

Abstract
Problem
Heightened maternal stress affects trophoblast function and increases risk for adverse pregnancy outcomes.
Methods of Study
Studies were performed using the first-trimester trophoblast cell line, Sw.71. Cytokines were quantified using qPCR and ELISA. Epigenetic regulation of cytokines was characterized by inhibiting histone deacetylation (1 μmol/L suberoylanilide hydroxamic acid [SAHA]) or methylation (5 μmol/L 5-azacytidine), or with chromatin immunoprecipitation (ChIP) with a pan-acetyl histone-3 antibody. Invasion assays used Matrigel chambers.
Results
Cortisol inhibited expression of CSF2 (GM-CSF) and CSF3 (G-CSF) in trophoblast cells. Cortisol-associated inhibition was dependent on DNA methylation and was not affected by acetylation. There was also a modest decrease in trophoblast invasion, not dependent on loss of CSFs.
Conclusion
In first-trimester trophoblast cells, the physiological glucocorticoid, cortisol, inhibited two cytokines with roles in placental development and decreased trophoblast invasion. Cortisol-associated changes in trophoblast function could increase the risk for immune-mediated abortion or other adverse pregnancy outcomes.
Keywords: cortisol, G-CSF, GM-CSF, methylation, stress, trophoblast
1 | INTRODUCTION
Women suffering with chronic stressors during pregnancy have increased risk for adverse pregnancy outcomes such as spontaneous abortion (SA),1–3 intrauterine growth restriction (IUGR),4,5 and preeclampsia (PE).4,6 Glucocorticoids regulate gene expression by binding the glucocorticoid receptor (GR) and affecting gene transcription.7 Placental trophoblast cells express high levels of the GR8,9 and are responsive to the stress hormone, cortisol. Therefore, stress-associated pregnancy failure could be due to changes in trophoblast signaling and/or function.
In healthy pregnancies, extravillous trophoblast cells invade the decidua and remodel uterine spiral arteries to increase maternal blood flow to the placenta. This invasion relies on expression of inflammatory cytokines and collagenases, such as matrix metalloproteinases (MMPs),10 but glucocorticoids can negatively affect these processes. For example, human first-trimester trophoblast cells exposed to the synthetic glucocorticoid, dexamethasone (DEX), exhibit reduced invasive capacity.10 In mice, DEX treatment affects the expression of genes associated with trophoblast growth and proliferation.11 Rats exposed to DEX during early pregnancy have altered placental blood vessel morphology and develop symptoms of PE.6 Furthermore, the physiological glucocorticoid in rodents, corticosterone, affects glucose transport mechanisms and amino acid synthesis in the placenta, resulting in IUGR.12
The trophoblast also regulates the maternal immune system to promote tolerance of the fetus and placenta. Early in pregnancy, invasive trophoblasts provide signals to ensure maternal immune cells remain tolerant of fetal cells.13–16 Maternal stress can affect the maternal-fetal immune cross talk and increase risk for SA.2 The role of stress in SA has also been studied in mice using the DBA/2-mated CBA/J mouse, a model for the study of immune-mediated abortions.17 Specifically in this model, maternal stress increased the number of abortions, which was associated with increased TNF-α, decreased TGF-β, and activation of cytotoxic NK cells.17,18 Interestingly, administration of the cytokine, granulocyte-macrophage colony-stimulating factor (GM-CSF), prevents immune-mediated abortions in mice and improves pregnancy rates in women with a history of recurrent abortions.17,19
The family of cytokines called colony-stimulating factors (CSFs) are best known as hematopoietic growth factors, but they are also expressed by endometrial and trophoblast cells.20–22 These include granulocyte-macrophage colony-stimulating factor (CSF2), granulocyte colony-stimulating factor (G-CSF; CSF3), and macrophage colony-stimulating factor (M-CSF; CSF1). In reproduction, the CSFs have roles in embryo development, placental development, and maternal immune tolerance.20–22
Granulocyte CSF induces expression of genes associated with implantation in the endometrium23 and invasion in first-trimester trophoblast cells.24 Furthermore, administration of G-CSF to women undergoing in vitro fertilization due to a thin endometrium resulted in higher pregnancy rates.19,20 Granulocyte-macrophage colony-stimulating factor (CSF2) also has important roles in embryo development.17,25 Human embryos, cultured in the presence of GM-CSF prior to implantation, had improved survival and pregnancy rates were increased, especially in women previously suffering recurrent miscarriages.19,20 The increased pregnancy rates following GM-CSF treatment is conserved in other species, as GM-CSF improves bovine embryo survival.26
Granulocyte-macrophage CSF also has roles in immune tolerance. Circulating GM-CSF is reportedly lower in the circulation of women suffering SA compared to those who maintained pregnancy.27 Furthermore, treatment with immunoglobulin as a means of preventing SA increased GM-CSF in women who delivered at term, but did not increase GM-CSF in those who suffered first-trimester abortions.27 In mice, administration of GM-CSF to DBA/2 × CBA/J resulted in CD8+ cell-mediated suppression of NK cell cytotoxicity that prevented abortions.17 In an endotoxin-induced abortion model, GM-CSF administration also decreased embryo resorptions and was associated with reduced (inflammatory) TNF-α and increased (protective) TNF-β.28
Previous studies demonstrate the important role of CSFs in pregnancy, and the potential for stress to increase risk for adverse pregnancy outcomes. Therefore, we hypothesize that cortisol, at physiological concentrations mimicking heightened stress in women, inhibits CSF expression in first-trimester trophoblasts. Furthermore, we propose these changes will be associated with changes in trophoblast functions, such as invasion capacity. While it is not known how cortisol regulates CSF gene expression in first-trimester trophoblast cells, in other tissues, CSFs are regulated by glucocorticoids via several different epigenetic mechanisms, such as histone acetylation or DNA methylation, depending on treatment and cell type.29,30 Therefore, we examined the epigenetic mechanism by which CSF2 and CSF3 are regulated by cortisol and we sought to determine how these changes related to trophoblast invasion.
2 | MATERIALS AND METHODS
2.1 | Cell culture and treatments
The first-trimester extravillous trophoblast cell line, Sw.71,31 was cultured in DMEM-F12 (Gibco, Life Technologies, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (Seradigm, VWR, Radnor, PA, USA) and 100 U/mL penicillin-streptomycin (Thermo Fisher, Waltham, MA, USA) at 37C with 5% CO2. Cortisol (CORT; Sigma-Aldrich, St. Louis, MO, USA) was prepared in 100% methanol. Cells were treated with 5, 10, or 20 ng/mL CORT in culture medium for 24, 48, or 72 hours. Control cells were supplemented with equivalent volumes of methanol. To maintain histone acetylation, SAHA (Sigma-Aldrich), a histone deacetylase inhibitor, was resuspended in DMSO and Sw.71 cells were cotreated with 20 ng/mL CORT and 1 μmol/L SAHA for 72 hours. To inhibit DNA methylation, cells were treated with 5-azacytidine, which was prepared immediately before each use (5-Aza-C; Sigma-Aldrich). Cells were cotreated with 20 ng/mL CORT and 5 μmol/L 5-Aza-C for 72 hours. Culture media was changed daily due to the short half-life of 5-Aza-C. Cells were collected and stored at −80°C until further use.
2.2 | Quantitative PCR
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s protocol, including an on-column DNAse treatment. Complimentary DNA (cDNA) was synthesized using the iScript cDNA synthesis Kit (Bio-Rad, Hercules, CA, USA), diluted 1:20 in nuclease-free water, and quantitative real-time PCR was carried out using the Sso Advanced SYBR Supermix (Bio-Rad) and gene-specific primers (Table 1) on the Applied Biosystems ViiA7 PCR machine. Cycling conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 30 seconds, 60°C for 1 minute. Values were normalized to GAPDH and calculated using delta delta Ct method: delta delta Ct = delta ct treated − delta Ct control; results are expressed as fold differences from controls.
TABLE 1.
Primer sequences for qPCR and chromatin immunoprecipitation-qPCR
| Gene/Locus | Forward primer (5′->3′) | Reverse primer (5′->3′) |
|---|---|---|
| CSF2 | CACTGCTGCTGAGATGAATGAAA | GTCTGTAGGCAGGTCGGCTC |
| CSF3 | ATAGCGGCCTTTTCCTCTACC | GCCATTCCCAGTTCTTCCAT |
| MMP-3 | ATTCCATGGAGCCAGGCTTTC | CATTTGCCTCAAACTCCAACTGTG |
| DNMT1 | GGTTTCCTTCCTCAGCTACTGCGA | CACTCATAGCCCATGCGGACC |
| DNMT3a | TCCACTGTGAATCATAAGCTG | GGAAACCAAATACCCTTTCCA |
| DNMT3b | GACTGCTTGGAATACAATAGG | AAAGCCAAAGATCCTTTCGAG |
| GAPDH | AGGGCTGCTTTTAACTCTGGT | CCCCACTTGATTTTGGAGGGA |
| CSF2-promoter | TGTCGGTTCTTGGAAAGGTTCA | TGTGGAATCTCCTGGCCCTTA |
| CSF2-intragenic | ATGGCAGTCACATGAGCTCCTT | TGAAGTGACCCCCACTTTACCA |
2.3 | Enzyme-linked immunosorbent assay (ELISA)
Cell-conditioned supernatants were collected, centrifuged for 5 minutes at 17 000 g to remove debris, and analyzed for GM-CSF and G-CSF concentrations using ELISA according to the manufacturer’s protocol (DGM00/DCS50; R&D Systems, Minneapolis, MN, USA). Total protein was quantified using BCA (Pierce, Waltham, MA, USA) to determine whether total cellular protein was affected by CORT treatment.
2.4 | Chromatin immunoprecipitation-qPCR
Sw.71 cells were plated at a density of 3 × 105 cells per 10-cm dish and treated with CORT as described above. At collection, cells were washed once with 1× PBS and fixed with 1% formaldehyde (Thermo Scientific) at room temperature. Cells were scraped from plates and stored at −80°C until use. One million cells were sonicated using BioRuptor (Diagenode, Denville, NJ, USA), and chromatin was isolated and immunoprecipitated using the Chromatin immunoprecipitation (ChIP) Kit (EMD Millipore, Billerica, MA, USA) and a mouse anti-rabbit pan-acetyl histone-3 antibody (EMD Millipore). Immunoprecipitated chromatin was reverse cross-linked and purified. Quantitative PCR was carried out as described above with primers spanning regulatory regions of CSF2 (Table 1).
2.5 | Trophoblast invasion
Trophoblast invasion was assessed using 8-μm Matrigel invasion chambers (Corning, Corning, NY, USA). Chambers were rehydrated for 2 hours at 37°C in DMEM-F12 1:1 (Sigma-Aldrich). To each chamber, 40 000 cells resuspended in 200 μL of DMEM-F12 1:1 supplemented with 2% FBS and the appropriate treatment (vehicle or 20 ng/mL CORT) were added. Chambers were added to wells containing DMEM-F12 1:1 supplemented with 10% FBS as the chemo-attractant. After 24 hours, Matrigel chambers were fixed with 70% ethanol and stained with 0.01% crystal violet. Each Matrigel was divided into four quadrants. A grid was superimposed onto each quadrant using ImageJ analysis software (NIH, Bethesda, MD, USA), and cells were counted in a 4 × 4 area within each quadrant.
2.6 | Statistics
Results of ELISA, qPCR, and ChIP were analyzed by independent t test (comparing two means) or one-way ANOVA (comparing more than two means) followed by Tukey’s test. Data are represented as means ± standard error of the mean. P-values equaling, or less than .05 were considered significantly different.
3 | RESULTS
3.1 | Cortisol inhibits CSF2 (GM-CSF) and CSF3 (G-CSF) in first-trimester trophoblast cells
We first determined how CSF2 (GM-CSF) and CSF3 (G-CSF) expression was affected in first-trimester trophoblast cells (Sw.71) when treated with physiological concentrations of cortisol (5, 10 and 20 ng/mL). Gene expression was quantified 24, 48, and 72 hours post-treatment, and we determined there was a time-and dose-dependent decrease in CSF2 and CSF3 mRNA with maximum downregulation at 72 hours (Figure 1A, ANOVA, P < .05; Figure 2B, ANOVA, P < .05). Both GM-CSF and G-CSF proteins were also reduced in trophoblast-conditioned medium following cortisol treatment by 48 hours (Figure 1C, t test, P < .05; Figure 1D, t test, P < .05). Total cellular protein did not differ between controls and CORT-treated cells; therefore, changes in secreted protein were not a result of a decline in total cellular protein (data not shown).
FIGURE 1.
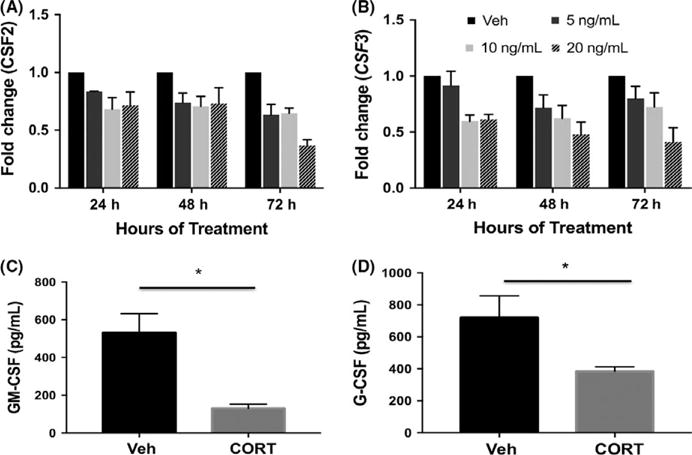
Cortisol downregulates CSF2 (GM-CSF) and CSF3 (G-CSF). (A, B) Sw.71 cells were treated with 5, 10, or 20 ng/mL cortisol (CORT) for 24, 48, or 72 h. Expression of CSF2 (A) and CSF3 (B) was quantified using qPCR (ANOVA, P < .05). (C, D) Cell-conditioned media was collected from Sw.71 cultured in the presence of 20 ng/mL CORT for 48 h. GM-CSF (C) and G-CSF (D) were quantified using ELISA (t test; GM-CSF, P < .01; G-CSF, *P < .05). qPCR data represent three to six independent replicates per treatment per time point. ELISA data represent four and five independent replicates for G-CSF and GM-CSF, respectively
FIGURE 2.
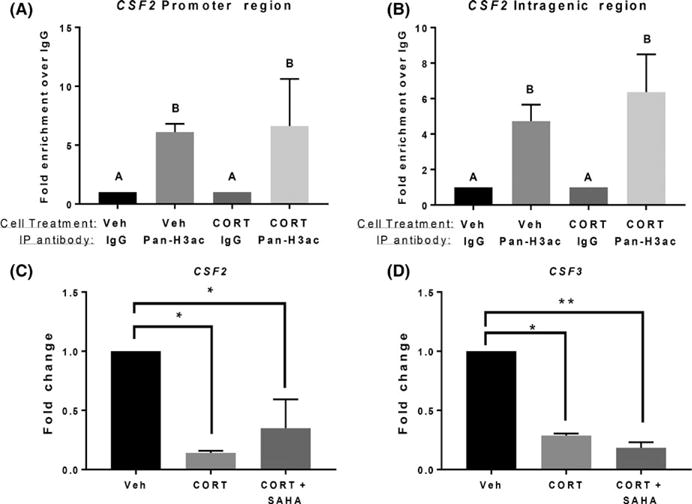
Cortisol-induced downregulation of CSF2 is independent of histone acetylation. (A, B) Histone (H3) acetylation was analyzed within the CSF2 promoter regions (A) and intragenic region (B) using chromatin immunoprecipitation (ChIP)-qPCR (ANOVA, A vs. B, P < .05). (C, D) Sw.71 cells were treated vehicle (Veh) or treated with cortisol (CORT; 20 ng/mL) and SAHA (1 μmol/L) for 72 h. CSF2 (C) and CSF3 (D) expression was quantified using qPCR (ANOVA, *P < .05, **P < .01). ChIP-qPCR data represent three independent experiments. qPCR data represent four and six independent replicates for CSF3 and CSF2, respectively
3.2 | Cortisol inhibition of CSF2 and CSF3 in trophoblast cells is independent of histone deacetylation
To characterize cortisol-associated inhibition of CSF2/3, we next determined whether cortisol inhibited CSF2 and CSF3 via histone deacetylation. First, we performed CHIP with an antibody directed against pan-acetylated histone-3, to quantify its occupancy at the CSF2 promotor and gene body. Interestingly, acetylated histone-3 did not change its occupancy at the CSF2 gene when trophoblast cells were treated with cortisol (Figure 2A,B, ANOVA, n.s.). Based on this result, we pre-treated trophoblast cells with SAHA, an HDAC inhibitor (1 μmol/L), followed by cortisol (20 ng/mL), and then quantified CSF2 and CSF3 expression. Confirming the previous finding, inhibiting HDACs did not affect cortisol-associated inhibition of CSF2/3 (Figure 2C,D, ANOVA, n.s.), suggesting cortisol does not regulate the CSFs by regulating gene acetylation.
3.3 | Cortisol inhibits CSF2 and CSF3 in trophoblast cells by affecting DNA methylation
Next, we determined whether cortisol affected DNA methylation of CSF2 and CSF3 in the trophoblast by treating cells with cortisol (20 ng/mL) in the presence of 5-Aza-C (5 μmol/L), a DNA methylation inhibitor. Interestingly, inhibition of DNA methylation prevented the cortisol-associated decrease in CSF2 and CSF3 expression (Figure 3A, ANOVA, P < .01; Figure 3B, ANOVA, P < .01). Cortisol did not affect expression of DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, or DNMT3B (Figure 3C; ANOVA, n.s.).
FIGURE 3.
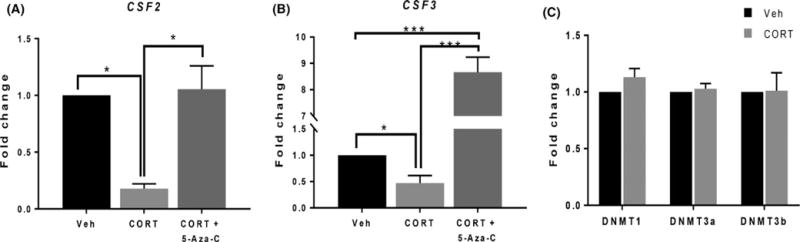
Cortisol inhibits CSF2 and CSF3 in trophoblasts via DNA methylation. (A, B) Sw.71 cells were treated with vehicle (Veh) or treated with cortisol (CORT; 20 ng/mL) in the presence of 5-Aza-C (1 μmol/L) for 72 h. CSF2 (A) and CSF3 (B) expression was quantified using qPCR (ANOVA; *P < .05, ***P < .001). (C) Sw.71 cells were treated with 20 ng/mL CORT for 72 h. Expression of DNA methyltransferases (DNMTs) was quantified using qPCR (ANOVA, n.s.). qPCR data represent four independent replicates for CSF2 and CSF3 and three independent replicates for DNMTs
3.4 | Cortisol regulates trophoblast invasion
Dexamethasone treatment can reduce first-trimester trophoblast cell invasion; therefore, we tested trophoblast invasion using the physiological glucocorticoid, cortisol. Physiological concentrations of cortisol did cause inhibition of trophoblast invasion in a Matrigel invasion assay (Figure 4A, t test, P < .02), although much more modest than reported with DEX-treated cells.10 This was associated with a significant decrease in MMP-3 (Figure 4B, t test, P < .01), but not MMP-9 as previously reported.10 To determine whether downregulation of GM-CSF was responsible for reduced trophoblast invasion, we tried to rescue the trophoblast invasion phenotype with exogenous GM-CSF, but trophoblast invasion was not affected by supplementation of GM-CSF (data not shown), suggesting a separate mechanism.
FIGURE 4.
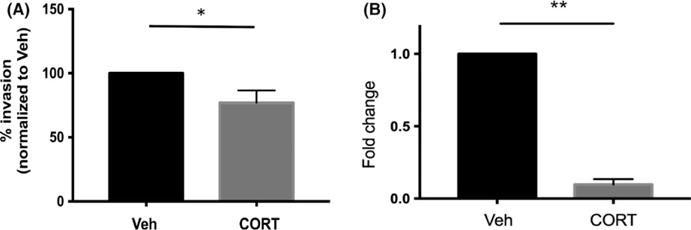
Cortisol regulates trophoblast invasion. (A) Sw.71 cells were treated with vehicle (Veh) or cortisol (CORT) for 72 h, and invasion capacity was analyzed using 8-μm Matrigel chambers (P < .03). (B) Sw.71 cells were treated with Veh or CORT for 72 h, and MMP-3 expression was quantified using qPCR (t test; *P = .02**P < .01). Invasion assay data represent three independent experiments. qPCR data represent three independent replicates for MMP-3
4 | DISCUSSION AND CONCLUSIONS
In this study, we determined that CORT inhibited CSF1 and CSF2 in first-trimester extravillous trophoblast. Cells were treated with CORT at a concentration that would be detected in pregnant women with elevated stress. CSF1 and CSF2 inhibition was independent of histone deacetylation and dependent on DNA methylation. We also determined that physiological concentrations of CORT inhibited trophoblast invasion, and this was not dependent on the inhibition of CSFs.
Women with heightened stress are at higher risk for early pregnancy complications, but little is known about how the physiological stress hormone, CORT, affects early pregnancy. Previous studies have shown the synthetic glucocorticoid, DEX, can significantly affect trophoblast gene expression, growth, and proliferation.10,11,30,32 Importantly, DEX has singular affinity for GR and affects gene expression differently than physiological glucocorticoids such as cortisol.30 Glucocorticoids such as cortisol also bind mineralocorticoid receptors, while DEX is specific for GR. Finally, CORT, but not DEX, is converted to its inactive form via 11β-HSD2, and can therefore be much less potent in trophoblast cells.30 Therefore, in vitro and in vivo DEX treatments are only reasonable models for the effects of synthetic glucocorticoid use during pregnancy. Multiple studies have also tested the role of DEX and other glucocorticoids on term placental cells.30 While many of these studies are strengthened by the use of primary trophoblast cells, this is only an appropriate model system for studying late pregnancy because term trophoblast cells are non-dividing and functionally unique from early trophoblasts. Therefore, to understand how elevated maternal stress hormone affects early pregnancy, in the present study we used the physiological glucocorticoid, CORT, with human first-trimester trophoblast cells. We determined that CORT did cause a modest decrease in trophoblast invasion, although the effect was more modest than previous reports using DEX.
Glucocorticoids can regulate CSF gene expression via several different epigenetic mechanisms.29 For example, DEX treatment of lung epithelial cells, following stimulation with IL-1β, results in recruitment of HDAC2 and loss of histone-4 acetylation at the CSF2 promoter.33 Interestingly, in human first-trimester trophoblast cells, histone acetylation/deacetylation was not involved in CORT inhibition of CSFs. Blocking histone deacetylation did not affect CSF2/3 expression, and pan-acetylated histone-3, a signature of transcriptionally active genes, was not decreased at the CSF2 locus in response to CORT.
Gene expression can also be affected by DNA methylation of CpG within regulatory elements of genes. Although there are no CpG islands within the CSF2 promoter, it still has several CpG sites that are differentially methylated in response to stimuli in other cell types.34 Interestingly, in human first-trimester trophoblast cells, CORT inhibition of CSFs was dependent on DNA methylation. The DNMTs are important regulators of DNA methylation, but CORT did not affect gene expression of DNMT1, DNMT3A, or DNMT3B. Therefore, it is likely CORT affects expression or recruitment of coregulatory factors that mediate DNMT functions. For example, CORT could inhibit factors with CXXC zinc finger domains, which can recruit histone methyltransferases that can maintain DNA in an unmethylated state.35
In conclusion, we studied the effect of the physiological glucocorticoid, cortisol, on the function of actively dividing, first-trimester human trophoblast cells. We determined that cortisol, at concentrations found in the circulation of pregnant women, inhibited invasive capacity of trophoblast cells and inhibited expression of CSFs via DNA methylation. Identifying how CSFs are regulated by cortisol in the trophoblast will help us identify other targets of the stress response. For example, we can now target genes that are methylated in response to cortisol. These approaches will improve our understanding of how stress affects the placenta in early pregnancy and how this results in aberrant pregnancy outcomes.
Acknowledgments
Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number T32HD087166. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Kwak-Kim J, Bao S, Lee SK, Kim JW, Gilman-Sachs A. Immunological modes of pregnancy loss: inflammation, immune effectors, and stress. Am J Reprod Immunol. 2014;72:129–140. doi: 10.1111/aji.12234. [DOI] [PubMed] [Google Scholar]
- 2.Nepomnaschy PA, Welch KB, McConnell DS, Low BS, Strassmann BI, England BG. Cortisol levels and very early pregnancy loss in humans. Proc Natl Acad Sci USA. 2006;103:3938–3942. doi: 10.1073/pnas.0511183103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nepomnaschy PA, Sheiner E, Mastorakos G, Arck PC. Stress, immune function, and women’s reproduction. Ann N Y Acad Sci. 2007;1113:350–364. doi: 10.1196/annals.1391.028. [DOI] [PubMed] [Google Scholar]
- 4.Ozmen A, Unek G, Korgun ET. Effect of glucocorticoids on mechanisms of placental angiogenesis. Placenta. 2017;52:41–48. doi: 10.1016/j.placenta.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Rondo PHC, Ferreira RF, Nogueira F, Ribeiro MCN, Lobert H, Artes R. Maternal psychological stress and distress as predictors of low birth weight, prematurity and intrauterine growth retardation. Eur J Clin Nutr. 2003;57:266–272. doi: 10.1038/sj.ejcn.1601526. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Liu H, Zeng J, et al. Glucocorticoid exposure in early placentation induces preeclampsia in rats via interfering trophoblast development. Gen Comp Endocrinol. 2016;225:61–70. doi: 10.1016/j.ygcen.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 7.Sapolsky R, Romero L, Munck A. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 8.Saif Z, Hodyl N, Stark M, et al. Expression of eight glucocorticoid receptor isoforms in the human preterm placenta vary with fetal sex and birthweight. Placenta. 2015;36:723–730. doi: 10.1016/j.placenta.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saif Z, Hodyl NA, Hobbs E, et al. The human placenta expresses multiple glucocorticoid receptor isoforms that are altered by fetal sex, growth restriction and maternal asthma. Placenta. 2014;35:260–268. doi: 10.1016/j.placenta.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 10.Librach C, Feigenbaum S, Bass K, et al. Interleukin 1B regulates human cytotrophoblast metalloproteinase activity and invasion in vitro. J Biol Chem. 1994;269:17125–17131. [PubMed] [Google Scholar]
- 11.Baisden B, Sonne S, Joshi RM, Ganapathy V, Shekhawat PS. Antenatal dexamethasone treatment leads to changes in gene expression in a murine late placenta. Placenta. 2007;28:1082–1090. doi: 10.1016/j.placenta.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaughan OR, Fisher HM, Dionelis KN, et al. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J Physiol. 2015;593:1307–1321. doi: 10.1113/jphysiol.2014.287177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrahams VM, Schaefer TM, Fahey JV, et al. Expression and secretion of antiviral factors by trophoblast cells following stimulation by the TLR-3 agonist, Poly(I: C) Hum Reprod. 2006;21:2432–2439. doi: 10.1093/humrep/del178. [DOI] [PubMed] [Google Scholar]
- 14.Abrahams VM, Straszewski-Chavez SL, Guller S, Mor G. First trimester trophoblast cells secrete Fas ligand which induces immune cell apoptosis. Mol Hum Reprod. 2004;10:55–63. doi: 10.1093/molehr/gah006. [DOI] [PubMed] [Google Scholar]
- 15.Abrahams VM, Visintin I, Aldo PB, Guller S, Romero R, Mor G. A role for TLRs in the regulation of immune cell migration by first trimester trophoblast cells. J Immunol. 2005;175:8096–8104. doi: 10.4049/jimmunol.175.12.8096. [DOI] [PubMed] [Google Scholar]
- 16.Mor G, Romero R, Aldo PB, Abrahams VM. Is the trophoblast an immune regulator? The role of Toll-like receptors during pregnancy. Crit Rev Immunol. 2005;25:375–388. doi: 10.1615/critrevimmunol.v25.i5.30. [DOI] [PubMed] [Google Scholar]
- 17.Clark D, Chaouat G, Mogil R, Wegmann T. Prevention of spontaneous abortion in DBA/2-mated CBA/J mice by GM-CSF involves CD8+ T cell-dependent suppression of natural effector cell cytotoxicity against trophoblast target cells. Cell Immunol. 1994;154:143–152. doi: 10.1006/cimm.1994.1064. [DOI] [PubMed] [Google Scholar]
- 18.Arck P, Troutt A, Clark D. Soluble receptors neutralizing TNF-a and IL-1 block stress-triggeredmurine abortion. Am J Reprod Immunol. 1997;37:262–266. doi: 10.1111/j.1600-0897.1997.tb00225.x. [DOI] [PubMed] [Google Scholar]
- 19.Ziebe S, Loft A, Povlsen BB, et al. A randomized clinical trial to evaluate the effect of granulocyte-macrophage colony-stimulating factor (GM-CSF) in embryo culture medium for in vitro fertilization. Fertil Steril. 2013;99:1600–1609. doi: 10.1016/j.fertnstert.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 20.Robertson SA. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor Rev. 2007;18:287–298. doi: 10.1016/j.cytogfr.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Sferruzzi-Perri AN, Macpherson AM, Roberts CT, Robertson SA. Csf2 null mutation alters placental gene expression and trophoblast glycogen cell and giant cell abundance in mice. Biol Reprod. 2009;81:207–221. doi: 10.1095/biolreprod.108.073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson SA, Roberts CT, Farr KL, Dunn AR, Seamark RF. Fertility impairment in granulocyte-macrophage colony-stimulating factor-deficient mice. Biol Reprod. 1999;60:251–261. doi: 10.1095/biolreprod60.2.251. [DOI] [PubMed] [Google Scholar]
- 23.Rahmati M, Petitbarat M, Dubanchet S, Bensussan A, Chaouat G, Ledee N. Granulocyte-colony stimulating factor related pathways tested on an endometrial ex-vivo model. PLoS ONE. 2014;9:e102286. doi: 10.1371/journal.pone.0102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furmento VA, Marino J, Blank VC, Roguin LP. The granulocyte colony-stimulating factor (G-CSF) upregulates metalloproteinase-2 and VEGF through PI3K/Akt and Erk1/2 activation in human trophoblast Swan 71 cells. Placenta. 2014;35:937–946. doi: 10.1016/j.placenta.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 25.Savion S, Zeldich E, Orenstein H, et al. Cytokine expression in the uterus of mice with pregnancy loss-effect of maternal immunopotentiation with GM CSF.pdf. Reproduction. 2002;123:399–409. doi: 10.1530/rep.0.1230399. [DOI] [PubMed] [Google Scholar]
- 26.Loureiro B, Bonilla L, Block J, Fear J, Bonilla A, Hansen PJ. Colony-stimulating factor 2 (CSF-2) improves development and post-transfer survival of bovine embryos. Endocrinology. 2009;150:5046–5054. doi: 10.1210/en.2009-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perricone R, De Carolis C, Giacomelli R, Guarino M, De Sanctis G, Fontana L. GM CSF and pregnancy- evidence of reduced concentrations in recurrent abortion reverted by immunoglobulin treatment.pdf. Am J Reprod Immunol. 2003;50:232–237. doi: 10.1034/j.1600-0897.2003.00083.x. [DOI] [PubMed] [Google Scholar]
- 28.Chaouat G, Menu E, Clark D, Dy M, Minkowski M, Wegmann T. Control of fetal survivial in CBA × DBA/2 mice by lymphokine therapy. J Reprod Fertil. 1990;89:447–458. doi: 10.1530/jrf.0.0890447. [DOI] [PubMed] [Google Scholar]
- 29.Monk C, Feng T, Lee S, Krupska I, Champagne FA, Tycko B. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am J Psychiat. 2016;173:705–713. doi: 10.1176/appi.ajp.2015.15091171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michael AE, Papageorghiou AT. Potential significance of physiological and pharmacological glucocorticoids in early pregnancy. Human Reprod Update. 2008;14:497–517. doi: 10.1093/humupd/dmn021. [DOI] [PubMed] [Google Scholar]
- 31.Straszewski-Chavez SL, Abrahams VM, Alvero AB, et al. The isolation and characterization of a novel telomerase immortalized first trimester trophoblast cell line, Swan 71. Placenta. 2009;30:939–948. doi: 10.1016/j.placenta.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guller S, Markiewicz L, Wozniak R, et al. Developmental regulation of glucocorticoid-mediated effects on extracellular matrix protein expression in the human placenta. Endocrinology. 1994;134:2064–2071. doi: 10.1210/endo.134.5.8156906. [DOI] [PubMed] [Google Scholar]
- 33.Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Ohms SJ, Shannon FM, Sun C, Fan JY. IL-2 and GM-CSF are regulated by DNA demethylation during activation of T cells, B cells and macrophages. Biochem Biophys Res Comm. 2012;419:748–753. doi: 10.1016/j.bbrc.2012.02.094. [DOI] [PubMed] [Google Scholar]
- 35.Blattler A, Farnham PJ. Cross-talk between site-specific transcription factors and DNA methylation states. J Biol Chem. 2013;288:34287–34294. doi: 10.1074/jbc.R113.512517. [DOI] [PMC free article] [PubMed] [Google Scholar]