical toxins that cause seizures. Neurotoxicology. 2012;33(6):1473–1475. doi: 10.1016/j.neuro.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Unattributed. Assad kills at least 85 with chemical weapons. The Economist. 2017.
- 6.Unattributed. OPCW Fact-Finding Mission Confirms Use of Chemical Weapons in Khan Shaykhun on 4 April 2017. Organization for the Prohibition of Chemical Weapons. 2017.
- 7.Unattributed. Syria: Thousands suffering neurotoxic symptoms treated in hospitals supported by MSF. Doctors Without Borders. 2013.
- 8.Beg JM, Anderson TD, Francis K, Meckley LM, Fitzhenry D, Foster T, Sukhtankar S, Kanes SJ, Moura LM. Burden of illness for super-refractory status epilepticus patients. J Med Econ. 2017;20(1):45–53. doi: 10.1080/13696998.2016.1223680. [DOI] [PubMed] [Google Scholar]
- 9.Meierkord H, Boon P, Engelsen B, Göcke K, Shorvon S, Tinuper P, Holtkamp M. EFNS guideline on the management of status epilepticus in adults. Eur J Neurol. 2010;17(3):348–355. doi: 10.1111/j.1468-1331.2009.02917.x. [DOI] [PubMed] [Google Scholar]
- 10.Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH. A definition and classification of status epilepticus—report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia. 2015;56(10):1515–1523. doi: 10.1111/epi.13121. [DOI] [PubMed] [Google Scholar]
- 11.Neligan A, Shorvon SD. Frequency and prognosis of convulsive status epilepticus of different causes: a systematic review. Arch Neurol. 2010;67(8):931–940. doi: 10.1001/archneurol.2010.169. [DOI] [PubMed] [Google Scholar]
- 12.Buckmaster PS, Abrams E, Wen X. Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J Comp Neurol. 2017;525(11):2592–2610. doi: 10.1002/cne.24226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sloviter RS. “Epileptic” brain damage in rats induced by sustained electrical stimulation of the perforant path. I. Acute electrophysiological and light microscopic studies. Brain Res Bull. 1983;10(5):675–697. doi: 10.1016/0361-9230(83)90037-0. [DOI] [PubMed] [Google Scholar]
- 14.Hocker S. Systemic complications of status epilepticus—an update. Epilepsy Behav. 2015;31(49):83–87. doi: 10.1016/j.yebeh.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 15.Penberthy LT, Towne A, Garnett LK, Perlin JB, DeLorenzo RJ. Estimating the economic burden of status epilepticus to the health care system. Seizure. 2005;14(1):46–51. doi: 10.1016/j.seizure.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Kapur J, Macdonald RL. Rapid seizure-induced reduction of benzodiazepine and Zn2 + sensitivity of hippocampal dentate granule cell GABAA receptors. The Journal of Neuroscience. 1997;17(19):7532–7540. doi: 10.1523/JNEUROSCI.17-19-07532.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodkin HP, Yeh J-L, Kapur J. SE increases the intracellular accumulation of GABAA receptors. J Neurosci. 2005;25(23):5511–5520. doi: 10.1523/JNEUROSCI.0900-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kittler JT, Chen G, Honing S, Bogdanov Y, McAinsh K, Arancibia-Carcamo IL, Jovanovic JN, Pangalos MN, Haucke V, Yan Z, Moss SJ. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc Natl Acad Sci USA. 2005;102(41):14871–14876. doi: 10.1073/pnas.0506653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandon NJ, Jovanovic JN, Moss SJ. Multiple roles of protein kinases in the modulation of γ-aminobutyric acid A receptor function and cell surface expression. Pharmacol Ther. 2002;94(1):113–122. doi: 10.1016/s0163-7258(02)00175-4. [DOI] [PubMed] [Google Scholar]
- 20.Terunuma M, Jang IS, Ha SH, Kittler JT, Kanematsu T, Jovanovic JN, Nakayama KI, Akaike N, Ryu SH, Moss SJ, Hirata M. GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. J Neurosci. 2004;24(32):7074–7084. doi: 10.1523/JNEUROSCI.1323-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, Haydon PG, Coulter DA, Moss SJ. Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J Neurosci. 2008;28(2):376–384. doi: 10.1523/JNEUROSCI.4346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckel R, Szulc B, Walker MC, Kittler JT. Activation of calcineurin underlies altered trafficking of α2 subunit containing GABA A receptors during prolonged epileptiform activity. Neuropharmacology. 2015;31(88):82–90. doi: 10.1016/j.neuropharm.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purves D, Augustine GJ, Fitzpatrick D, Katz LC, LaMantia AS, McNamara JO, Williams SM, editors. Glutamate receptors. In: Neuroscience, 2nd edn. Sunderland (MA): Sinauer Associates; 2001.
- 24.Carver CM, Wu X, Gangisetty O, Reddy DS. Perimenstrual-like hormonal regulation of extrasynaptic δ-containing GABAA receptors mediating tonic inhibition and neurosteroid sensitivity. J Neurosci. 2014;34(43):14181–14197. doi: 10.1523/JNEUROSCI.0596-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HH, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc Natl Acad Sci. 2015;112(11):3523–3528. doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niquet J, Baldwin R, Suchomelova L, Lumley L, Naylor D, Eavey R, Wasterlain CG. Benzodiazepine-refractory status epilepticus: pathophysiology and principles of treatment. Ann N Y Acad Sci. 2016;1378(1):166–173. doi: 10.1111/nyas.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Althaus AL, McCarren HS, Alqazzaz A, Jackson C, McDonough JH, Smith CD, Hoffman E, Hammond RS, Robichaud AJ, Doherty JJ. The synthetic neuroactive steroid SGE-516 reduces status epilepticus and neuronal cell death in a rat model of soman intoxication. Epilepsy Behav. 2017;31(68):22–30. doi: 10.1016/j.yebeh.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 28.McDonough JH, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci Biobehav Rev. 1997;21(5):559–579. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- 29.Savic I, Roland P, Sedvall G, Persson A, Pauli S, Widen L. In-vivo demonstration of reduced benzodiazepine receptor binding in human epileptic foci. The Lancet. 1988;332(8616):863–866. doi: 10.1016/s0140-6736(88)92468-3. [DOI] [PubMed] [Google Scholar]
- 30.Ribak CE, Harris AB, Vaughn JE, Roberts E. Inhibitory, GABAergic nerve terminals decrease at sites of focal epilepsy. Science. 1979;205(4402):211–214. doi: 10.1126/science.109922. [DOI] [PubMed] [Google Scholar]
- 31.Houser CR, Harris AB, Vaughn JE. Time course of the reduction of GABA terminals in a model of focal epilepsy: a glutamic acid decar ylase immunocytochemical study. Brain Res. 1986;383(1):129–145. doi: 10.1016/0006-8993(86)90014-4. [DOI] [PubMed] [Google Scholar]
- 32.Esclapez M, Trottier S. Changes in GABA-immunoreactive cell density during motor focal epilepsy induced by cobalt in the rat. Exp Brain Res. 1989;76(2):369–385. doi: 10.1007/BF00247895. [DOI] [PubMed] [Google Scholar]
- 33.Olney JW. Excitatory transmitters and epilepsy-related brain damage. Int Rev Neurobiol. 1985;27:337–362. doi: 10.1016/s0074-7742(08)60561-9. [DOI] [PubMed] [Google Scholar]
- 34.Naylor DE, Liu H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis. 2013;30(54):225–238. doi: 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajasekaran K, Joshi S, Kozhemyakin M, Todorovic MS, Kowalski S, Balint C, Kapur J. Receptor trafficking hypothesis revisited: plasticity of AMPA receptors during established status epilepticus. Epilepsia. 2013;54(s6):14–16. doi: 10.1111/epi.12266. [DOI] [PubMed] [Google Scholar]
- 36.Rajasekaran K, Todorovic M, Kapur J. Calcium-permeable AMPA receptors are expressed in a rodent model of status epilepticus. Ann Neurol. 2012;72(1):91–102. doi: 10.1002/ana.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.David M. Treiman, treatment of convulsive SE. Int Rev Neurobiol. 2007;81:273–285. doi: 10.1016/S0074-7742(06)81018-4. [DOI] [PubMed] [Google Scholar]
- 38.Grover EH, Nazzal Y, Hirsch LJ. Treatment of convulsive status epilepticus. Curr Treat Options Neurol. 2016;18(3):11. doi: 10.1007/s11940-016-0394-5. [DOI] [PubMed] [Google Scholar]
- 39.Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5(7):553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- 40.Beyreuther BK, Freitag J, Heers C, Krebsfänger N, Scharfenecker U, Stöhr T. Lacosamide: a review of preclinical properties. CNS Drug Rev. 2007;13(1):21–42. doi: 10.1111/j.1527-3458.2007.00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, Barsan W. Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med. 2012;366(7):591–600. doi: 10.1056/NEJMoa1107494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Unattributed. Cholinesterase inhibitors: including pesticides and chemical warfare nerve agents. Agency for Toxic Substances and Disease Registry. 2007.
- 43.Zorthian J. Switzerland’s new medical drones. Time. 2017;190(14). Available from: http://time.com/4960622/switzerlands-new-medical-drones/.
- 44.Inokuchi R, Ohashi-Fukuda N, Nakamura K, Wada T, Gunshin M, Kitsuta Y, Nakajima S, Yahagi N. Comparison of intranasal and intravenous diazepam on status epilepticus in stroke patients: a retrospective cohort study. Medicine. 2015;94(7):e555. doi: 10.1097/MD.0000000000000555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kälviäinen R. Intranasal therapies for acute seizures. Epilepsy Behav. 2015;31(49):303–306. doi: 10.1016/j.yebeh.2015.04.027. [DOI] [PubMed] [Google Scholar]
- 46.Lochhead J, Thorne R. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 47.Pardeshi CV, Belgamwar VS. Direct nose to brain drug delivery via integrated nerve pathways bypassing the blood–brain barrier: an excellent platform for brain targeting. Expert Opin Drug Deliv. 2013;10(7):957–972. doi: 10.1517/17425247.2013.790887. [DOI] [PubMed] [Google Scholar]
- 48.Price JL, Powell TP. Certain observations on the olfactory pathway. J Anat. 1971;110(Pt 1):105. [PMC free article] [PubMed] [Google Scholar]
- 49.Illig KR. Projections from orbitofrontal cortex to anterior piriform cortex in the rat suggest a role in olfactory information processing. J Comp Neurol. 2005;488(2):224–231. doi: 10.1002/cne.20595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iannilli E, Del Gratta C, Gerber JC, Romani GL, Hummel T. Trigeminal activation using chemical, electrical, and mechanical stimuli. Pain. 2008;139(2):376–388. doi: 10.1016/j.pain.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 51.Johnson NJ, Hanson LR, Frey WH. Trigeminal pathways deliver a low molecular weight drug from the nose to the brain and orofacial structures. Mol Pharm. 2010;7(3):884–893. doi: 10.1021/mp100029t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thorne RG, Pronk GJ, Padmanabhan V, Frey W. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127(2):481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 53.Kyrkanides S, Yang M, Tallents RH, Jen-nie HM, Brouxhon SM, Olschowka JA. The trigeminal retrograde transfer pathway in the treatment of neurodegeneration. J Neuroimmunol. 2009;209(1):139–142. doi: 10.1016/j.jneuroim.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 54.Carmichael ST, Clugnet MC, Price JL. Central olfactory connections in the macaque monkey. Journal of Comparative Neurology. 1994;346(3):403–434. doi: 10.1002/cne.903460306. [DOI] [PubMed] [Google Scholar]
- 55.Dhuria SV, Hanson LR, Frey WH. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 56.Kapoor M, Cloyd JC, Siegel RA. A review of intranasal formulations for the treatment of seizure emergencies. J Control Release. 2016;10(237):147–159. doi: 10.1016/j.jconrel.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 57.Behl CR, Pimplaskar HK, Sileno AP, Romeo VD. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv Drug Deliv Rev. 1998;29(1):89–116. doi: 10.1016/s0169-409x(97)00063-x. [DOI] [PubMed] [Google Scholar]
- 58.Kumar PR, Vijayaraghavan R, Singh M. Efficacy of atropine nasal aerosol spray against organophosphorous poisoning. Indian J Pharmacol. 2001;33(6):431–436. [Google Scholar]
- 59.Rech MA, Barbas B, Chaney W, Greenhalgh E, Turck C. When to pick the nose: out-of-hospital and emergency department intranasal administration of medications. Ann Emerg Med. 2017;70(2):203–211. doi: 10.1016/j.annemergmed.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 60.Bancke LL, Dworak HA, Rodvold KA, Halvorsen MB, Gidal BE. Pharmacokinetics, pharmacodynamics, and safety of USL261, a midazolam formulation optimized for intranasal delivery, in a randomized study with healthy volunteers. Epilepsia. 2015;56(11):1723–1731. doi: 10.1111/epi.13131. [DOI] [PubMed] [Google Scholar]
- 61.Upsher-Smith Laboratories. A Long-term Safety and Tolerability Study of USL261 in Patients With Seizure Clusters. National Institutes of Health Clinical Trials Database. 2016.
- 62.Siegel RA, Kapoor M, Cheryala N, Georg GI, Cloyd JC. Water-soluble benzodiazepine prodrug/enzyme combinations for intranasal rescue therapies. Epilepsy Behav. 2015;31(49):347–350. doi: 10.1016/j.yebeh.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 63.Kumar A, Pandey AN, Jain SK. Nasal-nanotechnology: revolution for efficient therapeutics delivery. Drug Deliv. 2016;23(3):671–683. doi: 10.3109/10717544.2014.920431. [DOI] [PubMed] [Google Scholar]
- 64.Alivisatos AP. Perspectives on the physical chemistry of semiconductor nanocrystals. J Phys Chem. 1996;100(31):13226–13239. [Google Scholar]
- 65.Chen J, Zhang C, Liu Q, Shao X, Feng C, Shen Y, Zhang Q, Jiang X. Solanum tuberosum lectin-conjugated PLGA nanoparticles for nose-to-brain delivery: in vivo and in vitro evaluations. J Drug Target. 2012;20(2):174–184. doi: 10.3109/1061186X.2011.622396. [DOI] [PubMed] [Google Scholar]
- 66.Xiao C, Davis FJ, Chauhan BC, Viola KL, Lacor PN, Velasco PT, Klein WL, Chauhan NB. Brain transit and ameliorative effects of intranasally delivered anti-amyloid-β oligomer antibody in 5XFAD mice. J Alzheimer’s Dis. 2013;35(4):777–788. doi: 10.3233/JAD-122419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Furrer E, Hulmann V, Urech DM. Intranasal delivery of ESBA105, a TNF-alpha-inhibitory scFv antibody fragment to the brain. J Neuroimmunol. 2009;215(1):65–72. doi: 10.1016/j.jneuroim.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 68.Wagenman KL, Blake TP, Sanchez SM, et al. Electrographic SE and long-term outcome in critically ill children. Neurology. 2014;82(5):396–404. doi: 10.1212/WNL.0000000000000082. [DOI] [PMC free article] [PubMed] [Google Scholar]