

We demonstrate mechanochemical regulation of platelet adhesion to von Willebrand factor in thrombosis and hemostasis.
Abstract
Force-dependent binding of platelet glycoprotein Ib (GPIb) receptors to plasma von Willebrand factor (VWF) plays a key role in hemostasis and thrombosis. Previous studies have suggested that VWF activation requires force-induced exposure of the GPIb binding site in the A1 domain that is autoinhibited by the neighboring A2 domain. However, the biochemical basis of this “mechanopresentation” remains elusive. From a combination of protein chemical, biophysical, and functional studies, we find that the autoinhibition is controlled by the redox state of an unusual disulfide bond near the carboxyl terminus of the A2 domain that links adjacent cysteine residues to form an eight-membered ring. Only when the bond is cleaved does the A2 domain bind to the A1 domain and block platelet GPIb binding. Molecular dynamics simulations indicate that cleavage of the disulfide bond modifies the structure and molecular stresses of the A2 domain in a long-range allosteric manner, which provides a structural explanation for redox control of the autoinhibition. Significantly, the A2 disulfide bond is cleaved in ~75% of VWF subunits in healthy human donor plasma but in just ~25% of plasma VWF subunits from heart failure patients who have received extracorporeal membrane oxygenation support. This suggests that the majority of plasma VWF binding sites for platelet GPIb are autoinhibited in healthy donors but are mostly available in heart failure patients. These findings demonstrate that a disulfide bond switch regulates mechanopresentation of VWF.
INTRODUCTION
Von Willebrand factor (VWF) is a plasma protein produced by vascular endothelial cells and megakaryocytes that chaperones blood coagulation cofactor factor VIII and tethers platelets to the injured blood vessel wall (1). It is a large glycoprotein that circulates as a series of multimers containing variable numbers of 500-kDa dimeric units. Pro-VWF dimers are assembled in the endoplasmic reticulum via disulfide bridges between cysteine residues located in the C-terminal domains of the 250-kDa monomers. These tail-to-tail linked homodimers are subsequently variably multimerized within the Golgi apparatus by formation of head-to-head disulfide bonds near the N termini.
The multimers can range in size from 500 to 20,000 kDa, and the largest multimers are more effective at capturing platelets in the shear forces of flowing blood. This is due to the polyvalent nature of the protein because each monomer contains binding sites for collagen and for platelet glycoprotein Ib (GPIb) and integrin αIIbβ3 (1). Platelet adhesion and thrombus formation are strictly dependent on VWF interaction with platelet GPIb at the high shear rates found in arterioles and stenosed vessels (2, 3). The binding site in VWF for GPIb is the A1 domain.
Hemodynamic drag forces stretch immobilized VWF that unfolds the molecule and exposes the A1 domain to platelet GPIb (4–6); this has been referred to as mechanopresentation. How VWF harnesses mechanical force to delicately regulate its activation to ensure a perfect balance between its hemostatic and thrombotic functions remains a biologically significant but elusive question. Plasma VWF has a limited binding potential for platelet GPIb in the circulation due to autoinhibitory mechanisms that mask the A1 binding site. The D′D3 domain (7), the N-terminal flanking region of A1 (residues 1238 to 1260) (8–10), and the A2 domain (11–13) have been implicated in autoinhibition.
There are three VWF A domains (A1, A2, and A3) about 200 amino acids in size, and each contains a pair of cysteine residues that form a disulfide bond in crystal structures (Fig. 1A). The disulfide bond in A1 and A3 links cysteines located at the N- and C-terminal ends of the domain. In contrast, the A2 domain disulfide links adjacent cysteines (Cys1669 and Cys1670) at the C terminus, forming an eight-membered ring that provides rigidity to the domain (14, 15). This disulfide bond is not found in any other VWA domains and has been suggested to act like a plug that, when dislodged by sufficient shear force, destabilizes the core and initiates unfolding of the domain (14).
Fig. 1. The reduced A2 domain binds to the A1 domain and masks interaction with platelet GPIb.
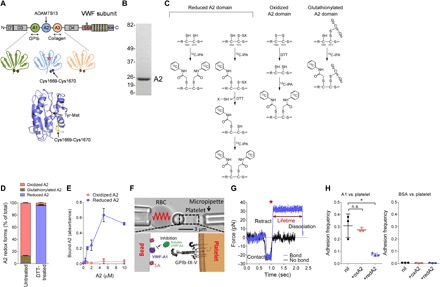
(A) The A1 domain binds the GPIb receptor on platelets, the A2 domain Tyr1605-Met1606 peptide bond is cleaved by ADAMTS13, and the A3 domain binds collagen exposed during vascular damage. The oxidized A2 crystal structure has a Protein Data Bank (PDB) identifier 3GXB. (B) Recombinant A2 (residues M1473 to L1675) produced in Escherichia coli and resolved on SDS–polyacrylamide gel electrophoresis (PAGE). The positions of molecular weight markers are indicated on the left. (C) Differential cysteine alkylation and mass spectrometry method of measuring the redox state of the A2 domain cysteines. Unpaired cysteine thiols in the A2 domain are alkylated with 12C-IPA, and the disulfide bonded cysteine thiols with 13C-IPA after reduction with DTT. Alternatively, unpaired cysteine thiols in the A2 domain were alkylated with 13C-IPA, the protein was digested with Glu-C, and peptides were analyzed by tandem mass spectrometry. A search for peptides labeled with 13C-IPA at Cys1669 and an unknown adduct at Cys1670 was undertaken. The only adduct detected was S-glutathionylation. (D) The recombinant A2 domain exists in oxidized (87%), glutathionylated (11%), and reduced (2%) forms. Treatment of the domain with DTT results in 96% reduced and 4% oxidized protein. (E) Reduced but not oxidized A2 domains bind to the A1 domain immobilized on plastic. A2 binding was corrected for nonspecific binding to bovine serum albumin (BSA)–blocked wells. (F) Biomembrane force probe (BFP) protein functionalization. The probe bead (bottom left) was coated with the A1 domain and streptavidin (SA) for attachment of the bead to a biotinylated red blood cell (RBC). A1 is the focus for interaction with the GPIb on an aspired platelet (bottom right). The soluble A2 domain competes for this interaction. (G) Force-time traces from two representative test cycles. A target was driven to approach a probe, contacted, and retracted. In a no-bond event (black), the cycle ended after the probe-target separation. In a bond event (blue), the target was held (marked by *) at a preset force until dissociation. Lifetime is measured from the point when the clamped force (30 pN) was reached to the point when the bond dissociated, signified by a force drop to zero. (H) The reduced but not oxidized A2 domain competes for binding of platelet GPIb to the A1 domain (left). There were very few interactions of platelets with BSA-coated beads in the absence or presence of A2 (right). The adhesion frequencies (mean ± SEM) were measured on BFP from three independent experiments (n = 3 donors for human platelets) in the absence or presence of the 5 μM A2 domain. For each experiment, three (BSA-coated beads) or five (A1-coated beads) random bead-platelet pairs with 50 touches were analyzed and averaged. *P < 0.05, unpaired, two-tailed Student’s t test. n.s., not significant.
Our studies reveal that A2 autoinhibition of the A1 function is controlled by the redox state of the A2 disulfide bond. Only when the bond is cleaved does A2 bind to the A1 domain and block platelet GPIb binding. Analysis of the redox state of the bond in healthy human donors and those receiving mechanical circulatory assist for heart failure indicates that the A2 disulfide is dynamically regulated in vivo.
RESULTS
The redox state of A2 domain Cys1669 and Cys1670 thiols determines affinity of A2 for the A1 domain
The binding of the A2 domain to A1 was investigated using purified recombinant domains and an enzyme-linked immunosorbent assay method. The recombinant A2 domain consisting of residues M1473 to L1675 was produced (Fig. 1B). No high-affinity binding of the native A2 domain to A1 was observed (Fig. 1E). This is in contrast to an earlier report of an A2-A1 interaction with a dissociation constant in the low micromolar range (11). The previous study used a truncated A2 domain (G1481-R1668) missing the Cys1669-Cys1670 disulfide bond that is unique to VWF type A domains. Because the A2 disulfide has been proposed to stabilize the hydrophobic core of the domain (14), its absence was predicted to influence structure and perhaps function. We reasoned that the truncated domain would resemble a full-length domain where the disulfide bond is cleaved. This hypothesis was tested by examining the interaction of the A1 domain with the oxidized and reduced A2 domains.
It was critical for this study to accurately quantify the redox state of the A2 disulfide bond in recombinant A2 fragment and native plasma VWF. This was achieved using differential cysteine alkylation and mass spectrometry (Fig. 1C) (16, 17). Reduced Cys1669-Cys1670 disulfide bond cysteines were alkylated with 2-iodo-N-phenylacetamide (12C-IPA), and oxidized disulfide bond cysteines were alkylated with a stable carbon-13 isotope of IPA (13C-IPA) after reduction with dithiothreitol (DTT). Two peptides encompassing the Cys1669-Cys1670 disulfide bond cysteines (LVLQRCCSGE and TLPREAPDLVLQRCCSGE) were resolved by mass spectrometry and quantified (fig. S1). Three redox forms of the A2 domain cysteines were identified: Cys1669 and Cys1670 in reduced dithiol form, Cys1669 as a free thiol and Cys1670 as a mixed disulfide with an unknown compound, and Cys1669 and Cys1670 forming a disulfide. The extent of alkylation of the A2 cysteines was effectively complete. No peptides containing Cys1669 and Cys1670 not labeled with either 12C-IPA or 13C-IPA were detected. To determine the nature of the small thiol conjugated to Cys1670, we alkylated the unpaired Cys1669 with 13C-IPA and digested the protein with Glu-C. The two peptides containing Cys1669 and Cys1670 were resolved and analyzed for conjugates attached to Cys1670. The only adduct detected was S-glutathionylation (fig. S1D). There were no detectable conjugates with nitric oxide or other small-molecule thiols present in plasma, including cysteine, cysteinylglycine, or homocysteine.
The recombinant A2 domain was a mix of three redox forms: the cysteine pair exist in the oxidized form in 87%, they exist as unpaired thiols in 2%, and Cys1669 is an unpaired thiol and Cys1670 is glutathionylated in 11%. Treatment with DTT resulted in a 96% reduced and 4% oxidized A2 domain (Fig. 1D). The reduced A2 domain bound the immobilized A1 domain with a dissociation constant of 2 to 4 μM, whereas the untreated, predominantly oxidized domain bound with >50-fold lower affinity (Fig. 1E).
To date, investigating VWF mechanopresentation with live platelets at a single-molecule scale remains a challenge, largely because of the lack of suitable experimental techniques to investigate molecular behaviors at high spatial and temporal resolution on the live-cell surface and partly because of the small size of the platelets and their propensity to be rapidly activated. Our recently developed BFP technology has enabled us to manipulate single platelets and characterize in situ A1-GPIb kinetics at a single-bond level (9, 18). To examine the functional effect of redox states of the A2 disulfide on the A1–platelet GPIb interaction, the BFP was functionalized with VWF-A1 on the bead (Fig. 1F) and brought into repetitive touches with an aspirated platelet with controlled contact duration. If a bond connects the bead and platelet during their separation, a tensile clamped force would result and be reported by RBC-bead deflection (Fig. 1G, blue); otherwise, no deflection is observed (Fig. 1G, black). The intermittent “touch and retract” cycles mimic VWF-GPIb–mediated platelet translocation behavior under shear (9, 19). The adhesion frequencies [the number of productive touches (Fig. 1G, blue) divided by the number of total touches] reflect the likelihood of finding a receptor-ligand bond or bonds at the time of detection. To assess competition of A2 for the binding of platelet GPIb to A1, we measured the A1–platelet GPIb adhesion frequencies in the absence or presence of A2 in solution (Fig. 1F). The reduced but not the oxidized A2 domain competitively inhibited the binding of platelet GPIb to the A1 domain (Fig. 1H). These findings indicate that the reduced (but not the oxidized) A2 domain binds A1 and competes for the binding of platelet GPIb to A1.
Oxidized A2 domain VWF is much more effective than reduced A2 domain VWF at engaging platelet GPIb under fluid shear conditions
VWF captures platelets from flowing blood to an injured vessel wall. To test the influence of the redox state of the A2 domain disulfide on this critical function of VWF, full-length recombinant wild-type or disulfide bond mutant VWF was expressed in mammalian cells and examined for platelet binding as a function of fluid shear stress. The A2 disulfide was ablated by replacing Cys1669 or both Cys1669 and Cys1670 with Ala. These mutants represent reduced VWF, were expressed and secreted from human embryonic kidney (HEK) 293 cells at comparable levels to wild-type protein, and have a comparable multimer size (Fig. 2A). Elimination of the disulfide by replacing both Cys with Gly also has no effect on expression and secretion of VWF from HEK293 cells (20). These results indicate that elimination of the A2 disulfide bond does not appreciably affect maturation of VWF.
Fig. 2. Oxidized VWF is more effective than reduced VWF at engaging platelet GPIb under fluid shear conditions.
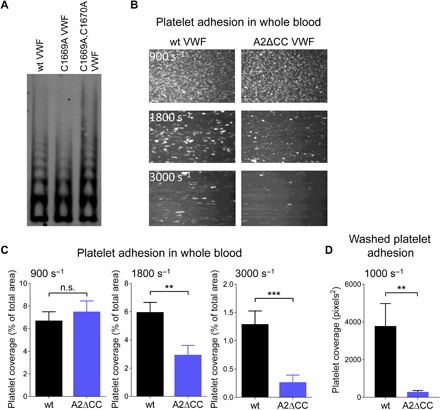
(A) HEK293 cells were transfected with wild-type (wt), C1669A, or C1669A,C1670A mutant VWF plasmids. Conditioned medium was collected after 3 days, and equal weights of protein were resolved by agarose gel electrophoresis and blotted with anti-VWF polyclonal antibodies. (B) Representative images of platelet adhesion on channels coated with wt or double Cys mutant (A2ΔCC) VWF at fluid shear rates of 900, 1800, and 3000 s−1. (C) Surface coverage of whole-blood platelets on channels coated with wt or A2ΔCC VWF at fluid shear rates of 900, 1800, and 3000 s−1. Bars and errors are mean ± SEM of three technical replicates of platelets from four healthy donors (n = 12 measurements). **P < 0.01 and ***P < 0.001. (D) Surface coverage of washed platelets on channels coated with wt or A2ΔCC VWF at a fluid shear rate of 1000 s−1. Bars and errors are mean ± SEM of three technical replicates of platelets from three healthy donors (n = 9 measurements). **P < 0.01.
Platelet adhesion to immobilized wild-type and double Cys mutant (A2ΔCC) VWF was measured in human whole blood at shear rates of 900, 1800 and 3000 s−1, which is in the range found in arterioles and stenosed vessels (Fig. 2, B and C) (2). Platelet adhesion and thrombus formation are dependent on VWF interaction with platelet GPIb at these shear rates (3, 21). At high fluid shear rates, platelet coverage on A2ΔCC VWF was significantly reduced compared to wild-type protein. VWF multimer size is reduced in the circulation by ADAMTS13 cleavage of the A2 domain Tyr1605-Met1606 peptide bond (22, 23). The proteolysis reduces the polyvalency of VWF and, therefore, avidity for platelets. This possible confounder in whole blood was tested by using washed platelets in the adhesion assay that depletes plasma ADAMTS13. A lower shear rate (1000 s−1) also prevents RBC compression and diminishes platelet activation (9). Consistent with the whole-blood assay, adhesion of washed platelets to A2ΔCC VWF was significantly reduced compared to wild-type protein (Fig. 2D).
Interaction of reduced and glutathionylated VWF with GPIb is characterized by low adhesion frequency and short bond lifetimes
The biophysical reasons for the impaired binding of platelets to reduced VWF were determined using single-molecule force spectroscopy. We functionalized the BFP with wild-type and A2ΔCC VWF and measured their direct interactions with platelet GPIb (Fig. 3A). As expected, the frequency of adhesion of platelets to A2ΔCC VWF was significantly lower than that to the wild-type protein (Fig. 3B). To investigate the force effect, we measured VWF–platelet GPIb bond lifetimes at multiple clamped forces. Both wild-type and A2ΔCC VWF displayed catch-bond behaviors in which force first decelerates and then accelerates bond dissociation. Consistent with our previous studies, this binding behavior plays an important role in VWF mechanopresentation (9, 19, 24, 25). However, from 10 to 60 pN, the bond lifetimes of A2ΔCC VWF were globally suppressed compared to those of wild-type VWF (Fig. 3C). These findings indicate that reduced VWF has a significantly lower affinity for platelet GPIb, which is in accordance with decreased platelet adhesion to A2ΔCC VWF under fluid shear conditions (Fig. 2, B to D).
Fig. 3. Interaction of reduced and glutathionylated VWF with GPIb is characterized by low collision frequency and short bond lifetimes.
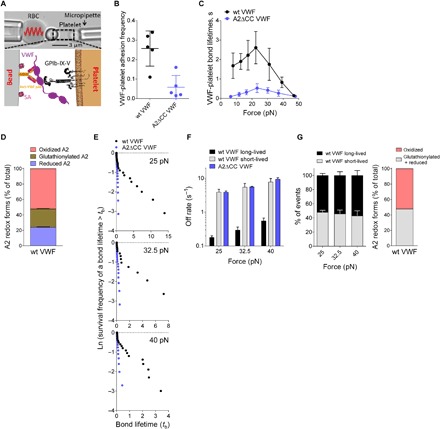
(A) BFP bright-field scheme (top) and protein functionalization (bottom). A micropipette-aspirated RBC with a probe bead attached to the apex (top left) was aligned against a human platelet held by an apposing micropipette (top right). The probe bead was covalently linked to a polyclonal antibody (pAb) for the capture of VWF and SA for the attachment of the bead to a biotinylated RBC (bottom left). VWF is the focus for interaction with the GPIb on an aspirated platelet (bottom right). (B) Adhesion frequencies of five bead-platelet pairs with 50 touches for each pair. Errors are mean ± SEM. (C) Lifetime of VWF-platelet GPIb bonds versus clamp force in BFP. Results represent mean ± SEM of >50 measurements per point. (D) Recombinant wt VWF exists in oxidized (52%), glutathionylated (24%), and reduced (24%) forms. (E) Lifetime distributions of VWF-platelet bonds measured by BFP were compared at three representative forces: 25, 32.5, and 40 pN. (F and G) A two-state single-bond dissociation model was fit to the wt VWF bond lifetime distribution to evaluate model parameters at the indicated force bin. The kinetic parameters are the slow (k1) and fast (k2) off rates of dissociation from the two dissociation states (G) and the fractions (w1, w2; w1+ w2 = 1) of bonds associated with the corresponding short-lived (high off rate) and long-lived (low off rate) states. The error bars represent ±95% confidence interval (CI) of the best-fit value. The fraction of oxidized (52%) and glutathionylated plus reduced (48%) forms of wt VWF corresponding to (E) is shown on the right.
Furthermore, mass spectrometry studies revealed that recombinant full-length VWF A2 domains exist in oxidized (52%), glutathionylated (24%), and reduced (24%) forms (Fig. 3D). To elucidate the relative contributions of the three redox forms in VWF-GPIb interactions, we analyzed the lifetime distributions of VWF-GPIb bonds. As expected, bond lifetimes of A2ΔCC VWF were distributed as single exponentials at all forces owing to the single redox state of VWF (Fig. 3E; straight lines in exponential decay plots). Lifetimes for wild-type VWF were distributed as two well-separated exponentials (Fig. 3E; folded lines in exponential decay plots) indicating two VWF bond species with distinct off rates. The off rate of the long-lived bond species (k1) was an order of magnitude smaller than that of the short-lived bond species (k2) at the three different forces (Fig. 3F). Notably, the off rate of the short-lived wild-type VWF bond matches that of the A2ΔCC VWF bond. The fractions of the long-lived (w1) and short-lived (w2) wild-type VWF bonds were 55 and 45% (Fig. 3G), respectively, which correlate with the fraction of oxidized (52%) and glutathionylated plus reduced (48%) forms of wild-type VWF (Fig. 3G). Together, these results suggest that glutathionylated and reduced VWF forms have the same low affinity for platelet GPIb as A2ΔCC VWF.
Reducing the vicinal disulfide bond in A2 has long-range allosteric effects
The structural reasons for the impaired binding of platelets to reduced VWF were investigated using molecular dynamics (MD). MD simulations of the oxidized and reduced A2 domains were performed. The oxidized and reduced A2 domains displayed highly divergent global collective motions (Fig. 4, A to C, and fig. S2). As seen in the projection onto the first two major collective modes of motion, obtained from PCA, the oxidized A2 domain (green) adopted structures similar to the starting structure (black), whereas the reduced domain (orange) substantially changed its conformation and stabilized around a structure dissimilar to the starting structure (Fig. 4B). In particular, loop L4, helix H5, loop L3-4, and the C terminus displayed the largest conformational changes upon reduction (Fig. 4C).
Fig. 4. The oxidized and reduced forms of the A2 domain have different dynamics and stresses.
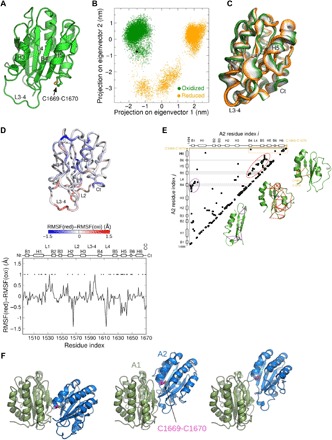
(A) Overall structure of the VWF A2 domain (from the x-ray structure, PDB identifier 3GXB) used as starting conformation for the MD simulations. The structure is shown in cartoon representation, highlighting the Cys1669-Cys1670 disulfide bond in yellow. (B) Dynamics of the oxidized (green) and reduced (orange) VWF A2 domain projected onto the first two eigenvectors (main modes of motion) obtained from principal components analysis (PCA). Each dot represents a conformation observed in the MD simulations. The black dot corresponds to the starting structure for both simulations. (C) Interpolation of the structures along the first PCA eigenvector (main mode of motion), ranging from the conformations sampled in the oxidized state (green) to the reduced state (orange) (going from the extreme left to the extreme right). Ct, C-terminal. (D) Per-residue difference of the root mean square fluctuations (RMSFs) between the reduced (red) and oxidized (oxi) forms of VWF A2. Statistically significant differences are highlighted with the * symbol. The secondary structure of the protein is presented on top. Nt, N-terminal. (E) Force distribution analysis (FDA). (Left) Residue pairs (i,j) with the time-averaged pairwise force of the reduced state minus that of the oxidized state larger than a cutoff value of 90 pN, |〈Fij(red)〉 − 〈Fij(oxi)〉| > 90 pN. Here, | | indicates absolute values. For the dependence on the cutoff value, see fig. S2. Secondary-structure elements of A2 are shown on both axes. To guide the eye, the regions corresponding to the β strands B4, B5, and B6 are shown in gray and the region corresponding to the Cys1669-Cys1670 bond is shown in orange. (Right) The three groups of residue pairs enclosed by circles, showing interactions between the β strands and their surrounding helices, loops, or the C terminus, are explicitly shown as lines connecting points in the A2 structure. (F) The three most favorable structural models of the VWF A1A2 complex predicted by molecular docking and MD simulations (13). A1 (green) and A2 (blue) domains are shown in cartoon representation. The Cys1669-Cys1670 bond (magenta and yellow) is shown in ball-stick representation.
Further local dynamical changes upon reduction of the A2 domain were quantified by the difference in their RMSFs, RMSF(reduced) − RMSF(oxidized), a quantity obtained by considering all positional fluctuations (Fig. 4D). The RMSF obtained from the simulations shows trends similar to the crystallographic B factors (fig. S3). The C-terminal region close to the Cys1669-Cys1670 disulfide bond was found less flexible in the reduced state than in the oxidized state (blue). The differences in RMSF were not only concentrated in this region but also spread over the entire A2 domain. The helix H5 motif with high conformational displacement upon reduction (Fig. 4C) also presented statistically significant less positional fluctuations in the reduced state than in the oxidized state (Fig. 4D), opposite to loop L1, which was found to be significantly more mobile in the reduced form. L2, L3-4, and L4 presented large but not statistically significant fluctuation differences.
Contrary to the helical elements and the loops, the β strands (in particular B4 where the peptide bond cleaved by ADAMTS13 is located) did not present appreciable dynamical changes depending on the redox state of the Cys1669-Cys1670 bond. However, an FDA is more sensitive in revealing allosteric communication through protein cores (26) and was used here to reveal residues drastically changing their time-averaged pairwise force in the reduced state compared to the oxidized state (Fig. 4E). Many of these pairs involved the β strands, thus implying that the internal stress between the β strands and their surrounding elements is altered upon reduction. Together, these results indicate that a change in redox state of the VWF A2 domain leads to subtle yet significant allosteric changes in dynamics and stress throughout the domain. The increased mobility of the reduced and glutathionylated A2 domain presumably underlies its ability to bind the adjacent A1 domain. Notably, docking studies of the A1 and A2 domains feature a domain-domain interface with the A2 disulfide in close proximity (Fig. 4F) (13).
The A2 domain in healthy donor VWF exists predominantly in a reduced dithiol and glutathionylated form
The following are two key questions regarding the biological significance of autoinhibitory control of VWF: What is the redox state of VWF in health? Does this change in disease? We first measured the redox state of the A2 disulfide in plasma VWF from healthy donors. In a cohort of 22 healthy donors, the VWF A2 domain cysteine pair exists as unpaired thiols in ~40% (95% CI, 37 to 42%), Cys1669 exists as an unpaired thiol and Cys1670 is glutathionylated in ~36% (95% CI, 33 to 37%), and the pair exists in an oxidized disulfide form in ~24% (95% CI, 23 to 28%) (Fig. 5A). The coefficient of variation for these VWF redox forms is 15.4, 12.5, and 21.7%, respectively. These results indicate that most plasma VWF binding sites for platelets are autoinhibited and that the relative proportions of the isoforms do not vary widely in healthy donors. The posttranslational modifications of the A2 domain cysteines are summarized in Fig. 5B.
Fig. 5. The VWF A2 domain exists predominantly in a reduced dithiol and glutathionylated form in healthy donors.
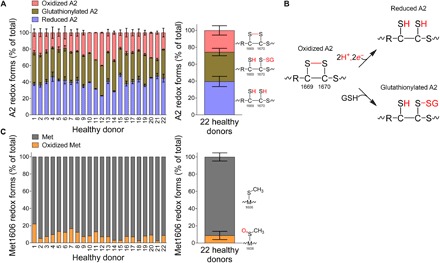
(A) Distribution of reduced, glutathionylated, and oxidized VWF A2 in 22 healthy donor plasmas. Mean and range for the two A2 domain cysteine-containing peptides of individual samples are shown on the left, and the mean and SD for the 22 samples are shown on the right. The VWF A2 domain cysteine pair exists as unpaired thiols in ~40%, Cys1669 exists as an unpaired thiol and Cys1670 is glutathionylated in ~36%, and the pair exists in an oxidized disulfide form in ~24%. (B) Posttranslational modifications of the A2 domain cysteines. (C) Distribution of unmodified and oxidized Met1606 in the 22 healthy donor plasmas. The mean and SD for the 22 samples are shown on the right. Only a minor fraction of VWF Met1606 is oxidized in the cohort of healthy donors.
We have observed that the levels of the different redox forms of VWF are independent of whether blood is collected into citrate, EDTA, heparin, or hirudin as an anticoagulant (fig. S4); freeze/thawing of plasma (at least three times); and plasma storage time (for at least 2 years at −80°C). On some occasions, blood was collected into an anticoagulant containing 5 mM 12C-IPA to freeze the redox state of VWF upon collection. This procedure had no obvious effect on the distribution of the VWF redox forms. On other occasions, plasma VWF was precipitated using polyethylene glycol (PEG) 4000 before collection on antibody-coated beads. This manipulation also had no obvious effect on the distribution of the VWF redox forms. These data indicate that the redox forms of VWF are relatively stable in plasma and are not appreciably influenced by differences in the collection or processing of blood.
The redox state of Met1606 of the Tyr1605-Met1606 peptide bond cleaved by ADAMTS13 provides an internal control for oxidative damage of the A2 domain. Neutrophil-derived hypochlorous acid can oxidize Met1606 that impairs ADAMTS13 cleavage of VWF and may contribute to a prothrombotic inflammatory environment (27). A peptide encompassing Met1606 (QAPNLVYMVTGNPASDE) in VWF was resolved by mass spectrometry and quantified for unmodified and oxidized residue (Fig. 5C). Only a small percentage of Met1606 was found to be oxidized in the cohort (mean, 8.8%; 95% CI, 6.7 to 10.9%).
The VWF A2 domain in heart failure patients receiving ECMO is markedly oxidized
We next sought to determine whether the redox state of VWF is fixed in the circulation or changes in response to disease or other stimulation. To answer this question, we studied plasma VWF of heart failure patients who have or have not received extracorporeal membrane oxygenation (ECMO) support. ECMO is used for patients with refractory heart failure. The circulating VWF in ECMO patients (n = 11) is markedly more oxidized than that in heart failure patients who have not received this support (n = 9) (P < 0.0001) (Fig. 6). The A2 disulfide bond is cleaved in just ~25% of plasma VWF subunits in the ECMO patients, which suggests that most plasma VWF binding sites for platelet GPIb are available.
Fig. 6. Plasma VWF in ECMO patients is markedly more oxidized than that in heart failure patients not implanted with these devices.
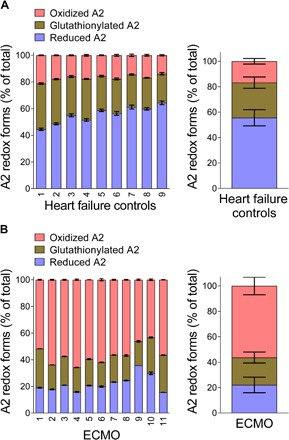
(A) Results from 9 patients with heart failure not treated with ECMO and (B) results from 11 patients who received ECMO.
DISCUSSION
Crystal structures of the A2 domain indicate that the Cys1669-Cys1670 disulfide stabilizes the hydrophobic core of the domain (14). The disulfide interacts with β4 strand residues Leu1603 and Tyr1605, the α1-β2 loop residue Met1528, the β2 strand residue Ile1535, and the N-terminal residue Met1495. The disulfide is at the C terminus of the α6 helix, and its rigidity is thought to set a high energy barrier for the early events in A2 unfolding. Dislodgement of the disulfide plug via sufficient shear forces has been proposed to allow water into the hydrophobic core of the domain, solvation of the Tyr1605 side chain, and disorder of the β4 strand. This unfolding of the A2 domain leads to exposure of the GPIb binding site in A1 through interdomain conformation changes that have yet to be defined. We show herein that this disulfide plug is intact in only a minority of plasma VWF subunits, and the absence of the plug is associated with the autoinhibition of the A1 domain by A2 and with the masking of the platelet binding site.
A conclusion from previous MD simulations and atomic force microscopy experiments (12, 13) was that A2 binds A1 under low shear conditions and sterically blocks platelet interaction with A1, whereas high shear displaces A2 from A1, allowing binding of GPIb. Our findings indicate that the autoinhibition of the A1 domain by A2 operates in VWF subunits containing the reduced and glutathionylated A2 domain, not subunits containing oxidized A2 domains. In addition, high shear did not appear to displace the autoinhibition in A2ΔCC VWF. These findings imply that VWF subunits exist in two functional forms in the circulation based on the redox state of the A2 domain: an oxidized form with high affinity for platelet GPIb when sufficient shear force unfolds the domain and a reduced plus glutathionylated form that has low affinity for platelet GPIb, irrespective of applied shear force.
Our MD simulations demonstrate that cleavage of the disulfide modifies the structure, dynamics, and molecular stresses of the A2 domain in a long-range allosteric manner. Not only were the changes observed near the two cysteines, but they also propagated to other distant regions of the protein, such as helices H3 and H5 and loops L2, L3-4, and L4. These conformational changes presumably underpin the interaction of the glutathionylated and reduced A2 domain with the adjacent A1 domain. Of interest, the energetically most favorable candidates for A1-A2 complexes from docking studies feature a domain-domain interface encompassing the A2 disulfide bond (13). It will be highly interesting to investigate how the redox state of Cys1669-Cys1670 modifies the rupture force of the A1-A2 complex. These findings serve as a structural explanation of the redox regulation of A1 inhibition by A2.
The A2 domain binds a calcium ion (28) and is glycosylated at N1574 (29) (both features have been proposed to stabilize its structure). Anticoagulation of blood with calcium chelators (citrate or EDTA) or direct thrombin inhibitors (heparin or hirudin) gave comparable results, indicating that depletion of calcium from VWF does not appreciably influence the redox state of the A2 cysteines (fig. S4). In addition, both the recombinant reduced A2 domain that is lacking the N-linked glycan and the reduced full-length VWF that is glycosylated bind to A1, which implies that glycosylation is not directly involved in autoregulation.
The finding that heart failure patients who have received ECMO support have markedly more oxidized VWF in the circulation may contribute to the hemostatic complications associated with this device. Both thrombosis and bleeding can occur with this intervention (30). It is notable that despite the variability in disease severity and therapeutic intervention in these patients, the coefficient of variation for VWF redox state measurements is only 2.6% for the heart failure cohort and 15.9% for the ECMO cohort. This suggests that oxidation of the A2 domain in the ECMO patients is a fundamental result of the blood shear associated with the device and not noticeably influenced by disease or therapy variations. This result indicates that the A2 redox forms can interchange in the circulation, which has implications for hemostatic disorders in general.
The redox state of A2 domain cysteines also influences ADAMTS13 regulation of VWF multimer size. A previous structure/function study of the A2 domain showed that A2ΔCC VWF is a much more efficient substrate for ADAMTS13 than wild-type VWF (20). The glutathionylated A2 domain is also likely efficiently cleaved by ADAMTS13, but this has not been directly tested. These findings have implications for ADAMTS13 regulation of VWF function. Only a minority of plasma VWF subunits (the oxidized fraction) are effective platelet adhesives, and this fraction is resistant to ADAMTS13 proteolysis at the shear rates tested. The reason that ADAMTS13 preferentially cleaves the poorly adhesive majority of plasma VWF subunits remains to be elucidated. It is possible that the ADAMTS13-resistant fraction of VWF represents a naturally occurring regulatory mechanism to ensure stable platelet plug formation at sites of vascular injury. Conversely, most of the circulating VWF is autoinhibited and is ADAMTS13-susceptible. This could be a protective mechanism to maintain VWF as lower-molecular weight multimers in plasma and thus avoid unwanted thrombosis in the circulation.
Notably, thrombospondin-1 (31), N-acetylcysteine (32), and ADAMTS13 (33) have been reported to cleave a disulfide bond or bonds in VWF and reduce multimer size. Our findings may provide a mechanistic basis for these observations. Other future questions are how the distribution of the different redox forms of VWF changes in the circulation during normal and pathological hemostasis and thrombosis, and whether they are involved in other VWF functions such as FVIII binding.
MATERIALS AND METHODS
Experimental design
VWF and A2 domain
All procedures involving collection of human blood were in accordance with the Human Research Ethics Committee (HREC) at the University of Sydney, the Alfred Hospital Ethics and the Monash University Standing Committee for Research in Humans, and the Helsinki Declaration of 1983. Blood from healthy donors, patients at a single center who had ECMO support, or patients with cardiac comorbidities including severe congestive cardiac failure was collected by venipuncture into evacuated tubes containing EDTA or citrate as anticoagulant and plasma prepared by centrifugation. Patients with ECMO support received anticoagulation and/or antiplatelet medications at the clinicians’ discretion or based on institutional guidelines where patients typically commenced on warfarin target an international normalized ratio of 2 to 3, with bridging heparin infusion and aspirin therapy, as well as dipyridamole for those who are considered high risk for thrombosis.
The recombinant A1 domain (residues Q1238 to P1471) produced in E. coli was a gift from M. A. Cruz. The recombinant A2 domain (residues M1473 to L1675) was produced in E. coli Bl21 (DE3) as an N-terminal 6×His-ZZ fusion protein with a thrombin cleavage site. Recombinant full-length human wild-type and C1669A and C1669A,C1670A (A2ΔCC VWF) mutant VWF (20) were transiently expressed in HEK293 cells using polyethylenimine HCl MAX (PEI MAX; molecular weight, 4000) with a PEI/DNA w/w ratio of 4:1 as previously described (34). Three days after transfection, culture media were collected, clarified by centrifugation at 1500g for 10 min, and concentrated using 3.5K molecular weight cutoff dialysis tubing (Thermo Fisher Scientific) and PEG 20000. VWF was resolved by agarose gel electrophoresis and Western blotted as previously described (Fig. 2A) (31, 35). Native human VWF was purified from ~0.5 to 1 ml of fresh or fresh-frozen plasma. Double-spun plasma was incubated with polyclonal anti-VWF antibody-coated (Dako) Dynabeads (MyOne Streptavidin T1, Life Technologies) (1 mg of beads per milliliter of plasma) on a rotating wheel for 1 hour at 22°C. The unbound plasma was discarded, and the beads were washed three times with 1 ml of phosphate-buffered saline (PBS; pH 7.2) (fig. S5). Further details of protein purification can be found in the Supplementary Materials.
Redox states of the VWF Cys1669-Cys1670 disulfide bond
The redox states of the A2 domain Cys1669-Cys1670 disulfide bond were measured in plasma and recombinant VWF and recombinant A2 domain. For VWF, unpaired cysteine thiols in the bead-bound protein were alkylated with 5 mM 12C-IPA (Cambridge Isotopes) for 1 hour at 22°C in the dark. The beads were washed three times with 1 ml of PBS, resuspended in 60 μl of PBS containing lithium dodecyl sulfate buffer (Life Technologies) and 1 mM DTT, and incubated at 60°C for 30 min. The protein was resolved on SDS-PAGE and stained with colloidal Coomassie (Sigma). The VWF was run under reducing conditions to enable excision of the monomer from the gel. For the A2 domain, purified protein (10 μg) was alkylated with 5 mM 12C-IPA (Cambridge Isotopes) for 1 hour at 22°C in the dark, and the protein was resolved on SDS-PAGE and stained with colloidal Coomassie (Sigma). The VWF or A2 band was excised, destained, dried, incubated with 40 mM DTT, and washed. The fully reduced protein was alkylated with 5 mM IPA where all six carbon atoms of the phenyl ring had a mass of 13 (13C-IPA, Cambridge Isotopes). The gel slice was washed and dried before digestion of VWF with Glu-C (14 ng/μl; Roche) in 25 mM NH4CO2 overnight at 30°C. Peptides were eluted from the slices with 5% formic acid and 50% acetonitrile. Liquid chromatography, mass spectrometry, and data analysis were performed as previously described (36, 37). Two peptides encompassing the Cys1669-Cys1670 disulfide bond cysteines (LVLQRCCSGE and TLPREAPDLVLQRCCSGE) were resolved and quantified. The levels of the different redox forms of the A2 domain cysteines were calculated from the relative ion abundance of peptides containing 12C-IPA and 13C-IPA. To calculate the ion abundance of peptides, extracted ion chromatograms were generated using the Xcalibur Qual Browser Software (v2.1.0; Thermo Fisher Scientific). The area was calculated using the automated peak detection function built into the software.
S-glutathionylation of Cys1670 was determined by alkylation of VWF with 13C-IPA for 1 hour at room temperature and by digestion of VWF with Glu-C (14 ng/μl) in 25 mM NH4CO2 overnight at 22°C under fluid shear conditions (32 dynes/cm2) by shaking at 2500 rpm in a MixMate orbital shaker (Eppendorf) (38). This was to facilitate proteolysis of VWF by Glu-C. Peptides were recovered in the solution with 5% formic acid and 50% acetonitrile solution and subjected to liquid chromatography, mass spectrometry, and data analysis as described above.
Binding of the A2 domain to the A1 domain
The A1 domain [2.5 μg/ml in 65 mM phosphate buffer (pH 6.5)] or buffer alone was incubated in MaxiSorp plates (Nunc 442404) for 1 hour at 37°C. The wells were then washed three times with 200 μl of PBS and blocked with 200 μl of 3% (w/v) BSA in tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 hour at 37°C. Afterward, the wells were washed three times with 200 μl of TBS-T [before the addition of untreated or reduced A2 in TBS-T (0 to 10 μM)] and incubated for 1 hour at 37°C. The wells were then washed three times with 200 μl of TBS-T and incubated with anti-A2 domain antibody MAB2764 (0.1 μg/ml; R&D Systems) for 1 hour at 37°C. This was followed by washing the wells three times with 200 μl of TBS-T and incubation with 1:2000 dilution of goat anti-mouse horseradish peroxidase for 1 hour at 37°C. The wells were then washed four times with TBS-T, and 100 μl of tetramethylbenzidine (T444; Sigma) was added. The reaction was finally quenched with 100 μl of 0.16 M H2SO4, and absorbance at 450 nm was measured. Binding of A2 to adsorbed A1 was corrected for nonspecific binding to BSA-coated wells at each A2 concentration.
BFP assays
All procedures involving collection of human blood were in accordance with HREC (project number 2014/244) at the University of Sydney. Human blood was obtained from healthy volunteers who had not received any antiplatelet medication in the preceding 2 weeks. Blood was anticoagulated with acid-citrate-dextrose (ACD) anticoagulant (13 mM sodium citrate, 1 mM citric acid, 20 mM dextrose, and 10 mM theophylline). Washed human platelets and RBCs were prepared as previously described (39). To avoid preactivation, human washed platelets were kept at 37°C in platelet-washing buffer [4.3 mM K2HPO4, 4.3 mM Na2HPO4, 24.3 mM NaH2PO4, 113 mM NaCl, 5.5 mM glucose, and 0.5% BSA (pH 6.5)] containing theophylline (10 mM) and apyrase (0.04 U/ml) and then resuspended in Tyrode’s buffer [10 mM Hepes, 12 mM NaHCO3, 137 mM NaCl, 2.7 mM KCl, and 5 mM glucose (pH 7.4)] before use in adhesion studies.
The BFP was assembled as previously described (40). In brief, to assemble the BFP as a picoforce transducer (spring constant, ~0.3 pN/nm), a VWF-A1 domain (Fig. 1F, bottom left), full-length VWF (Fig. 3A, bottom left), or control BSA-coated bead was glued via biotin-SA interaction to the apex of a micropipette-aspirated human RBC. On the other side, a human resting platelet (discoid shape) was aspirated by another apposing micropipette (Fig. 1F, top). In a BFP test cycle, the platelet is driven with subnanometer precision to touch the probe bead, allowing A1 to interact with GPIb on the platelet. Upon retraction of the platelet, whether a bond is absent (Fig. 1G, black) or present (Fig. 1G, blue) is detected by the RBC-bead deflection, monitored at a temporal and spatial precision of 0.7 ms and 3 nm, respectively.
The force spectroscopy traces (force versus time) were obtained by measuring the RBC-bead deflection from the bead edge tracking as previously described (9, 25, 40). Bond formation/dissociation and force application were enabled and monitored in controlled BFP touch cycles (~2.5 s each). In each cycle, an aspirated platelet was driven to approach and contact the probe (a VWF-bearing bead) with a 20-pN compressive force for a certain contact time (0.2 s) that allowed for bond formation and then retracted at a constant speed (3.3 μm/s) for bond detection. During the retraction phase, a “bond” event was signified by a tensile force. Conversely, no tensile force indicated a “no-bond” event. For the adhesion frequency assay, bond and no-bond events were enumerated to calculate an adhesion frequency in 50 repeated cycles for each bead-platelet pair. The force-clamp assay was used to measure bond lifetimes (9, 18, 25). In a similar BFP cycle to the adhesion frequency assay, upon detection of a bond event, a feedback loop controls the retraction so that it would be paused at a desired clamped force (5 to 50 pN) until bond dissociation. Afterward, the aspirated platelet was returned to the original position to complete the cycle. Lifetimes were measured from the instant the force reached the desired level to the point of bond dissociation.
Analysis of lifetime distributions with a two-state model
For all measured lifetime events at an indicated force bin, the survival frequency as a function of bond lifetime tb was calculated as the fraction of events with lifetime < tb. A2ΔCC VWF bonds were assumed to dissociate from a single state so that the pooled bond lifetimes were analyzed by a single exponential distribution (Eq. 1).
| (1) |
Given that the log linearizes the right-hand side of Eq. 1, the semi-log survival frequency versus bond lifetime plot is predicted to be linear. The negative slope of the line is used as an estimate for the off rate, koff.
For the wild-type VWF bonds, the ln(survival frequency) versus tb plots were no longer linear (see Fig. 4F). To analyze such data, we extended the single-state model to a two-state model that assumes a slow dissociation state coexisting with a fast dissociation state (9). The survival frequency data for each force bin were fitted by Eq. 2, which superimposes two exponential decays
| (2) |
where ki and wi are the off rate and the associated fraction of the ith state (i = 1, 2), which satisfy the constraints w1 + w2 = 1 and k1 > k2, respectively.
Platelet adhesion assays in flow chambers
Blood samples were collected from healthy volunteers in accordance with the St George Hospital HREC (12/252). Microchannels of Vena8 Fluoro Biochip (Cellix Ltd.) were coated with 10 μl of recombinant wild-type or A2ΔCC VWF [either whole-blood assay (5 μg/ml) or washed platelet assay (50 μg/ml)] overnight at 4°C in a humidified box, blocked with 10 μl of 0.1% BSA in PBS for 1 hour at room temperature, and washed with 40 μl of PBS. For the whole-blood assay, blood from healthy donors was drawn into ACD-A tubes (BD Vacutainer) and labeled with calcein (1 μg/ml; Thermo Fisher Scientific). Blood was injected via Mirus Nanopump into the channels at fluid shear stresses of 900, 1800, and 3000 s−1 (flow rate, 592, 1185, and 2222 nl/s, respectively) within 3 hours of collection. For the washed platelet assay, whole blood from healthy donors was drawn into ACD-A tubes (BD Vacutainer), and platelet-rich plasma was collected by centrifugation at 200g for 20 min at room temperature. After the addition of 1 μM prostaglandin E1, platelets were collected by centrifugation at 800g for 20 min, washed with Hepes Tyrode’s glucose buffer [20 mM Hepes, 134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 1 mM MgCl2, and 5 mM glucose (pH 7.4)], resuspended in the same buffer at a concentration of 300,000 to 400,000 per microliter, and labeled with calcein (1 μg/ml; Thermo Fisher Scientific). Platelets were injected into the channels at a shear stress of 1000 s−1 (flow rate, 333 nl/s) within 3 hours from collection. Adhesion of platelets was monitored in real time with images captured via an Exi Blue CCD camera (Q imaging) connected to an Axio Observer A1 Inverted Epi-Fluorescence microscope (Zeiss). Images were captured using the accompanying VenaFlux 2.3 imaging software, and videos were captured at a rate of 1 frame per 5 s (30 frames). Images were analyzed at positions 2, 4, and 6 (located at 6, 14, and 22 mm from the entry site of blood) of the microchannels at the 3-min time point (since initiation of flow) using the ImagePro Premier 64-bit software. These positions are representative of flow nearest, midway, and furthest from the entry of blood into the channel. Data were exported into Excel, and area coverage by platelets was calculated for each position. Whole-blood results were from four donors, and washed platelet results were from three donors for each experiment.
MD simulations
We used the Amber99sb-ildn* force field for all energy minimizations and MD simulations of the VWF A2 domain in both oxidized and reduced forms. All simulations were carried out using the GROMACS 4.5.5 package. Initial structures of the protein in both redox states were taken from the crystal structure of the human VWF A2 domain (PDB identifier 3GXB) (14). For the reduced state, hydrogens were added to the cysteines. Acetyl and N-methyl groups were placed on N and C termini, respectively, to cap the termini. Both structures, oxidized and reduced, were immersed in a cubic box containing approximately 23,600 TIP3P water molecules, with sodium and chloride ions added at a concentration of 150 mM, and few sodium counterions to maintain neutrality of the simulation box. Parameters for the ions were taken from Joung and Cheatham. Before MD simulations, both systems were minimized using the steepest descent method for 10,000 steps, which was followed by 2 ns of MD simulations, during which harmonic positional restraints were applied on protein heavy atoms (harmonic elastic constant of 1000 kJ mol−1 nm−2). Finally, unrestrained equilibrium MD simulations were carried out for 200 ns, of which the last 150 ns was used for subsequent analysis. The temperature was kept constant at 300 K by using a velocity rescaling thermostat with a coupling time of 0.5 ps. The pressure was kept constant at 1 bar using isotropic coupling to a Parrinello-Rahman barostat with a coupling time of 5.0 ps. An integration time step of 4 fs was used. In all simulations, the long-range electrostatic interactions were treated with the particle mesh Ewald method. All bonds were constrained using the LINCS (Linear Constraint Solver) algorithm, and angular degrees of freedom of hydrogen atoms were replaced by using virtual interaction sites. For the water molecules, both bond lengths and angular vibrations were constrained using the SETTLE algorithm. To investigate the conformational dynamics of both redox states of the VWF A2 domain, the MD trajectories were subjected to PCA. This analysis consists of the calculation and diagonalization of the covariance matrix of the atomic positions (here, of the backbone atoms), enabling filtering relevant large-amplitude collective motions from local fluctuations. The covariance matrix, along with its eigenvalues and eigenvectors, was calculated from a merged trajectory resulting from the concatenation of both the oxidized and the reduced trajectories. Subsequently, MD trajectories were projected onto the first two eigenvectors, representing the two collective motions with the largest variance, with each protein conformation represented by a point in this two-dimensional projection. These two motional modes accounted for almost 50% of the total motion of the protein backbone. Protein structures were visualized with PyMOL and UCSF (University of California, San Francisco) Chimera. Changes in the internal stress of the protein upon reduction were quantified by carrying out an FDA. In brief, time-averaged scalar pairwise forces 〈Fij〉 were computed for all residue pairs (i,j) of the A2 domain (excluding the solvent) in the oxidized [〈Fij(oxidized)〉] and the reduced [〈Fij(reduced)〉] states. All “force-field” terms, including electrostatic, van der Waals, bonds, bond angles, and dihedral interactions, were considered for the calculation of 〈Fij〉. The absolute value of the difference |DFij| = |〈Fij(reduced)〉 − 〈Fij(oxidized)〉| was subsequently calculated as a measure of the change in internal stress upon reduction. Here, | | indicates absolute values. Residue pairs showing differences larger than 90 pN were selected. MD trajectories were postprocessed to compute 〈Fij(red)〉 and 〈Fij(oxi)〉 by using the FDA tools available at https://github.com/HITS-MBM/gromacs-fda.
Statistical analysis
Parametric unpaired two-tailed t test was used to evaluate differences between groups. Statistical significance was defined as P < 0.05. In the MD simulations, the P values were obtained by applying a Wilcoxon test on n = 3 RMSF estimates, per redox state, resulting from trajectory fragments of 50 ns.
Supplementary Material
Acknowledgments
J. Hamilton assisted with the platelet adhesion studies and J. Wong contributed to the analysis of the mass spectrometry data. The pTAZH vector was provided by C. Romero Ramos (BioTick). Funding: This study was supported by grants from the National Health and Medical Research Council of Australia (P.J.H.), Royal College of Pathologists Foundation Kanematsu/Novo Nordisk Research Award (F.P. and L.J.), Diabetes Australia Research Trust grant G179720 and Sydney Medical School Early-Career Researcher Kickstart Grant (L.J.), National Heart Foundation of Australia Postdoctoral Fellowship (101285) (L.J.) and British Heart Foundation Intermediate Basic Science Research Fellowship (FS/11/51/28920) (B.M.L.), Deutsche Forschungsgemeinschaft (research unit FOR 1543 to C.A.-S., C.B., and F.G.), the Center for Modelling and Simulation in the Biosciences postdoctoral program of the Heidelberg University (A.B.), and the Klaus Tschira Foundation (F.G.). B.L. was supported by the Dutch Thrombosis Foundation through grant number 2016-03. Author contributions: D.B., F.P., L.J., C.A.-S., and A.B. performed the experiments and contributed to the experimental design. K.M.C., H.W., E.G., A.K.D., D.A.M., B.M.L., C.B., S.J., R.A., and F.G. contributed intellectually to the study and provided key reagents. P.J.H. conceived the study and wrote the manuscript. Competing interests: P.J.H. is an inventor on a Patent Cooperation Treaty (PCT) patent application related to this work filed by the University of Sydney (no. PCT/AU2017/050621, filed 20 June 2017). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaaq1477/DC1
Expression and purification of the recombinant A2 domain
fig. S1. Differential cysteine alkylation and mass spectrometry analysis of the VWF Cys1669-Cys1670 disulfide bond.
fig. S2. FDA of the VWF A2 domain.
fig. S3. Concordance between RMSF for the simulated A2 redox states and crystallographic B factors.
fig. S4. Anticoagulation of blood with calcium chelators (citrate or EDTA) or direct thrombin inhibitors (heparin or hirudin) does not appreciably influence the redox state of the A2 domain cysteines.
fig. S5. Plasma VWF collected on antibody-coated beads, resolved on reducing SDS-PAGE, and stained with colloidal Coomassie.
REFERENCES AND NOTES
- 1.Sadler J. E., Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 67, 395–424 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Kroll M. H., Hellums J. D., McIntire L. V., Schafer A. I., Moake J. L., Platelets and shear stress. Blood 88, 1525–1541 (1996). [PubMed] [Google Scholar]
- 3.Al-Tamimi M., Tan C. W., Qiao J., Pennings G. J., Javadzadegan A., Yong A. S. C., Arthur J. F., Davis A. K., Jing J., Mu F.-T., Hamilton J. R., Jackson S. P., Ludwig A., Berndt M. C., Ward C. M., Kritharides L., Andrews R. K., Gardiner E. E., Pathologic shear triggers shedding of vascular receptors: A novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood 119, 4311–4320 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Barg A., Ossig R., Goerge T., Schneider M. F., Schillers H., Oberleithner H., Schneider S. W., Soluble plasma-derived von Willebrand factor assembles to a haemostatically active filamentous network. Thromb. Haemost. 97, 514–526 (2007). [PubMed] [Google Scholar]
- 5.Schneider S. W., Nuschele S., Wixforth A., Gorzelanny C., Alexander-Katz A., Netz R. R., Schneider M. F., Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. U.S.A. 104, 7899–7903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu H., Jiang Y., Yang D., Scheiflinger F., Wong W. P., Springer T. A., Flow-induced elongation of von Willebrand factor precedes tension-dependent activation. Nat. Commun. 8, 324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulrichts H., Udvardy M., Lenting P. J., Pareyn I., Vandeputte N., Vanhoorelbeke K., Deckmyn H., Shielding of the A1 domain by the D’D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J. Biol. Chem. 281, 4699–4707 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Auton M., Sowa K. E., Behymer M., Cruz M. A., N-terminal flanking region of A1 domain in von Willebrand factor stabilizes structure of A1A2A3 complex and modulates platelet activation under shear stress. J. Biol. Chem. 287, 14579–14585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ju L., Dong J.-f., Cruz M. A., Zhu C., The N-terminal flanking region of the A1 domain regulates the force-dependent binding of von Willebrand factor to platelet glycoprotein Ibα. J. Biol. Chem. 288, 32289–32301 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng W., Wang Y., Druzak S. A., Healey J. F., Syed A. K., Lollar P., Li R., A discontinuous autoinhibitory module masks the A1 domain of von Willebrand factor. J. Thromb. Haemost. 15, 1867–1877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin C., Morales L. D., Cruz M. A., Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibα. J. Thromb. Haemost. 5, 1363–1370 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Posch S., Aponte-Santamaría C., Schwarzl R., Karner A., Radtke M., Gräter F., Obser T., König G., Brehm M. A., Gruber H. J., Netz R. R., Baldauf C., Schneppenheim R., Tampé R., Hinterdorfer P., Mutual A domain interactions in the force sensing protein von Willebrand factor. J. Struct. Biol. 197, 57–64 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Aponte-Santamaría C., Huck V., Posch S., Bronowska A. K., Grässle S., Brehm M. A., Obser T., Schneppenheim R., Hinterdorfer P., Schneider S. W., Baldauf C., Gräter F., Force-sensitive autoinhibition of the von Willebrand factor is mediated by interdomain interactions. Biophys. J. 108, 2312–2321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q., Zhou Y. F., Zhang C. Z., Zhang X., Lu C., Springer T. A., Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc. Natl. Acad. Sci. U.S.A. 106, 9226–9231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marti T., Rösselet S. J., Titani K., Walsh K. A., Identification of disulfide-bridged substructures within human von Willebrand factor. Biochemistry 26, 8099–8109 (1987). [DOI] [PubMed] [Google Scholar]
- 16.Pasquarello C., Sanchez J. C., Hochstrasser D. F., Corthals G. L., N-t-butyliodoacetamide and iodoacetanilide: Two new cysteine alkylating reagents for relative quantitation of proteins. Rapid Commun. Mass Spectrom. 18, 117–127 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Bekendam R. H., Bendapudi P. K., Lin L., Nag P. P., Pu J., Kennedy D. R., Feldenzer A., Chiu J., Cook K. M., Furie B., Huang M., Hogg P. J., Flaumenhaft R., A substrate-driven allosteric switch that enhances PDI catalytic activity. Nat. Commun. 7, 12579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ju L., Chen Y., Xue L., Du X., Zhu C., Cooperative unfolding of distinctive mechanoreceptor domains transduces force into signals. Elife 5, e15447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yago T., Lou J., Wu T., Yang J., Miner J. J., Coburn L., López J. A., Cruz M. A., Dong J. F., McIntire L. V., McEver R. P., Zhu C., Platelet glycoprotein Ibα forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J. Clin. Invest. 118, 3195–3207 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luken B. M., Winn L. Y. N., Emsley J., Lane D. A., Crawley J. T. B., The importance of vicinal cysteines, C1669 and C1670, for von Willebrand factor A2 domain function. Blood 115, 4910–4913 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savage B., Saldívar E., Ruggeri Z. M., Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 84, 289–297 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Furlan M., Robles R., Lammle B., Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood 87, 4223–4234 (1996). [PubMed] [Google Scholar]
- 23.Levy G. G., Nichols W. C., Lian E. C., Foroud T., McClintick J. N., McGee B. M., Yang A. Y., Siemieniak D. R., Stark K. R., Gruppo R., Sarode R., Shurin S. B., Chandrasekaran V., Stabler S. P., Sabio H., Bouhassira E. E., Upshaw J. D. Jr, Ginsburg D., Tsai H. M., Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 413, 488–494 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Ju L., Chen Y., Zhou F., Lu H., Cruz M. A., Zhu C., Von Willebrand factor-A1 domain binds platelet glycoprotein Ibα in multiple states with distinctive force-dependent dissociation kinetics. Thromb. Res. 136, 606–612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ju L., Lou J., Chen Y., Li Z., Zhu C., Force-induced unfolding of leucine-rich repeats of glycoprotein Ibα strengthens ligand interaction. Biophys. J. 109, 1781–1784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seifert C., Gräter F., Force distribution reveals signal transduction in E. coli Hsp90. Biophys. J. 103, 2195–2202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen J., Fu X., Wang Y., Ling M., McMullen B., Kulman J., Chung D. W., López J. A., Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood 115, 706–712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu A. J., Springer T. A., Calcium stabilizes the von Willebrand factor A2 domain by promoting refolding. Proc. Natl. Acad. Sci. U.S.A. 109, 3742–3747 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch C. J., Lane D. A., N-linked glycan stabilization of the VWF A2 domain. Blood 127, 1711–1718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy D. A., Hockings L. E., Andrews R. K., Aubron C., Gardiner E. E., Pellegrino V. A., Davis A. K., Extracorporeal membrane oxygenation-hemostatic complications. Transfus. Med. Rev. 29, 90–101 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Xie L., Chesterman C. N., Hogg P. J., Control of von Willebrand factor multimer size by thrombospondin-1. J. Exp. Med. 193, 1341–1349 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen J., Reheman A., Gushiken F. C., Nolasco L., Fu X., Moake J. L., Ni H., López J. A., N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J. Clin. Invest. 121, 593–603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh H. C., Zhou Z., Choi H., Tekeoglu S., May W. III, Wang C., Turner N., Scheiflinger F., Moake J. L., Dong J. F., Disulfide bond reduction of von Willebrand factor by ADAMTS-13. J. Thromb. Haemost. 8, 2778–2788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKinnon T. A., Chion A. C., Millington A. J., Lane D. A., Laffan M. A., N-linked glycosylation of VWF modulates its interaction with ADAMTS13. Blood 111, 3042–3049 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Ganderton T., Wong J. W., Schroeder C., Hogg P. J., Lateral self-association of VWF involves the Cys2431-Cys2453 disulfide/dithiol in the C2 domain. Blood 118, 5312–5318 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Chiu J., Wong J. W., Hogg P. J., Redox regulation of methionine aminopeptidase 2 activity. J. Biol. Chem. 289, 15035–15043 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cook K. M., McNeil H. P., Hogg P. J., Allosteric control of βII-tryptase by a redox active disulfide bond. J. Biol. Chem. 288, 34920–34929 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han Y., Xiao J., Falls E., Zheng X. L., A shear-based assay for assessing plasma ADAMTS13 activity and inhibitors in patients with thrombotic thrombocytopenic purpura. Transfusion. 51, 1580–1591 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nesbitt W. S., Tovar-Lopez F. J., Westein E., Harper I. S., Jackson S. P., A multimode-TIRFM and microfluidic technique to examine platelet adhesion dynamics. Methods Mol. Biol. 1046, 39–58 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Chen Y., Liu B., Ju L., Hong J., Ji Q., Chen W., Zhu C., Fluorescence biomembrane force probe: Concurrent quantitation of receptor-ligand kinetics and binding-induced intracellular signaling on a single cell. J. Vis. Exp. e52975 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/2/eaaq1477/DC1
Expression and purification of the recombinant A2 domain
fig. S1. Differential cysteine alkylation and mass spectrometry analysis of the VWF Cys1669-Cys1670 disulfide bond.
fig. S2. FDA of the VWF A2 domain.
fig. S3. Concordance between RMSF for the simulated A2 redox states and crystallographic B factors.
fig. S4. Anticoagulation of blood with calcium chelators (citrate or EDTA) or direct thrombin inhibitors (heparin or hirudin) does not appreciably influence the redox state of the A2 domain cysteines.
fig. S5. Plasma VWF collected on antibody-coated beads, resolved on reducing SDS-PAGE, and stained with colloidal Coomassie.