

Abstract
Lysosomes serve dual roles in cancer metabolism, executing catabolic programs (i.e. autophagy and macropinocytosis), while promoting mTORC1-dependent anabolism. Antimalarial compounds such as chloroquine or quinacrine have been used as lysosomal inhibitors, but fail to inhibit mTOR signaling. Further, the molecular target of these agents has not been identified. We report a screen of novel dimeric antimalarials that identifies dimeric quinacrines (DQs) as potent anticancer compounds, which concurrently inhibit mTOR and autophagy. Central nitrogen methylation of the DQ linker enhances lysosomal localization and potency. An in situ photoaffinity pulldown identified palmitoyl-protein thioesterase 1 (PPT1) as the molecular target of DQ661. PPT1 inhibition concurrently impairs mTOR and lysosomal catabolism through the rapid accumulation of palmitoylated proteins. DQ661 inhibits the in vivo tumor growth of melanoma, pancreatic, and colorectal cancer mouse models and can be safely combined with chemotherapy. Thus, lysosome-directed PPT1 inhibitors represent a new approach to concurrently targeting mTORC1 and lysosomal catabolism in cancer.
Keywords: Lysosome, autophagy, mTOR, palmitoylation, macropinocytosis
Introduction
The lysosome is the only organelle that can degrade other organelles and proteins due to its acid-dependent degradative enzymes. Because of this specialized function, lysosomes receive cargo via multiple pathways, including macroautophagy (autophagy hereafter) and macropinocytosis, which allows for scavenging of intracellular and extracellular nutrients, respectively (1). Autophagy and macropinocytosis are both regulated by the mammalian target of rapamycin complex 1 (mTORC1) (2). In turn, the lysosome membrane directly regulates mTORC1 by housing key mTOR regulators. The pentameric Ragulator protein complex resides on the lysosomal surface and serves as a docking site for Rag GTPases. When amino acids (AAs) are present, Rag GTPases directly recruit mTORC1 to the lysosomal surface (3, 4), where it can be fully activated by Rheb (5). Therefore, the lysosome serves critical catabolic and growth signaling roles.
Aberrant autophagic-lysosomal activity (6) and dysregulated mTORC1 signaling (7) allow tumor cells to resist the stresses of chemotherapy and targeted therapy. However, attempts to address these intertwined pro-tumorigenic mechanisms independently have produced few durable responses in the clinic (8, 9). The combination of a rapamycin derivative with the lysosome inhibitor hydroxychloroquine (HCQ) has been tested clinically with an encouraging safety profile. However, response rates were low, and pharmacokinetic (PK)-pharmacodynamic (PD) studies performed in patients receiving the highest FDA-allowed dosage of HCQ demonstrated only modest autophagy inhibition (10). Furthermore, the mechanism underlying how CQ derivatives inhibit autophagy-lysosome function remains unclear. These results underscore the need to develop more potent lysosomal inhibitors and to define the molecular target responsible for lysosomal inhibition.
We previously reported Lys05, a dimeric chloroquine (CQ) that is 10-fold more potent than monomeric CQ derivatives (11). Given the recent appreciation of the lysosomal regulation of mTORC1, we hypothesized that more potent and specific inhibitors of the lysosome could concurrently inhibit lysosomal catabolism and mTORC1 signaling. Towards this goal, we modified the Lys05 structure, ultimately yielding a series of potent dimeric quinacrines (DQs). We find that methylation of the central nitrogen of the linker impacts DQ subcellular localization and enhances DQ function against autophagy. This approach has led to the identification of a class of compounds that that can be targeted specifically to the lysosome. Using these more potent and lysosome-specific inhibitors, we have identified the molecular target of DQ as palmitoyl-protein thioesterase 1 (PPT1), resulting in the impairment of mTOR signaling via disruption of the interaction between mTORC1 and its master activator Rheb, even in the setting of amino acid-replete medium. The concurrent inhibition of mTORC1 and lysosomal catabolism, one of the key resistance mechanisms to existing mTOR inhibitors, leads to a more complete abrogation of protein translation and anti-cancer activity relative to what is achievable with allosteric or catalytic mTORC1 inhibitors. Our findings reveal an unanticipated role of PPT1 in regulating both autophagy and mTORC1 localization and activity in cancer.
Results
DQ has superior anticancer efficacy amongst dimeric antimalarials
In an effort to improve the activity of Lys05, we extended the triamine linker of Lys05 using the naturally occurring triamine, spermidine. The anti-cancer activity of the resulting compound, DC340, the name of which was derived as described below, was assessed in BRAF-mutant melanoma and KRAS-mutant pancreatic cancer cell lines, as cancers driven by these oncogenes depend on autophagy/lysosome function (12, 13). DC340 inhibited the growth of a panel of melanoma and pancreatic cancer cell lines significantly better than Lys05 (Figure 1A and 1B). We next developed a series of dimeric compounds linked by the spermidine linker, whereby the 7-chloroquinoline heterocycles of CQ were replaced with the heterocyclic rings present in other antimalarials, such as primaquine (PQ), mefloquine (MQ) and QN (quinacrine, Figure 1A). We describe these resulting novel structures as DC (dimeric CQ), DM (dimeric MQ), DP (dimeric PQ) or DQ (dimeric QN), followed by three digits (m, n, R) where m and n signify the number of carbons to the left and right of the central nitrogen of the triamine linker, respectively, and the R signifies whether the central nitrogen of the linker is methylated (1) or not (0) (Figure 1A). Anti-proliferative activity was improved between 3-10 fold for each dimeric compound that contained CQ, MQ, or PQ, relative to its monomeric parent. In contrast, dimerization of QN resulted in ≥ 60-fold improvement in anti-proliferative activity relative to monomeric QN (Figure 1C).
Figure 1. DQs have superior anti-cancer efficacy amongst dimeric anti-malarials.
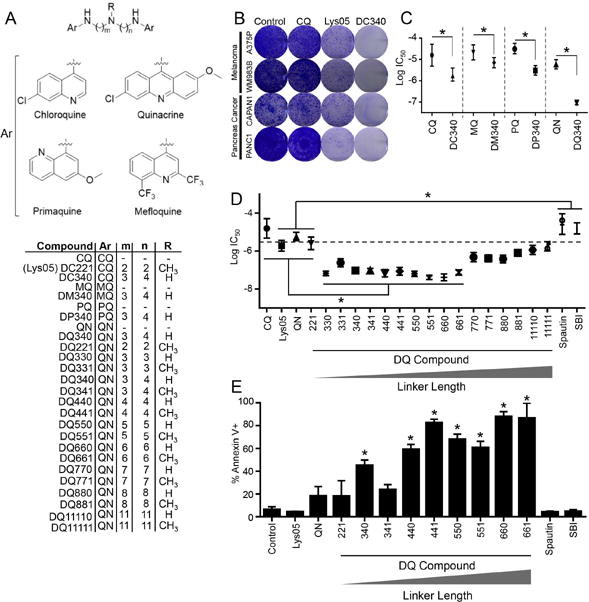
(A) Chemical structures of dimeric antimalarials. Ar: aromatic ring; CQ (chloroquine), QN (quinacrine); PQ (primaquine); MQ (mefloquine). (B) Cells were treated with compounds shown (2 weeks, 3 μM) in colony formation assays. (C) Calculated Log IC50s from 72-hour MTT assays in A375P (melanoma) cells treated with compounds shown. (D) Calculated Log IC50s of indicated compounds (72 hr, 1 nM - 30 μM) from MTT assays in A375P cells. (E) PANC1 cells were treated as indicated (24 hr, 3 μM), stained with Annexin-V and analyzed using flow cytometry. (B-D) for all graphs, mean ± standard deviation (s.d.) for N = 3 independent experiments are presented; *p<0.05.
We next generated a focused library of DQs with triamine linkers of increasing length, ranging from 2-11 carbons between the 9-aminoacridine and the central nitrogen of the triamine linker. Both unmethylated (R=0) and methylated (R=1) derivatives of each analog were prepared to determine the role of central nitrogen methylation on biological activity. Treatment of A375P and PANC1 cells with this DQ library further established the superior anti-proliferative potency of DQs compared to their monomeric counterpart (QN) (Figure 1D, Supplemental Figure S1A). While, DQ221, the acridine dimer with the same linker as Lys05 (DC221 using this nomenclature system), showed the same potency as its corresponding monomer with no benefit from dimerization, a clear relationship between linker length and potency emerged amongst the DQs with increasing linker length, whereby DQs with 3-6 carbons between linker nitrogens possessed significant anti-proliferative potency. Further extension of the distance between the two QN heterocycles resulted in incrementally decreased anti-proliferative activity (Figure 1D, Supplemental Figure S1A). Pharmacological agents that specifically inhibit autophagy, such as Spautin-1, which promotes degradation of Beclin (14), and SBI-0206965 (SBI), an unc-like kinase 1 (ULK1) inhibitor (14, 15), exhibited significantly less potency relative to DQs (Figure 1D, Supplemental Figure S1A). The compounds in the series DQ330–DQ661 produce significantly greater long-term growth suppression compared to monomeric QN and DQ221 (Supplemental Figure S1B). Unlike Spautin-1 and SBI, which did not induce appreciable levels of apoptosis, the majority of DQs trigger significantly greater levels of apoptosis, which directly correlates with increasing linker length, relative to monomeric QN (Figure 1E).
Central nitrogen methylation directs subcellular localization of DQs
We next interrogated the specificity of DQs as lysosomal inhibitors, as the parent monomer, QN, both binds to DNA in the nucleus and accumulates in the lysosome (16). The inherent fluorescence of DQs was exploited to study their subcellular localization. The most potent longer-linked DQs (DQ550, DQ551, DQ660, DQ661) fluoresce in both the red and green channels, under both neutral (pH = 7) and acidic (pH = 4) conditions (Figure 2A). Under neutral conditions, methylated DQs demonstrated increased green fluorescence compared to unmethylated DQs. However, there were no significant differences in red fluorescence observed between unmethylated and methylated DQs. Acidic conditions led to partial quenching of green fluorescence, but had minimal effects on red fluorescence in both methylated (DQ551, DQ661) and unmethylated (DQ550, DQ660) DQs. These findings indicate that red fluorescence is more effective to assess lysosomal localization of these compounds. Surprisingly, each unmethylated DQ (DQ550, DQ660) displayed no detectable red fluorescence, whereas each methylated DQ (DQ551, DQ661) possessed strong red fluorescence in the lysosomal compartment (Figure 2B, Supplemental Figure S2A). No colocalization with mitochondria was observed for DQ551 or DQ661 (Supplemental Figure S2B). To rule out the possibility that the addition of a methyl group to the central nitrogen was simply increasing the basicity of the DQ, thereby trapping the methylated derivatives in the acidic lysosome, the pKa’s of each DQ were calculated (Supplemental Table 1). This analysis demonstrated that the pKa of each pair of methylated and unmethylated DQ did not differ significantly, and therefore differential basicity could not account for the difference in subcellular localization. Having established the role of central nitrogen methylation as a determinant of lysosomal localization, we next characterized how DQs impact autophagy. Changes in autophagic vesicle (AV) levels were interrogated by measuring Atg8/LC3B (LC3-I, LC3-II hereafter) protein levels, as LC3-II is the most reliable protein marker of completed autophagosomes (17). Increasing DQ linker length was associated with increasing LC3II/LC3I ratios (Figure 2C), reflecting an accumulation of AVs. Interestingly, there was also a relationship between central nitrogen methylation and LC3II/LC3I ratios, in which compounds with central nitrogen methylation (DQXX1) had a significantly higher LC3II/LC3I ratio relative to their unmethylated (DQXX0) counterparts. Due to the inherent fluorescence of QN, spectral overlap with mCherry-eGFP-LC3 expressing cells (18) prevented the use of this approach to characterize autophagic flux. The effects of these compounds on autophagic flux were therefore determined by a bafilomycin (baf) clamp experiment (19). Treatment with DQs lacking central nitrogen methylation (DQXX0) resulted in a further increase in the LC3II/LC3I ratio in bafilomycin (baf)-treated cells compared with control cells, confirming that all DQs lacking a centrally methylated nitrogen (DQXX0) significantly increase autophagic flux (Figure 2D). In contrast, every DQ with central nitrogen methylation (DQXX1) did not demonstrate an increase in LC3II/LC3I ratio in baf-treated cells compared to control (Figure 2D). This difference in autophagy modulation between unmethylated and methylated DQs was also evidenced by unmethylated DQs producing striking degradation of the autophagy substrates p62 and NBR1, whereas treatment with methylated DQs did not (Figure 2E). Taken together, these data indicate that DQs without central nitrogen methylation function as autophagy inducers, whereas DQs with central nitrogen methylation modulate autophagy in a more pronounced fashion, acting as autophagy inhibitors. To further confirm that central nitrogen methylation dictates subcellular localization of DQs, lysosomal fractionation was performed in cells treated with either DQ660 or DQ661. Quantification of DQ660 and DQ661 concentrations in subcellular compartments revealed a 3.5-fold difference in the concentration of DQ661 compared to DQ660 in lysosomal fractions (Figure 2F).
Figure 2. Central nitrogen methylation status directs effects upon autophagy, induction versus inhibition.
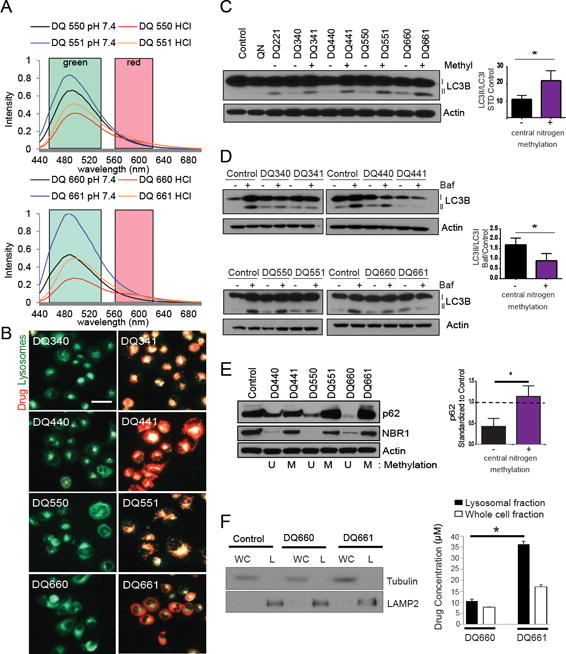
(A) Fluorescence spectroscopy emission spectra are shown (excitation 424 nm) (B) Fluorescent microscopy of PANC1 (pancreas cancer) cells treated as indicated (6 hr, 3 μM) and co-stained with LysoTracker deep red (shown green); arrows: co-localization of DQ compound (red) with LysoTracker (green). (C) LC3B immunoblot in lysates from C8161 (melanoma cells) treated as indicated (4 hr, 3 μM). (D) Bafilomycin clamp (100 nM) to measure autophagic flux was performed on A375P cells treated with DQ compounds (4 hr, 3 μM). Lysate was immunoblotted for LC3B. (E) PANC1 cells were treated with compounds shown (6 hr, 3 μM). Lysates were immunoblotted and change in p62 densitometry levels were quantified and are depicted in the graph to the right; *p<0.05. (F) A375P cells were treated with DQ660 or DQ661 (6 hr, 3 μM) and lysosomal and cytosolic fractions were subsequently isolated. Lysates were immunoblotted for LAMP2 and Tubulin (left panel). Bio-orthogonal fluorescence of DQ660 and DQ661 was measured on a fluorescent plate reader to quantify the lysosomal quantity of DQ660 and DQ661.
DQs cause either significant DNA damage or lysosomal membrane permeabilization depending on the central nitrogen methylation status
We next determined whether DQs cause DNA damage, since QN is known to intercalate DNA (20). A striking relationship between nitrogen methylation and DNA damage emerged, whereby unmethylated DQs induced significantly greater levels of DNA damage relative to their methylated counterparts (Figure 3A). Furthermore, the difference in DNA damage induced by unmethylated DQs versus the corresponding methylated analogs increased with increasing linker length for each pair of compounds. The integrity of the lysosomal membrane was interrogated by detection of lectin galactoside binding soluble 3 (LGALS3)/galectin-3 localization, which was recently demonstrated to rapidly translocate to damaged lysosomes (21). When lysosomes are healthy, galectin-3 is uniformly dispersed throughout the cell. However, when the lysosome is damaged, galectin-3 rapidly localizes to the site of damaged lysosomes, appearing as puncta. Only methylated DQs (DQXX1) caused lysosomal membrane permeabilization (LMP), as seen by the formation of galectin-3+ puncta (Figure 3B). The differential ability to induce LMP was further demonstrated by the observation of mature cathepsin D in the extra-lysosomal fraction of cells treated with DQ661 (Figure 3C).
Figure 3. Central nitrogen methylation status dictates DNA damage versus lysosomal membrane permeability.
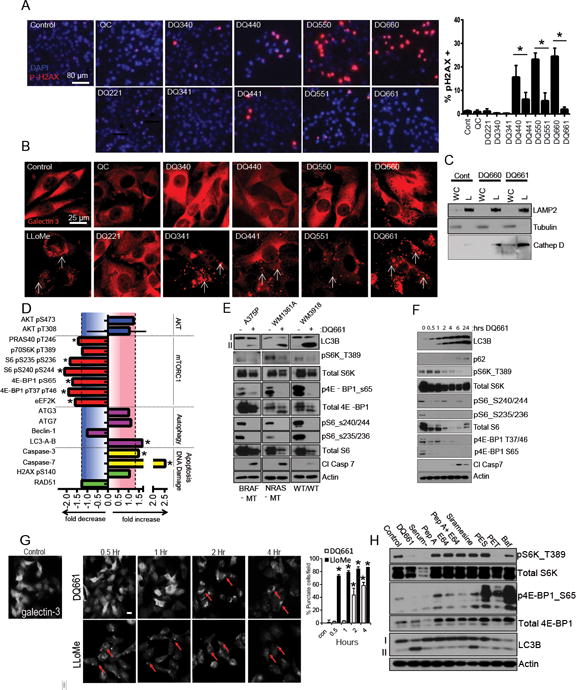
(A) IF microscopy of A375P cells treated as indicated (6 hr, 3 μM) and stained for phospo-H2AX (red; arrows) and DAPI (blue); and scored using ImageJ (mean ± SD); *p<0.05. (B) IF microscopy of A375P cells treated as in (A) with the addition of the positive control LLoMe (3 hr, 2 mM). IF against galectin-3 is shown. White arrows: galectin-3 punctae, reflecting lysosomal membrane permeabilization. (C) Lysosomal sub-fractionation and immunoblotting in A375P cells treated as indicated (6 hr, 3 μM. WC: whole lysate, L: lysosomal fraction. (D) A375P cells were treated with DQ661 (24 hr, 3 μM) and lysates were subjected to reverse phase protein array (RPPA; see supplemental figure S3A for complete dataset). Bar graphs show fold change of a selected panel of proteins at 24 hrs. *p<0.05. (E) Cells treated with DQ661 (6 hr, 3 μM) and lysate was immunoblotted. (F) A375P cells were treated with DQ661 (0 – 24 hr; 3 μM) and lysate was immunoblotted. (G) A375P cells were treated with DQ661 (0 – 4 hr; 3 μM) or Llome (0 – 24 hr; 2 mM) and subsequently stained for galect-3 and imaged by IF microscopy. Below: quantitation of percentage of galectin-3 puncta positive cells. (H) A375P cells were treated with DQ661 (6 hr, 3 μM), PBS, Pepstatin A (10 μG/mL), E64 (10 μG/mL), Pepstatin A + E64, Siramesine (8 μM), PES (10 μM), PET (10 μM), Bafilomycin A1 (100 nM), or Bafilomycin A1 + DQ661. Lysate was immunoblotted against phospho-S6K T389, total S6K, phospho-4E-BP1 S65, total 4E-BP1, LC3B and Actin. (I) A375P cells were treated with a CQ, Lys05, QC, DQ660 and DQ661 (6 hr, 3 μM) and lysate was subjected to RPPA. Shown are graphs reflecting fold decrease of mTORC1 substrates. (A – B) a Cy5 secondary antibody was used for pH2AX and galectin-3 to avoid spectral overlap with DQ661. Scale bars: 80 μm in (A) and 25 μm in (B).
DQ661 emerged as the lead lysosomal inhibitor in this series of compounds, capable of most potently inducing DNA damage-independent apoptosis, blocking autophagic flux and triggering LMP. To determine the underlying mechanism of action of DQ661, treated cells were assayed for >290 proteins or phospho-proteins spanning key signaling programs commonly perturbed in cancer cells using reverse phase protein arrays (RPPA). The proteins most significantly and reproducibly inhibited by DQ661 were downstream of mTORC1 signaling (Figure 3D, Supplemental Figure S3A). These included reduced phosphorylation of PRAS40, p70S6K, S6, and 4E-BP1. Immunoblotting confirmed the RPPA results, i.e., the ability of DQ661 to inhibit autophagy and mTORC1 activity, and to induce apoptosis in BRAFV600E, NRASQ61R, and BRAF-WT/NRAS-WT melanoma cell lines (Figure 3E). To understand the kinetics of pathway inhibition, immunoblotting revealed that mTORC1 activity was inhibited within 30 minutes of treatment with DQ661, before appreciable accumulation of AVs was observed, as seen by LC3 lipidation (Figure 3F). Autophagy modulation and dephosphorylation of a number of mTOR target substrates was sustained even at the 24-hour timepoint. Cleavage of caspase 7 did not occur until mTORC1 and autophagy were both significantly inhibited. A kinetic analysis of LMP induction was also performed, revealing LMP does not occur until 2-4 hours after treatment with DQ661, suggesting the rapid mTORC1 inhibition associated with DQ661 is independent of LMP (Figure 3G). Further, Leu-Leu methyl ester hydrobromide (LloMe), a prototypical lysosomal membrane permeabilizing agent, did not inhibit mTORC1 activity at the 30-minute timepoint when LloMe-induced LMP is clearly evident, demonstrating that DQ661-induced LMP is not responsible for mTOR inhibition (Supplemental Figure S3B).
To determine whether the anti-mTORC1 activity of DQ661 could be observed with other agents that impair lysosomal deacidification, produce LMP, or inhibit lysosomal enzymes, A375P cells were treated with either amino-acid/serum starvation, aspartyl protease inhibitor pepstatin A, cysteine protease inhibitor E64, Pepstatin A + E64, acid sphingomyelinase inhibitor siramesine (22), HSP70 inhibitors PES or PET (23), baf, or baf + DQ661 (Figure 3H). Immunoblotting confirmed DQ661 inhibits mTORC1 activity to a similar level as serum starvation, and to a greater extent than any other lysosomal agent tested. PET inhibited S6K phosphorylation, but both PES and PET treatment led to the hyperphosphorylation of 4E-BP1. Baf had minimal effects on phospho-S6K, and produced hyper-phosphorylation of 4E-BP1. DQ661 had the greatest ability to inhibit autophagy amongst the entire panel of lysosomal inhibitors, as seen by the greatest accumulation of LC3II/LC3I ratio. At the doses and timepoints tested, DQ661 appears to be unique in its ability to inhibit both mTORC1 signaling and autophagy (Figure 3H). In contrast to these effects observed with DQ661, RPPA data and Western blot analysis demonstrated that neither QC nor DQ660 had a significant impact on mTORC1 signaling at equimolar doses (Supplemental Figure S3A and S3C).
MTORC1 inhibition not only upregulates autophagy, but also macropinocytosis, a process by which nutrient-limited, Ras-driven cancer cells endocytose, envelop extracellular protein, and release AAs through lysosome-mediated degradation, paradoxically fueling growth of cancer cells (24). We next asked whether or not DQ661 would increase lysosomal catabolism of extracellular proteins since it inhibits mTORC1. We confirmed that DQ661 blocked autophagy and mTOR in G43 mouse pancreatic cancer cells (Supplemental Figure S3D) derived from the LSL-KrasLSL-G12D/+, LSL-p53LSL-R172H/+, Pdx1-Cre [KPC] mouse model) (25). Neither torin-1 nor DQ661 significantly changed the level of macropinocytic uptake of albumin in glutamine-limited G43 cells (Supplemental Figure 3E). Torin-1 treatment significantly increased lysosomal degradation of externally supplied protein, as displayed by the unquenched fluorescence of DQ-BSA. In contrast, treatment with DQ661 did not produce increased lysosomal degradation of external proteins when G43 cells were grown in similar conditions (Supplemental Figure 3E). Further, torin-1 significantly increased the proliferation of glutamine-limited G43 cells supplemented with 2% albumin, relative to cells grown in low glutamine with 2% albumin treated with vehicle control (Supplemental Figure 3F). DQ661 decreased the proliferation of G43 cells to the levels observed by cells cultured in low glutamine in the absence of 2% albumin.
To directly measure the effect of DQ661 on catabolism of extracellular protein by pancreatic cells, we devised an isotope tracer-based method to monitor protein scavenging. This method enables the separate quantification of (i) AAs added to the medium as monomers and (ii) AAs derived from degradation of extracellular protein taken up by macropinocytosis. Another cell line derived from a KPC mouse tumor, KRPC-A, which is capable of robust proliferation despite AA-deficiency in BSA-supplemented medium, was cultured in medium -containing fully 13C-labeled essential AAs for 5 doublings, such that intracellular protein and AAs in these cells was isotopically labeled. These cells were then switched to AA-free medium containing 50 g/L BSA. Under these conditions, cells can substitute AA monomers with scavenged extracellular protein, but because BSA contains unequal levels of AAs relative to their cellular requirements, serum protein catabolism produces some of these AAs in excess of these requirements and secretes them into the medium, providing a quantitative read-out for protein scavenging rate. Using this assay, we observe that DQ661 significantly inhibits protein scavenging in a dose-dependent fashion (Supplemental Figure 3G). Altogether, these results demonstrate that DQ661 is not only capable of blocking autophagy, but also other lysosomal catabolic programs such as macropinocytosis.
Palmitoyl-Protein Thioesterase 1 (PPT1) is a molecular target of DQ661
Given that LMP did not explain the mTOR inhibition observed with DQ661, we leveraged the increased potency and lysosomal localization of DQ661 to perform an in situ chemical pulldown experiment, in order to find the putative molecular target of DQ661 within the lysosome (Figure 4A). A photoprobe consisting of the DQ661 warhead attached to benzophenone (as a photoaffinity label), which was in turn covalently linked to desthiobiotin was synthesized (Figure 4B; see Methods for chemical strategy). Treatment of A375P cells with the DQ661 photoprobe produced accumulation of LC3II and decreased phosphorylation of mTOR substrates in A375P cells, demonstrating that the photoprobe was cell-permeable, localized to the lysosome, and retained activity (Figure 4C). Next A375P cells were treated with the DQ661 photoprobe with or without UV irradiation, and with or without competition with an excess of free DQ661 ligand immediately prior to UV irradiation. Cell lysis was followed by pulldown with neutravidin beads, and elution with biotin. Eluents were subjected to proteomic analysis (see Methods). Candidate protein targets were identified using the criteria of proteins that had >10-fold intensity ratios of +UV/-UV and –competition/+competition samples (Figure 4D). Of these 15 proteins, cathepsin D and PPT1 had the highest signal intensity for the +UV sample. Unlike cathepsin D, however, treatment with DQ661 competition completely abrogated the ability of the photolabel probe to pulldown PPT1 as detected by MS. Immunoblotting against PPT1 in a separate experiment using the DQ661-photoprobe and 10X concentration of free DQ661 ligand as competition confirmed that the DQ661-photoprobe specifically bound to, and could be UV-conjugated to PPT1 in cells (Supplemental Figure S4A). Cathepsin D enzymatic activity was not significantly impaired in cell lysates from A375P cells treated with vehicle, DQ660 and DQ661 (Figure 4E). In contrast DQ661, but not DQ660 significantly impaired PPT1 enzymatic activity in cells (Figure 4F). The inability to inhibit cathepsin D enzymatic activity demonstrates the selectivity of DQ661 for PPT1, further supporting a specific rather than generic mechanism of action for DQ661 in the lysosome. Finally, differential scanning calorimetry (DSC) of recombinant PPT1 protein in the absence or presence of a 4-fold molar excess of DQ661 revealed protein melting temperatures of 67.16 and 64.60 °C, respectively (Figure 4G). This statistically significant difference of >2.5 °C in protein melting temperature, as a function of added DQ661, is consistent with direct DQ661 binding to PPT1.
Figure 4. PPT1 is a target of DQ661.
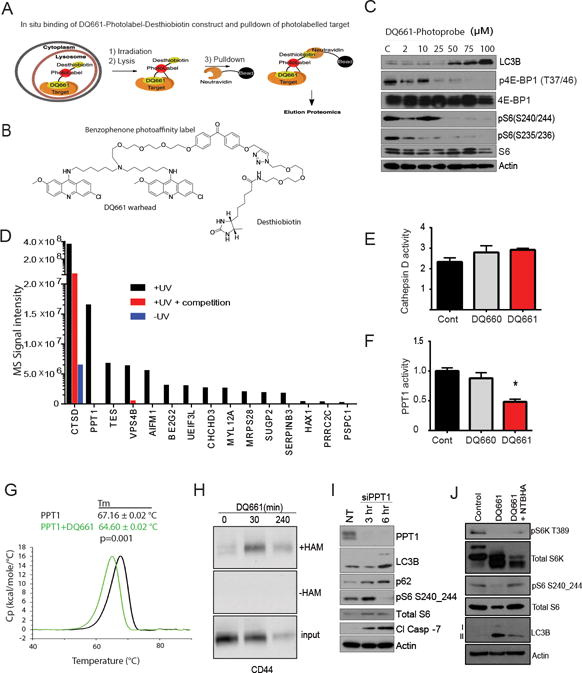
(A) Schematic demonstrating the pulldown strategy. (B) Chemical structure of the DQ661-photoprobe. (C) A375P cells were treated with the DQ661-photoprobe (24 hr, 0 – 100 μM) and lysates were immunoblotted. (D) Graph depicting the mass spectrometry analysis of lysate from pulldown with the DQ661-photoprobe. Conditions analyzed were cells that were treated with the DQ661-photoprobe ± UV and ± 10X concentration of DQ661 competition. (E) Cathepsin activity in A375P cells following treatment with DQ660 or DQ661 (3 hr, 3 μM). (F) PPT1 enzymatic activity in A375P cells following treatment with DQ660 or DQ661 (3 hr, 3 μM). (G) In vitro binding of DQ661 to PPT1. Differential Scanning Calorimetry of 1 mg/mL PPT1 (29.4 μM) in the absence (black) or presence of DQ661 (100 μM, green). Tm: melting temperature. (H) A375P cells were treated with DQ661 (0 – 240 minutes, 3 μM). CD44 palmitoylation measured using the acyl-biotinyl exchange (ABE) assay increases with DQ661 treatment compared with control. Samples not treated with hydroxylamine (-HAM) serve as a negative control. (I) A375P cells were treated with 25 nM PPT1 siRNA or non-targeting (siNT). Cells were transfected overnight in the absence of serum. Upon serum restimulation, cells were collected 3 or 6 hr thereafter and lysate was immunoblotted. (J) A375P cells were treated with DQ661 (1 hr, 3 μM) in the presence or absence of NTBHA (2 mM). Lysate was immunoblotted.
PPT1 is the only lysosomal enzyme that cleaves thioester bonds from palmitoylated proteins, liberating these proteins from membranes to facilitate proteolysis. An acyl-biotinyl exchange assay to measure protein palmitoylation demonstrated that accumulation of palmitoylated proteins occurred as early as 30 minutes and was sustained after 2 hours of treatment with DQ661 (Supplemental Figure S4B). As proof of principle, the palmitoylated form of the prototypically palmitoylated protein CD44 accumulated after 30 minutes of DQ661 treatment (Figure 4H). SiRNA knockdown of PPT1 phenocopied DQ661 effects, as evidenced by accumulation of lipidated LC3 and p62, the abrogation of S6 phosphorylation, the inhibition of cellular proliferation, the cleavage of caspase-7 and induction of cancer cell death (Figure 4I, Supplemental FigureS4C, S4D). The palmitate mimetic hexadecylsulfonyl fluoride (HDSF; Supplemental Figure S4E), a low potency tool compound that serves as an irreversible inhibitor of PPT1 (26), also concurrently inhibited mTOR and autophagy (Supplemental Figure S4F). Importantly, co-treatment of melanoma cells with DQ661 and the thioesterase mimetic N-(tert-butyl) hydroxylamine (NtBuHA) partially rescued mTORC1 signaling and autophagy levels (Figure 4J). SiRNA knockdown of PPT1 also partially blunted the ability of DQ661 to inhibit mTORC1 and autophagy (Supplemental Figure S4G). These results further demonstrate the biological activity of DQ661 is mediated through inhibition of PPT1-dependent depalmitoylation of palmitoylated proteins.
DQ661 ejects mTOR from the lysosome membrane, disrupting mTORC1-Rheb interactions
We hypothesized that PPT1 inhibition by DQ661 could lead to mTOR inhibition through an “inside out” signaling pathway connecting V-ATPase and Ragulator interactions. Recently the regulation of V-ATPase localization by PPT1 was described in neurons (27). The amino acid-dependent physical interaction between vATPase and the Ragulator complex facilitates the lysosomal recruitment of mTORC1 by Rag GTPases (28). This recruitment to the lysosome surface is critical for mTOR-Rheb interaction, allowing Rheb to phosphorylate and activate mTOR (Figure 5A). Membrane fractionation of cells treated with DQ661 revealed a significant displacement of multiple components of the vATPase from the membrane fraction to the cytoplasmic fraction (Figure 5B). The physical interaction between subunit V1A (ATP6V1A) and the Ragulator component p18 was also disrupted by DQ661 as seen by proximity ligation assay (PLA) (Figure 5C). Immunofluorescence microscopy revealed that p18 colocalization with lysosomes was not impacted by DQ661 (Figure 5D). In contrast, DQ661 treatment significantly displaced RagC (Figure 5E) and mTORC1 (Figure 5F) from the lysosomal surface. Immunoblotting of lysosome fractions taken from A375P cells treated with vehicle or DQ661 further corroborated the DQ661-induced displacement of RagC and mTOR from the lysosome and into the extralysosome fraction (Figure 5G). In addition to displacement, DQ661 also breaks the physical interaction between Rag GTPases and mTOR, as seen by PLA (Figure 5H) and immunoprecipitation of RagB in cells expressing FLAG-RagB, followed by immunoblotting for mTOR (Figure 5I). In contrast, neither rapamycin nor torin-1 could displace mTOR from the lysosome (Supplemental Figure S5A). As Rheb is the master activator of mTORC1, we next interrogated whether DQ661, existing lysosome-targeting agents or existing mTORC1 inhibitors could impact the physical interaction between mTORC1 and Rheb. Neither rapamycin nor torin-1 was able to disrupt the physical interaction of mTOR and Rheb, as seen by PLA (Figure 5J). Similarly, while neither baf nor the LMP-inducer siramesine disrupted the mTOR-Rheb interaction, which is critical for mTORC1 growth signaling, this interaction was completely disrupted by DQ661 (Figure 5J). The unique ability of DQ661 to disrupt the mTOR-Rheb interaction is therefore not shared by allosteric or catalytic inhibitors of mTORC1, nor by other prototypical lysosomally directed cationic amphiphilic drugs (CADs). Next, incubation of cells with O-propargyl-puromycin followed by click chemistry-enabled attachment of a fluorophore was used to compare the effects of mTOR and lysosomal inhibitors on de novo protein translation (29). Catalytic inhibition of mTORC1 decreased protein translation to a significantly greater degree than allosteric inhibition of mTORC1. Meanwhile, lysosomal inhibition alone with baf had no effect on protein translation. In contrast, concurrent inhibition of mTORC1 and lysosomal catabolism with DQ661 was the only approach that produced near complete inhibition of protein translation (Supplemental Figure S5B).
Figure 5. DQ661 functionally inhibits mTORC1.
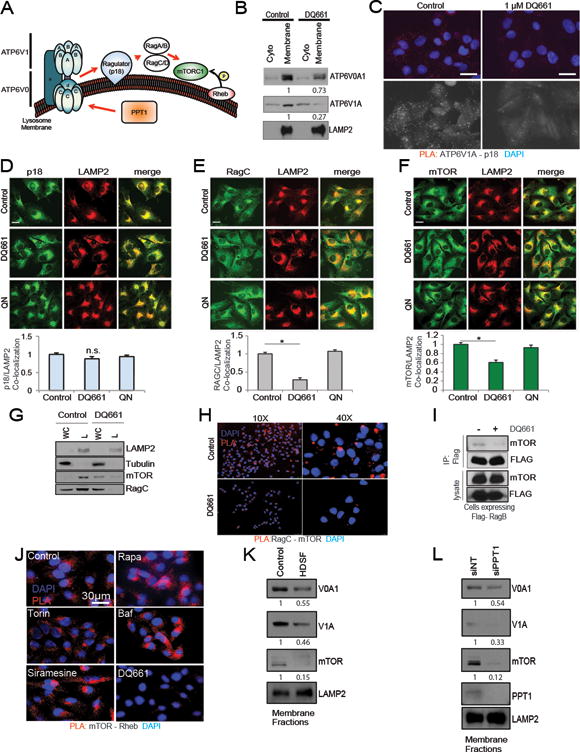
(A) Schematic depicting interactions between vATPase/Ragulator/Rag/mTORC1 and Rheb on the lysosome surface. (B) A375P cells were treated with DQ661 (6 hr, 3 μM) and membrane fractions were immunoblotted for vATPase machinery. Densitometry shown below corresponding blot. (C) PLA was performed on A375P cells treated with DQ661 (6 hr, 1 μM) for the p18 (Ragulator) – V1A physical interaction. (D) A375P cells were treated with DQ661 or QN (6 hr, 3 μM) and IF microscopy was performed to detect changes in p18/LAMP2 localization or (E) RagC/LAMP2 localization or (F) mTOR/LAMP2 localization. Below each panel is the respective co-localization analysis. Data are represented as mean ± SD. N = 50 cells per condition (ANOVA with Dunnett’s multiple comparison test). *p<0.0001 versus Untreated group. (G) A375P cells were treated with DQ661 (6 hr, 3 μM) and lysosome fractions were isolated and immunoblotted. (H) A375P cells were treated with DQ661 (3 hr, 3 μM) and PLA was performed. Blue represents DAPI and red fluorescence represents mTOR-RagC interactions. (I) HEK293T cells expressing FLAG-RagB were treated with DQ661 (4 hr, 3 μM) and immunoprecipitation lysates were probed for mTOR. (J) PLA was performed on A375P cells were treated with rapamycin (3 μM), torin-1 (3 μM), baf (100 nM), siramesine (8 μM), or DQ661 (1 μM) for 3 hr. Blue is DAPI and red fluorescence reflects mTOR-Rheb interaction. (K) A375P cells were treated with HDSF (6 hr, 40 μM) and membrane fractions were isolated and immunoblotted. (L) A375P cells were treated with NT or PPT1 siRNA for 24 hours. Membranes were subsequently fractionated and immunoblotted.
We observed striking differences in the anti-proliferative activity of catalytic/allosteric mTOR inhibitors compared to DQ661. While torin-1 displays significantly greater anti-proliferative activity relative to rapamycin, the growth impairment reaches a plateau at 1 μM whereby a residual population of cancer cells persists despite increasing dosages (Supplemental Figure S5C). In contrast, DQ661 (≥ 1 μM) treatment completely inhibited viability of cancer cells with a variety of genotypes and therapy resistance profiles (Supplemental Figure S5C). Importantly, HDSF treatment phenocopied DQ661-mediated displacement of V0A1, V1A, and mTOR from membranes (Figure 5K). Further, siPPT1 also displaced V0A1, V1A and mTOR (Figure 5L), corroborating the necessity of PPT1 for the proper lysosomal localization of vATPase components and mTOR.
In vivo lysosomal inhibition and anti-tumor activity of DQ661 in a melanoma xenograft model
To determine whether DQ661 possesses in vivo activity, 1205Lu (BRAFV600E mutant, HCQ-resistant (11)) melanoma xenografts were established on the flanks of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. After 1-2 weeks, tumors were palpable and mice were treated intraperitoneally (i.p.) with vehicle control (water), QN, DQ660 or DQ661 (8 mg/kg) (Figure 6A). QN had no significant effect on tumor growth compared to vehicle control. In contrast, DQ660 treatment resulted in a modest growth impairment of tumors compared to control or QN treated tumors. DQ661 inhibited tumor growth to a significantly greater extent than DQ660, despite differing in structure by only one methyl group. Further, DQ661 was the only compound to produce a nearly 0% growth rate (Figure 6B). Immunohistochemistry of tumor slides revealed the presence of γH2AX only in the DQ660-treated mice (Figure 6C). Electron microscopy demonstrated DQ661 alone produced visible accumulation of AVs in tumor cells (Figure 6D). At the 8 mg/kg dose, however, DQ661 caused weight loss greater than 10%, and by day 7 all mice were euthanized due to lethargy and bowel distension (Supplemental Figure S6A). Histological analysis of the intestines demonstrated Paneth cell dysfunction only in DQ661-treated mice, similar to that observed in mice treated with the highest doses of Lys05 (11) (Supplemental Figure S6B). Therefore, another 1205Lu xenograft experiment was performed in which mice were treated with vehicle control (water) or DQ661 4 mg/kg i.p. daily for a period of 14 days. Treatment with DQ661 resulted in a significant reduction in tumor volume compared to control mice without significantly affecting mouse weight (Figure 6E, Supplemental Figure S6C). Mice treated with 4 mg/kg DQ661 showed significantly slower rates of tumor growth (Figure 6F). Evidence of in vivo mTORC1 and autophagy inhibition was observed through immunoblotting of lysate from treated tumors (Figure 6G). To ensure the effects of DQ661 were not due to a predisposition of 1205Lu cells to mTOR or autophagy inhibition, genetic knockdown with siRNA against ATG5 or mTOR was performed and no significant impact on viability was observed (Supplemental Figure S6D and S6E). In vivo evidence of DQ661-induced disruption of the mTOR-Rheb interaction was demonstrated by PLA (Figure 6H).
Figure 6. DQ661 has significant single-agent in vivo activity in melanoma xenograft model.
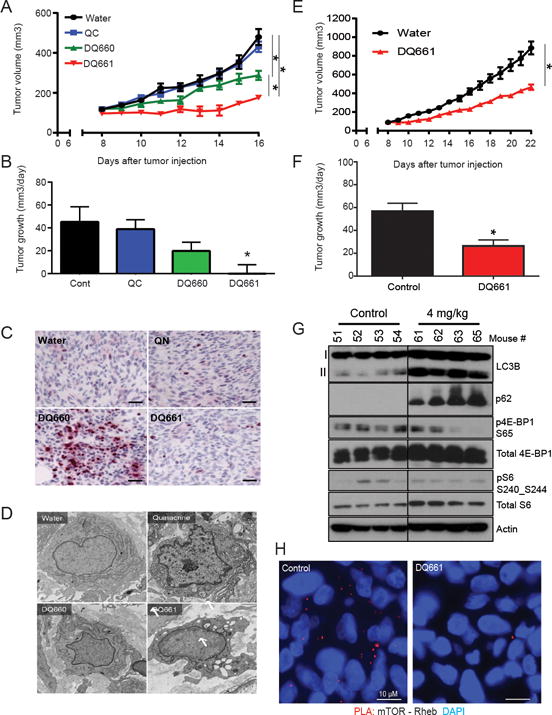
(A) 1205Lu cells were injected subcutaneously in the flanks of NSG-mice (2 × 106 cells/mouse) and grown until tumors were palpable. Mice (n=8/arm) were treated with vehicle control (water, i.p.), quinacrine (8 mg/kg, i.p.), DQ660 (8 mg/kg, i.p.) or DQ661 (8 mg/kg, i.p.). Treatments were given as shown by the black arrows, (2 days on, 2 days off). Mean +/− SEM is presented; *P<0.05. A linear mixed-effect model was used to test the difference of the tumor growth trends among treatment groups. (B) Mean +/− SD tumor growth rate. (C) Tumor tissues stained for phopsho-H2AX Ser139. Scale bar: 40 μm. (D) Representative electron micrographs of tumors harvested from mice after 2 days of treatment with each agent. Arrows: autophagic vesicles. Scale bars represent 730 nm. (E) 1205Lu cells were injected subcutaneously in the flanks of NSG-mice (2 × 106 cells/mouse) and grown until tumors were palpable (1 – 2 weeks). Mice (n=8 per arm) were treated with water (i.p.) or DQ661 (4 mg/kg, i.p.). Black arrows: treatment schedule (2 days on, 2 days off) Mean +/− SEM are presented for daily tumor volumes; *p<0.05 (F) Average tumor growth rate. (G) Protein lysate from mouse tumors at the end of the experiment was immunoblotted as indicated. (H) In vivo PLA in mice treated with vehicle control or 4 mg/kg DQ661. Red fluorescence indicates mTOR-Rheb interaction, blue represents DAPI staining of nuclei. Depicted below is quantitation of PLA signal intensity. The graph reflects mean intensity, N=200 cells were quantified from 2 mice in each arm.
DQ661 improves survival in a colon cancer model and has antitumor activity in an immunotherapy-resistant pancreatic syngeneic model
Autophagy serves a critical role in the survival of colorectal cancer cells under hypoxic conditions as well as a resistance mechanism to anti-angiogenic therapy (30). A recent phase I clinical trial of FOLFOX, bevacizumab and hydroxychloroquine in colon cancer showed significant activity (O’Hara, manuscript under revision), leading to additional clinical trials in colon cancer involving lysosomal autophagy inhibition. To determine whether DQ661 possessed activity in a colorectal mouse model, HT-29 cells were subcutaneously injected into the flanks of NSG mice. DQ661 treatment significantly decreased tumor volumes compared to control (Figure 7A). Treatment with DQ661 was well tolerated and significantly increased the survival of mice (Supplemental Figure S7A and Figure 7B). Tumor growth rate was significantly impaired with DQ661 treatment (Figure 7C).
Figure 7. DQ661 improves survival in colon cancer model and potentiates activity of gemcitabine in KPC pancreatic cancer syngeneic model.
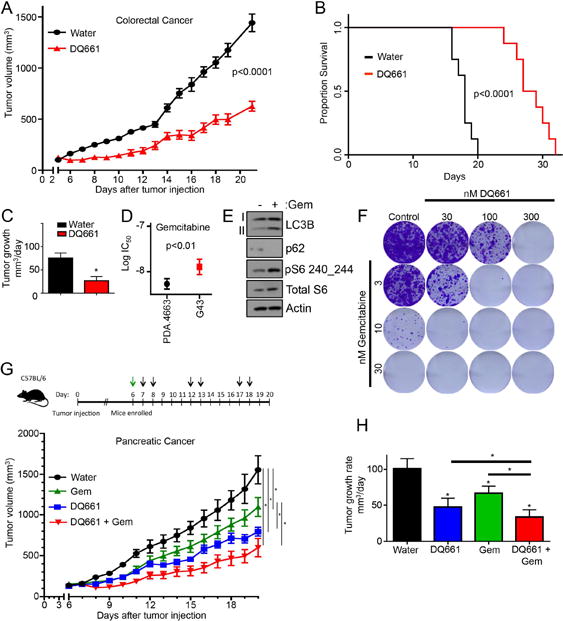
(A) HT-29 cells were injected s.c. into the flanks of NSG-mice and grown until tumor were palpable. Mice (n=8/arm) were treated with water or DQ661 (4 mg/kg) i.p. DQ661 treated mice were treated 2 days on, 2 days off. Mean +/− SEM is presented. (B) Survival curve for (A) displaying the time it took for mice to reach death (defined as time when tumor volume exceeded 1000mm3). (C) A linear mixed-effect model was used to test the difference of the tumor growth trends among treatment groups (A) Mean +/− SEM tumor growth rate. (D) MTT assay of KPC cell lines 4662 and G43 treated with gemcitabine (72 hr, 3 – 30 nM). *p<0.05. (E) G43 cells were treated with gemcitabine (24 hr, 10 nM) and lysate was immunoblotted. (F) G43 cells were treated chronically for 2 weeks with gemcitabine (3 – 30 nM) in the presence or absence of DQ661 in colony formation assays. Cells were stained with crystal violet and imaged. (G) G43 cells were injected subcutaneously into the flanks of C57BL/6 mice (2 × 106 cells/mouse. Once palpable, mice (n=8 mice per treatment arm) were treated with vehicle (PBS), gemcitabine (120 mg/kg, i.p.), DQ661 (4 mg/kg, i.p., 2 days on, 3 days off) or a combination of gemcitabine and DQ661. (H) A linear mixed-effect model was used to test the difference of the tumor growth trends among treatment groups (D) Mean +/− SEM tumor growth rate.
Pancreatic ductal adenocarcinoma (PDA) requires high levels of autophagy-lysosome function to maintain metabolism and resist therapy (12) (31). The combination of gemcitabine and HCQ has been shown to be synergistic in an orthotopic model of KPC pancreas cancer. Further, clinical translation of this finding was performed in a neoadjuvant trial of gemcitabine and HCQ in locally advanced pancreas cancer with encouraging results (32). We next determined whether DQ661 has activity against treatment-naïve, as well as immunotherapy/radiation-resistant, pancreas cancer cells. Two mouse cancer cell lines were derived from the KPC genetically engineered mouse model (GEMM): the PDA.4662 cell line (parental) and the G43 cell line. The latter was generated from a PDA.4662 tumor that grew in the face of anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA4) blockade combined with radiation therapy (25). The immunotherapy-resistant G43 cells were found to be resistant to gemcitabine, relative to the parental treatment-naïve PDA.4662 cells (Figure 7D). Gemcitabine was found to concurrently increase mTORC1 activity and autophagy as resistance mechanisms (Figure 7E). To determine whether DQ661 would potentiate the effects of gemcitabine, G43 cells were treated with gemcitabine in the presence or absence of DQ661 in long-term colony formation assays. DQ661 significantly enhanced the anti-tumor activity of gemcitabine against G43 cells (Figure 7F). To determine whether DQ661 would significantly enhance the in vivo activity of gemcitabine, G43 cells were implanted in the flanks of syngeneic mice treated with vehicle control or with a single dose of gemcitabine (120 mg/kg), combined with repeated doses of either additional vehicle or with DQ661 (4 mg/kg) (Figure 7G). Both gemcitabine alone and DQ661 alone significantly impaired tumor growth. However, compared to DQ661 alone, or gemcitabine alone, the combination of DQ661 and gemcitabine produced a significantly lower tumor growth rate and was well tolerated (Figure 7H and Supplemental Figure S7B). These results demonstrate DQ661 has activity in colorectal xenografts and immunocompetent mouse models of pancreatic cancer, and has activity in cancers that have developed resistance to immunotherapy/radiation therapy.
Discussion
Here we perform the first ever anti-cancer screen of novel dimers of antimalarial compounds. We have discovered an unprecedented correlation between chemical structure and subcellular localization. This provides a chemical strategy to more specifically target compounds to the lysosome. The potency of the optimized compound was leveraged to identify the protein target of this compound, PPT1. PPT1 inhibition results not only in autophagy inhibition, but also mTOR inhibition, through the disruption of mTOR recruitment to the lysosome under both nutrient-limiting and nutrient-replete conditions.
DQ661 was developed by systematic modification of linker length, aromatic heterocycle, and central nitrogen methylation of our previously described compound Lys05. Extending the triamine linker of Lys05 with the spermidine linker led to DC340, which demonstrated significantly increased anticancer activity. Replacing the CQ rings of DC340 with MQ, PQ or QN revealed that the strategy of dimerization significantly improved the anticancer efficacy with all antimalarial heterocycles tested. Our results show that the historically oldest antimalarial substance (QN) has the greatest anti-cancer activity on dimerization. The effectiveness of these dimeric compounds in cancer models suggests that they should be investigated in other disorders where CQ has shown efficacy, including rheumatoid arthritis (33), malaria (34), prion disease (35) and viral disorders (36).
Extension of our DQ library using longer linkers established an optimal distance between the two QN moieties of nine to fifteen atoms. Furthermore, linker length correlated directly with the ability of DQs to induce apoptosis and modulate autophagy. The limited potency of DQ221, which has the same linker length as Lys05, could be attributed to the increased steric demands of the tricyclic acridine heterocycle in quinacrine (QN) compared to the bicyclic quinoline moiety of chloroquine (CQ). QN possesses both lysosomal and extra-lysosomal functions in the cell (37). Methylation of the central nitrogen plays a critical steering role in localizing the DQs to the lysosome in a pH-dependent manner, triggering striking levels of LMP. In contrast, DQs lacking central nitrogen methylation do not induce LMP and instead produce DNA damage. Linker length correlated directly with the level of induced DNA damage, as well as with the induction of autophagic flux amongst the unmethylated DQs. With the methylated DQs, linker length directly correlated with the induction of LMP and with autophagy inhibition. The clear dependence of biological activity on linker length strongly supported a specific interaction of DQ661 with a protein target, and not with a general deacidification mechanism, as suggested for the monomeric compounds and other previously described CADs. Furthermore, the remarkable selectivity in localization as a function of nitrogen methylation cannot be attributed to a simple difference in pKa. The differences observed between the unmethylated and methylated amines could be attributed to Schiff base formation, which is available only to the unmethylated secondary amines.
The DQs are CADs, which have been reported to possess anti-cancer activity, neutralize lysosomal pH, induce lysosomal leakage, inhibit autophagy and blunt drug resistance(38). However, CADs often have multiple effects within the cell and can in some cases induce or inhibit autophagy. CADs such as amiodarone, fluoxetine, imipramine and amitriptyline all have been shown to induce autophagy and have no effect on mTOR signaling (39-42). Our study demonstrates that incorporating a specific structural motif (central nitrogen methylation of a triamine linker) in a dimeric compound can enhance lysosomal localization and increase the specificity of CADs as anti-lysosomal agents. Since the unmethylated compounds tested (DQ330, DQ440, DQ550 and DQ660) are all CADs, this was the most stringent demonstration that not all CADs share these biological properties, and that lysosomal localization and specificity with respect to mechanism of action can be optimized through chemical modification.
DQ661 is the first example of a lysosome-targeted agent that can mechanistically inhibit mTORC1 in the context of nutrient-rich conditions via disruption of mTOR. One previous study demonstrated that CADs as well as lysosome deacidifying agents possess anti-mTORC1 activity specifically due to lysosome impairment (43). The compounds interrogated in that study included siramesine, concanamycin A, bafilomycin A and ammonium chloride. Siramesine inhibits lysosome function by inhibiting acid sphingomyelinase (ASM) (22). However no connection between mTORC1 signaling and ASM inhibition was made in this previous report, and our data indicates that siramesine in nutrient-replete conditions cannot break the mTOR-Rheb interaction in the same manner as DQ661.
Deacidification of the lysosome with concanamycin A, baf and NH4Cl results in robust inhibition of mTORC1 only in the context of limited nutrients when cells are heavily reliant on lysosomal recycling for survival. Studies from the laboratories of Thompson and Sabatini have confirmed that, in the setting of AA-replete media, agents that target lysosomal acidification and/or the vacuolar ATPase fail to inhibit mTORC1 (24, 44) (28). DQ661 is the first example of a lysosomally-targeted agent that can impair mTORC1 signaling in the presence of AAs.
The vacuolar H+-adenosine triphosphatase ATPase (v-ATPase), known to be responsible for the acidification of the lysosomal lumen, also aids in the lysosomal recruitment of mTORC1 through AA-dependent physical interactions with the Ragulator complex (28). Our mechanistic studies demonstrate that the inhibition of PPT1 initiates a cascade, beginning with the lysosomal displacement of vATPase subunits, which disrupts the function of Ragulator and Rag GTPase machinery, impairing lysosomal recruitment of mTORC1, and preventing Rheb-dependent activation of mTOR. Further study is warranted to determine the roles of the mTOR recruitment machinery (regulator-Rag complex), the vacuolar ATPase, and the TFE3/TFEB family of transcriptional regulators of lysosomal biogenesis in DQ661 activity and resistance.
Targeting mTORC1, which is constitutively activated in >70% of all cancers, with small molecule inhibitors remains a challenge due to limited efficacy and the onset of resistance and toxicity (7, 45). Acquired resistance mechanisms to existing mTOR inhibitors include mTOR mutations. A third-generation mTOR inhibitor, RapaLink-1, can overcome these acquired resistance mechanisms (46). However, lysosome-dependent catabolic programs such as autophagy and macropinocytosis represent mutation-independent adaptive resistance mechanisms to mTOR inhibition (12) (24). By targeting PPT1, DQ661 concurrently inactivates mTORC1 function and overcomes these adaptive resistance mechanisms.
Finally, this work has identified PPT1 as an important new target in cancer. Germline mutations in PPT1 can result in infantile neuronal ceroid lipofuscinosis (INCL or CLN1), a fatal disease in which children develop normally in utero, but develop neuronal degeneration in early childhood. While lipofuscinosis has many of the hallmarks of defective autophagy, to date the characterization of autophagic flux and mTOR signaling in INCL cells has not been reported. The fact that PPT1 modulates protein palmitoylation, and DQ661 has now been shown to impair this enzymatic function, is important, because many of the critical regulators of autophagy and mTOR signaling are predicted to be palmitoylated proteins (47). Studies investigating the link between protein palmitoylation and lysosomally-controlled signaling are ongoing.
Methods
Biological Methods
Chemical Methods
Please see supplemental methods.
Cell culture and reagents
Melanoma cell lines A375P, 451Lu 1205Lu, C8161, WM1361A, and WM3918 were obtained from Meenhard Herlyn, Wistar Institute between 2008-2015. HT29 (ATCC HTB-38) were purchased from ATCC 2015 and authenticated by the ATCC utilizing short tandem repeat profiling. CAPAN1 and PANC-1 were obtained from Tetralogics Pharmaceuticals 2012. These cell lines are biannually tested for mycoplasma and authenticated using short tandem repeat fingerprinting. The majority of cell lines are maintained in RPMI-1640 (Invitrogen, 11875) supplemented with 10% fetal bovine serum (Sigma-Aldrich, F6178) and 25 mmol/l HEPES in the presence of 5% CO2 at 37°CThe KRPC, PDA.4662 and G43 pancreatic cancer cell lines were established from KrasLSL-G12D/+, p53LSL-R172H/+, Pdx1-Cre mice as previously described (25). PDA.4662 and G43 cells were cultured in DMEM + GlutaMAX (Gibco, Life Technologies) supplemented with 10% FBS, 1% L-glutamine and 1% gentamycin. Commercially purchased compounds: Rapamycin, Torin-1, Spautin-1, SBI-0206965, NTBHA, gemcitabine, and Bafilomycin-A1 (Sigma-Aldrich); HDSF and LLoMe (Santa Cruz); HCQ, CQ and QN (Spectrum Chemicals). Knockdown of Rheb, mTOR and ATG5 expression was performed using Cell Signaling Technology SignalSilence® Rheb siRNA I (#14267) and siRNA II (#14284), mTOR siRNA (#6381S), and ATG5 siRNA (#6345), along with Control siRNA (#6568) per manufacturers protocol.
Electron Microscopy
Electron microscopy was performed as previously described (48).
Immunoblotting, lysosome fractionation and fluorescence microscopy
Immunoblotting was performed on whole-cell lysates, lysosomal extracts as previously described (11). Cell Signaling Technology antibodies: Rheb (#13879), β-actin (#3700), 4E-BP1 (#9644), phospho-4E-BP1 S65 (#13443), S6K (#2708), phospho-S6K T389 (#9206), S6 (#2317), phospho-S6 S240_S244 (#5364), p18 (#8975), RagA (#4357), RagC (#3360), mTOR (#2983), ATG5 (#2630), Cathepsin D (#2284), Caspase-7 (#12827) and phospho-H2AX (#9718). Galectin-3. LAMP2 was purchased from Santa Cruz. LC3B antibody was generated as described previously (48). Lysosomal and extra-lysosomal fractions were purified according to the manufacturer’s instructions (Sigma #LYSISO1).
PLA
PLA (Sigma #DUO92008) was performed as previously described (48).
MTT assay, and clonogenic assay
For MTT assays, cells were plated in 96-well plates (2,000 cells/well), allowed to adhere overnight, and treated. The clonogenic assay was performed as previously described (49). Briefly, cells were suspended in appropriate culture media, and plated in 6-well plates (2 × 103 cells/well). Medium was changed with fresh drug every 3-4 days for 2 weeks. Colonies were stained with crystal violet and quantified.
RPPA and analysis methods
RPPA was conducted as previously described(50).
Macropinocytosis experiments
KPC pancreatic cancer cells plated (100,000 cells/well) upon glass cover slips and cultured in DMEM media containing 10% dialyzed FBS (Gemini Bio-Products), 0.2 mM glutamine (0.2Q) and 2% albumin (0.2Q + Alb). To measure macropinocytic uptake and lysosomal catabolism of ingested protein, cells were either fed BSA-647 (Thermo Fisher Scientific A34785) or DQ-BSA (Thermo Fisher Scientific D12051) for 3 hours. Cells were subsequently washed 4 times with cold PBS, fixed with 4% paraformaldehyde. KRPC cells were cultured for 5 doublings in DMEM containing fully 13C-labeled essential histidine, lysine, phenylalanine, threonine, tyrosine, and valine, such that intracellular protein and AAs in the resulting cells were mostly labeled. After one PBS wash, these cells were switched to AA-free medium supplemented with 5% unlabeled BSA. Excretion of unlabeled AAs into the medium was monitored after 8 hours. AAs were extracted from the medium and subjected to LC-MS analysis as described previously (51).
Fluorimetry
Small molecule fluorescent properties were observed on a Tecan M1000 fluorescent plate reader. UV-Vis spectroscopy was collected in triplicate and 420 nm was found to be the average most red shifted absorption across the library of dimeric quinacrines. Emission spectra were collected at 1 uM, which was confirmed to be in the linear range of fluorescence. Emission data was processed with a five point floating average to reduce instrument noise. The data was then normalized to the fluorescence maximum of the library, DQ661.
Measurement of drug concentration in subcellular fractions
The fluorescence emission intensity (ex. 424, em. 495) of each inhibitor, DQ660 or DQ661, was measured at multiple concentrations in PBS. From these data, a separate linear calibration curve was made for each drug. Lysosomal and whole cell fractions were diluted 1:100 in PBS, while nuclear and cytosolic fractions were diluted 1:10 in PBS. The fluorescence emission intensity (ex 424, em 495) of each of these diluted samples was measured and converted into a concentration by utilizing each inhibitors respective calibration curve, and corrected for dilution. The resulting drug concentration values of each cellular extract were then normalized for protein content and compared. Error is reported as the standard deviation of each fluorescence measurement.
Protein Translation
Cells were treated as described and nascent protein synthesis was detected utilizing IF microscopy according to the manufacturer’s instructions (Click-iT Plus OPP Protein Synthesis Assay Kit C10458).
Membrane fractionation
Membranes were fractioned out according to the manufacturers instructions (Subcellular Protein Fractionation Kit, Thermo, #78840).
Proteomics
DQ661 (DQ) pulldowns from cell lysates were separated for 0.5 cm on 10% NuPage gels (Life Technologies, NP0303), stained using colloidal Coomassie (Life Technologies, LC6025), and the entire stained region was digested with trypsin, prior to LC-MS/MS analysis using nanoACQUITY UPLC (Waters, Milford, MA) in-line with a QExactive Plus mass spectrometer (Thermo Fisher Scientific) using conditions similar to those previously described (Goldman A et al. Mol Cell Proteomics 2016). Proteins were identified using MaxQuant version 1.5.2.8 (http://www.maxquant.org) (52) with the UniProt human sequence database (August 29, 2016; 176,545 sequences; 61,529,373 residues) and an appended expected contaminants database. Protein and peptide false discovery rates were set at 1% and other critical MaxQuant parameters were as described recently (53). Proteins identified by at least two unique peptides were further analyzed using label free quantitation values to determine the ratios of protein intensities in DQ+UV relative to the two controls (DQ No UV and DQ+Competitor+UV) for each protein. If DQ+UV protein intensity was 0 the ratio was reported as N. D. (not detected) whereas if the protein intensity in a control was 0, the ratio was reported as an arbitrary value of 1,000 to avoid division by zero. Proteins showing a ratio of greater than 10-fold were reported as potential interactors.
Purification of PPT1
Full length PPT1 plasmid was obtained from Addgene (Plasmid #25205). Sf9 cells grown at a density of 3×10^6 cells per milliliter of media were infected with 10 mL/L of P2 viral stock. Cell culture medium was collected following 96 hours of incubation at 27 °C. Final cell viability was 48%. The cultured medium was centrifuged at 4000 ×g for 30 minutes. Secreted PPT1 protein was collected using ammonium sulfate precipitation at 60% saturation. Media was stirred with ammonium sulfate for 1 hour at 4 °C and precipitated protein was collected by filtration. The precipitate was resuspended in Resuspension Buffer (20 mM HEPES pH 7, 150 mM NaCl) and purified on Ni-NTA resin. Following washing (Resuspension Buffer plus 20 mM imidazole) and elution (Resuspension Buffer plus 300 mM imidazole), the protein was further purified on an s200 gel filtration column (20 mM Bis-Tris pH 6, 150 mM NaCl).
Differential Scanning Calorimetry (DSC)
Protein was concentrated to 1 mg/mL (29.4 μM) and dialyzed for 18 hours into 20 mM NaOAc pH 5, 50 mM NaCl. DSC experiments used a MicroCal VP-Capillary DSC with and without the addition of 100 μM DQ661. Scans were run from 10 °C to 90 °C using a scan rate of 60 °C/hour and a filter period of 10 seconds.
PPT1 and Cathepsin D Enzyme assays
PPT1 enzymatic activity was measured as previously described (54). Cathepsin D enzyme assay was performed according to the manufacturer’s instructions (Abcam ab65302).
Acyl-biotinyl exchange (ABE) assay
The protocol is adapted from (55). Cells were harvested in lysis buffer (50mM HEPES pH 7.4, 1% Triton X-100, 150mM NaCl, 5mM EDTA, 50mM N-ethyl-maleimide (NEM), 1μg/ml leupeptin, 1μg/ml aprotinin, 2μg/ml pepstatin A). Lysates were clarified by centrifugation at 15,000 RPM for 10 minutes. The lysate was methanol/chloroform (m/c) precipitated and the dried pellet was resuspended in 40μl 4%SDS buffer+50mM NEM and incubated at room temperature for 1 hour. The samples were m/c precipitated twice then resuspended in 80μl 4%SDS buffer. The samples were split in half and 160μl of hydroxylamine buffer (0.7M hydroxylamine pH 7.4, 50mM HEPES pH 7.4, 0.2% Triton X-100, 150mM NaCl, 5M EDTA) was added to one half of the sample and control 0.2% Triton X-100 buffer (50mM HEPES pH 7.4, 0.2% Triton X-100, 150mM NaCl, 5mM EDTA) was added to the remaining sample and incubated at room temperature for 1hour. The samples were m/c precipitated and resuspended in 40μl 4%SDS buffer containing 10μM Biotin-HPDP. 160μl of 0.2% Triton X-100 buffer +10μM Biotin-HPDP was added and incubated at RT for 1hour. The samples were m/c precipitated and resuspended in 20μl of 4% SDS buffer followed by addition of 180μl of 1% Triton X-100 buffer (20μl removed for analysis as “input”). The sample was further diluted with 600μl of 1% Triton X-100 buffer and 30μl of streptavidin agarose beads were added to the samples and incubated overnight at 4°C rotating. The samples were washed in 1% Triton-X100 buffer and analyzed by SDS-PAGE.
In vivo mouse studies
NOD-scid gamma (NSG) mice (Jackson Laboratory) were used for xenograft studies. Tumor generation, tumor measurement and tumor harvesting performed as previously described (48). For the pancreatic cancer model, 2 × 106 G43 cells were subcutaneously injected with an equal volume of matrigel (BD) over the right flank of C57BL/6 mice (Jackson Laboratory) and tumors were treated when they became palpable. Tumors were measured using electronic calipers and volume was calculated as L × W2 × 0.5. Tumor harvesting procedure and immunohistochemistry and EM were performed as previously described (48). All animal experiments were performed in accordance to the protocols approved by the Institute of Animal Care and Use Committee of the University of Pennsylvania.
Statistics
Statistical significance was determined by using Student’s unpaired, 2-tailed t test. For in vivo longitudinal growth data, linear mixed-effect models were used to test the difference of the tumor growth trends among treatment groups. A P value less than 0.05 was considered as significantly different from the null hypothesis.
Supplementary Material
Statement of Significance.
This study identifies chemical features of dimeric compounds that increase their lysosomal specificity, and a new molecular target for these compounds, reclassifying these compounds as targeted therapies. Targeting PPT1 blocks mTOR signaling in a manner distinct from catalytic inhibitors, while concurrently inhibiting autophagy, thereby providing a new strategy for cancer therapy.
Acknowledgments
We would like to thank Elise Arbeille for assistance with imaging. We would like to thank Andy J. Minn and Robert H. Vonderheide for providing the G43 cell line. We acknowledge the assistance of the Wistar Institute Proteomics and Metabolomics Core.
Funding: This work was entirely supported by NIH grants R01CA169134; P01 CA114046; P30 CA016520; SPORE P50 CA174523; 1R01CA198015; CA016672; P30CA010815.
Footnotes
Conflict of Interest Statement: RA and JW are inventors on 3 patent applications related to this work. One patent has been licensed to a biotech company to promote clinical development of Lys05 derivatives.
References
- 1.Jiang X, Overholtzer M, Thompson CB. Autophagy in cellular metabolism and cancer. J Clin Invest. 2015;125:47–54. doi: 10.1172/JCI73942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krishna S, Palm W, Lee Y, Yang W, Bandyopadhyay U, Xu H, et al. PIKfyve Regulates Vacuole Maturation and Nutrient Recovery following Engulfment. Developmental cell. 2016;38:536–47. doi: 10.1016/j.devcel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll B, Maetzel D, Maddocks OD, Otten G, Ratcliff M, Smith GR, et al. Control of TSC2-Rheb signaling axis by arginine regulates mTORC1 activity. eLife. 2016:5. doi: 10.7554/eLife.11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klempner SJ, Myers AP, Cantley LC. What a tangled web we weave: emerging resistance mechanisms to inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov. 2013;3:1345–54. doi: 10.1158/2159-8290.CD-13-0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korfel A, Schlegel U, Herrlinger U, Dreyling M, Schmidt C, von Baumgarten L, et al. Phase II Trial of Temsirolimus for Relapsed/Refractory Primary CNS Lymphoma. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.64.9897. [DOI] [PubMed] [Google Scholar]
- 9.Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. The oncologist. 2014;19:637–8. doi: 10.1634/theoncologist.2014-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–402. doi: 10.4161/auto.29119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAfee Q, Zhang Z, Samanta A, Levi SM, Ma XH, Piao S, et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc Natl Acad Sci U S A. 2012;109:8253–8. doi: 10.1073/pnas.1118193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–5. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strohecker AM, White E. Targeting mitochondrial metabolism by inhibiting autophagy in BRAF-driven cancers. Cancer Discov. 2014;4:766–72. doi: 10.1158/2159-8290.CD-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–34. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59:285–97. doi: 10.1016/j.molcel.2015.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jennings BR, Ridler PJ. Interaction of chromosomal stains with DNA. An electrofluorescence study. Biophysics of structure and mechanism. 1983;10:71–9. doi: 10.1007/BF00535543. [DOI] [PubMed] [Google Scholar]
- 17.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke JJ, Sokal DC, Cancel AM, Campen DB, Gudi R, Wagner VO, et al. Re-evaluation of the mutagenic potential of quinacrine dihydrochloride dihydrate. Mutation research. 2001;494:41–53. doi: 10.1016/s1383-5718(01)00178-4. [DOI] [PubMed] [Google Scholar]
- 21.Aits S, Kricker J, Liu B, Ellegaard AM, Hamalisto S, Tvingsholm S, et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11:1408–24. doi: 10.1080/15548627.2015.1063871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen NH, Olsen OD, Groth-Pedersen L, Ellegaard AM, Bilgin M, Redmer S, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–93. doi: 10.1016/j.ccr.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell. 2015;162:259–70. doi: 10.1016/j.cell.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–7. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Potts MB, McMillan EA, Rosales TI, Kim HS, Ou YH, Toombs JE, et al. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nature chemical biology. 2015;11:401–8. doi: 10.1038/nchembio.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, et al. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nature communications. 2017;8:14612. doi: 10.1038/ncomms14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slomnicki LP, Malinowska A, Kistowski M, Palusinski A, Zheng JJ, Sepp M, et al. Nucleolar Enrichment of Brain Proteins with Critical Roles in Human Neurodevelopment. Molecular & cellular proteomics : MCP. 2016;15:2055–75. doi: 10.1074/mcp.M115.051920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy inhibition sensitizes colon cancer cells to antiangiogenic and cytotoxic therapy. Clin Cancer Res. 2013;19:2995–3007. doi: 10.1158/1078-0432.CCR-12-1542. [DOI] [PubMed] [Google Scholar]
- 31.Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014;4:905–13. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann Surg Oncol. 2015;22:4402–10. doi: 10.1245/s10434-015-4566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keyszer G, Keysser C, Keysser M. Efficacy and safety of a combination therapy of methotrexate, chloroquine and cyclophosphamide in patients with refractory rheumatoid arthritis: results of an observational study with matched-pair analysis. Clinical rheumatology. 1999;18:145–51. doi: 10.1007/s100670050073. [DOI] [PubMed] [Google Scholar]
- 34.Wangchuk S, Drukpa T, Penjor K, Peldon T, Dorjey Y, Dorji K, et al. Where chloroquine still works: the genetic make-up and susceptibility of Plasmodium vivax to chloroquine plus primaquine in Bhutan. Malaria journal. 2016;15:277. doi: 10.1186/s12936-016-1320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barret A, Tagliavini F, Forloni G, Bate C, Salmona M, Colombo L, et al. Evaluation of quinacrine treatment for prion diseases. Journal of virology. 2003;77:8462–9. doi: 10.1128/JVI.77.15.8462-8469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bishop BM. Potential and emerging treatment options for Ebola virus disease. The Annals of pharmacotherapy. 2015;49:196–206. doi: 10.1177/1060028014561227. [DOI] [PubMed] [Google Scholar]
- 37.Salas E, Roy S, Marsh T, Rubin B, Debnath J. Oxidative pentose phosphate pathway inhibition is a key determinant of antimalarial induced cancer cell death. Oncogene. 2016;35:2913–22. doi: 10.1038/onc.2015.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellegaard AM, Dehlendorff C, Vind AC, Anand A, Cederkvist L, Petersen NH, et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine. 2016;9:130–9. doi: 10.1016/j.ebiom.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin CW, Chen YS, Lin CC, Chen YJ, Lo GH, Lee PH, et al. Amiodarone as an autophagy promoter reduces liver injury and enhances liver regeneration and survival in mice after partial hepatectomy. Sci Rep. 2015;5:15807. doi: 10.1038/srep15807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowie M, Pilie P, Wulfkuhle J, Lem S, Hoffman A, Desai S, et al. Fluoxetine induces cytotoxic endoplasmic reticulum stress and autophagy in triple negative breast cancer. World J Clin Oncol. 2015;6:299–311. doi: 10.5306/wjco.v6.i6.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shchors K, Massaras A, Hanahan D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell. 2015;28:456–71. doi: 10.1016/j.ccell.2015.08.012. [DOI] [PubMed] [Google Scholar]
- 42.Villanueva-Paz M, Cordero MD, Pavon AD, Vega BC, Cotan D, De la Mata M, et al. Amitriptyline induces mitophagy that precedes apoptosis in human HepG2 cells. Genes Cancer. 2016;7:260–77. doi: 10.18632/genesandcancer.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostenfeld MS, Hoyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy. 2008;4:487–99. doi: 10.4161/auto.5774. [DOI] [PubMed] [Google Scholar]
- 44.Cheong H, Lindsten T, Wu J, Lu C, Thompson CB. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108:11121–6. doi: 10.1073/pnas.1107969108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, Versus Everolimus in Patients With Metastatic Renal Cell Carcinoma. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.64.8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534:272–6. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanders SS, Martin DD, Butland SL, Lavallee-Adam M, Calzolari D, Kay C, et al. Curation of the Mammalian Palmitoylome Indicates a Pivotal Role for Palmitoylation in Diseases and Disorders of the Nervous System and Cancers. PLoS Comput Biol. 2015;11:e1004405. doi: 10.1371/journal.pcbi.1004405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma XH, Piao SF, Dey S, McAfee Q, Karakousis G, Villanueva J, et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. The Journal of clinical investigation. 2014 doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebecca VW, Massaro RR, Fedorenko IV, Sondak VK, Anderson AR, Kim E, et al. Inhibition of autophagy enhances the effects of the AKT inhibitor MK-2206 when combined with paclitaxel and carboplatin in BRAF wild-type melanoma. Pigment cell & melanoma research. 2014 doi: 10.1111/pcmr.12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016;126:1834–56. doi: 10.1172/JCI82661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer research. 2015;75:544–53. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 53.Goldman AR, Bitler BG, Schug Z, Conejo-Garcia JR, Zhang R, Speicher DW. The Primary Effect on the Proteome of ARID1A-mutated Ovarian Clear Cell Carcinoma is Downregulation of the Mevalonate Pathway at the Post-transcriptional Level. Mol Cell Proteomics. 2016;15:3348–60. doi: 10.1074/mcp.M116.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Diggelen OP, Keulemans JL, Winchester B, Hofman IL, Vanhanen SL, Santavuori P, et al. A rapid fluorogenic palmitoyl-protein thioesterase assay: pre- and postnatal diagnosis of INCL. Mol Genet Metab. 1999;66:240–4. doi: 10.1006/mgme.1999.2809. [DOI] [PubMed] [Google Scholar]
- 55.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–84. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.