

Endorsed by the Asia Pacific Heart Rhythm Society (APHRS), the European Heart Rhythm Association (EHRA), the Heart Rhythm Society (HRS), and the Latin American Society of Cardiac Pacing and Electrophysiology (Sociedad Latinoamericana de Estimulacifin Cardíaca y Electrofisiología [SOLAECE])
Preamble
The J-wave syndromes (JWSs), consisting of the Brugada syndrome (BrS) and early repolarization syndrome (ERS), have captured the interest of the cardiology community over the past 2 decades following the identification of BrS as a new clinical entity by Pedro and Josep Brugada in 1992.1 The clinical impact of ERS was not fully appreciated until 2008.2–4 Consensus conferences dedicated to BrS were held in 2000 and 2004,5,6 but a consensus conference specifically focused on ERS has not previously been convened other than that dealing with terminology, and guidelines for both syndromes were last considered in 2013.7 A great deal of new information has emerged since. The present forum was organized to evaluate new information and highlight emerging concepts with respect to differential diagnosis, prognosis, cellular and ionic mechanisms, and approaches to therapy of the JWSs. Leading experts, including members of the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA), and the Asian-Pacific Heart Rhythm Society (APHRS), met in Shanghai, China, in April 2015. The Task Force was charged with a review of emerging concepts and assessment of new evidence for or against particular diagnostic procedures and treatments. Every effort was made to avoid any actual, potential, or perceived conflict of interest that might arise as a result of outside relationships or personal interest. This consensus report is intended to assist health care providers in clinical decision-making. The ultimate judgment regarding care of a particular patient, however, must be made by the health care provider based on all of the facts and circumstances presented by the patient.
Members of this Task Force were selected to represent professionals involved with the medical care of patients with the JWSs, as well as those involved in research into the mechanisms underlying these syndromes. These selected experts in the field undertook a comprehensive review of the literature. Critical evaluation of methods of diagnosis, risk stratification, approaches to therapy, and mechanistic insights was performed, including assessment of the risk- to-benefit ratio. The level of evidence and the strength of the recommendation of particular management options were weighed and graded. Recommendations with class designations are taken from HRS, EHRA, APHRS, and/or European Society of Cardiology (ESC) consensus statements or guidelines.8,9 Recommendations without class designations are derived from unanimous consensus of the authors. The consensus recommendations in this document use the commonly used Class I, IIa, IIb, and III classifications and the corresponding language: “is recommended” for a Class I consensus recommendation; “can be useful” or “is reasonable” for a Class IIa consensus recommendation; “may be considered” for a Class IIb consensus recommendation; and “is not recommended” for a Class III consensus recommendation.
Introduction
The appearance of prominent J waves in the electrocardiogram (ECG) have long been reported in cases of hypothermia10–12 and hypercalcemia.13,14 More recently, accentuation of the J wave has been associated with life-threatening ventricular arrhythmias.15 Under these circumstances, the accentuated J wave typically may be so broad and tall as to appear as an ST-segment elevation, as in cases of BrS. In humans, the normal J wave often appears as a J-point elevation, with part of the J wave buried inside the QRS. An early repolarization pattern (ERP) in the ECG, consisting of a distinct J-wave or J-point elevation, or a notch or slur of the terminal part of the QRS with and without an ST-segment elevation, has traditionally been viewed as benign.16,17 The benign nature of an ERP was challenged in 200018 based on experimental data showing that this ECG manifestation predisposes to the development of polymorphic ventricular tachycardia (VT) and ventricular fibrillation (VF) in coronary-perfused wedge preparations.15,18–20 Validation of this hypothesis was provided 8 years later by Haissaguerre et al,2 Nam et al,3 and Rosso et al.4 These seminal studies together with numerous additional case-control and population-based studies have provided clinical evidence for an increased risk for development of life-threatening arrhythmic events and sudden cardiac death (SCD) among patients presenting with an ERP, particularly in the inferior and inferolateral leads. The lack of agreement regarding the terminology relative to early repolarization (ER) has led to a great deal of confusion and inconsistency in reporting.21–23 A recent expert consensus report that focused on the terminology of ER recommends that the peak of an end QRS notch and/or the onset of an end QRS slur be designated as Jp and that Jp should exceed 0.1 mV in ≥2 contiguous inferior and/or lateral leads of a standard 12-lead ECG for ER to be present.24 It was further recommended that the start of the end QRS notch or J wave be designated as Jo and the termination as Jt.
ERS and BrS are thought to represent 2 manifestations of the JWSs. Both syndromes are associated with vulnerability to development of polymorphic VT and VF leading to SCD1–3,15 in young adults with no apparent structural heart disease and occasionally to sudden infant death syndrome.25–27 The region generally most affected in BrS is the anterior right ventricular outflow tract (RVOT); in ERS, it is the inferior region of the left ventricle (LV).2,4,28–32 As a consequence, BrS is characterized by accentuated J waves appearing as a coved-type ST-segment elevation in the right precordial leads V1–V3, whereas ERS is characterized by J waves, Jo elevation, notch or slur of the terminal part of the QRS, and ST segment or Jt elevation in the lateral (type 1), inferolateral (type 2), or inferolateral + anterior or right ventricular (RV) leads (type III).15 An ERP is often encountered in ostensibly healthy individuals, particularly in young males, black individuals, and athletes. ERP is also observed in acquired conditions, including hypothermia and ischemia.15,33,34 When associated with VT/VF in the absence of organic heart disease, ERP is referred to as ERS.
The prevalence of BrS with a type 1 ECG in adults is higher in Asian countries, such as Japan (0.15%–0.27%)35,36 and the Philippines (0.18%),37 and among Japanese-Americans in North America (0.15%)38 than in western countries, including Europe (0%–0.017%)39–41 and North America (0.005%–0.1%).42,43 In contrast, the prevalence of an ERP in the inferior and/or lateral leads with a J-point elevation ≥0.1 mV ranges between 1% and 24% and for J-point elevation ≥0.2 mV ranges between 0.6% to 6.4%.44–46 No significant regional differences in the prevalence of an ERP have been reported.47 However, ERP is significantly more common in blacks than in Caucasians. Little in the way of regional differences in the manifestation of ERS has been reported. ERP appears to be more common in Aboriginal Australians than in Caucasian Australians.48
Updates on the diagnosis of BrS
According to the 2013 consensus statement on inherited cardiac arrhythmias8 and the 2015 guidelines for the management of patients with ventricular arrhythmias and prevention of SCD9: “BrS is diagnosed in patients with ST- segment elevation with type 1 morphology ≥2 mm in ≥1 lead among the right precordial leads V1, V2, positioned in the 2nd, 3rd or 4th intercostal space occurring either spontaneously or after provocative drug test with intravenous administration of Class I antiarrhythmic drugs. BrS is diagnosed in patients with type 2 or type 3 ST-segment elevation in ≥1 lead among the right precordial leads V1, V2 positioned in the 2nd, 3rd or 4th intercostal space when a provocative drug test with intravenous administration of Class I antiarrhythmic drugs induces a type I ECG morphology.”
The present Task Force is concerned that this could result in overdiagnosis of BrS, particularly in patients displaying a type 1 ECG only after a drug challenge. Data suggest the latter population is at very low risk and that the presumed false- positive rate of pharmacologic challenge is not trivial.49 Although a rigorous process was undertaken to establish the preceding guidelines, there remains no gold standard for establishing a diagnosis, particularly in patients with weak evidence of disease. Accordingly, we recommend adoption of the following diagnostic criteria and score system for BrS. Consistent with the recommendation of the 2013 and 2015 guidelines, only a type 1 (“coved-type”) ST-segment elevation is considered diagnostic of BrS (Figure 1), and BrS is characterized by ST-segment elevation ≥2 mm (0.2 mV) in ≥1 right precordial leads (V1–V3) positioned in the 4th, 3rd, or 2nd intercostal space. However, as a departure from the guidelines, this consensus report recommends that when a type 1 ST-segment elevation is unmasked using a sodium channel blocker (Table 1), diagnosis of BrS should require that the patient also present with 1 of the following: documented VF or polymorphic VT, syncope of probable arrhythmic cause, a family history of SCD at o45 years old with negative autopsy, coved-type ECGs in family members, or nocturnal agonal respiration. Inducibility of VT/VF with 1 or 2 premature beats supports the diagnosis of BrS under these circumstances.50
Figure 1.

Three types of ST-segment elevation associated with Brugada syndrome. Only type 1 is diagnostic of Brugada syndrome.
Table 1.
Drugs used to unmask the Brugada ECG
| Drug | Dose | Administration |
|---|---|---|
| Ajmaline | 1 mg/kg over 10 minutes | Intravenous |
| Flecainide | 2 mg/kg over 10 minutes | Intravenous |
| 200–300 mg | Oral (41 hour) | |
| Procainamide | 10 mg/kg over 10 minutes | Intravenous |
| Pilsicainide | 1 mg/kg over 10 minutes | Intravenous |
A type 2 (“saddle-back type”) or type 3 ST-segment elevation cannot substitute for a type 1, unless converted to type 1 with fever or sodium drug challenge. A drug challenge–induced type 1 can be used to diagnose BrS only if accompanied by 1 of the criteria specified above. Type 2 is characterized by ST-segment elevation ≥0.5 mm (generally ≥2 mm in V2) in ≥1 right precordial lead (V1–V3), followed by a convex ST. The ST segment is followed by a positive T wave in V2 and variable morphology V1. Type 3 is characterized by either a saddleback or coved appearance with an ST-segment elevation o1 mm. Placement of the right precordial leads in more cranial positions (in the 3rd or 2nd intercostal space) in a 12-lead resting ECG or 12-lead Holter ECG increases the sensitivity of ECG.51–53 It is recommended that ECG recordings be obtained in the standard and superior positions for the V1 and V2 leads. Veltman et al.54 showed that RVOT localization using magnetic resonance imaging (MRI) correlates with type 1 ST-segment elevation in BrS and that lead positioning according to RVOT location improves the diagnosis of BrS. Interestingly, in most cases a type I pattern was found in the 3rd intercostal space in the sternal and left parasternal positions.54 In reviewing ECGs of a large cohort of BrS patients, Richter et al.55 concluded that lead V3 does not yield diagnostic information in BrS.
A proposed diagnostic score system for BrS, referred to as the Proposed Shanghai BrS Score, is presented in Table 2. These recommendations are based on the available literature and the clinical experience of the Task Force members.8,56–60 Weighting of variables is based on expert opinion informed by cohort studies that typically do not include all variables presented. Thus, rigorous, objectively weighted coefficients were not derived from large-scale risk factor and outcome- informed datasets. Nonetheless, the authors believed that some inferential weighting would be of benefit when applied to patients. As with all such recommendations, they will need to undergo initial and ongoing validation in future studies.
Table 2.
Proposed Shanghai Score System for diagnosis of Brugada syndrome
| Points | |
|---|---|
| I. ECG (12-Lead/Ambulatory) | |
| A. Spontaneous type 1 Brugada ECG pattern at nominal or high leads | 3.5 |
| B. Fever-induced type 1 Brugada ECG pattern at nominal or high leads | 3 |
| C. Type 2 or 3 Brugada ECG pattern that converts with provocative drug challenge | 2 |
| *Only award points once for highest score within this category. One item from this category must apply. | |
| II. Clinical History* | |
| A. Unexplained cardiac arrest or documented VF/ polymorphic VT | 3 |
| B. Nocturnal agonal respirations | 2 |
| C. Suspected arrhythmic syncope | 2 |
| D. Syncope of unclear mechanism/unclear etiology | 1 |
| E. Atrial flutter/fibrillation in patients <30 years without alternative etiology | 0.5 |
| *Only award points once for highest score within this category. | |
| III. Family History | |
| A. First- or second-degree relative with definite BrS | 2 |
| B. Suspicious SCD (fever, nocturnal, Brugada aggravating drugs) in a first- or second-degree relative | 1 |
| C. Unexplained SCD <45 years in first- or second- degree relative with negative autopsy | 0.5 |
| *Only award points once for highest score within this category. | |
| IV. Genetic Test Result | |
| A. Probable pathogenic mutation in BrS susceptibility gene | 0.5 |
| Score (requires at least 1 ECG finding) | |
| ≥3.5 points: Probable/definite BrS | |
| 2–3 points: Possible BrS | |
| <2 points: Nondiagnostic | |
BrS = Brugada syndrome; SCD = sudden cardiac death; VF = ventricular fibrillation; VT = ventricular tachycardia.
Pharmacologic tests and other diagnostic tools
When there is clinical suspicion of BrS in the absence of spontaneous type 1 ST-segment elevation, a pharmacologic challenge using a sodium channel blocker is recommended. A list of agents used for this purpose is presented in Table 1 (also see www.brugadadrugs.org). The test is considered positive only if a type 1 ECG pattern is obtained, and it should be discontinued in case of frequent ventricular extrasystoles or other arrhythmias, or widening of the QRS 4130% over the baseline value.6 As an alternative, the “full stomach test” has been proposed for diagnosing BrS.61 In this case, ECGs are performed before and after a large meal. The use of “high electrodes” increases the sensitivity for recognizing spontaneous type I ST-segment elevation at night or after heavy meals.62 A type 1 ST-segment elevation recorded using a Holter is a spontaneous type 1, and it is reasonable to assume that a spontaneous type 1 recorded by Holter at night or after a large meal has more value—both diagnostic and prognostic—than a drug-induced type 1.
Drug challenge is not indicated in asymptomatic patients displaying the type 1 ECG under baseline conditions because of the lack of the additional diagnostic value. These provocative drug tests are also not recommended in cases in which fever has been documented to induce a type I ECG, other than for research purposes. Much debate has centered around the definition of a false-positive sodium channel block challenge.63 The consensus is that a false-positive is difficult to define because of the lack of a gold standard. The development of a type 1 ST-segment elevation in response to sodium block challenge should be considered as probabilistic, rather than binary, in nature. As will be discussed later, a similar approach is recommended in evaluating the ability of genetic variants to promote the BrS phenotype.
Asymptomatic patients with a family history of BrS or SCD should be informed of the availability of a sodium channel blocker challenge test to provide a more definitive diagnosis of BrS. However, patients should be advised that no therapy may be recommended regardless of the outcome because the long- term risk of patients with BrS diagnosed by this test is significantly lower than the risk of patients with spontaneous type 1. Patients also should be informed about the risk of the test and about the emotional consequences of having a positive test not followed by definitive therapy. The decision as to whether to undergo the drug challenge ultimately should be left up to the well-informed patient.
Performing an ajmaline test in children is problematic for 2 reasons. First, the test is apparently less sensitive in children than in adults. In fact, in 1 study, a repeat ajmaline challenge performed after puberty unmasked BrS in 23% of relatives with a previously negative drug test performed during childhood.64 Second, the test is associated with greater risk than in adults. In 1 series, 10% of children undergoing the ajmaline test, including 3% of the asymptomatic subgroup, developed sustained VT.64,65 Caution also should be exercised when performing a sodium blocker challenge in adults with a known pathogenic sodium channel mutation or in patients with prolonged PR intervals, pointing to a carrier of such a mutation.66
Differential diagnosis
Other causes of ST-segment elevation should be excluded before establishing the diagnosis of BrS (Table 3). Artifacts secondary to low-pass filtering should be ruled out.67
Table 3.
Differential diagnosis and modulating factors in Brugada syndrome
| A. Differential diagnosis | B. Modulating factors |
|---|---|
|
|
Circumstances that produce a type 1 Brugada-like ECG include right bundle branch block (RBBB), pectus excavatum, arrhythmogenic right ventricular cardiomyopathy (ARVC), and occlusion of the left anterior descendent artery or the conus branch of the right coronary artery, which supplies the RVOT (Table 3A).
Discrimination between BrS and ARVC is particularly challenging. Although debate continues as to the extent to which structural abnormalities are present in BrS, most investigators consider BrS to be a channelopathy. Concealed structural abnormalities, such as histologic myocardial fibrosis of the RVOT, which may not become evident using conventional imaging techniques, have been proposed to account for or contribute to delayed conduction and ventricular arrhythmias in BrS. MRI and electron beam computed tomographic studies of BrS patients consistently show subtle abnormalities, including wall motion abnormalities and reduced contractile function of the RV and, to a lesser extent, of the LV, and dilation of the RVOT.68–71 In the only study that discriminated between patients with and those without SCN5A mutations, no difference was observed in RVOT dimensions or RV ejection fraction between these patients. Slightly greater depressions of LV dimensions and ejection fraction were observed in patients with SCN5A mutations. Significant differences were observed in RV and LV dimensions and ejection fraction compared to healthy controls.72 Cardiac dilation and reduced contractility in all of these studies were attributed to structural changes (fibrosis, fatty degeneration). However, as noted by van Hoorn et al,72 virtually no signs of fibrosis or fatty degeneration could be detected, perhaps because the spatial resolution of the imaging used was too low to detect such subtle changes.
Antzelevitch and colleagues have long suggested an alternative explanation.31,73,74 Loss of the action potential (AP), which has been shown in experimental models to create the arrhythmogenic substrate in BrS, leads to contractile changes that could explain the wall motion abnormalities observed. The all-or-none repolarization at the end of phase 1 of the epicardial AP responsible for loss of the dome causes the calcium channel to inactivate very soon after it activates. As a consequence, calcium channel current is dramatically reduced, the cell becomes depleted of calcium, and contractile function ceases in those cells. This is expected to lead to wall motion abnormalities, particularly in the RVOT, dilation of the RVOT region, and reduced ejection fraction observed in patients with BrS. It has also been proposed that the loss of the AP dome, because it creates a hibernation-like state, may, over long periods of time, lead to mild structural changes, including intracellular lipid accumulation, vacuolization, and connexin 43 redistribution. These structural changes may, in turn, contribute to the arrhythmogenic substrate of BrS, although they are very different from those encountered in arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D).31,75 This hypothesis would predict that some of the changes observed by recent studies may be the result of, rather than the cause of, the BrS phenotype.76
In a recent study, Nademanee et al.76 reported additional evidence pointing to pathologic changes in the RVOT of patients with BrS that have proved undetectable by echocardiography or MRI.
In contrast, imaging techniques in ARVC clearly display morphologic and functional changes (e.g., dilation, bulging/ aneurysms, wall motion abnormalities). ARVC is an inherited cardiac disease resulting from genetically defective desmosomal (DS) proteins,77,78 characterized by fibrofatty myocardial replacement predisposing to scar-related ventricular arrhythmias that may lead to SCD, mostly in young people and athletes.79 Life-threatening ventricular arrhythmias may occur early, during the “concealed phase” of the disease, before overt structural changes.77,78,80 Recent experimental studies demonstrated that loss of expression of DS proteins may induce electrical ventricular instability by causing sodium channel dysfunction and current reduction as a consequence of the cross-talk between these molecules at the intercalated discs, which predisposes to sodium current-dependent lethal arrhythmias, similar to those leading to SCD in patients with J-wave syndromes.80–82 Further evidence of the overlap between phenotypic manifestation of ARVC and BrS comes from (1) clinicopathologic studies showing that a subset of ARVC patients may share ECG changes and patterns of ventricular arrhythmias with BrS;83 and (2) genotype–phenotype correlation studies demonstrating that PKP2 mutation may cause a Brugada phenotype in the human heart by reducing sodium current.84 These findings support the concept that specific DS gene mutations involved in the pathogenesis of ARVC can lead to a decreased depolarization reserve that manifests as J-wave/BrSs. Thus, ARVC and J wave syndromes are not completely different conditions but are the ends of a spectrum of structural myocardial abnormalities and sodium current deficiency that share a common origin as diseases of the connexome.84 The ECG abnormalities in ARVC are not dynamic and display a constant T-wave inversion, epsilon waves, and, in the progressive stage, reduction of the R amplitude. End-stage ARVC is usually associated with monomorphic VT with left bundle branch morphology and is precipitated by catecholamines,85 whereas BrS is associated with polymorphic VT predominantly during sleep or rest.86 A positive ajmaline challenge has been reported in 16% of patients with ARVC.87,88
Modulating factors
Sympathovagal balance, hormones, metabolic factors, and pharmacologic agents are thought to modulate not only ECG morphology but also explain the development of ventricular arrhythmias under certain conditions.89 Any of these modulating factors, if present, should be promptly corrected (Table 3B).
Acquired Brugada pattern and phenocopies
The Brugada ECG is often concealed and can be unmasked with a wide variety of drugs and conditions, including a febrile state, vagotonic agents and maneuvers, α-adrenergic agonists, β-adrenergic blockers, Class IC antiarrhythmic drugs, tricyclic or tetracyclic antidepressants, hyperkalemia, hypokalemia, hypercalcemia, and alcohol and cocaine toxicity.90–100 Preexcitation of RV can unmask the BrS phenotype in cases of RBBB.101 An up-to-date list of agents known to unmask the Brugada ECG that should be avoided by patients with BrS can be found at www.brugadadrugs.org.89
Environmental factors leading to the appearance of an ECG similar or identical to a type 1 BrS pattern in the absence of any apparent genetic dysfunction has been suggested to represent a Brugada ECG phenocopy.102 Features of the Brugada phenocopies include (1) Brugada- like ECG pattern; (2) presence of an identifiable underlying condition; (3) disappearance of the ECG pattern after resolution of the condition; (4) absence of family history of sudden death in relatively young first-degree relatives (≤45 years) or of type 1 BrS pattern; (5) absence of symptoms such as syncope, seizures, or nocturnal agonal respiration; and (6) a negative sodium channel blocker challenge test. Debate continues as to the appropriateness of this terminology given that it is very difficult to rule out a genetic predisposition, which is a prerequisite for designating the ECG manifestation as a phenocopy. Designation of these conditions as acquired forms of Brugada ECG pattern or BrS may be more appropriate and better aligned with the terminology used in the long QT syndrome.
Update on the diagnosis of ERS
ERS is generally diagnosed in patients who display ER in the inferior and/or lateral leads presenting with aborted cardiac arrest, documented VF, or polymorphic VT. Consistent with the recent consensus report on ERP,24 ER is recognized if (1) there is an end QRS notch (J wave) or slur on the downslope of a prominent R wave with and without ST-segment elevation; (2) the peak of the notch or J wave (Jp) ≥0.1 mV in ≥2 contiguous leads of the 12-lead ECG, excluding leads V1–V3; and (3) QRS duration (measured in leads in which a notch or slur is absent) o120 ms. Table 4 lists the exclusion criteria in the differential diagnosis of ERS.
Table 4.
Differential diagnosis of early repolarization pattern
| Other causes of early repolarization pattern include the following: |
|---|
|
STEMI = ST segment elevation myocardial infarction.
A proposed diagnostic score system for ERS, referred to as the Proposed Shanghai ERS Score, is presented in Table 5. The scoring system is based on evidence available in the literature to date. As in BrS, weighting of variables is based on expert opinion informed by cohort studies that do not include all variables presented. Thus, rigorous, objectively weighted coefficients were not derived from large-scale risk factor- and outcome-informed datasets. Nonetheless, the authors believed that some inferential weighting would be of benefit when applied to patients. As with all such recommendations, they will need to undergo initial and ongoing validation in future studies.
Table 5.
Proposed Shanghai Score System for diagnosis of early repolarization syndrome
| Points | |
|---|---|
| I. Clinical History | |
| A. Unexplained cardiac arrest, documented VF or polymorphic VT | 3 |
| B. Suspected arrhythmic syncope | 2 |
| C. Syncope of unclear mechanism/unclear etiology | 1 |
| *Only award points once for highest score within this category | |
| II. Twelve-Lead ECG | |
| A. ER ≥0.2 mV in ≥2 inferior and/or lateral ECG leads with horizontal/descending ST segment | 2 |
| B. Dynamic changes in J-point elevation (≥0.1 mV) in ≥2 inferior and/or lateral ECG leads | 1.5 |
| C. ≥0.1 mV J-point elevation in at least 2 inferior and/or lateral ECG leads | 1 |
| *Only award points once for highest score within this category | |
| III. Ambulatory ECG Monitoring | |
| A. Short-coupled PVCs with R on ascending limb or peak of T wave | 2 |
| IV. Family History | |
| A. Relative with definite ERS | 2 |
| B. ≥2 first-degree relatives with a II.A. ECG pattern | 2 |
| C. First-degree relative with a II.A. ECG pattern | 1 |
| D. Unexplained sudden cardiac death <45 years in a first- or second-degree relative | 0.5 |
| *Only award points once for highest score within this category | |
| V. Genetic Test Result | |
| A. Probable pathogenic ERS susceptibility mutation | 0.5 |
| Score (requires at least 1 ECG finding) | |
| ≥5 points: Probable/definite ERS | |
| 3–4.5 points: Possible ERS | |
| <3 points: Nondiagnostic |
ER = early repolarization; ERS = early repolarization syndrome; PVC = premature ventricular contraction; VF = ventricular fibrillation; VT = ventricular tachycardia.
Similarities and difference between BrS and ERS
BrS and ERS display several clinical similarities, suggesting similar pathophysiology (Table 6).19,21,103–105 Males predominate in both syndromes, with BrS presenting in 71%–80% among Caucasians and 94%–96% among Japanese.106,107 In the setting of ERP, VF occurred mainly in males (72%) when studied in an international cohort2 but in a much higher percentage in a report by Japanese investigators.108 BrS and ERS patients may be totally asymptomatic until they present with cardiac arrest. In both syndromes, the highest incidence of VF or SCD occurs in the third decade of life, perhaps related to testosterone levels in males.109 In both syndromes, the appearance of accentuated J waves and ST-segment elevation is generally associated with bradycardia or pauses.110,111 This can explain why VF in both syndromes often occurs during sleep or during a low level of physical activities.108,112 The QT interval is relatively short in patients with ERS,2,113 and BrS who carry mutations in calcium channel genes.114
Table 6.
Similarities and differences between Brugada and early repolarization syndromes and possible underlying mechanisms
| BrS | ERS | Possible Mechanism(s) | |
|---|---|---|---|
| Similarities between BrS and ERS | |||
| Male predominance | Yes (>75%) | Yes (>80%) | Testosterone modulation of ion currents underlying the epicardial AP notch |
| Average age of first event | 30–50 | 30–50 | |
| Associated with mutations or rare variants in KCNJ8, CACNA1C, CACNB2, CACNA2D, SCN5A, ABCC9, SCN10A | Yes | Yes | Gain of function in outward currents (IK-ATP) or loss of function in inward currents (ICa or INa) |
| Relatively short QT intervals in subjects with Ca channel mutations | Yes | Yes | Loss of function of ICa |
| Dynamicity of ECG | High | High | Autonomic modulation of ion channel currents underlying early phases of the epicardial AP |
| VF often occurs during sleep or at a low level of physical activity | Yes | Yes | Higher level of vagal tone and higher levels of Ito at the slower heart rates |
| VT/VF trigger | Short-coupled PVC | Short-coupled PVC | Phase 2 reentry |
| Ameliorative response to quinidine and bepridil | Yes | Yes | Inhibition of Ito and possible vagolytic effect |
| Ameliorative response to isoproterenol denopamine and milrinone | Yes | Yes | Increased ICa and faster heart rate |
| Ameliorative response to cilostazol | Yes | Yes | Increased ICa, reduced Ito and faster heart rate |
| Ameliorative response to pacing | Yes | Yes | Reduced availability of Ito due to slow recovery from inactivation |
| Vagally mediated accentuation of ECG pattern | Yes | Yes | Direct effect to inhibit ICa and indirect effect to increase Ito (due to slowing of heart rate) |
| Effect of sodium channel blockers on unipolar epicardial electrogram | Augmented J waves | Augmented J wave | Outward shift of balance of current in the early phases of the epicardial AP |
| Fever | Augmented J waves | Augmented J waves (rare) | Accelerated inactivation of INa and accelerated recovery of Ito from inactivation. |
| Hypothermia | Augmented J waves mimicking BrS | Augmented J waves | Slowed activation of ICa, leaving Ito unopposed. Increased phase 2 reentry but reduced pVT due to prolongation of APD358 |
| Differences between BrS and ERS | |||
| Region most involved | RVOT | Inferior LV wall | Higher levels of Ito and/or differences in conduction |
| Leads affected | V1–V3 | II, II a, VF, V4, V5, V6; I, aVL, Both: inferolateral | |
| Regional difference in prevalence | Europe: BrS = ERS | ||
| Asia: BrS > ERS | |||
| Incidence of late potential in signal- averaged ECG | Higher | Lower | |
| Prevalence of atrial fibrillation | Higher | Lower | |
| Effect of sodium channel blockers on surface ECG | Increased J-wave manifestation | Reduced J-wave manifestation | Reduction of J wave in the setting of ER is thought to be due largely to prolongation of QRS. Accentuation of repolarization defects predominates in BrS, whereas accentuation of depolarization defects predominates in ERS. |
| Structural changes, including mild fibrosis and reduced expression of Cx43 in RVOT or fibrofatty infiltration in cases of arrhythmogenic right ventricular cardiomyopathy. Imaging studies have also revealed wall motion abnormalities and mild dilation in the region of the RVOT. | Higher in some forms of the syndrome | Unknown | Some investigators have hypothesized that some of these changes may be the result of, rather than the cause of the BrS substrate, which may create a hibernation-like state due to loss of contractility in the RVOT secondary to loss of the AP dome. |
AP = action potential; APD = action potential duration; BrS = Brugada syndrome; ERS = early repolarization syndrome; RVOT = right ventricular outflow tract; PVC = premature ventricular contraction; pVT=polymorphic ventricular tachycardia; VF = ventricular fibrillation; VT = ventricular tachycardia.
As will be discussed in more detail later, ERS and BrS also share similarities with respect to the response to pharmacologic therapy. In both, electrical storms and associated J-wave manifestations can be suppressed using β- adrenergic agonists.115–118 Chronic oral pharmacologic therapy using quinidine,119,120 bepridil,117 denopamine,115,121 and cilostazol115,117,121–125 is reported to suppress the development of VT/VF in both ERS and BrS secondary to inhibition of Ito, augmentation of ICa, or both.3,122,126
Differences between the 2 syndromes include (1) the region of the heart most affected (RVOT vs inferior LV); (2) the presence of (discrete) structural abnormalities in BrS but not in ERS; (3) the incidence of late potentials in signal- averaged ECGs (BrS 60% 4 ERS 7%)108; and (4) greater elevation of Jo, Jp, or Jt (ST-segment elevation) in response to sodium channel blockers in BrS vs ERS and higher prevalence of atrial fibrillation in BrS vs ERS.127 Early studies suggested a different pathophysiologic basis for ERS and BrS based on the observation that sodium channel blockers unmask or accentuate J-wave manifestation in BrS but reduces the amplitude in ERS.108 However, the recent study by Nakagawa et al.357 showed that J waves recorded using unipolar LV epicardial leads introduced into the left lateral coronary vein in ERS patients are indeed augmented, even though J waves recorded in the lateral precordial leads are diminished, due principally to engulfment of the surface J wave by the widened QRS.29,108 The case report of Nakagawa et al. has recently been supplemented with additional cases in which this technique was used; 2 of these 3 cases showed pilsicainide-induced accentuation of the J waves in electrograms recorded from the epicardial surface of the LV (H. Morita, unpublished observations). Also in support of the thesis that these ECG patterns and syndromes are closely related are reports of cases in which ERS transitions into ERS plus BrS.105,128
The principal difference between BrS and ERS is related to the region of the ventricle most affected. Epicardial mapping studies in BrS patients report accentuated J waves and fragmented and/or late potentials in the epicardial region of the RVOT,129–131 whereas in ERS only accentuated J waves, particularly in the inferior wall of LV, are observed.29 Fractionated electrogram activity and late potentials have been observed in experimental models of ERS30 but have not yet been reported clinically. Noninvasive mapping electroanatomic studies have reported very steep localized repolarization gradients across the inferior/lateral regions of LV of ERS patients, preceded by normal ventricular activation,132 whereas in BrS both slow discontinuous conduction and steep dispersion of repolarization are present in the RVOT.133 Another presumed difference is the presence of structural abnormalities in BrS, which have not yet been described in ERS.76
Although J waves are accentuated or induced by both hypothermia and fever,33,34,134–139 the development of arrhythmias in ERS is much more sensitive to hypothermia, and arrhythmogenesis in BrS appears to be promoted only by fever.33,34,138,139 Hypothermia has been reported to increase the risk of VF in ERS,33,34,134,135,140 and fever is well recognized as a major risk factor in BrS.138,139 It is noteworthy that hypothermia can diminish the manifestation of a BrS ECG when already present.141,142
An ERP is associated with an increased risk for VF in patients with acute myocardial infarction143 and hypothermia.33,144 A concomitant ERP in the inferolateral leads has also been reported to be associated with an increased risk of arrhythmic events in patients with BrS. Kawata et al.145 reported that the prevalence of ER in inferolateral leads was high (63%) in BrS patients with documented VF.
Genetics
BrS has been associated with variants in 18 genes (Table 7). To date, more than 300 BrS-related variants in SCN5A have been described21,146–148Figure 2 shows the overlap syndromes attributable to genetic defects in SCN5A. Loss-of- function mutations in SCN5A contribute to the development of both BrS and ERS, as well as to a variety of conduction diseases, Lenegre disease, and sick sinus syndrome. The available evidence suggests that the presence of a prominent Ito determines whether loss-of-function mutations resulting in a reduction in INa will manifest as BrS/ERS or as conduction disease.59,149–151
Table 7.
Gene defects associated with the early repolarization syndrome (ERS) and Brugada (BrS) syndrome
| Genetic Defects Associated with ERS | ||||
|---|---|---|---|---|
| Locus | Gene/protein | Ion channel | Percent of Probands | |
| ERS1 | 12p11.23 | KCNJ8, Kir6.1 | ↑IK-ATP | Rare |
| ERS2 | 12p13.3 | CACNA1C, Cav1.2 | ↓ ICa | 4.1% |
| ERS3 | 10p12.33 | CACNB2b, Cavß2b | ↓ ICa | 8.3% |
| ERS4 | 7q21.11 | CACNA2D1, Cavα2δ1 | ↓ ICa | 4.1% |
| ERS5 | 12p12.1 | ABCC9, SUR2A | ↑ IK-ATP | Rare |
| ERS6 | 3p21 | SCN5A, Nav1.5 | ↓ INa | Rare |
| ERS7 | 3p22.2 | SCN10A, Nav1.8 | ↓ INa | Rare |
| Genetic Defects Associated with BrS | ||||
| BrS1 | 3p21 | SCN5A, Nav1.5 | ↓ INa | 11%–28% |
| BrS2 | 3p24 | GPD1L | ↓ INa | Rare |
| BrS3 | 12p13.3 | CACNA1C, Cav1.2 | ↓ ICa | 6.6% |
| BrS4 | 10p12.33 | CACNB2b, Cavß2b | ↓ ICa | 4.8% |
| BrS5 | 19q13.1 | SCN1B, Navß1 | ↓ INa | 1.1% |
| BrS6 | 11q13-14 | KCNE3, MiRP2 | ↑ Ito | Rare |
| BrS7 | 11q23.3 | SCN3B, Navß3 | ↓ INa | Rare |
| BrS8 | 12p11.23 | KCNJ8, Kir6.1 | ↑ IK-ATP | 2% |
| BrS9 | 7q21.11 | CACNA2D1, Cavα2δ1 | ↓ ICa | 1.8% |
| BrS10 | 1p13.2 | KCND3, Kv4.3 | ↑ Ito | Rare |
| BrS11 | 17p13.1 | RANGRF, MOG1 | ↓ INa | Rare |
| BrS12 | 3p21.2-p14.3 | SLMAP | ↓ INa | Rare |
| BrS13 | 12p12.1 | ABCC9, SUR2A | ↑ IK-ATP | Rare |
| BrS14 | 11q23 | SCN2B, Navß2 | ↓ INa | Rare |
| BrS15 | 12p11 | PKP2, Plakophillin-2 | ↓ INa | Rare |
| BrS16 | 3q28 | FGF12, FHAF1 | ↓ INa | Rare |
| BrS17 | 3p22.2 | SCN10A, Nav1.8 | ↓ INa | 5%–16.7% |
| BrS18 | 6q | HEY2 (transcriptional factor) | ↑ INa | Rare |
Listed in chronologic order of their discovery.
Figure 2.

Schematic showing overlap syndromes resulting from genetic defects resulting in loss of function of sodium channel current (INa) or gain of function in Late INa. In the absence of prominent Ito or IK-ATP, loss-of-function mutations in the inward currents result in various manifestations of conduction disease. In the presence of prominent Ito or IK-ATP, loss-of-function mutations in inward currents cause conduction disease as well as the J-wave syndromes (Brugada and early repolarization syndromes). Early repolarization syndrome is believed to be caused by loss-of-function mutations of inward current in the presence of prominent Ito in certain regions of the left ventricle, particularly the inferior wall of the left ventricle. The genetic defects that contribute to Brugada syndrome and early repolarization syndrome can also contribute to the development of long QT and conduction system disease, in some cases causing multiple expressions of these overlap syndromes. In some cases, structural defects contribute to the phenotype. PVT = polymorphic ventricular tachycardia; VF = ventricular fibrillation.
Variants in CACNA1C (Cav1.2), CACNB2b (Cavβ2b), and CACNA2D1 (Cavα2δ) have been reported in up to 13% of probands.152–155 Mutations in glycerol-3-phophate dehydrogenase 1-like enzyme gene (GPD1L), SCN1B (β1 subunit of Na channel), KCNE3 (MiRP2), SCN3B (β3 subunit of Na channel), KCNJ8 (Kir6.1), KCND3 (Kv4.3), RANGRF (MOG1), SLMAP, ABCC9 (SUR2A), (Navβ2), PKP2 (plakophillin-2), FGF12 (FHAF1), HEY2, and SEMA3A (semaphorin) are relatively rare.156–176 An association of BrS with SCN10A, a neuronal sodium channel, was recently reported.167,177,178 A wide range of yields of variants was reported by the 2 studies that examined the prevalence of pathogenic SCN10A mutations and rare variants (5%– 16.7%).177–179 Mutations in these genes lead to loss of function in sodium (INa) and calcium (ICa) channel currents, as well as to a gain of function in transient outward potassium current (Ito) or ATP-sensitive potassium current (IK-ATP).178
New susceptibility genes recently proposed and awaiting confirmation include the transient receptor potential melastatin protein-4 gene (TRPM4)180 and the KCND2 gene. The mutation uncovered in KCND2 in a single patient was shown to cause a gain of function in Ito when heterologously expressed.181
Variants in KCNH2, KCNE5, and SEMA3A, although not causative, have been identified as capable of modulating the substrate for the development of BrS.182–185 Loss-of-function mutations in HCN4 causing a reduction in the pacemaker current If can unmask BrS by reducing heart rate.186
An ERP in the ECG has been shown to be familial.187–189 ERP and ERS have been associated with variants in 7 genes. Consistent with the findings that IK-ATP activation can generate an ERP in canine ventricular wedge preparations, variants in KCNJ8 and ABCC9, responsible for the pore- forming and ATP-sensing subunits of the IK-ATP channel, have been reported in patients with ERS.156,158,190 Loss-of- function variations in the α1, β2, and α2δ subunits of the cardiac L-type calcium channel (CACNA1C, CACNB2, CACNA2D1) and the α1 subunit of NaV1.5 and NaV1.8 (SCN5A, SCN10A) have been reported in patients with ERS.113,152,177
It is important to point out that only a small fraction of identified genetic variants in the genes associated with BrS and ERS have been examined using functional expression studies to ascertain causality and establish a plausible contribution to pathogenesis. Only a handful have been studied in genetically engineered animal models, and very few have been studied in native cardiac cells or in induced pluripotent stem cell-derived cardiac myocytes isolated from ERS and BrS patients. Computational strategies developed to predict the functional consequences of mutations are helpful, but these methods have not been rigorously tested. The lack of functional or biologic validation of mutation effects remains the most severe limitation of genetic test interpretation, as recently highlighted by Schwartz et al.191
Recent technological advances have resulted in expansion of disease-specific panels.192 Large public databases of genetic variation from next-generation sequencing programs such as the 1000 Genomes Project, the National Heart Lung and Blood Institute Grand Opportunity Exome Sequencing Project (GO-ESP), and the Exome Aggregation Consortium (ExAC), have challenged drastically our understanding of the “normal” burden and extent of background genetic variation within cardiac channelopathy susceptibility genes.193–195
Although SCN5A variants account for 18%–28% of BrS,196SCN5A genetic testing is complicated by an approximately 3%–5% “benign” variant frequency in the general population.194 Therefore, even in the most common genetic cause of BrS, 1 in 10 “positive” tests could be a “false- positive” even if found in an individual with a robust BrS phenotype. To date, there are more than 20 JWS susceptibility genes.146,195,197 However, these additional genes have only magnified the issues of interpretation by adding to the overall “genetic noise” without significantly increasing the true mutation yield.178,198–200 In fact, 1 study revealed that 1:23 individuals in the GO-ESP population possess a previously published BrS-associated variant that would prompt a “positive” genetic test had it been identified in a patient.201
These issues reinforce the necessity to interpret JWS genetic test results as strictly probabilistic, rather than binary/deterministic, in nature. Additional lines of evidence202 can be amassed to aid in the probabilistic interpretation of variants in JWS susceptibility genes, such as case phenotype,203 segregation, functional studies,204in silico predictions,205–208 variant type and location,194 and variant frequency in cases and control databases.193 Despite these aids, a large number of variants remain in “genetic purgatory,” and this number will only increase as exome/genome sequencing becomes more utilized. This then demands the development and utilization of a uniform variant repository that would include clinical assertions and evidence for variant classification. Even with these issues, the emergence of exome/genome sequencing holds promise for the opportunity to study genetic variation like never before, holding the promise of improvements in diagnostic, prognostic, and therapeutics for the JWSs and the other heritable cardiac channelopathies. Kapplinger et al.209 recently reported the synergistic use of up to 7 in silico tools to help promote or demote a variant's pathogenic status and alter its relegation to genetic purgatory.
It is noteworthy that in a recent study, Le Scouarnec et al.199 estimated the burden of rare coding variation in arrhythmia susceptibility genes among 167 BrS index patients and compared that with 167 individuals ≥65 years old with no history of cardiac arrhythmia. The authors concluded that, except for SCN5A, rare coding variations in all previously reported BrS susceptibility genes do not contribute significantly to the occurrence of BrS in a population of European ancestry, emphasizing that caution should be taken when interpreting genetic variations in these other genes because rare coding variants are observed to a similar extent in both cases and controls.199 Similar data were obtained and a similar conclusion was reached by Kapplinger et al.209 by analyzing the prevalence of rare variants in the BrS susceptibility genes in the publicly available ExAC exomes.
Collectively, these data suggest the possibility that. in the individual patient, BrS and the susceptibility to VF and SCD may not be due to a single mutation (classic mendelian view) but rather to inheritance of multiple BrS susceptibility variants (oligogenic) acting in concert through one or more mechanistic pathways.167 This also fits with the findings of Probst et al.210 that in 5 of 13 large families with a putative SCN5A mutation, the genotype did not co-segregate with the phenotype. In addition to the multifactorial nature of the genetics, expressivity of the syndrome may be multifactorial in that the genetic predisposition can be modulated by hormonal (testosterone,211,212 thyroxine213) and other environmental factors, as well as morphologic changes (fibrosis).76
Update on the ionic and cellular mechanisms underlying BrS and ERS
The JWSs are so named because they involve accentuation of the ECG J wave. Experimental evidence indicates that the J wave is inscribed as a consequence of a transmural voltage gradient caused by the manifestation of an AP notch in epicardium but not endocardium due to a heterogeneous transmural distribution of Ito.104 An end of QRS notch, resembling a J wave, has been proposed to be due to intraventricular conduction delays. The 2 ECG manifestations can be distinguished based on their response to rate, with the latter showing accentuation at faster rates.24,59
The cellular mechanisms underlying JWS have long been a matter of debate.214,215 In the case of BrS, 2 principal hypotheses have been advanced. (1) The repolarization hypothesis asserts that an outward shift in the balance of currents in RV epicardium can lead to repolarization abnormalities resulting in the development of phase 2 reentry, which generates closely coupled premature beats capable of precipitating VT/VF. (2) The depolarization hypothesis suggests that slow conduction in the RVOT, secondary to fibrosis and reduced Cx43 leading to discontinuities in indeterminate conduction, plays a primary role in the development of the ECG and arrhythmic manifestations of the syndrome. Conduction slowing is not necessarily limited to the RVOT area. Some investigators have postulated that changes in ion channel current responsible for BrS (i.e., loss of function INa and ICa and gain of function of Ito) can alter AP morphology so as to reduce the safety of conduction at high-resistance junctions, such as regions of extensive fibrosis.216,217 Others have argued that this is highly unlikely because conduction at critical junctions of current-to-load mismatch is exquisitely sensitive to changes in rate. The typical behavior of patients with BrS to acceleration of rate is diminution of ST-segment elevation, opposite to that expected at a site of discontinuous conduction. The diminution of ST-segment elevation is consistent with the reduced availability of Ito at the faster rate due to slow recovery of the current from inactivation.59,214 The repolarization and depolarization theories are not necessarily mutually exclusive and may indeed be synergistic.
The most compelling apparent evidence in support of the depolarization hypothesis derives from the seminal studies of Nademanee et al.129 showing that radiofrequency ablation (RFA) of epicardial sites displaying late potentials and fractionated bipolar electrograms in the RVOT of patients with BrS significantly reduced the arrhythmia vulnerability as well as the ECG manifestation of the syndrome. Similar results were reported by Brugada et al.131 and by Sacher et al,130 who also observed in an isolated case that accentuation of the Brugada ECG by ajmaline was associated with an increased area of low-voltage and fragmented electrogram activity. A wider area of low-voltage activity was associated with a more prominent ST-segment elevation.131 These authors concluded that the late potentials and fractionated electrogram activity are due to conduction delays within the RVOT/RV anterior wall and that ablation of the sites of slow conduction is the basis for the ameliorative effect of ablation therapy.129–131 In a direct test of this hypothesis, Szel and Antzelevitch150 provided evidence for an alternative mechanism using an experimental model of BrS. The low-voltage fractionated electrogram activity was shown to develop as a result of regional desynchronization in the appearance of the second AP upstroke, secondary to accentuation of the epicardial AP notch, and high-frequency late potentials to develop in the RV epicardium secondary to concealed phase 2 reentry. Delayed conduction of the primary beat was never observed in a wide variety of BrS models created by exposing canine RV wedge preparations to drugs mimicking the different genetic defects known to give rise to BrS.150 In more recent studies, ablation of the RV epicardium was shown to diminish the manifestation of J waves and ST-segment elevation and to abolish all arrhythmic activity by destroying the cells with the most prominent AP notch, thus eliminating the cells responsible for the repolarization abnormalities that give rise to phase 2 reentry and VT/VF.15,218 Confirmation of all of these results in in vivo animal models is desirable. In an attempt to create such a model, Park et al.149 recently genetically engineered Yucatan minipigs to heterozygously express a nonsense mutation in SCN5A (E558X) originally identified in a child with BrS. Patch clamp analysis of atrial myocytes isolated from the SCN5AE558X/+ pigs showed a loss of function of INa. Conduction abnormalities consisting of prolongation of P wave, QRS complex, and PR interval were observed, but a BrS phenotype was not observed, not even after administration of flecainide. These observations are expected because of the lack of Ito in the pig, which is a prerequisite for the development of the repolarization abnormalities associated with BrS. Some have argued that the absence of a BrS phenotype is due to the young age of the minipigs (22 months).219 However, it is difficult to reconcile why the minipigs manifest major conduction delays at this age but not a BrS phenotype, if indeed the latter depends on the former. Finally, it is noteworthy that monophasic APs recorded from the epicardial and endocardial surfaces of the RVOT of a patient with BrS are nearly identical to transmembrane APs recorded from the epicardial and endocardial surfaces of the wedge model of BrS.220,221 These differences were not observed in an isolated heart explanted from a BrS patient after transplantation of a new heart. However, the epicardium of this heart was very depressed, perhaps as a result of the 129 shocks delivered by the implantable cardioverter-defibrillator (ICD) in an attempt to control the multiple electrical storms.32
Zhang et al.133 recently performed noninvasive electrocardiographic imaging (ECGI) on 25 BrS and 6 RBBB patients. The authors concluded that both slow discontinuous conduction and steep dispersion of repolarization are present in the RVOT of patients with BrS. ECGI was able to differentiate between BrS and RBBB. Unlike BrS, RBBB showed delayed activation in the entire RV, without ST- segment elevation, fractionation, or repolarization abnormalities showing on the electrograms. Importantly, the response to an increase in rate was studied in 6 BrS patients. Increasing rate increased fractionation of the electrogram but reduced ST-segment elevation, indicating that the conduction impairment was not the principal cause of the BrS ECG.
The congruence between BrS and ERS with respect to clinical manifestations and response to therapy lends further support to the repolarization hypothesis. Using an experimental model of ERS, Koncz et al.30 recently provided evidence in support of the hypothesis that, similar to the mechanism operative in BrS, an accentuation of transmural gradients in the LV wall are responsible for the repolarization abnormalities underlying ERS, giving rise to J-point elevation, distinct J waves, or slurring of the terminal part of the QRS. The repolarization defect is accentuated by cholinergic agonists and reduced by quinidine, isoproterenol, cilostazol, and milrinone, accounting for the ability of these agents to reverse the repolarization abnormalities responsible for ERS.30,222 Higher intrinsic levels of Ito in the inferior LV were also shown to underlie the greater vulnerability of the inferior LV wall to VT/VF.30 The advent and implementation of ECGI by Rudy and colleagues provided additional evidence for repolarization abnormalities by identifying abnormally short activation–recovery intervals in the inferior and lateral regions of LV and a marked dispersion of repolarization.132 More recent studies involving ECGI mapping in an ERS patient during VF have demonstrated VF rotors anchored in the inferior lateral LV wall.22
Conduction delay is known to give rise to notching of the QRS complex. When it occurs on the rising phase of the R wave, it is due to a conduction defect within the ventricle. When it occurs at the terminal portion of the QRS, thus masquerading as a J wave, it may be due to either a conduction defect or a repolarization defect.21,223 The response to prematurity or to an increase in rate can differentiate between the two.59 Delayed conduction invariably becomes more exaggerated at faster rates or during premature beats, thus leading to accentuation of the QRS notch, whereas repolarization defects usually are mitigated, resulting in diminution of the J wave at faster rates. Although typical J waves usually are accentuated with bradycardia or long pauses, the opposite has also been described.224,225 J waves are often seen in young males with no apparent structural heart diseases, whereas intraventricular conduction delay is often observed in older individuals or those with a history of myocardial infarction or cardiomyopathy.223,224 The prognostic value of a fragmented QRS has been demonstrated in BrS,49,226 although fragmentation of the QRS is not associated with increased risk in the absence of cardiac disease.227 Factors that may aid in the differential diagnosis of J wave vs intraventricular conduction delay (IVCD)-mediated syndromes are summarized in Table 8.
Table 8.
Differential diagnosis of J wave vs intraventricular conduction defect–mediated notch syndromes (IVCD)
| J wave | IVCD-induced end QRS notch | |
|---|---|---|
| Male predominance | Yes | No |
| Average age at initial presentation | Young adults | Older adults |
| Most common morphology | Dome-like smooth appearance | Relatively sharp appearance |
| Response to change in heart rate | Bradycardia- and pause-dependent augmentation of J wave, which may be accompanied by T-wave inversion | Tachycardia and prematurity-dependent augmentation of the notch |
| Structural heart diseases | Rare | Common |
| History of myocardial infarction and/or cardiomyopathy |
Risk stratification
J-wave syndromes
A great deal of attention has been devoted to risk assessment for the development of life-threatening arrhythmias in BrS and ERS.1,228 The incidental discovery of a J wave on routine screening should not be interpreted as a marker of “high risk” for SCD because the odds for this fatal disease are approximately 1:10,000.229 Rosso et al. indicated that the presence of a J wave on the ECG increases the probability of VF from 3.4:100,000 to 11:100,000.4,230 However, careful attention needs to be paid to subjects with “high-risk” ER or J waves. Figure 3 illustrates the various ECG manifestations of ER. Figure 4 shows a graphic representation of the prevalence and arrhythmic risk associated with the appearance of ECG J waves and clinical manifestations of BrS and ERS. Tables 9, 10, and 11 list the available data from studies designed to identify patients at high risk for BrS and ERS. Among these risk stratifiers, some are highly predictive, including (1) history of cardiac events or syncope likely due to VT/VF and (2) prominent J waves in global leads including type 1 ST-segment elevation in the right precordial leads (Figure 5).
Figure 3.
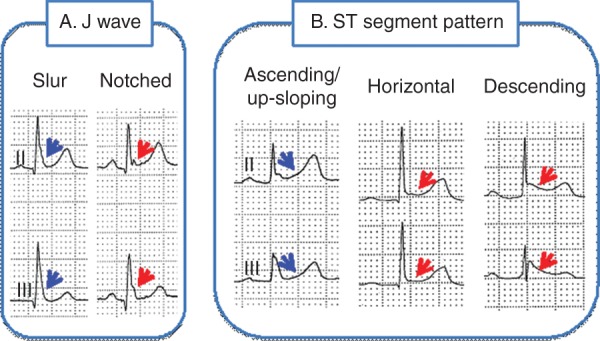
Different manifestations of early repolarization. A: The J wave may be distinct or appear as a slur. In the latter case, part of the J wave is buried inside the QRS, resulting in an elevation of Jo. Patients with a distinct J wave have a worse prognosis than do patients with a slurred J wave. B: The ST segment may be upsloping, horizontal, or descending. Horizontal and descending ST segments are associated with a worse prognosis.
Figure 4.

Prevalence and arrhythmic risk associated with the appearance of ECG J waves and clinical manifestations of Brugada and early repolarization syndromes. Yellow highlighted region estimates the prevalence of the J-wave syndromes. J waves in the lateral ECG leads have a high prevalence but are associated with a very low arrhythmic risk in a relatively small fraction of the cohort of individuals displaying J waves. On the other extreme, J waves appearing globally in the ECG have a very low prevalence but are associated with a very high level of arrhythmic risk in a large fraction of the cohort presenting with J waves. Likewise, individuals displaying rapidly ascending ST-segment elevation have a high prevalence but low risk, whereas subjects resuscitated from cardiac arrest have a very low prevalence but the highest level of arrhythmic risk.
Table 9.
Clinical variables associated with an increased risk of major arrhythmic events in Brugada syndrome*
| Variable | No. patients | Prevention | Study endpoint | Multivariable analysis [hazard ratio (95% confidence interval), P value] | Reference |
|---|---|---|---|---|---|
| History | |||||
| Previous VF | 93 | P/S | SD, cardiac arrest, or sustained VT/VF (N = 25) | N/A (N/A), .005 | Makimoto292 |
| Cardiac arrest | 1029 | P/S | SD (N = 7), appropriate ICD shocks (N = 44), or sustained VT/VF (N = 0) | 11 (4.8–24.3), .001 | Probst359 |
| Syncope or cardiac arrest | 460 | P/S | VF or SD (N = 38) | 12.7 (4.5–53.4), <.0001 | Takagi360 |
| Syncope of unknown origin | 547 | P | SD (N = 16), VF (N = 29) | 2.5 (1.2–5.3), .017 | Brugada361 |
| Syncope | 44 | P/S | SCD (N = 5), polymorphic VT or VF (recorded by ECG, Holter, or ICD) (N = 7), or syncope of unknown etiology (11) | 3.6 (1.09–11.7), .035 | Huang362 |
| Syncope + spontaneous type 1 ECG | 200 | P/S | VF or SD from birth (N = 22) | 6.4 (1.9–21), <.002 | Priori363 |
| Syncope of probable arrhythmic origin | 1029 | P/S | SD (N = 7), appropriate ICD shocks (N = 44), or sustained VT/VF (N = 0) | 3.4 (1.6–7.4), .002 | Kamakura364 |
| Syncope | 320 | P | SD (N = 3), appropriate ICD shocks (N = 14), or sustained VT/VF (N = 0) |
2.8 (1.1–8.1), .03 | Delise262 |
| Syncope + spontaneous type 1 ECG | 308 | P | VF (N = 1) or appropriate ICD intervention (N = 13) | 4.2 (1.4–12.8), .012 | Priori49 |
| Ventricular refractoriness | |||||
| Ventricular refractory period <200 ms | 308 | P | VF (N = 1) or appropriate ICD intervention (N = 13) | 3.9 (1.03–12.8), .045 | Priori49 |
| ECG characteristics | |||||
| Spontaneous type 1 ECG | 1029 | P/S | SD (N = 7), appropriate ICD shocks (N = 44), or sustained VT/VF (N = 0) | 1.8 (1.03–3.3), .04 | Probst247 |
| Spontaneous type 1 ECG | 320 | P | SD (N = 3), appropriate ICD shocks (N = 14) or sustained VT/VF (N = 0) |
6.2 (1.8–40), .002 | Delise262 |
| QRS fragmentation (≥2 spikes within the QRS complex in leads V1–V3) | 308 | P | VF (N = 1) or appropriate ICD intervention (N = 13) | 4.9 (1.5–1.8), .007 | Priori49 |
| Family history of sudden cardiac death at age 45 years | 330 | P | VF (N = 56), syncope (N = 67), or asymptomatic (N = 207) | 3.28 (1.4–7.6), .005 | Kamakura364 |
| J wave in inferior and lateral leads | 330 | P | VF (N = 56), syncope (N = 67), or asymptomatic (N = 207) | 2.66 (1.1–6.7), .005 | Kamakura364 |
| QRS duration >90 ms in lead V2 | 460 | P/S | VF or SD (N = 38) | 3.6 (1.4–12.2), .007 | Takagi360 |
| Horizontal ST segment after J wave (ST-segment elevation ≤0.1 mV within 100 ms after J point and continued as a flat ST segment until onset of T wave in ≥1 lead with J wave) | 460 | P/S | VF or SD (N = 38) | >10 (1.9–20.2), .02 | Takagi360 |
| Late potentials (root mean square voltage of terminal 40 ms of filtered QRS complex <20 μV + duration of low-amplitude signals <40 μV of QRS in terminal filtered QRS complex >38 ms) | 44 | P/S | SCD (N = 5), polymorphic VT or VF (recorded by ECG, Holter, or ICD) (N = 7), or syncope of unknown etiology (11) | 10.9 (1.1–104), .038 | Huang362 |
| ST-segment augmentation at early recovery of exercise test (ST-segment amplitude increase ≥0.05 mV in at least 1 of V1–V3 leads at 1–4 minutes of recovery compared with ST-segment amplitude at pre-exercise) | 93 | P/S | SD, cardiac arrest, or sustained VT/VF (N = 25). | N/A (N/A), .007 | Makimoto292 |
ICD = implantable cardioverter-defibrillator; N/A = not available; P = primary prevention patients only; P/S = primary and secondary prevention patients; SD = sudden death, VF = ventricular fibrillation, VT = ventricular tachycardia
*The list includes predictor variables that have been associated with an increased risk of major arrhythmic events (i.e., SCD, appropriate ICD interventions, or ICD therapy on fast VT/VF) in at least 1 published multivariable analysis in prospective studies.
Table 10.
Prognostic value of programmed ventricular stimulation resulting from multivariate analysis in large multicenter studies on Brugada syndrome
| No. | Prevention | Inducibility | Multivariable hazard ratio (95% confidence interval), P value | Reference |
|---|---|---|---|---|
| 408 | P | 40% | 5.88 (2.0–16.7), <.001 | Brugada364 |
| 308 | P | 41% | 0.89 (0.3–2.6), .84 | Priori49 |
| 638 | P/S | 62% | N/A, 0.48 | Probst247 |
| 245 | P | 39% | Not tested on multivariable analysis, but an analysis based on C–statistics demonstrated that results of electrophysiologic studies in combination with other risk factors provided additional value for risk stratification | Delise262 |
| 334 | P/S | 67% | 0.63 (0.3–1.3), .20* | Takagi360 |
| 1312† | P/S | 42% | 2.66 (1.44–4.92), .002 | Sroubek50 |
N/A = not available, P = primary prevention patients only, P/S = primary and secondary prevention patients.
*Univariate analysis; not included in multivariable analysis.
†Pooled analysis from 8 international databases.
Table 11.
Early repolarization patterns associated with idiopathic ventricular fibrillation, cardiac death, or all-cause mortality
| Study design | Patient population | Early repolarization patterns | Endpoint | Odds/Hazard ratio (95% confidence intervals) P value* | Reference |
|---|---|---|---|---|---|
| Case-control | 206 idiopathic VF 412 matched controls |
J-point elevation ≥0.1 mV | Idiopathic VF | 10.9 (6.3–18.9) | Haissaguerre2 |
| Case-control | 45 idiopathic VF 124 matched controls 121 noncompetitive athletes |
J-point elevation in inferior leads | Idiopathic VF | 3.2 (1.4–7.5), P = .006 | Rosso4 |
| J-point elevation in I/aVL | Idiopathic VF | 16.9 (2.0–140), P = .009 | |||
| J-point elevation in V4–V6 | Idiopathic VF | NS | |||
| Case-control | 45 idiopathic VF 124 matched controls 121 noncompetitive athletes |
J-point elevation | Idiopathic VF | 4.0 (2.0–7.9) | Rosso245 |
| J-point elevation + horizontal ST segment | Idiopathic VF | 13.8 (5.1–37.2) | |||
| Case-control | 21 athletes with idiopathic VF 365 controls athletes |
J-point elevation ≥0.1 mV in inferolateral leads | Idiopathic VF | 4.63 (1.67–12.9), P = .007 | Cappato365 |
| QRS slurring in any lead | Idiopathic VF | 4.81 (1.73–13.4), P = .007 | |||
| Prospective | 10,864 middle-aged people enrolled in the Finnish Social Insurance Institution's Coronary Heart Disease Study (CHD study) between 1966 and 1972 | J-point elevation ≥0.1 mV in inferior leads | Death from cardiac causes | 1.28 (1.04–1.59), P = .03 | Tikkanen44 |
| Death from arrhythmias | 1.43 (1.06–1.94), P = .03 | ||||
| J-point elevation ≥0.2 mV in inferior leads | Death from any cause | 1.54 (1.06–2.24), P = .03 | |||
| Death from cardiac causes | 2.98 (1.85–4.92), P <.001 | ||||
| Death from arrhythmias | 2.92 (1.45–5.89), P = .01 | ||||
| Prospective | 10,864 middle-aged people enrolled in the Finnish Social Insurance Institution's Coronary Heart Disease Study (CHD study) between 1966 and 1972 | J-point elevation ≥0.1 mV and horizontal/descending ST segment | Sudden death | 1.43 (1.05–1.94) | Tikkanen237 |
| J-point elevation ≥0.1 mV and upsloping ST segment | Sudden death | NS | |||
| Prospective | 1161 middle-aged people enrolled in the third French Monitoring Trends and Determinants in Cardiovascular Disease (MONICA) Project between 1994 and 1997 | J-point elevation ≥0.1 mV | Total mortality | 2.45 (1.44–4.15), P = .001 | Rollin236 |
| Cardiovascular mortality | 5.6 (2.27–11.8), P = .001 | ||||
| J-point elevation ≥0.2 mV | Total mortality | NS | |||
| Cardiovascular mortality | 5.14 (1.72–15.4), P = .004 | ||||
| J-point elevation ≥0.1 mV in inferior leads | Total mortality | 2.85 (1.62–5.02), P = .001 | |||
| Cardiovascular mortality | 5.28 (1.96–14.2), P = .001 | ||||
| J-point elevation ≥0.1 mV in lateral leads | Total mortality | NS | |||
| Cardiovascular mortality | 6.27 (1.85–21.3), P = .003 | ||||
| J-point elevation ≥0.1 mV and horizontal ST-segment elevation | Total mortality | 3.04 (1.71–5.41), P = .001 | |||
| Cardiovascular mortality | 6.93 (2.75–17.4), P = .001 | ||||
| J-point elevation ≥0.1 mV and ascending ST-segment elevation | Total mortality | NS | |||
| Cardiovascular mortality | NS | ||||
| J-point elevation ≥0.1 mV with notching pattern | Total mortality | 3.11 (1.72–5.6), P = .001 | |||
| Cardiovascular mortality | 8.32 (3.32–20.8), P = .001 | ||||
| J-point elevation ≥0.1 mV with slurring pattern | Total mortality | NS | |||
| Cardiovascular mortality | NS | ||||
| Prospective | 15,792 middle-aged biracial people enrolled in the US Atherosclerosis Risk in Communities (ARIC) between 1987 and 1989 | J-point elevation ≥0.1 mV in white men | Sudden death | NS | Olson238 |
| Coronary events | NS | ||||
| All cause mortality | NS | ||||
| J-point elevation ≥0.1 mV in white women | Sudden death | 8.77 (3.19–24.13) | |||
| Coronary events | NS | ||||
| All cause mortality | NS | ||||
| J-point elevation ≥0.1 mV in black men | Sudden death | NS | |||
| Coronary events | NS | ||||
| All cause mortality | NS | ||||
| J-point elevation ≥0.1 mV in black women | Sudden death | NS | |||
| Coronary events | 1.47 (1.03–2.09) | ||||
| All cause mortality | NS | ||||
| Prospective | 29,281 subjects evaluated at the Palo Alto Veterans Affairs Hospital | J-point elevation >0.1 mV in black individuals | Cardiovascular death | NS | Perez-Riera367 |
| J-point elevation >0.1 mV in non-black individuals | Cardiovascular death | 1.6, P = .02 |
NS = not significant, VF = ventricular fibrillation.
*Adjusted odds ratio/hazard ratio reported when available.
Figure 5.
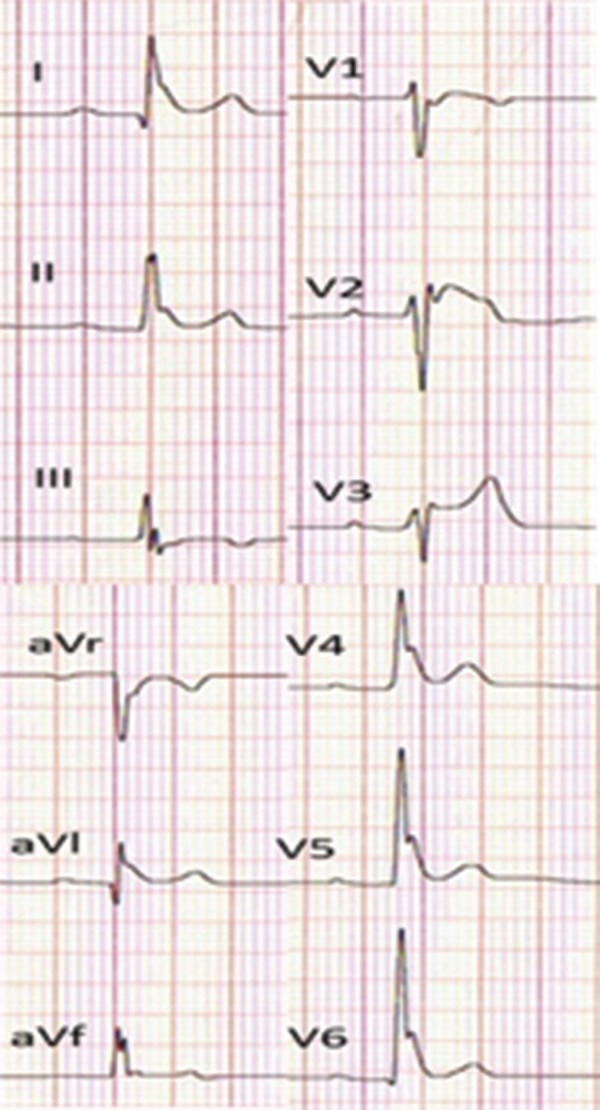
Global early repolarization (type 3 early repolarization). J waves are apparent in the inferior, lateral, and anterior (right precordial) leads.
Early repolarization syndrome
The majority of the studies using the criteria of Haissaguerre et al.2 for diagnosing the ERP have shown that ER, especially in the inferior ECG leads, predicts cardiac and arrhythmic death. Negative studies are few and may be attributable to the exclusion criteria used (e.g., atrial fibrillation, flutter, acute coronary syndrome), a relatively short follow-up period,231,232 or different definitions of ERP.233 The recent consensus paper by Macfarlane et al.24 dealing with the terminology of J-wave–related phenomena in the setting of ER should enable us to avoid such confusion in the future. The inclusion of Africans or African-Americans, in whom ER is prevalent but apparently not associated with high risk, may alter outcomes as well.234
Huikuri and colleagues reported in a series of seminal papers the results of a population-based study in Finland involving long-term prognosis of subjects with an ERP in the ECG.44 Tikkanen et al.44 showed that J-point elevation ≥0.1 mV was present in 5.8% of the population and that only 0.3% of the population had significant J-point elevation ≥0.2 mV. J-point elevation ≥0.1 mV in the inferior leads was associated with cardiovascular death (relative risk [RR] 1.28) and arrhythmic death (RR 1.43), and J-point elevation ≥0.2 mV had a markedly elevated risk of death from cardiac causes (RR 2.92) and from arrhythmia (RR 2.92). Subsequent studies confirmed the association of J wave or ER with death from all causes, death from cardiovascular disease, sudden/unexpected death, and death from arrhythmias.44–46,127,235–238 A horizontal or descending ST segment is associated with a worse prognosis than is an ascending ST segment (Figure 3).237,239 Individuals with a high-amplitude J wave ≥0.2 mV followed by a horizontal or descending ST segment in the inferior/inferolateral leads have a higher risk of lethal arrhythmias than do those with a lower-amplitude J wave, especially those with a rapidly ascending ST segment following the J wave.
The appearance of J wave or ER is now recognized to predispose to the development of arrhythmogenesis when associated with other cardiac disorders, such as ischemia, heart failure, and hypothermia. The J wave might predict prognosis of cardiac events in various heart diseases, and the appearance of a new J wave during acute ischemia seems to be a messenger of VF.240,241
Family history of sudden death in subjects with ERP has been identified as a risk factor.189,242 The presence of coexisting Brugada ECG pattern (J waves in V1–V3) or short QT intervals in subjects with ER also suggests a more malignant nature.243,244
ER is commonly observed in the young, especially in fit and highly trained athletes, with a prevalence ranging up to 40%. In the majority of cases, the ensuing ST segment is rapidly ascending, suggesting that this is a benign ECG manifestation.237,245
Mahida et al.246 recently reported that electrophysiologic (EP) study using programmed stimulation protocols does not enhance risk stratification in ERS.
Brugada syndrome
Numerous studies consistently show that clinical presentation is the strongest predictor of risk in BrS, overshadowing all other risk factors. The risk of recurrent VF among patients presenting with cardiac arrest is considerable: ≈35% at 4 years,247,248 44% at 7 years,249 and 48% at 10 years.250 Fortunately, only a minority of patients with BrS (6% in Europe247 but 18% in Japan248) diagnosed today have a history of cardiac arrest.
Approximately one-third of contemporary BrS cohorts present with syncope.247 Their risk of arrhythmic events during follow-up is intermediate: approximately 4 times higher than the risk of asymptomatic patients49,247,251 but 4 times lower than that of patients diagnosed after cardiac arrest.247 One explanation for this observation is that the syncope population consists of 2 different groups, one with arrhythmic syncope and bad prognosis, and a second with vagal syncope and good prognosis. Although a detailed clinical history may be of great value in differentiating between these 2 groups, it is not infallible.252 In reviewing the records of 342 BrS patients, Olde Nordkamp et al.60 concluded that arrhythmic and nonarrhythmic syncope can be distinguished by clinical characteristics, including the absence of prodromes and specific triggers. Compared to patients with suspected nonarrhythmic syncope, patients presenting with presumed arrhythmic syncope were more likely to be male (RR 2.1) and to have urinary incontinence (RR 4.6) and were less likely to report prodromes. They also were older at first event (45 vs 20 years), and their syncope was never triggered by hot/crowded surroundings, pain or other emotional stress, sight of blood, or prolonged standing as in the case of nonarrhythmic syncope. During follow-up, all of the spontaneous arrhythmic events occurred in patients who originally presented with presumed arrhythmic syncope; patients with benign syncope had an excellent long-term prognosis.
Today asymptomatic patients represent a majority (∼63%) of newly diagnosed Brugada patients.247,248 Their risk of developing symptoms is relatively low (0.5% per year).247,248 Unfortunately, for most the first symptom is cardiac arrest or SCD. Therefore, risk stratification of asymptomatic patients is of utmost importance, and strategies for doing so are discussed later. In cardiac arrest patients or patients with presumed arrhythmic syncope, these strategies are of little benefit because these patients are recognized to be at high risk.
Age and gender
Mean age at the time of cardiac arrest in Brugada patients is 39–48 years, and the vast majority develop symptoms between 20 and 65 years of age.247–249 Asymptomatic elderly patients with BrS are thought to be at relatively low risk for future cardiac events.253 BrS in children is very rare, but sudden death in this population has been described.65,254,255 As in adults, cardiac arrest survivors are at high risk for recurrence, but data regarding risk stratification of asymptomatic children are limited. In a large series, 64%–94% of patients with BrS who presented with cardiac arrest were male.247,248,250 Males are also at increased risk for displaying a spontaneous type I Brugada ECG and for having inducible VF during EP studies.106 Nevertheless, because the majority of asymptomatic patients are also male, gender is not an independent predictor of arrhythmic events.106,248
Familial and genetic background
Neither family history of SCD nor the presence of a mutation (of any type) in the SCN5A gene has consistently been demonstrated to be of value in risk stratification.247–249 However, 1 study has shown that SCN5A mutations resulting in protein truncation do confer greater risk.256 Certain rare variants and polymorphisms in SCN5A and in other genes have also been associated with prognosis.167,257–261 Nevertheless, the data are limited, and genetic testing is not generally used for risk stratification at this time.
Spontaneous vs drug-induced type I Brugada pattern
A consistent finding in nearly all BrS series is that patients with spontaneous type I ECG at the time of diagnosis have a greater risk of arrhythmic events than do patients who develop such an ECG pattern only when challenged with a sodium channel blocker.247,262 This observation is true for asymptomatic patients247 as well as for patients who present with syncope49,247,251,263 and remains an independent predictor of arrhythmic events in multivariate analysis. The problem is that only a minority of patients have a consistent spontaneous type 1 pattern when repeated ECGs are analyzed.264,265 Therefore, caution is needed when using a single ECG for risk stratification.
Ventricular arrhythmias are rarely induced during a sodium channel block challenge65,262,266,267 and are likely to be dose dependent (i.e., will not occur when the infusion is stopped for safety reasons before the full dose is reached).
Their long-term prognostic significance remains unclear. Extra caution should be exercised when administering sodium channel blockers to patients with significant conduction disease.
Placing the right precordial leads over the 2nd and 3rd intercostal spaces in addition to the standard 4th intercostal space increases the sensitivity for detecting the coved ST- segment elevation of the type 1 Brugada pattern. Available data suggest that this increased detection of spontaneous type 1 pattern does not affect the value of this parameter for predicting VF.52,268,269
EP studies with programmed ventricular stimulation
The role of programmed ventricular stimulation during EP studies continues to be passionately debated.270–272 The question is not whether VF inducibility correlates with arrhythmic risk. In all series, the VF inducibility rate is highest for survivors of cardiac arrest, intermediate for those with syncope, and lowest for those who were asymptomatic at presentation.273 The central question is whether the prognostic information provided by VF inducibility is robust enough for clinical decision-making. Some studies suggest that this is the case,262,274,275 but others do not.248,249 One central issue suggested as an explanation for the discrepancy between studies is the extrastimulation protocol used. Specifically, the prognostic impact of site (RV apex vs RV apex + RVOT)49,270,276 and the number of extrastimuli (2 vs 3)49,276 has been analyzed but with inconsistent results. The only prospective study specifically designed to examine the prognostic yield of EP studies in BrS was the PRELUDE registry.49 It did not show that sustained VF induction identifies high-risk patients but did demonstrate that a short effective ventricular refractory period (o200 ms) is a risk marker.
Other risk markers
Several ECG markers have been associated with risk in BrS. These include (1) fragmentation of the QRS,226,277,278 (2) the concomitant finding of a type 1 Brugada pattern and an ERP in inferolateral leads,145,248,279–281 and (3) dynamic changes in the manifestation of prominent J waves or ST-segment elevation.282
Other markers associated with increased risk but with limited or inconsistent data include (1) late potentials recorded using signal-averaged ECG,283–285 (2) microscopic T-wave alternans,286 (3) macroscopic T-wave alternans during a sodium blocker challenge test,285,287,288 (4) increased QRS width,49,248,277,289,290 (5) prominent R wave in aVR,289–291 and (6) augmented ST-segment elevation of a type 1 Brugada pattern during the recovery phase of an exercise test.292 Prolonged Tpeak-Tend293–296 and relatively steep QT/RR slope have been associated with higher risk in cases of BrS.297,298 Combining several risk factors (e.g. fragmented QRS + ERP277) appears to confer an additive risk; however, data supporting this are limited.
Update on approaches to therapy for BrS and ERS
Figures 6 and 7 graphically present recommendations for the management of BrS and ERS as modified from the 2013 HRS/ EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes and the 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD.8,9 Those recommendations are based on the available literature and on the clinical experience of the Task Force members. As with all such recommendations, they will need to undergo continuous validation in future studies.
Figure 6.

Indications for therapy of patients with Brugada syndrome. Recommendations with class designations are taken from Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932–1963, and Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2757–9. Recommendations without class designations are derived from unanimous consensus of the authors. ES = extrastimuli at right ventricular apex; ICD = implantable cardioverter-defibrillator; ILR = implantable loop recorder; NAR = nocturnal agonal respiration; RVOT = right ventricular outflow tract; VF = ventricular fibrillation; VT = ventricular tachycardia.
Figure 7.

Indications for therapy of patients with early repolarization syndrome. Recommendations with Class designations are taken from Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932–1963, and Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36: 2757–9. Recommendations without Class designations are derived from unanimous consensus of the authors. ER = early repolarization; ICD = implantable cardioverter-defibrillator; ILR = implantable loop recorder; NAR = nocturnal agonal respiration; VT = ventricular tachycardia.
Education and lifestyle changes for the prevention of arrhythmias are critical in BrS. Patients should be informed of the various modulators and precipitating factors that could cause malignant arrhythmias. A prominent S-wave in Lead I has recently been identified as an ECG marker of BrS.298A Fever should be treated aggressively with antipyretics, and contraindicated substances should be avoided (see www.brugadadrugs.org).89 Referral for ECG is recommended during high fever. Family members may be referred for cardiopulmonary resuscitation training and advised to consider purchasing an automatic external defibrillator for home use. Because malignant ventricular arrhythmias are infrequent in asymptomatic patients with BrS247 or ERP44 and usually unrelated to physical activity, the presence of these patterns does not contraindicate participation in sports.
It is noteworthy, however, that the Brugada pattern is accentuated immediately after exercise, presumably because of an increase in vagal tone.292,299,300 In reviewing 98 case of BrS studies dealing with exercise, Masrur et al.299 concluded that there are insufficient data on the risks of exercise in BrS to make recommendations for exercise.
Implantable cardioverter-defibrillator
The only proven effective therapeutic strategy for the prevention of SCD in high-risk BrS and ERS patients is an ICD.301,302 It is important to recognize that ICDs are associated with complications, especially in young active individuals.249,303 At 10 years postimplantation, the rates of inappropriate shock and lead failure are 37% and 29%, respectively. Remote monitoring can identify lead failure and prevent inappropriate shocks.304 Subcutaneous ICDs are thought to represent the future for this indication because they are expected to be associated with fewer complications over a lifetime.305
Implantation of an ICD is first-line therapy for JWS patients presenting with aborted SCD or documented VT/VF with or without syncope (Class I recommendation).301,306 ICDs can be useful (Class IIa) in symptomatic BrS patients with type 1 pattern, in whom syncope was likely caused by VT/VF. The HRS/EHRA/APHRS expert consensus states that the ICD may be considered (Class IIb) in asymptomatic patients with inducible VF during programmed electrical stimulation (PES).8 Some studies suggest that the predictive value of EP studies may be improved by limiting the PES protocol to 2 extrastimuli,50,276 but that observation is not supported by other studies.49,307 Similarly, some studies advocate that PES should be limited to the RVA and credit this limited PES strategy for a very high positive predictive value found is some series.275 Again, that observation is not confirmed by other studies.276
The current Task Force proposes that ICDs are reasonable (Class IIa) in symptomatic BrS patients with type 1 pattern but that implantation be considered on a case-by-case basis by an electrophysiologist experienced in BrS, taking into consideration age, gender, clinical presentation, ECG characteristics (QRS fragmentation, Jp amplitude), and patient preference. The current Task Force also proposes that EP study may be considered in asymptomatic individuals with spontaneous type 1 Brugada pattern. If VT/VF is inducible, an ICD should be considered.7 More recent studies argue in favor of using ≤2 extrastimuli to induce VT/VF.50,276 ICDs are not indicated in asymptomatic patients without any of these characteristics. At present, there is no clear role for PES in patients with ERS.
Pacemaker therapy
Arrhythmic events and SCD in both BrS and ERS generally occur during sleep or at rest and are associated with slow heart rates. These observations notwithstanding, a potential therapeutic role for cardiac pacing remains largely unexplored.308 A few case reports are available.309,310
RFA therapy
Nademanee et al.129 showed that RFA of epicardial sites displaying late potentials and fractionated bipolar electrograms in the RVOT of BrS patients can significantly reduce arrhythmia vulnerability and the ECG manifestation of the disease. Ablation at these sites was reported to render VT/VF noninducible and to normalize the Brugada ECG pattern in the vast majority of patients over a period of weeks or months. Long-term follow-up (20–6 months) showed no recurrent VT/VF, with only 1 patient on medical therapy with amiodarone. Case reports in support of these effects have been published.311 Additional evidence in support of the effectiveness of epicardial substrate ablation was provided by Sacher et al.130 and Shah et al.312
More recently, Brugada et al.131 used flecainide to identify the full extent of low-voltage electrogram activity in the anterior RV and RVOT and targeted this region for RFA. In all 14 BrS patients, RFA eliminated abnormal bipolar electrograms, normalized ST-segment elevation on right precordial leads of ECG, and VT/VF was no longer inducible. Ablation therapy can be lifesaving in otherwise uncontrollable cases. RF ablation may be considered (Class IIb recommendation) in BrS patients with frequent appropriate ICD shocks due to recurrent electrical storms.7 There are no clinical reports of ablation of the LV substrate in patients with ERS. In patients in whom BrS combines with ERS, ablation of the anterior RV epicardium (including the RVOT) is not ameliorative.
Pharmacologic approach to therapy
Brugada syndrome
ICD implantation may be problematic in infants or young children because of the high complication rate. ICDs are also economically out of reach for patients in some regions of the world. A pharmacologic approach to therapy, based on a rebalancing of currents active during the early phases of the epicardial AP in the RV so as to reduce the magnitude of the AP notch and/or restore the AP dome, has been a focus of basic and clinical research in recent years. Antiarrhythmic agents such as amiodarone and beta-blockers have been shown to be ineffective.313 Class IC antiarrhythmic drugs (e.g., flecainide, propafenone) and Class IA agents (e.g., procainamide) are contraindicated because of their effects of unmasking BrS and inducing arrhythmogenesis. Disopyramide is a Class IA antiarrhythmic that has been demonstrated to normalize ST-segment elevation in some Brugada patients but to unmask the syndrome in others.314
Because the presence of a prominent Ito is a prerequisite for the development of both BrS and ERS, partial inhibition of this current is thought to be effective regardless of the ionic or genetic basis for the disease. Unfortunately, cardioselective and Ito-specific blockers are not available.
The only agent with significant Ito-blocking properties available in the United States and around the world is quinidine.19,73 Experimental studies have shown that quinidine is effective in restoring the epicardial AP dome, thus normalizing the ST segment and preventing phase 2 reentry and polymorphic VT in a variety of different experimental models of BrS.19,150,315–317A recent experimental study suggests that quinidine, because of its effect of blocking Ito, can also exert a protective effect against hypothermia- induced VT/VF in a JWS model.144 It is noteworthy that, historically, quinidine was used to prevent VF in patients who required hypothermia for surgical procedures.317
Clinical evidence for the effectiveness of quinidine in normalizing ST-segment elevation and/or preventing arrhythmic events in patients with BrS has been reported in numerous studies and case reports.117,119,120,124,318–331 Hermida et al.119 reported 76% efficacy in prevention of VF-induced by PES. Belhassen et al.332 recently reported a 90% efficacy in prevention of VF induction after treatment with quinidine despite the use of very aggressive protocols of extrastimulation. Furthermore, no arrhythmic events occurred among BrS patients treated with quinidine during a mean follow-up period of 10 years.
In a recent trial conducted at 2 French centers, 44 asymptomatic BrS patients with inducible VT/VF were enrolled (47–10 years, 95% male).333 Of these patients, 34 (77%) were no longer inducible while treated with 600 mg/day hydroquinidine for 6.2–3 years. Among the 10 other patients (22%) who remained inducible and received ICD (group PVS+), none received appropriate therapy during mean follow-up of 7.7–2 years.
A prospective registry of empiric quinidine for asymptomatic BrS has been established. The study appears at the National Institutes of Health website (ClinicalTrials.gov) and can be accessed at http://clinicaltrials.gov/ct2/show/NCT00789165?term_brugada&rank_2. Doses between 600 and 900 mg were recommended, if tolerated.322
Quinidine may be considered (Class IIb indication) in BrS patients presenting with electrical storms and in patients implanted with an ICD who are experiencing repeated appropriate shocks. Quinidine can also be useful in asymptomatic BrS patients displaying a spontaneous type I ECG, if they qualify for an ICD and the device is refused or is contraindicated (Class IIa recommendation).
Agents that augment the L-type calcium channel current, such as beta-adrenergic agents (e.g., isoproterenol, denopamine, orciprenaline) are useful as well.19,117,121,327,334,335 Isoproterenol, at times in combination with quinidine, has been used successfully to control VF storms and normalize ST elevation, particularly in children.93,115–118,279,319,320,325,331,336–343 Spontaneous VF in patients with BrS is often related to increases in vagal tone and is amenable to treatment by an increase of sympathetic tone via isopro-terenol administration. Administration of isoproterenol is a Class IIa recommendation for BrS patients presenting with electrical storms.7
Another promising pharmacologic approach for BrS is the administration of the phosphodiesterase III inhibitor cilostazol,117,121,123 which normalizes the ST segment, most likely by augmenting calcium current (ICa) as well as by reducing Ito secondary to an increase in cAMP and heart rate.344 Other effects of cilostazol may contribute to its actions (e.g., adenosine, NO, mitochondrial IKATP345). Its efficacy in combination with bepridil in preventing VF episodes was recently reported by Shinohara et al.125 The failure of cilostazol in the treatment of BrS has been described in a single case report.346
Milrinone is another phosphodiesterase III inhibitor recently identified as a more potent alternative to cilostazol in suppressing ST elevation and arrhythmogenesis in an experimental model of BrS.150,347 No clinical reports have yet been published.
Wenxin Keli, a traditional Chinese medicine, has recently been shown to inhibit Ito and thus to suppress polymorphic VT in experimental models of BrS when combined with low concentrations of quinidine (5 μM).316
Agents that augment peak and late INa, including bepridil and dimethyl lithospermate B, are suggested to be of value in BrS. Bepridil has been reported to suppress VT/VF in several studies of patients with BrS.117,297,298,348 The drug's action are thought to be mediated by (1) inhibition of Ito; (2) augmentation of INa via up-regulation of the sodium channels349; and (3) prolongation of QT interval at slow rates thus increasing the QT/RR slope.297,298 Dimethyl lithospermate B, an extract of Danshen, a traditional Chinese herbal remedy, has been reported to slow inactivation of INa, thus increasing INa during the early phases of the AP and suppressing arrhythmogenesis in experimental models of BrS.350
Because malignant ventricular arrhythmias are infrequent in asymptomatic patients with BrS247 or ERP44 and usually unrelated to physical activity, the presence of these patterns does not contraindicate participation in sports, although, as previously discussed, insufficient data are currently available to make definitive recommendations for participation in sports.
Early repolarization syndrome
It is not surprising that the approach to therapy of ERS is similar to that of BrS, because the mechanisms underlying the 2 syndromes are potentially similar. Quinidine, phosphodiesterase III inhibitors, and isoproterenol have all been shown to exert an ameliorative effect in preventing or quieting arrhythmias associated with ERS. Isoproterenol has been shown to be effective in quieting electrical storms developing in patients with either BrS117,338 or ERS.190 Isoproterenol has been shown to act by reversing the repolarization abnormalities responsible for the disease phenotype secondary to restoration of the epicardial AP dome in experimental models of both BrS19,315 and ERS.30 This action of the beta-adrenergic agonist is expected because of its actions to potently increase ICa.
The phosphodiesterase III inhibitor cilostazol has been reported to reduce the ECG and arrhythmic manifestations of ERS.122 Phosphodiesterase inhibitors are known to activate ICa secondary to an increase in cAMP.121,344,351–355 The augmentation of ICa is thought to prevent arrhythmias associated with JWS by reversing the repolarization defects and restoring electrical homogeneity across the ventricular wall secondary to restoration of the epicardial AP dome in both BrS347 and ERS.144 Cilostazol has been hypothesized to also block Ito. Augmentation of ICa together with inhibition of Ito are expected to produce an inward shift in the balance of currents active during the early phases of the epicardial AP that should be especially effective in suppressing J-wave activity. The effectiveness of bepridil in ERS has been reported in a single patient thus far.356
No clinical data are available regarding the effectiveness of RFA in the setting of ERS, despite the fact that low- voltage fractionated electrogram activity and high-frequency late potentials are observed in the LV of patients with ERS357 and in experimental models of ERS (Yoon and Antzelevitch, unpublished data). Nakagawa et al.357 reported the results of a study in which they recorded epicardial electrograms directly from the LV of patients diagnosed with ERS by introducing a multipolar catheter into the left lateral (marginal) coronary vein, anterior interventricular vein, and middle cardiac vein via the coronary sinus. The authors reported late potentials in the bipolar electrograms recorded from the LV epicardium of the ERS patients.357
Acknowledgments
We are grateful to Buchang Pharmaceutical for providing the funds to host the J-Wave Consensus Conference. We are also grateful to Dr. Mina Chung for helpful discussions and advice and to Ms. Donna Loyle for proofing the manuscript.
References
- 1. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol 1992;20:1391–1396. [DOI] [PubMed] [Google Scholar]
- 2. Haissaguerre M, Derval N, Sacher F et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016–2023. [DOI] [PubMed] [Google Scholar]
- 3. Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. N Engl J Med 2008;358:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosso R, Kogan E, Belhassen B, Rozovski U, Scheinman MM, Zeltser D, Halkin A, Steinvil A, Heller K, Glikson M, Katz A, Viskin S. J-point elevation in survivors of primary ventricular fibrillation and matched control subjects: incidence and clinical significance. J Am Coll Cardiol 2008;52:1231–1238. [DOI] [PubMed] [Google Scholar]
- 5. Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation 2002;106:2514–2519. [DOI] [PubMed] [Google Scholar]
- 6. Antzelevitch C, Brugada P, Borggrefe M et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005;111:659–670. [DOI] [PubMed] [Google Scholar]
- 7. Priori SG, Wilde AA, Horie M et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;15:1389–1406. [DOI] [PubMed] [Google Scholar]
- 8. Priori SG, Wilde AA, Horie M et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013;10:1932–1963. [DOI] [PubMed] [Google Scholar]
- 9. Priori SG, Blomstrom-Lundqvist C, Mazzanti A et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2757–2759. [DOI] [PubMed] [Google Scholar]
- 10. Clements SD, Hurst JW. Diagnostic value of ECG abnormalities observed in subjects accidentally exposed to cold. Am J Cardiol 1972;29:729–734. [DOI] [PubMed] [Google Scholar]
- 11. Thompson R, Rich J, Chmelik F, Nelson WL. Evolutionary changes in the electrocardiogram of severe progressive hypothermia. J Electrocardiol 1977;10:67–70. [DOI] [PubMed] [Google Scholar]
- 12. Eagle K. Images in clinical medicine. Osborn waves of hypothermia. N Engl J Med 1994;10:680. [DOI] [PubMed] [Google Scholar]
- 13. Kraus F. Ueber die wirkung des kalziums auf den kreislauf 1. Dtsch Med Wochenschr 1920;46:201–203. [Google Scholar]
- 14. Sridharan MR, Horan LG. Electrocardiographic J wave of hypercalcemia. Am J Cardiol 1984;54:672–673. [DOI] [PubMed] [Google Scholar]
- 15. Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm 2010;7:549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wasserburger RH, Alt WJ. The normal RS-T segment elevation variant. Am J Cardiol 1961;8:184–192. [DOI] [PubMed] [Google Scholar]
- 17. Mehta MC, Jain AC. Early repolarization on scalar electrocardiogram. Am J Med Sci 1995;309:305–311. [DOI] [PubMed] [Google Scholar]
- 18. Gussak I, Antzelevitch C. Early repolarization syndrome: clinical characteristics and possible cellular and ionic mechanisms. J Electrocardiol 2000;33:299–309. [DOI] [PubMed] [Google Scholar]
- 19. Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation 1999;100:1660–1666. [DOI] [PubMed] [Google Scholar]
- 20. Shu J, Zhu T, Yang L, Cui C, Yan GX. ST-segment elevation in the early repolarization syndrome, idiopathic ventricular fibrillation, and the Brugada syndrome: cellular and clinical linkage. J Electrocardiol 2005;38:26–32. [DOI] [PubMed] [Google Scholar]
- 21. Antzelevitch C. J wave syndromes: molecular and cellular mechanisms. J Electrocardiol 2013;46:510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mahida S, Derval N, Sacher F et al. History and clinical significance of early repolarization syndrome. Heart Rhythm 2015;12:242–249. [DOI] [PubMed] [Google Scholar]
- 23. Wellens HJ, Schwartz PJ, Lindemans FW et al. Risk stratification for sudden cardiac death: current status and challenges for the futuredagger. Eur Heart J 2014;35:1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Macfarlane P, Antzelevitch C, Haissaguerre M, Huikuri HV, Potse M, Rosso R, Sacher F, Tikkanen J, Wellens H, Yan GX. The early repolarization pattern: consensus paper. J Am Coll Cardiol 2015;66:470–477. [DOI] [PubMed] [Google Scholar]
- 25. Kanter RJ, Pfeiffer R, Hu D, Barajas-Martinez H, Carboni MP, Antzelevitch C. Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation 2012;125:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antzelevitch C. Molecular biology and cellular mechanisms of Brugada and long QT syndromes in infants and young children. J Electrocardiol 2001;34:177–181. [DOI] [PubMed] [Google Scholar]
- 27. Wedekind H, Smits JP, Schulze-Bahr E et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation 2001;104:1158–1164. [DOI] [PubMed] [Google Scholar]
- 28. Nagase S, Kusano KF, Morita H, Fujimoto Y, Kakishita M, Nakamura K, Emori T, Matsubara H, Ohe T. Epicardial electrogram of the right ventricular outflow tract in patients with the Brugada syndrome: using the epicardial lead. J Am Coll Cardiol 2002;39:1992–1995. [DOI] [PubMed] [Google Scholar]
- 29. Nakagawa K, Nagase S, Morita H, Ito H. Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm 2014;11:314–317. [DOI] [PubMed] [Google Scholar]
- 30. Koncz I, Gurabi Z, Patocskai B, Panama BK, Szel T, Hu D, Barajas-Martinez H, Antzelevitch C. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol 2014;68C:20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Antzelevitch C. Brugada syndrome. Pacing ClinElectrophysiol 2006;29:1130–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Coronel R, Casini S, Koopmann TT et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005;112:2769–2777. [DOI] [PubMed] [Google Scholar]
- 33. Bastiaenen R, Hedley PL, Christiansen M, Behr ER. Therapeutic hypothermia and ventricular fibrillation storm in early repolarization syndrome. Heart Rhythm 2010;7:832–834. [DOI] [PubMed] [Google Scholar]
- 34. Federman NJ, Mechulan A, Klein GJ, Krahn AD. Ventricular fibrillation induced by spontaneous hypothermia in a patient with early repolarization syndrome. J Cardiovasc Electrophysiol 2013;24:586–588. [DOI] [PubMed] [Google Scholar]
- 35. Sakabe M, Fujiki A, Tani M, Nishida K, Mizumaki K, Inoue H. Proportion and prognosis of healthy people with coved or saddle-back type ST segment elevation in the right precordial leads during 10 years follow-up. EurHeart J 2003;24:1488–1493. [DOI] [PubMed] [Google Scholar]
- 36. Tsuji H, Sato T, Morisaki K, Iwasaka T. Prognosis of subjects with Brugada- type electrocardiogram in a population of middle-aged Japanese diagnosed during a health examination. Am J Cardiol 2008;102:584–587. [DOI] [PubMed] [Google Scholar]
- 37. Gervacio-Domingo G, Isidro J, Tirona J, Gabriel E, David G, Amarillo ML, Morales D, Dans A. The Brugada type 1 electrocardiographic pattern is common among Filipinos. J Clin Epidemiol 2008;61:1067–1072. [DOI] [PubMed] [Google Scholar]
- 38. Ito HYK, Chen R, He Q, Curb JD. The prevalence and prognosis of a Brugada- type electrocardiogram in a population of middle-aged Japanese-American men with follow-up of three decades. Am J Med Sci 2006;331:4. [DOI] [PubMed] [Google Scholar]
- 39. Letsas KP, Weber R, Astheimer K, Kalusche D, Arentz T. Tpeak-Tend interval and Tpeak-Tend/QT ratio as markers of ventricular tachycardia inducibility in subjects with Brugada ECG phenotype. Europace 2010;12:271–274. [DOI] [PubMed] [Google Scholar]
- 40. Gallagher MM, Forleo GB, Behr ER, Magliano G, De LL, Morgia V, De LF, Romeo F. Prevalence and significance of Brugada-type ECG in 12,012 apparently healthy European subjects. Int J Cardiol 2008;130:44–48. [DOI] [PubMed] [Google Scholar]
- 41. Pecini R, Cedergreen P, Theilade S, Haunso S, Theilade J, Jensen GB. The prevalence and relevance of the Brugada-type electrocardiogram in the Danish general population: data from the Copenhagen City Heart Study. Europace 2010;12:982–986. [DOI] [PubMed] [Google Scholar]
- 42. Patel SS, Anees SS, Ferrick KJ. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin Electrophysiol 2009;32:704–708. [DOI] [PubMed] [Google Scholar]
- 43. Lee C, Soni A, Tate RB, Cuddy TE. The incidence and prognosis of Brugada electrocardiographic pattern in the Manitoba Follow-Up Study. Can J Cardiol 2005;21:1286–1290. [PubMed] [Google Scholar]
- 44. Tikkanen JT, Anttonen O, Junttila MJ, Aro AL, Kerola T, Rissanen HA, Reunanen A, Huikuri HV. Long-term outcome associated with early repolarization on electrocardiography. NEnglJ Med 2009;361:2529–2537. [DOI] [PubMed] [Google Scholar]
- 45. Sinner MF, Reinhard W, Muller M et al. Association of early repolarization pattern on ECG with risk of cardiac and all-cause mortality: a population-based prospective cohort study (MONICA/KORA). PLoS Med 2010;7:e1000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haruta D, Matsuo K, Tsuneto A, Ichimaru S, Hida A, Sera N, Imaizumi M, Nakashima E, Maemura K, Akahoshi M. Incidence and prognostic value of early repolarization pattern in the 12–lead electrocardiogram. Circulation 2011;123:2931–2937. [DOI] [PubMed] [Google Scholar]
- 47. Hayashi M, Shimizu W, Albert CM. The spectrum of epidemiology underlying sudden cardiac death. Circ Res 2015;116:1887–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brosnan MJ, Kumar S, LaGerche A, Brown A, Stewart S, Kalman JM, Prior DL. Early repolarization patterns associated with increased arrhythmic risk are common in young non-Caucasian Australian males and not influenced by athletic status. Heart Rhythm 2015;12:1576–1583. [DOI] [PubMed] [Google Scholar]
- 49. Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, Giordano U, Pappone C, Mascioli G, Rossetti G, De Nardis R, Colombo M. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. Journal of the American College of Cardiology 2012;59:37–45. [DOI] [PubMed] [Google Scholar]
- 50. Sroubek J, Probst V, Mazzanti A et al. Programmed Ventricular Stimulation for Risk Stratification in the Brugada Syndrome: A Pooled Analysis. Circulation 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. Right ventricular electrocardiographic leads for detection of Brugada syndrome in sudden unexplained death syndrome survivors and their relatives. ClinCardiol 2001;24:776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miyamoto K, Yokokawa M, Tanaka K, Nagai T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, Shimizu W. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol 2007;99:53–57. [DOI] [PubMed] [Google Scholar]
- 53. Nagase S, Hiramatsu S, Morita H, Nishii N, Murakami M, Nakamura K, Kusano KF, Ito H, Ohe T. Electroanatomical correlation of repolarization abnormalities in Brugada syndrome: detection of type 1 electrocardiogram in the right ventricular outflow tract. J Am Coll Cardiol 2010;56:2143–2145. [DOI] [PubMed] [Google Scholar]
- 54. Veltmann C, Papavassiliu T, Konrad T, Doesch C, Kuschyk J, Streitner F, Haghi D, Michaely HJ, Schoenberg SO, Borggrefe M, Wolpert C, Schimpf R. Insights into the location of type I ECG in patients with Brugada syndrome: correlation of ECG and cardiovascular magnetic resonance imaging. Heart Rhythm 2012;9:414–421. [DOI] [PubMed] [Google Scholar]
- 55. Richter S, Sarkozy A, Paparella G, Henkens S, Boussy T, Chierchia GB, Brugada R, Brugada J, Brugada P. Number of electrocardiogram leads displaying the diagnostic coved-type pattern in Brugada syndrome: a diagnostic consensus criterion to be revised. EurHeart J 2010;31:1357–1364. [DOI] [PubMed] [Google Scholar]
- 56. Veerakul G, Nademanee K. Brugada syndrome: two decades of progress. Circ J 2012;76:2713–2722. [DOI] [PubMed] [Google Scholar]
- 57. Brugada P, Brugada J, Roy D. Brugada syndrome 1992–2012: 20 years of scientific excitement, and more. Eur Heart J 2013;34:3610–3615. [DOI] [PubMed] [Google Scholar]
- 58. Huikuri HV, Juhani Junttila M. Clinical aspects of inherited J-wave syndromes. Trends Cardiovasc Med 2015;25:24–30. [DOI] [PubMed] [Google Scholar]
- 59. Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm 2015;12:1852–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Olde Nordkamp LR, Vink AS, Wilde AA, de Lange FJ, de Jong JS, Wieling W, van Dijk N, Tan HL. Syncope in Brugada syndrome: prevalence, clinical significance, and clues from history taking to distinguish arrhythmic from nonarrhythmic causes. Heart Rhythm 2015;12:367–375. [DOI] [PubMed] [Google Scholar]
- 61. Ikeda T, Abe A, Yusa S, Nakamura K, Ishiguro H, Mera H, Yotsukura M, Yoshino H. The full stomach test as a novel diagnostic technique for identifying patients at risk for Brugada Syndrome. J Cardiovasc Electrophysiol 2006;17:602–607. [DOI] [PubMed] [Google Scholar]
- 62. Shimeno K, Takagi M, Maeda K, Tatsumi H, Doi A, Yoshiyama M. Usefulness of multichannel Holter ECG recording in the third intercostal space for detecting type 1 Brugada ECG: comparison with repeated 12–lead ECGs. J Cardiovasc Electrophysiol 2009;20:1026–1031. [DOI] [PubMed] [Google Scholar]
- 63. Viskin S, Rosso R, Friedensohn L, Havakuk O, Wilde AA. Everybody has Brugada syndrome until proven otherwise? Heart Rhythm 2015;12:1595–1598. [DOI] [PubMed] [Google Scholar]
- 64. Conte G, de Asmundis C, Ciconte G, Julia J, Sieira J, Chierchia GB, Brugada P. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA 2014;312:2039–2041. [DOI] [PubMed] [Google Scholar]
- 65. Conte G, Dewals W, Sieira J et al. Drug-induced Brugada syndrome in children: clinical features, device-based management and long-term follow-up. J Am Coll Cardiol 2014;63:2272–2279. [DOI] [PubMed] [Google Scholar]
- 66. Gandjbakhch E, Fressart V, Duthoit G, Marquie C, Deharo JC, Pousset F, Hebert JL, Simon F, Himbert C, Klug D, Charron P, Hidden-Lucet F. Malignant response to ajmaline challenge in SCN5A mutation carriers: experience from a large familial study. Int J Cardiol 2014;172:256–258. [DOI] [PubMed] [Google Scholar]
- 67. Kligfield P, Gettes LS, Bailey JJ et al. Recommendations for the standardization and interpretation of the electrocardiogram: part I: the electrocardiogram and its technology a scientific statement from the American Heart Association Electrocardiography and Arrhythmias Committee, Council on Clinical Cardiology; the American College of Cardiology Foundation; and the Heart Rhythm Society endorsed by the International Society for Computerized Electrocardiology. Journal of the American College of Cardiology 2007;49:1109–1127. [DOI] [PubMed] [Google Scholar]
- 68. Papavassiliu T, Wolpert C, Fluchter S, Schimpf R, Neff W, Haase KK, Duber C, Borggrefe M. Magnetic resonance imaging findings in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2004;15:1133–1138. [DOI] [PubMed] [Google Scholar]
- 69. Papavassiliu T, Veltmann C, Doesch C, Haghi D, Germans T, Schoenberg SO, van Rossum AC, Schimpf R, Brade J, Wolpert C, Borggrefe M. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm 2010;7:1790–1796. [DOI] [PubMed] [Google Scholar]
- 70. Takagi M, Aihara N, Kuribayashi S, Taguchi A, Kurita T, Suyama K, Kamakura S, Takamiya M. Abnormal response to sodium channel blockers in patients with Brugada syndrome: augmented localised wall motion abnormalities in the right ventricular outflow tract region detected by electron beam computed tomography. Heart 2003;89:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Catalano O, Antonaci S, Moro G et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. EurHeart J 2009;30:2241–2248. [DOI] [PubMed] [Google Scholar]
- 72. van Hoorn F, Campian ME, Spijkerboer A, Blom MT, Planken RN, van Rossum AC, de Bakker JM, Wilde AA, Groenink M, Tan HL. SCN5A mutations in Brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PLoS One 2012;7:e42037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin JA. Clinical approaches to tachyarrhythmias. The Brugada syndrome. Vol 10 Armonk, NY: Futura Publishing Company, Inc; 1999. [Google Scholar]
- 74. Antzelevitch C. Brugada syndrome: historical perspectives and observations. EurHeart J 2002;23:676–678. [DOI] [PubMed] [Google Scholar]
- 75. Slezak J, Tribulova N, Okruhlicova L, Dhingra R, Bajaj A, Freed D, Singal P. Hibernating myocardium: pathophysiology, diagnosis, and treatment. Can J Physiol Pharmacol 2009;87:252–265. [DOI] [PubMed] [Google Scholar]
- 76. Nademanee K, Raju H, de Noronha SV et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J Am Coll Cardiol 2015;66:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/ dysplasia: clinical impact of molecular genetic studies. Circulation 2006;113:1634–1637. [DOI] [PubMed] [Google Scholar]
- 78. Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart 2011;97:530–539. [DOI] [PubMed] [Google Scholar]
- 79. Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. The New England journal of medicine 1988;318:129–133. [DOI] [PubMed] [Google Scholar]
- 80. Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res 2010;107:700–714. [DOI] [PubMed] [Google Scholar]
- 81. Delmar M, Makita N. Cardiac connexins, mutations and arrhythmias. Curr OpinCardiol 2012;27:236–241. [DOI] [PubMed] [Google Scholar]
- 82. Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, Hund T, Birchmeier W, Mohler P, van Veen TA, van Rijen HV, Delmar M. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res 2012;95:460–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation 2001;103:710–717. [DOI] [PubMed] [Google Scholar]
- 84. Cerrone M, Lin X, Zhang M et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Corrado D, Leoni L, Link MS et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003;108:3084–3091. [DOI] [PubMed] [Google Scholar]
- 86. Matsuo K, Kurita T, Inagaki M, Kakishita M, Aihara N, Shimizu W, Taguchi A, Suyama K, Kamakura S, Shimomura K. The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. EurHeart J 1999;20:465–470. [DOI] [PubMed] [Google Scholar]
- 87. Peters S, Trummel M, Denecke S, Koehler B. Results of ajmaline testing in patients with arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int J Cardiol 2004;95:207–210. [DOI] [PubMed] [Google Scholar]
- 88. Peters S, Trummel M, Koehler B. QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Heart Rhythm 2008;5:1417–1421. [DOI] [PubMed] [Google Scholar]
- 89. Postema PG, Wolpert C, Amin AS, Probst V, Borggrefe M, Roden DM, Priori SG, Tan HL, Hiraoka M, Brugada J, Wilde AA. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website. < www.brugadadrugs.org>. Heart Rhythm 2009;6:1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shimizu W. Acquired forms of the Brugada syndrome. J Electrocardiol 2005;38(Suppl):22–25. [DOI] [PubMed] [Google Scholar]
- 91. Brugada P, Brugada J, Brugada R. Arrhythmia induction by antiarrhythmic drugs. Pacing Clin Electrophysiol 2000;23:291–292. [DOI] [PubMed] [Google Scholar]
- 92. Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, Brugada P. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000;101:510–515. [DOI] [PubMed] [Google Scholar]
- 93. Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol 1996;27:1061–1070. [DOI] [PubMed] [Google Scholar]
- 94. Babaliaros VC, Hurst JW. Tricyclic antidepressants and the Brugada syndrome: an example of Brugada waves appearing after the administration of desipramine. Clin Cardiol 2002;25:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Goldgran-Toledano D, Sideris G, Kevorkian JP. Overdose of cyclic antidepressants and the Brugada syndrome. N Engl J Med 2002;346:1591–1592. [DOI] [PubMed] [Google Scholar]
- 96. Tada H, Sticherling C, Oral H, Morady F. Brugada syndrome mimicked by tricyclic antidepressant overdose. J Cardiovasc Electrophysiol 2001;12: 275–275. [DOI] [PubMed] [Google Scholar]
- 97. Ortega-Carnicer J, Bertos-Polo J, Gutierrez-Tirado C. Aborted sudden death, transient Brugada pattern, and wide QRS dysrrhythmias after massive cocaine ingestion. J Electrocardiol 2001;34:345–349. [DOI] [PubMed] [Google Scholar]
- 98. Nogami A, Nakao M, Kubota S et al. Enhancement of J-ST-segment elevation by the glucose and insulin test in Brugada syndrome. Pacing Clin Electrophysiol 2003;26:332–337. [DOI] [PubMed] [Google Scholar]
- 99. Araki T, Konno T, Itoh H, Ino H, Shimizu M. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J 2003;67:93–95. [DOI] [PubMed] [Google Scholar]
- 100. Pastor A, Nunez A, Cantale C, Cosio FG. Asymptomatic Brugada syndrome case unmasked during dimenhydrinate infusion. J Cardiovasc Electrophysiol 2001;12:1192–1194. [DOI] [PubMed] [Google Scholar]
- 101. Chiale PA, Garro HA, Fernandez PA, Elizari MV. High-degree right bundle branch block obscuring the diagnosis of Brugada electrocardiographic pattern. Heart Rhythm 2012;9:974–976. [DOI] [PubMed] [Google Scholar]
- 102. Baranchuk A, Nguyen T, Ryu MH, Femenia F, Zareba W, Wilde AA, Shimizu W, Brugada P, Perez-Riera AR. Brugada phenocopy: new terminology and proposed classification. Ann Noninvasive Electrocardiol 2012;17:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nam GB. Idiopathic ventricular fibrillation, early repolarization and other J wave-related ventricular fibrillation syndromes. Circ J 2012;76:2723–2731. [DOI] [PubMed] [Google Scholar]
- 104. Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation 1996;93:372–379. [DOI] [PubMed] [Google Scholar]
- 105. McIntyre WF, Perez-Riera AR, Femenia F, Baranchuk A. Coexisting early repolarization pattern and Brugada syndrome: recognition of potentially overlapping entities. J Electrocardiol 2012;45:195–198. [DOI] [PubMed] [Google Scholar]
- 106. Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, Arzamendi D, Berne P, Brugada R, Brugada P, Brugada J. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol 2008;52:1567–1573. [DOI] [PubMed] [Google Scholar]
- 107. Kamakura T, Kawata H, Nakajima I et al. Significance of non-type 1 anterior early repolarization in patients with inferolateral early repolarization syndrome. J Am Coll Cardiol 2013;62:1610–1618. [DOI] [PubMed] [Google Scholar]
- 108. Kawata H, Noda T, Yamada Y, Okamura H, Satomi K, Aiba T, Takaki H, Aihara N, Isobe M, Kamakura S, Shimizu W. Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and Brugada syndrome. Heart Rhythm 2012;9:77–83. [DOI] [PubMed] [Google Scholar]
- 109. Matsumoto AM. Fundamental aspects of hypogonadism in the aging male. Rev Urol 2003;5(Suppl 1):S3–S10. [PMC free article] [PubMed] [Google Scholar]
- 110. Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: a Brugada syndrome variant? J Cardiovasc Electrophysiol 2000;11:95–98. [DOI] [PubMed] [Google Scholar]
- 111. Aizawa Y, Sato A, Watanabe H et al. Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J Am Coll Cardiol 2012;59:1948–1953. [DOI] [PubMed] [Google Scholar]
- 112. Nademanee K. Sudden unexplained death syndrome in southeast Asia. Am J Cardiol 1997;79(6A):10–11. [DOI] [PubMed] [Google Scholar]
- 113. Watanabe H, Nogami A, Ohkubo K et al. Electrocardiographic characteristics and SCN5A mutations in idiopathic ventricular fibrillation associated with early repolarization. Circ Arrhythm Electrophysiol 2011;4:874–881. [DOI] [PubMed] [Google Scholar]
- 114. Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST- segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007;115:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Shimizu W, Kamakura S. Catecholamines in children with congenital long QT syndrome and Brugada syndrome. J Electrocardiol 2001;34(Suppl):173–175. [DOI] [PubMed] [Google Scholar]
- 116. Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:1277–1280. [DOI] [PubMed] [Google Scholar]
- 117. Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, Ohe T, Shimizu W. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007;4:695–700. [DOI] [PubMed] [Google Scholar]
- 118. Watanabe A, Fukushima KK, Morita H, Miura D, Sumida W, Hiramatsu S, Banba K, Nishii N, Nagase S, Nakamura K, Sakuragi S, Ohe T. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J 2006;27:1579–1583. [DOI] [PubMed] [Google Scholar]
- 119. Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, Guicheney P, Di Fusco S, Rey JL, Cauchemez B, Leenhardt A. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol 2004;43:1853–1860. [DOI] [PubMed] [Google Scholar]
- 120. Belhassen B, Glick A, Viskin S. Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation 2004;110:1731–1737. [DOI] [PubMed] [Google Scholar]
- 121. Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2002;13:698–701. [DOI] [PubMed] [Google Scholar]
- 122. Iguchi K, Noda T, Kamakura S, Shimizu W. Beneficial effects of cilostazol in a patient with recurrent ventricular fibrillation associated with early repolarization syndrome. Heart Rhythm 2013;10:604–606. [DOI] [PubMed] [Google Scholar]
- 123. Agac MT, Erkan H, Korkmaz L. Conversion of Brugada type I to type III and successful control of recurrent ventricular arrhythmia with cilostazol. Arch Cardiovasc Dis 2014;107:476–478. [DOI] [PubMed] [Google Scholar]
- 124. Hasegawa K, Ashihara T, Kimura H, Jo H, Itoh H, Yamamoto T, Aizawa Y, Horie M. Long-term pharmacological therapy of Brugada syndrome: is J-wave attenuation a marker of drug efficacy? Intern Med 2014;53:1523–1526. [DOI] [PubMed] [Google Scholar]
- 125. Shinohara T, Ebata Y, Ayabe R, Fukui A, Okada N, Yufu K, Nakagawa M, Takahashi N. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J-wave syndromes. Heart Rhythm 2014;11:1441–1445. [DOI] [PubMed] [Google Scholar]
- 126. Haissaguerre M, Sacher F, Nogami A et al. Characteristics of recurrent ventricular fibrillation associated with inferolateral early repolarization role of drug therapy. J Am Coll Cardiol 2009;53:612–619. [DOI] [PubMed] [Google Scholar]
- 127. Junttila MJ, Tikkanen JT, Kentta T, Anttonen O, Aro AL, Porthan K, Kerola T, Rissanen HA, Knekt P, Huikuri HV. Early repolarization as a predictor of arrhythmic and nonarrhythmic cardiac events in middle-aged subjects. Heart Rhythm 2014;11:1701–1706. [DOI] [PubMed] [Google Scholar]
- 128. Nam GB, Ko KH, Kim J, Park KM, Rhee KS, Choi KJ, Kim YH, Antzelevitch C. Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J 2010;31:330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, Likittanasombat K, Bhuripanyo K, Ngarmukos T. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011;123:1270–1279. [DOI] [PubMed] [Google Scholar]
- 130. Sacher F, Jesel L, Jais P, Haissaguerre M. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm 2014;11:732–734. [DOI] [PubMed] [Google Scholar]
- 131. Brugada J, Pappone C, Berruezo A, Vicedomini G, Manguso F, Ciconte G, Giannelli L, Santinelli V. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ Arrhythm Electrophysiol 2015;8:1373–1381. [DOI] [PubMed] [Google Scholar]
- 132. Ghosh S, Cooper DH, Vijayakumar R, Zhang J, Pollak S, Haissaguerre M, Rudy Y. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm 2010;7:534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhang J, Sacher F, Hoffmayer K et al. Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015;131:1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kowalczyk E, Kasprzak JD, Lipiec P. Giant J-wave and Brugada-like pattern in a patient with severe hypothermia. Acta Cardiol 2014;69:66–67. [DOI] [PubMed] [Google Scholar]
- 135. RuDusky BM. The electrocardiogram in hypothermia-the J wave and the Brugada syndrome. Am J Cardiol 2004;93:671–672. [DOI] [PubMed] [Google Scholar]
- 136. Adler A, Topaz G, Heller K, Zeltser D, Tzioni-Ohayon T, Rozovski U, Halkin A, Rosso R, Ben-Shachar S, Antzelevitch C, Viskin S. Fever-induced Brugada pattern: how common is it and what does it mean? Heart Rhythm 2013;10:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ansari E, Cook JR. Profound hypothermia mimicking a Brugada type ECG. J Electrocardiol 2003;36:257–260. [DOI] [PubMed] [Google Scholar]
- 138. Amin AS, Meregalli PG, Bardai A, Wilde AA, Tan HL. Fever increases the risk for cardiac arrest in the Brugada syndrome. Ann Intern Med 2008;149:216–218. [DOI] [PubMed] [Google Scholar]
- 139. Rattanawong P, Vutthikraivit W, Charoensri A, Jongraksak T, Prombandankul A, Kanjanahattakij N, Rungaramsin S, Wisaratapong T, Ngarmukos T. Fever- induced Brugada syndrome is more common than previously suspected: a cross- sectional study from an endemic area. Ann Noninvasive Electrocardiol 2016;21:136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Noda T, Shimizu W, Tanaka K, Chayama K. Prominent J wave and ST segment elevation: serial electrocardiographic changes in accidental hypothermia. J Cardiovasc Electrophysiol 2003;14:223. [DOI] [PubMed] [Google Scholar]
- 141. Tan HL, Meregalli PG. Lethal ECG changes hidden by therapeutic hypothermia. Lancet 2007;369:78. [DOI] [PubMed] [Google Scholar]
- 142. Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nakama Y, Maruhashi T, Kagawa E, Dai K, Aokage T, Matsushita J, Ikenaga H. Therapeutic hypothermia after out-of-hospital cardiac arrest due to Brugada syndrome. Resuscitation 2008;79:332–335. [DOI] [PubMed] [Google Scholar]
- 143. Patel RB, Ng J, Reddy V, Chokshi M, Parikh K, Subacius H, Sheikh-Ali AA, Nguyen T, Link MS, Goldberger JJ, Ilkhanoff L, Kadish AH. Early repolarization associated with ventricular arrhythmias in patients with chronic coronary artery disease. Circ Arrhythm Electrophysiol 2010;3:489–495. [DOI] [PubMed] [Google Scholar]
- 144. Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced VT/VF in the setting of early repolarization and the protective effect of quinidine, cilostazol and milrinone. Circ Arrhythm Electrophysiol 2014;7:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kawata H, Morita H, Yamada Y et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: a novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm 2013;10:1161–1168. [DOI] [PubMed] [Google Scholar]
- 146. Antzelevitch C. Genetic, molecular and cellular mechanisms underlying the J wave syndromes. Circ J 2012;76:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Kapplinger JD, Tester DJ, Alders M et al. An international compendium of mutations in the SCN5A encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ackerman MJ, Priori SG, Willems S et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011;8:1308–1339. [DOI] [PubMed] [Google Scholar]
- 149. Park DS, Cerrone M, Morley G et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest 2015;125:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Szel T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol 2014;63:2037–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Patocskai B, Szel T, Yoon N, Antzelevitch C. Cellular mechanisms underlying the fractionated and late potentials on epicardial electrograms and the ameliorative effect of epicardial radiofrequency ablation in an experimental model of Brugada syndrome. In: Program and Abstracts of the 24th Annual Upstate New York Cardiac Electrophysiology Society Meeting, November 3, 2014, Buffalo, NY, 2014Abstact 006. [Google Scholar]
- 152. Burashnikov E, Pfeiffer R, Barajas-Martinez H et al. Mutations in the cardiac L-type calcium channel associated J wave sydnrome and sudden cardiac death. Heart Rhythm 2010;7:1872–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Cordeiro JM, Marieb M, Pfeiffer R, Calloe K, Burashnikov E, Antzelevitch C. Accelerated inactivation of the L-type calcium due to a mutation in CACNB2b due to a mutation in CACNB2b underlies Brugada syndrome. J Mol Cell Cardiol 2009;46:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Antzelevitch C, Pollevick GD, Cordeiro JM et al. Loss-of-function mutations in the cardiac calcium channel underline a new clinical entity characterized by ST segment elevation, short QT intervals, and sudden cardiac death. Circ Res 2006;99:1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gurnett CA, De WM, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron 1996;16:431–440. [DOI] [PubMed] [Google Scholar]
- 156. Barajas-Martinez H, Hu D, Ferrer T et al. Molecular genetic and functional association of Bugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm 2012;9:548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Delaney JT, Muhammad R, Blair MA, Kor K, Fish FA, Roden DM, Darbar D. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace 2012;14:1428–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Medeiros-Domingo A, Tan BH, Crotti L et al. Gain-of-function mutation S422L in the KCNJ8–encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm 2010;7:1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hu D, Barajas-Martinez H, Medeiros-Domingo A et al. Novel mutations in the sodium channel 2 subunit gene (SCN2B) associated with Brugada syndrome and atrial fibrillation. Circulation 2012;126:A16521. [Google Scholar]
- 160. Riuro H, Beltran-Alvarez P, Tarradas A et al. A missense mutation in the sodium channel ß2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013;34:961–966. [DOI] [PubMed] [Google Scholar]
- 161. Giudicessi JR, Ye D, Tester DJ, Crotti L, Mugione A, Nesterenko VV, Albertson RM, Antzelevitch C, Schwartz PJ, Ackerman MJ. Transient outward current (Ito) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011;8:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Delpón E, Cordeiro JM, Nœæez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Burashnikov A, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythm Electrophysiol 2008;1:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Olesen MS, Jensen NF, Holst AG, Nielsen JB, Tfelt-Hansen J, Jespersen T, Sajadieh A, Haunso S, Lund JT, Calloe K, Schmitt N, Svendsen JH. A novel nonsense variant in Nav1.5 cofactor MOG1 eliminates its sodium current increasing effect and may increase the risk of arrhythmias. Can J Cardiol 2011;27: 523–523. [DOI] [PubMed] [Google Scholar]
- 164. Kattygnarath D, Maugenre S, Neyroud N et al. MOG1: a new susceptibility gene for Brugada syndrome. Circ Cardiovasc Genet 2011;4:261–268. [DOI] [PubMed] [Google Scholar]
- 165. Cerrone M, Lin X, Zhang M et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2013;129:1092–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hennessey JA, Marcou CA, Wang C, Wei EQ, Wang C, Tester DJ, Torchio M, Dagradi F, Crotti L, Schwartz PJ, Ackerman MJ, Pitt GS. FGF12 is a candidate Brugada syndrome locus. Heart Rhythm 2013;10:1886–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Bezzina CR, Barc J, Mizusawa Y et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 2013;45:1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Boukens BJ, Sylva M, de Gier-de VC, Remme CA, Bezzina C, Christoffels VM, Coronel R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ Res 2013;113:137–141. [DOI] [PubMed] [Google Scholar]
- 169. Hartman ME, Liu Y, Zhu WZ, Chien WM, Weldy CS, Fishman GI, Laflamme MA, Chin MT. Myocardial deletion of transcription factor CHF1/Hey2 results in altered myocyte action potential and mild conduction system expansion but does not alter conduction system function or promote spontaneous arrhythmias. FASEB J 2014;28:3007–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, Park JE, Makiyama T, Horie M, Arimura T, Makita N, Kimura A. Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ J 2013;77:959–967. [DOI] [PubMed] [Google Scholar]
- 171. Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2009;2:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Watanabe H, Koopmann TT, S. LS et al. Sodium channel ß1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J Clin Invest 2008;118:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Valdivia CR, Ueda K, Ackerman MJ, Makielski JC. GPD1L links redox state to cardiac excitability by PKC-dependent phosphorylation of the sodium channel SCN5A. Am J Physiol Heart Circ Physiol 2009;297:H1446–H1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shy D, Gillet L, Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta 2013;1833:886–894. [DOI] [PubMed] [Google Scholar]
- 175. Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3. Circulation 2002;105:707–713. [DOI] [PubMed] [Google Scholar]
- 176. Hu D, Barajas-Martinez H, Medeiros-Domingo A et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Na (v)1.5 and K(v)4.3 channel currents. Heart Rhythm 2012;9:760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hu D, Barajas-Martinez H, Pfeiffer R et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014;64:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Behr ER, Savio-Galimberti E, Barc J et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res 2015;106:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Antzelevitch C. Cardiac repolarization. The long and short of it. Europace 2005;7(Suppl 2):3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Liu H, Chatel S, Simard C et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PLoS One 2013;8:e54131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Perrin MJ, Adler A, Green S et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J wave syndrome associated with sudden cardiac death. Circ Cardiovasc Genet 2014;7:782–789. [DOI] [PubMed] [Google Scholar]
- 182. Verkerk AO, Wilders R, Schulze-Bahr E et al. Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome 1. Cardiovasc Res 2005;68:441–453. [DOI] [PubMed] [Google Scholar]
- 183. Wilders R, Verkerk AO. Role of the R1135H KCNH2 mutation in Brugada syndrome. Int J Cardiol 2010;144:149–151. [DOI] [PubMed] [Google Scholar]
- 184. Ohno S, Zankov DP, Ding WG et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ Arrhythm Electrophysiol 2011;4:352–361. [DOI] [PubMed] [Google Scholar]
- 185. Boczek NJ, Ye D, Johnson EK et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ Res 2014;115:460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Ueda K, Nakamura K, Hayashi T et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem 2004;279:27194–27198. [DOI] [PubMed] [Google Scholar]
- 187. Noseworthy PA, Tikkanen JT, Porthan K et al. The early repolarization pattern in the general population clinical correlates and heritability. J Am Coll Cardiol 2011;57:2284–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Reinhard W, Kaess BM, Debiec R, Nelson CP, Stark K, Tobin MD, Macfarlane PW, Tomaszewski M, Samani NJ, Hengstenberg C. Heritability of early repolarization: a population-based study. Circ Cardiovasc Genet 2011;4:134–138. [DOI] [PubMed] [Google Scholar]
- 189. Nunn LM, Bhar-Amato J, Lowe MD, Macfarlane PW, Rogers P, McKenna WJ, Elliott PM, Lambiase PD. Prevalence of J-point elevation in sudden arrhythmic death syndrome families. J Am Coll Cardiol 2011;58:286–290. [DOI] [PubMed] [Google Scholar]
- 190. Haissaguerre M, Chatel S, Sacher F et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol 2009;20:93–98. [DOI] [PubMed] [Google Scholar]
- 191. Schwartz PJ, Ackerman MJ, George AL Jr., Wilde AA. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 2013;62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Raffan E, Semple RK. Next generation sequencing: implications for clinical practice. Br Med Bull 2011;99:53–71. [DOI] [PubMed] [Google Scholar]
- 193. Refsgaard L, Holst AG, Sadjadieh G, Haunso S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet 2012;20:905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 2009;120:1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, Haunso S, Holst AG, Svendsen JH, Olesen MS. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol 2013;29:1104–1109. [DOI] [PubMed] [Google Scholar]
- 196. Kapplinger J, Tester D, Alders M et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Hu D, Barajas-Martinez H, Terzic A et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol 2014;171:431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Alfares AA, Kelly MA, McDermott G et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med 2015;17:8808–8888. [DOI] [PubMed] [Google Scholar]
- 199. Le Scouarnec S, Karakachoff M, Gourraud JB et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet 2015;24:2757–2763. [DOI] [PubMed] [Google Scholar]
- 200. Veeramah KR, Karafet TM, Wolf D, Samson RA, Hammer MF. The KCNJ8– S422L variant previously associated with J-wave syndromes is found at an increased frequency in Ashkenazi Jews. Eur J Hum Genet 2014;22:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunso S, Sadjadieh A, Winkel BG, Olesen M, Tfelt-Hansen J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 2013;84:489–495. [DOI] [PubMed] [Google Scholar]
- 202. Campuzano O, Allegue C, Fernandez A, Iglesias A, Brugada R. Determining the pathogenicity of genetic variants associated with cardiac channelopathies. Sci Rep 2015;5:7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Crotti L, Marcou CA, Tester DJ, Castelletti S, Giudicessi JR, Torchio M, Medeiros-Domingo A, Simone S, Will ML, Dagradi F, Schwartz PJ, Ackerman MJ. Spectrum and prevalence of mutations Involving BrS1– through BrS12– susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J Am Coll Cardiol 2012;60:1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Song W, Shou W. Cardiac sodium channel Nav1.5 mutations and cardiac arrhythmia. Pediatr Cardiol 2012;33:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Ruklisa D, Ware JS, Walsh R, Balding DJ, Cook SA. Bayesian models for syndrome- and gene-specific probabilities of novel variant pathogenicity. Genome medicine 2015;7:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Juang JM, Lu TP, Lai LC et al. Utilizing multiple in silico analyses to identify putative causal SCN5A variants in Brugada syndrome. Sci Rep 2014;4:3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Walsh R, Peters NS, Cook SA, Ware JS. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet 2014;51:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Kapplinger JD, Giudicessi JR, Tester DJ, Callis TE, Ackerman MJ. Enhanced classification of non-synonymous single nucleotide variants in the SCN5A- encoded Nav1.5 cardiac sodium channel. Heart Rhythm 2012;9:1912. [Google Scholar]
- 209. Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR, Makielski JC, Wilde AA, Ackerman MJ. Enhanced classification of Brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded Nav1.5 cardiac sodium channel. Circ Cardiovasc Genet 2015;8:582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Probst V, Wilde AA, Barc J et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet 2009;2:552–557. [DOI] [PubMed] [Google Scholar]
- 211. Matsuo K, Akahoshi M, Seto S, Yano K. Disappearance of the Brugada-type electrocardiogram after surgical castration: a role for testosterone and an explanation for the male preponderance? Pacing Clin Electrophysiol 2003;26:1151–1153. [DOI] [PubMed] [Google Scholar]
- 212. Antzelevitch C. Androgens and male predominance of the Brugada syndrome phenotype. Pacing Clin Electrophysiol 2003;26:1429–1431. [DOI] [PubMed] [Google Scholar]
- 213. Korte AK, Derde L, van Wijk J, Tjan DH. Sudden cardiac arrest as a presentation of Brugada syndrome unmasked by thyroid storm. BMJ Case Rep 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, Antzelevitch C. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol 2010;49:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Morita H, Zipes DP, Wu J. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm 2009;6:S34–S43. [DOI] [PubMed] [Google Scholar]
- 216. Hoogendijk MG, Opthof T, Postema PG, Wilde AA, de Bakker JM, Coronel R. The Brugada ECG pattern: a marker of channelopathy, structural heart disease, or neither? Toward a unifying mechanism of the Brugada syndrome. Circ Arrhythm Electrophysiol 2010;3:283–290. [DOI] [PubMed] [Google Scholar]
- 217. Hoogendijk MG, Potse M, Linnenbank AC et al. Mechanism of right precordial ST-segment elevation in structural heart disease: excitation failure by current-to- load mismatch. Heart Rhythm 2010;7:238–248. [DOI] [PubMed] [Google Scholar]
- 218. Patocskai B, Antzelevitch C. Expert opinion on orphan drugs. Expert Opin Orphan Drugs 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Wilde AA, Postema PG. Bringing home the bacon? The next step in cardiac sodium channelopathies. J Clin Invest 2015;125:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, Perez Riera AR. Brugada syndrome: a decade of progress. Circ Res 2002;91:1114–1119. [DOI] [PubMed] [Google Scholar]
- 221. Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, Aihara N, Kamakura S, Kobayashi J, Kosakai Y. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. J Am Coll Cardiol 2002;40:330–334. [DOI] [PubMed] [Google Scholar]
- 222. Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ Arrhythm Electrophysiol 2014;7:134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Huikuri HV. Separation of benign from ,alignant J waves. Heart Rhythm 2015;12:384–385. [DOI] [PubMed] [Google Scholar]
- 224. Aizawa Y, Sato M, Kitazawa H, Aizawa Y, Takatsuki S, Oda E, Okabe M, Fukuda K. Tachycardia-dependent augmentation of “notched J waves” in a general patient population without ventricular fibrillation or cardiac arrest: Not a repolarization but a depolarization abnormality? Heart Rhythm 2015;12:376–383. [DOI] [PubMed] [Google Scholar]
- 225. Badri M, Patel A, Yan G. Cellular and ionic basis of J-wave syndromes. Trends Cardiovasc Med 2015;25:12–21. [DOI] [PubMed] [Google Scholar]
- 226. Morita H, Kusano KF, Miura D, Nagase S, Nakamura K, Morita ST, Ohe T, Zipes DP, Wu J. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation 2008;118:1697–1704. [DOI] [PubMed] [Google Scholar]
- 227. Terho HK, Tikkanen JT, Junttila JM, Anttonen O, Kentta TV, Aro AL, Kerola T, Rissanen HA, Reunanen A, Huikuri HV. Prevalence and prognostic significance of fragmented QRS complex in middle-aged subjects with and without clinical or electrocardiographic evidence of cardiac disease. Am J Cardiol 2014;114:141–147. [DOI] [PubMed] [Google Scholar]
- 228. Osher HL, Wolff L. Electrocardiographic patern simulating acute myocardial injury. Am J Med Sci 1953;226:541–545. [PubMed] [Google Scholar]
- 229. Viskin S, Adler A, Halkin A, Rosso R. Reply: is the J wave or the ST slope malignant…or neither? J Am Coll Cardiol 2014;63:1812–1813. [DOI] [PubMed] [Google Scholar]
- 230. Rosso R, Adler A, Halkin A, Viskin S. Risk of sudden death among young individuals with J waves and early repolarization: putting the evidence into perspective. Heart Rhythm 2011;8:923–929. [DOI] [PubMed] [Google Scholar]
- 231. Aagaard P, Shulman E, Di Biase L, Fisher JD, Gross JN, Kargoli F, Kim SG, Palma EC, Ferrick KJ, Krumerman A. Prognostic value of automatically detected early repolarization. Am J Cardiol 2014;114:1431–1436. [DOI] [PubMed] [Google Scholar]
- 232. Uberoi A, Jain NA, Perez M, Weinkopff A, Ashley E, Hadley D, Turakhia MP, Froelicher V. Early repolarization in an ambulatory clinical population. Circulation 2011;124:2208–2214. [DOI] [PubMed] [Google Scholar]
- 233. Muramoto D, Yong CM, Singh N, Aggarwal S, Perez M, Ashley E, Hadley D, Froelicher V. Patterns and prognosis of all components of the J-wave pattern in multiethnic athletes and ambulatory patients. Am Heart J 2014;167:259–266. [DOI] [PubMed] [Google Scholar]
- 234. Perez MV, Uberoi A, Jain NA, Ashley E, Turakhia MP, Froelicher V. The prognostic value of early repolarization with ST-segment elevation in African Americans. Heart Rhythm 2012;9:558–565. [DOI] [PubMed] [Google Scholar]
- 235. Klatsky AL, Oehm R, Cooper RA, Udaltsova N, Armstrong MA. The early repolarization normal variant electrocardiogram: correlates and consequences. Am J Med 2003;115:171–177. [DOI] [PubMed] [Google Scholar]
- 236. Rollin A, Maury P, Bongard V, Sacher F, Delay M, Duparc A, Mondoly P, Carrie D, Ferrieres J, Ruidavets JB. Prevalence, prognosis, and identification of the malignant form of early repolarization pattern in a population-based study. Am J Cardiol 2012;110:1302–1308. [DOI] [PubMed] [Google Scholar]
- 237. Tikkanen JT, Junttila MJ, Anttonen O, Aro AL, Luttinen S, Kerola T, Sager SJ, Rissanen HA, Myerburg RJ, Reunanen A, Huikuri HV. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation 2011;123:2666–2673. [DOI] [PubMed] [Google Scholar]
- 238. Olson KA, Viera AJ, Soliman EZ, Crow RS, Rosamond WD. Long-term prognosis associated with J-point elevation in a large middle-aged biracial cohort: the ARIC study. Eur Heart J 2011;32:3098–3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Rosso R, Halkin A, Viskin S. J waves and early repolarization: do not confuse me with the facts! Heart Rhythm 2012;9:1603–1604. [DOI] [PubMed] [Google Scholar]
- 240. Demidova MM, Martin-Yebra A, van der Pals J, Koul S, Erlinge D, Laguna P, Martinez JP, Platonov PG. Transient and rapid QRS-widening associated with a J-wave pattern predicts impending ventricular fibrillation in experimental myocardial infarction. Heart Rhythm 2014;11:1195–1201. [DOI] [PubMed] [Google Scholar]
- 241. Shinde R, Shinde S, Makhale C, Grant P, Sathe S, Durairaj M, Lokhandwala Y, Di Diego JM, Antzelevitch C. Occurrence of “J waves” in 12–lead ECG as a marker of acute ischemia and their cellular basis. Pacing Clin Electrophysiol 2007;30:817–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Gourraud JB, Le Scouarnec S, Sacher F et al. Identification of large families in early repolarization syndrome. J Am Coll Cardiol 2013;61:164–172. [DOI] [PubMed] [Google Scholar]
- 243. Watanabe H, Makiyama T, Koyama T et al. High prevalence of early repolarization in short QT syndrome. Heart Rhythm 2010;7:647–652. [DOI] [PubMed] [Google Scholar]
- 244. Letsas KP, Charalampous C, Korantzopoulos P, Tsikrikas S, Bramos D, Kollias G, Efremidis M, Sideris A. Novel indexes of heterogeneity of ventricular repolarization in subjects with early repolarization pattern. Europace 2012;14:877–878. [DOI] [PubMed] [Google Scholar]
- 245. Rosso R, Glikson E, Belhassen B, Katz A, Halkin A, Steinvil A, Viskin S. Distinguishing “benign” from “malignant early repolarization”: The value of the ST-segment morphology. Heart Rhythm 2012;9:225–229. [DOI] [PubMed] [Google Scholar]
- 246. Mahida S, Derval N, Sacher F et al. Role of electrophysiological studies in predicting risk of ventricular arrhythmia in early repolarization syndrome. J Am Coll Cardiol 2015;65:151–159. [DOI] [PubMed] [Google Scholar]
- 247. Probst V, Veltmann C, Eckardt L et al. Long-term prognosis of patients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643. [DOI] [PubMed] [Google Scholar]
- 248. Kamakura S, Ohe T, Nakazawa K et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1–V3. Circ Arrhythm Electrophysiol 2009;2:495–503. [DOI] [PubMed] [Google Scholar]
- 249. Conte G, Sieira J, Ciconte G et al. Implantable cardioverter-defibrillator therapy in Brugada syndrome: a 20-year single-center experience. J Am Coll Cardiol 2015;65:879–888. [DOI] [PubMed] [Google Scholar]
- 250. Sacher F, Probst V, Maury P et al. Outcome after implantation of a cardioverter- defibrillator in patients with Brugada syndrome: a multicenter study: part 2. Circulation 2013;128:1739–1747. [DOI] [PubMed] [Google Scholar]
- 251. Priori SG, Gasparini M, Napolitano C, Della BP, Ottonelli AG, Sassone B, Giordano U, Pappone C, Mascioli G, Rossetti G, De NR, Colombo M. Risk stratification in Brugada syndrome: results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol 2012;59:37–45. [DOI] [PubMed] [Google Scholar]
- 252. Take Y, Morita H, Toh N, Nishii N, Nagase S, Nakamura K, Kusano KF, Ohe T, Ito H. Identification of high-risk syncope related to ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm 2012;9:752–759. [DOI] [PubMed] [Google Scholar]
- 253. Conte G, De Asmundis C, Sieira J, Levinstein M, Chierchia GB, DiGiovanni G, Baltogiannis G, Ciconte G, Saitoh Y, Casado-Arroyo R, Pappaert G, Brugada P. Clinical characteristics, management, and prognosis of elderly patients with Brugada syndrome. J Cardiovasc Electrophysiol 2014;25:514–519. [DOI] [PubMed] [Google Scholar]
- 254. Priori SG, Napolitano C, Glordano U, Collisani G, Memmi M. Brugada syndrome and sudden cardiac death in children. Lancet 2000;355:808–809. [DOI] [PubMed] [Google Scholar]
- 255. Probst V, Denjoy I, Meregalli PG et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007;115:2042–2048. [DOI] [PubMed] [Google Scholar]
- 256. Meregalli PG, Tan HL, Probst V et al. Type of SCN5A mutation determines clinical severity and degree of conduction slowing in loss-of-function sodium channelopathies. Heart Rhythm 2009;6:341–348. [DOI] [PubMed] [Google Scholar]
- 257. Cordeiro JM, Barajas-Martinez H, Hong K et al. Compound heterozygous mutations P336L and I1660 V in the human cardiac sodium channel associated with the Brugada syndrome. Circulation 2006;114:2026–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Nunez L, Barana A, Amoros I et al. p.D1690N Nav1.5 rescues p.G1748D mutation gating defects in a compound heterozygous Brugada syndrome patient. Heart Rhythm 2013;10:264–272. [DOI] [PubMed] [Google Scholar]
- 259. Poelzing S, Forleo C, Samodell M et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 2006;114:368–376. [DOI] [PubMed] [Google Scholar]
- 260. Viswanathan PC, Benson DW, Balser JR. A common SCN5A polymorphism modulates the biophysical effects of an SCN5A mutation. J Clin Invest 2003;111:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Sommariva E, Pappone C, Martinelli BF, Di RC, Rosaria CM, Salvi E, Vergara P, Sala S, Cusi D, Ferrari M, Benedetti S. Genetics can contribute to the prognosis of Brugada syndrome: a pilot model for risk stratification. EurJ Hum Genet 2013;21:911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Delise P, Allocca G, Marras E et al. Risk stratification in individuals with the Brugada type 1 ECG pattern without previous cardiac arrest: usefulness of a combined clinical and electrophysiologic approach. Eur Heart J 2011;32:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Yokokawa M, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, Shimizu W. Neurally mediated syncope as a cause of syncope in patients with Brugada electrocardiogram. J Cardiovasc Electrophysiol 2010;21:186–192. [DOI] [PubMed] [Google Scholar]
- 264. Richter S, Sarkozy A, Veltmann C, Chierchia GB, Boussy T, Wolpert C, Schimpf R, Brugada J, Brugada R, Borggrefe M, Brugada P. Variability of the diagnostic ECG pattern in an ICD patient population with Brugada syndrome. J Cardiovasc Electrophysiol 2009;20:69–75. [DOI] [PubMed] [Google Scholar]
- 265. Veltmann C, Schimpf R, Echternach C, Eckardt L, Kuschyk J, Streitner F, Spehl S, Borggrefe M, Wolpert C. A prospective study on spontaneous fluctuations between diagnostic and non-diagnostic ECGs in Brugada syndrome: implications for correct phenotyping and risk stratification. Eur Heart J 2006;27:2544–2556. [DOI] [PubMed] [Google Scholar]
- 266. Rolf S. The ajmaline challenge in Brugada syndrome: diagnostic impact, safety, and recommended protocol. Eur Heart J 2003;24:1104–1112. [DOI] [PubMed] [Google Scholar]
- 267. Veltmann C, Wolpert C, Sacher F, Mabo P, Schimpf R, Streitner F, Brade J, Kyndt F, Kuschyk J, Le MH, Borggrefe M, Probst V. Response to intravenous ajmaline: a retrospective analysis of 677 ajmaline challenges. Europace 2009;11:1345–1352. [DOI] [PubMed] [Google Scholar]
- 268. Okamura H, Kamakura T, Morita H et al. Risk stratification in patients with Brugada syndrome without previous cardiac arrest: prognostic value of combined risk factors. Circ J 2015;79:310–317. [DOI] [PubMed] [Google Scholar]
- 269. Govindan M, Batchvarov VN, Raju H, Shanmugam N, Bizrah M, Bastiaenen R, Kiotsekoglou A, Camm J, Behr ER. Utility of high and standard right precordial leads during ajmaline testing for the diagnosis of Brugada syndrome. Heart 2010;96:1904–1908. [DOI] [PubMed] [Google Scholar]
- 270. Brugada J, Brugada R, Brugada P. Electrophysiologic testing predicts events in Brugada syndrome patients. Heart Rhythm 2011;8:1595–1597. [DOI] [PubMed] [Google Scholar]
- 271. Wilde AA, Viskin S. EP testing does not predict cardiac events in Brugada syndrome. Heart Rhythm 2011;8:1598–1600. [DOI] [PubMed] [Google Scholar]
- 272. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: a cardiac ticking time-bomb? Europace 2007;9:707–710. [DOI] [PubMed] [Google Scholar]
- 273. Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AA, Breithardt G, Eckardt L. Role of programmed ventricular stimulation in patients with Brugada syndrome: a meta-analysis of worldwide published data. Eur Heart J 2007;28:2126–2133. [DOI] [PubMed] [Google Scholar]
- 274. Brugada P, Brugada R, Brugada J. Patients with an asymptomatic Brugada electrocardiogram should undergo pharmacological and electrophysical testing. Circulation 2005;112:279–285. [DOI] [PubMed] [Google Scholar]
- 275. Sieira J, Conte G, Ciconte G et al. Prognostic value of programmed electrical stimulation in Brugada syndrome: 20 years experience. Circ Arrhythm Electrophysiol 2015;8:777–784. [DOI] [PubMed] [Google Scholar]
- 276. Makimoto H, Kamakura S, Aihara N et al. Clinical impact of the number of extrastimuli in programmed electrical stimulation in patients with Brugada type 1 electrocardiogram. Heart Rhythm 2012;9:242–248. [DOI] [PubMed] [Google Scholar]
- 277. Tokioka K, Kusano KF, Morita H, Miura D, Nishii N, Nagase S, Nakamura K, Kohno K, Ito H, Ohe T. Electrocardiographic parameters and fatal Arrhythmic events in patients with Brugada syndrome: combination of depolarization and repolarization abnormalities. J Am Coll Cardiol 2014;63:2131–2138. [DOI] [PubMed] [Google Scholar]
- 278. Take Y, Morita H. Fragmented QRS: What is the meaning? Indian Pacing Electrophysiol J 2012;12:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Kaneko Y, Horie M, Niwano S et al. Electrical storm in patients with Brugada syndrome is associated with early repolarization. Circ Arrhythm Electrophysiol 2014;7:1122–1128. [DOI] [PubMed] [Google Scholar]
- 280. Takagi M, Aonuma K, Sekiguchi Y, Yokoyama Y, Aihara N, Hiraoka M. Japan Idiopathic Ventricular Fibrillation Study I. The prognostic value of early repolarization (J wave) and ST-segment morphology after J wave in Brugada syndrome: multicenter study in Japan. Heart Rhythm 2013;10:533–539. [DOI] [PubMed] [Google Scholar]
- 281. Rollin A, Sacher F, Gourraud JB et al. Prevalence, characteristics, and prognosis role of type 1 ST elevation in the peripheral ECG leads in patients with Brugada syndrome. Heart Rhythm 2013;10:1012–1018. [DOI] [PubMed] [Google Scholar]
- 282. Marquez MF, Bisteni A, Medrano G, De Micheli A, Guevara M, Iturralde P, Colin L, Hermosillo G, Cardenas M. Dynamic electrocardiographic changes after aborted sudden death in a patient with Brugada syndrome and rate- dependent right bundle branch block. J Electrocardiol 2005;38:256–259. [DOI] [PubMed] [Google Scholar]
- 283. Kasanuki H, Ohnishi S, Ohtuka M, Matsuda N, Nirei T, Isogai R, Shoda M, Toyoshima Y, Hosoda S. Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 1997;95:2277–2285. [DOI] [PubMed] [Google Scholar]
- 284. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, Tunsanga K, Kuasirikul S, Malasit P, Tansupasawadikul S, Tatsanavivat P. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation 1997;96:2595–2600. [DOI] [PubMed] [Google Scholar]
- 285. Ikeda T, Takami M, Sugi K, Mizusawa Y, Sakurada H, Yoshino H. Noninvasive risk stratification of subjects with a Brugada-type electrocardiogram and no history of cardiac arrest. Ann Noninvasive Electrocardiol 2005;10:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Yoshioka K, Amino M, Zareba W, Shima M, Matsuzaki A, Fujii T, Kanda S, Deguchi Y, Kobayashi Y, Ikari Y, Kodama I, Tanabe T. Identification of high- risk Brugada syndrome patients by combined analysis of late potential and T-wave amplitude variability on ambulatory electrocardiograms. Circ J 2013;77:610–618. [DOI] [PubMed] [Google Scholar]
- 287. Tada T, Kusano KF, Nagase S, Banba K, Miura D, Nishii N, Watanabe A, Nakamura K, Morita H, Ohe T. Clinical significance of macroscopic T-wave alternans after sodium channel blocker administration in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2008;19:56–61. [DOI] [PubMed] [Google Scholar]
- 288. Uchimura-Makita Y, Nakano Y, Tokuyama T et al. Time-domain T-wave alternans is strongly associated with a history of ventricular fibrillation in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2014;25:1021–1027. [DOI] [PubMed] [Google Scholar]
- 289. Junttila MJ, Brugada P, Hong K, Lizotte E, de Zutter M, Sarkozy A, Brugada J, Benito B, Perkiomaki JS, Makikallio TH, Huikuri HV, Brugada R. Differences in 12–lead electrocardiogram between symptomatic and asymptomatic Brugada syndrome patients. J Cardiovasc Electrophysiol 2008;19:380–383. [DOI] [PubMed] [Google Scholar]
- 290. Maury P, Rollin A, Sacher F et al. Prevalence and prognostic role of various conduction disturbances in patients with the Brugada syndrome. Am J Cardiol 2013;11:1384–1389. [DOI] [PubMed] [Google Scholar]
- 291. Bigi MA, Aslani A, Shahrzad S. Clinical predictors of atrial fibrillation in Brugada syndrome. Europace 2007;9:947–950. [DOI] [PubMed] [Google Scholar]
- 292. Makimoto H, Nakagawa E, Takaki H, Yamada Y, Okamura H, Noda T, Satomi K, Suyama K, Aihara N, Kurita T, Kamakura S, Shimizu W. Augmented ST-segment elevation during recovery from exercise predicts cardiac events in patients with Brugada syndrome. J Am Coll Cardiol 2010;56:1576–1584. [DOI] [PubMed] [Google Scholar]
- 293. Castro Hevia J, Antzelevitch C, Tornes Barzaga F, Dorantes Sanchez M, Dorticos Balea F, Zayas Molina R, Quinones Perez MA, Fayad Rodriguez Y. Tpeak-Tend and Tpeak-Tend dispersion as risk factors for ventricular tachycardia/ventricular fibrillation in patients with the Brugada syndrome. J Am Coll Cardiol 2006;47:1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Karim Talib A, Sato N, Sakamoto N, Tanabe Y, Takeuchi T, Saijo Y, Kawamura Y, Hasebe N. Enhanced transmural dispersion of repolarization in patients with J wave syndromes. J Cardiovasc Electrophysiol 2012;23:1109–1114. [DOI] [PubMed] [Google Scholar]
- 295. Lambiase PD. Tpeak-Tend interval and Tpeak-Tend/QT ratio as markers of ventricular tachycardia inducibility in subjects with Brugada ECG phenotype. Europace 2010;12:158–159. [DOI] [PubMed] [Google Scholar]
- 296. Maury P, Sacher F, Gourraud JB et al. Increased Tpeak-Tend interval is highly and independently related to arrhythmic events in Brugada syndrome. Heart Rhythm 2015;12:2469–2476. [DOI] [PubMed] [Google Scholar]
- 297. Sugao M, Fujiki A, Nishida K, Sakabe M, Tsuneda T, Iwamoto J, Mizumaki K, Inoue H. Repolarization dynamics in patients with idiopathic ventricular fibrillation: pharmacological therapy with bepridil and disopyramide. J Cardiovasc Pharmacol 2005;45:545–549. [DOI] [PubMed] [Google Scholar]
- 298. Aizawa Y, Yamakawa H, Takatsuki S et al. Efficacy and safety of bepridil for prevention of ICD shocks in patients with Brugada syndrome and idiopathic ventricular fibrillation. Int J Cardiol 2013;168:5083–5085. [DOI] [PubMed] [Google Scholar]
- 298A. Calò L, Giustetto C, Martino A, Sciarra L, Cerrato N, Marziali M, Rauzino J, Carlino G, deRuvo E, Guerra F, Rebecchi M, Lanzillo C, Anselmino M, Castro A, Turreni F, Penco M, Volpe M, Capucci A, Gaita F. J Am Coll Cardiol 2016 Mar 29;67(12):1427–1440. [DOI] [PubMed] [Google Scholar]
- 299. Masrur S, Memon S, Thompson PD. Brugada syndrome, exercise, and exercise testing. Clin Cardiol 2015;38:323–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Amin AS, de Groot EA, Ruijter JM, Wilde AA, Tan HL. Exercise-induced ECG changes in Brugada syndrome. Circ Arrhythm Electrophysiol 2009;2:531–539. [DOI] [PubMed] [Google Scholar]
- 301. Brugada J, Brugada R, Brugada P. Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol 2000;33(Suppl):41–47. [DOI] [PubMed] [Google Scholar]
- 302. Nademanee K, Veerakul G, Mower M, Likittanasombat K, Krittayapong R, Bhuripanyo K, Sitthisook S, Chaothawee L, Lai MY, Azen SP. Defibrillator Versus beta-Blockers for Unexplained Death in Thailand (DEBUT): a randomized clinical trial. Circulation 2003;107:2221–2226. [DOI] [PubMed] [Google Scholar]
- 303. Sacher F, Probst V, Maury P et al. Outcome after implantation of cardioverter- defibrillator in patients with Brugada syndrome: a multicenter study—part 2. Circulation 2013;128:1739–1747. [DOI] [PubMed] [Google Scholar]
- 304. Sacher F, Probst V, Bessouet M et al. Remote implantable cardioverter defibrillator monitoring in a Brugada syndrome population. Europace 2009;11:489–494. [DOI] [PubMed] [Google Scholar]
- 305. De Maria E, Olaru A, Cappelli S. The entirely subcutaneous defibrillator (S- ICD): state of the art and selection of the ideal candidate. Curr Cardiol Rev 2015;11:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Brugada P, Brugada R, Brugada J, Geelen P. Use of the prophylactic implantable cardioverter defibrillator for patients with normal hearts. Am J Cardiol 1999;83:98D–100D. [DOI] [PubMed] [Google Scholar]
- 307. Takagi M, Tatsumi H, Yoshiyama M. Approach to the asymptomatic patients with Brugada syndrome. Indian Pacing Electrophysiol J 2007;7:73–76. [PMC free article] [PubMed] [Google Scholar]
- 308. van Den Berg MP, Wilde AA, Viersma TJW, Brouwer J, Haaksma J, van der Hout AH, Stolte-Dijkstra I, Bezzina TCR, Van Langen IM, Beaufort-Krol GC, Cornel JH, Crijns HJ. Possible bradycardic mode of death and successful pacemaker treatment in a large family with features of long QT syndrome type 3 and Brugada syndrome. J Cardiovasc Electrophysiol 2001;12:630–636. [DOI] [PubMed] [Google Scholar]
- 309. Bertomeu-Gonzalez V, Ruiz-Granell R, Garcia-Civera R, Morell-Cabedo S, Ferrero A. Syncopal monomorphic ventricular tachycardia with pleomorphism, sensitive to antitachycardia pacing in a patient with Brugada syndrome. Europace 2006;8:1048–1050. [DOI] [PubMed] [Google Scholar]
- 310. Lee KL, Lau C, Tse H, Wan S, Fan K. Prevention of ventricular fibrillation by pacing in a man with Brugada syndrome. J Cardiovasc Electrophysiol 2000;11:935–937. [DOI] [PubMed] [Google Scholar]
- 311. Cortez-Dias N, Placido R, Marta L, Bernardes A, Sobral S, Carpinteiro L, de Sousa J. Epicardial ablation for prevention of ventricular fibrillation in a patient with Brugada syndrome. Rev Port Cardiol 2014;33:305–305. [DOI] [PubMed] [Google Scholar]
- 312. Shah AJ, Hocini M, Lamaison D, Sacher F, Derval N, Haissaguerre M. Regional substrate ablation abolishes Brugada syndrome. J Cardiovasc Electrophysiol 2011;22:1290–1291. [DOI] [PubMed] [Google Scholar]
- 313. Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3. A marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998;97:457–460. [DOI] [PubMed] [Google Scholar]
- 314. Chinushi M, Aizawa Y, Ogawa Y, Shiba M, Takahashi K. Discrepant drug action of disopyramide on ECG abnormalities and induction of ventricular arrhythmias in a patient with Brugada syndrome. J Electrocardiol 1997;30:133–136. [DOI] [PubMed] [Google Scholar]
- 315. Minoura Y, Di Diego JM, Barajas-Martinez H, Zygmunt AC, Hu D, Sicouri S, Antzelevitch C. Ionic and cellular mechanisms underlying the development of acquired Brugada syndrome in patients treated with antidepressants. J Cardiovasc Electrophysiol 2012;23:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Minoura Y, Panama BK, Nesterenko VV, Betzenhauser M, Barajas-Martinez H, Hu D, Di Diego JM, Antzelevitch C. Effect of Wenxin Keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm 2013;10:1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Johnson P, Lesage A, Floyd WL, Young WG Jr, Sealy WC. Prevention of ventricular fibrillation during profound hypothermia by quinidine. Ann Surg 1960;151:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Belhassen B, Shapira I, Shoshani D, Paredes A, Miller H, Laniado S. Idiopathic ventricular fibrillation: inducibility and beneficial effects of class I antiarrhythmic agents. Circulation 1987;75:809–816. [DOI] [PubMed] [Google Scholar]
- 319. Belhassen B, Viskin S, Antzelevitch C. The Brugada syndrome: is an implantable cardioverter defibrillator the only therapeutic option? Pacing Clin Electrophysiol 2002;25:1634–1640. [DOI] [PubMed] [Google Scholar]
- 320. Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1420–1422. [DOI] [PubMed] [Google Scholar]
- 321. Belhassen B, Viskin S. Pharmacologic approach to therapy of Brugada syndrome: quinidine as an alternative to ICD therapy? In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada Syndrome: From Bench to Bedside. Oxford: Blackwell Futura; 2004:202–211. [Google Scholar]
- 322. Viskin S, Wilde AA, Tan HL, Antzelevitch C, Shimizu W, Belhassen B. Empiric quinidine therapy for asymptomatic Brugada syndrome: time for a prospective registry. Heart Rhythm 2009;6:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Belhassen B, Glick A, Viskin S. Excellent long-term reproducibility of the electrophysiologic efficacy of quinidine in patients with idiopathic ventricular fibrillation and Brugada syndrome. Pacing Clin Electrophysiol 2009;32:294–301. [DOI] [PubMed] [Google Scholar]
- 324. Marquez MF, Bonny A, Hernandez-Castillo E, De SA, Gomez-Flores J, Nava S, Hidden-Lucet F, Iturralde P, Cardenas M, Tonet J. Long-term efficacy of low doses of quinidine on malignant arrhythmias in Brugada syndrome with an implantable cardioverter-defibrillator: a case series and literature review. Heart Rhythm 2013;9:1995–2000. [DOI] [PubMed] [Google Scholar]
- 325. Pellegrino PL, Di BM, Brunetti ND. Quinidine for the management of electrical storm in an old patient with Brugada syndrome and syncope. Acta Cardiol 2013;68:201–203. [DOI] [PubMed] [Google Scholar]
- 326. Probst V, Allouis M, Sacher F et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J Cardiovasc Electrophysiol 2006;17:270–275. [DOI] [PubMed] [Google Scholar]
- 327. Schweizer PA, Becker R, Katus HA, Thomas D. Successful acute and long-term management of electrical storm in Brugada syndrome using orciprenaline and quinine/quinidine. Clin Res Cardiol 2010;99:467–470. [DOI] [PubMed] [Google Scholar]
- 328. Marquez MF, Rivera J, Hermosillo AG, Iturralde P, Colin L, Moragrega JL, Cardenas M. Arrhythmic storm responsive to quinidine in a patient with Brugada syndrome and vasovagal syncope. Pacing Clin Electrophysiol 2005;28:870–873. [DOI] [PubMed] [Google Scholar]
- 329. Viskin S, Antzelevitch C, Marquez MF, Belhassen B. Quinidine: a valuable medication joins the list of “endangered species”. Europace 2007;12:1105–1106. [DOI] [PubMed] [Google Scholar]
- 330. Rosso R, Glick A, Glikson M et al. Outcome after implantation of cardioverter defribrillator in patients with Brugada syndrome: a multicenter Israeli study (ISRABRU). Isr Med Assoc J 2008;10:435–439. [PubMed] [Google Scholar]
- 331. Mok NS, Chan NY, Chi-Suen CA. Successful use of quinidine in treatment of electrical storm in Brugada syndrome. Pacing Clin Electrophysiol 2004;27:821–823. [DOI] [PubMed] [Google Scholar]
- 332. Belhassen B, Rahkovich M, Michowitz Y, Glick A, Viskin S. Management of Brugada syndrome: a 33-year experience using electrophysiologically-guided therapy with Class 1A antiarrhythmic drugs. Circ Arrhythm Electrophysiol 2015;6:1393–1402. [DOI] [PubMed] [Google Scholar]
- 333. Bouzeman A, Traulle S, Messali A, Extramiana F, Denjoy I, Narayanan K, Marijon E, Hermida JS, Leenhardt A. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace 2014;16:572–577. [DOI] [PubMed] [Google Scholar]
- 334. Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol 2001;12:268–272. [DOI] [PubMed] [Google Scholar]
- 335. Kyriazis K, Bahlmann E, van der Schalk H, Kuck KH. Electrical storm in Brugada syndrome successfully treated with orciprenaline: effect of low-dose quinidine on the electrocardiogram. Europace 2009;11:665–666. [DOI] [PubMed] [Google Scholar]
- 336. Shimizu W, Matsuo K, Takagi M, Tanabe Y, Aiba T, Taguchi A, Suyama K, Kurita T, Aihara N, Kamakura S. Body surface distribution and response to drugs of ST segment elevation in Brugada syndrome: clinical implication of eighty-seven-lead body surface potential mapping and its application to twelve- lead electrocardiograms. J Cardiovasc Electrophysiol 2000;11:396–404. [DOI] [PubMed] [Google Scholar]
- 337. Tanaka H, Kinoshita O, Uchikawa S et al. Successful prevention of recurrent ventricular fibrillation by intravenous isoproterenol in a patient with Brugada syndrome. Pacing Clin Electrophysiol 2001;24:1293–1294. [DOI] [PubMed] [Google Scholar]
- 338. Maury P, Couderc P, Delay M, Boveda S, Brugada J. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace 2004;6:130–133. [DOI] [PubMed] [Google Scholar]
- 339. Maury P, Hocini M, Haissaguerre M. Electrical storms in Brugada syndrome: review of pharmacologic and ablative therapeutic options. Indian Pacing Electrophysiol J 2005;5:25–34. [PMC free article] [PubMed] [Google Scholar]
- 340. Jongman JK, Jepkes-Bruin N, Ramdat Misier AR, Beukema WP, Delnoy PP, Oude LH, Dambrink JH, Hoorntje JC, Elvan A. Electrical storms in Brugada syndrome successfully treated with isoproterenol infusion and quinidine orally. Neth Heart J 2007;15:151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Sharif-Kazemi MB, Emkanjoo Z, Tavoosi A, Kafi M, Kheirkhah J, Alizadeh A, Sadr-Ameli MA. Electrical storm in Brugada syndrome during pregnancy. Pacing Clin Electrophysiol 2011;34:e18–e21. [DOI] [PubMed] [Google Scholar]
- 342. Furniss G. Isoprenaline and quinidine to calm Brugada VF storm. BMJ Case Rep 2012;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Roten L, Derval N, Sacher F et al. Heterogeneous response of J wave syndromes to beta-adrenergic stimulation. Heart Rhythm 2012;9:1970–1976. [DOI] [PubMed] [Google Scholar]
- 344. Kanlop N, Chattipakorn S, Chattipakorn N. Effects of cilostazol in the heart. J Cardiovasc Med (Hagerstown) 2011;12:88–95. [DOI] [PubMed] [Google Scholar]
- 345. Bai Y, Muqier, Murakami H, Iwasa M, Sumi S, Yamada Y, Ushikoshi H, Aoyama T, Nishigaki K, Takemura G, Uno B, Minatoguchi S. Cilostazol protects the heart against ischaemia reperfusion injury in a rabbit model of myocardial infarction: focus on adenosine, nitric oxide and mitochondrial ATP-sensitive potassium channels. Clin Exp Pharmacol Physiol 2011;38:658–665. [DOI] [PubMed] [Google Scholar]
- 346. Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J Cardiovasc Electrophysiol 2006;17:210–212. [DOI] [PubMed] [Google Scholar]
- 347. Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to supress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm 2013;10:1720–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Murakami M, Nakamura K, Kusano KF et al. Efficacy of low-dose bepridil for prevention of ventricular fibrillation in patients with Brugada syndrome with and without SCN5A mutation. J Cardiovasc Pharmacol 2010;56:389–395. [DOI] [PubMed] [Google Scholar]
- 349. Kang L, Zheng MQ, Morishima M, Wang Y, Kaku T, Ono K. Bepridil up- regulates cardiac Na+ channels as a long-term effect by blunting proteasome signals through inhibition of calmodulin activity. Br J Pharmacol 2009;157:404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Fish JM, Welchons DR, Kim YS, Lee SH, Ho WK, Antzelevitch C. Dimethyl lithospermate B, an extract of danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation 2006;113:1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Kanlop N, Shinlapawittayatorn K, Sungnoon R, Weerateerangkul P, Chattipakorn S, Chattipakorn N. Cilostazol attenuates ventricular arrhythmia induction and improves defibrillation efficacy in swine. Can J Physiol Pharmacol 2010;88:422–428. [DOI] [PubMed] [Google Scholar]
- 352. Endoh M, Yanagisawa T, Taira N, Blinks JR. Effects of new inotropic agents on cyclic nucleotide metabolism and calcium transients in canine ventricular muscle. Circulation 1986;73:III117–III133. [PubMed] [Google Scholar]
- 353. Rapundalo ST, Grupp I, Grupp G, Abdul MM, Solaro RJ, Schwartz A. Myocardial actions of milrinone: characterization of its mechanism of action. Circulation 1986;73:III134–III144. [PubMed] [Google Scholar]
- 354. Atarashi H, Endoh Y, Saitoh H, Kishida H, Hayakawa H. Chronotropic effects of cilostazol, a new antithrombotic agent, in patients with bradyarrhythmias. J Cardiovasc Pharmacol 1998;31:534–539. [DOI] [PubMed] [Google Scholar]
- 355. Matsui K, Kiyosue T, Wang JC, Dohi K, Arita M. Effects of pimobendan on the L-type Ca2+ current and developed tension in guinea-pig ventricular myocytes and papillary muscle: comparison with IBMX, milrinone, and cilostazol. Cardiovasc Drugs Ther 1999;13:105–113. [DOI] [PubMed] [Google Scholar]
- 356. Aizawa Y, Chinushi M, Hasegawa K et al. Electrical storm in idiopathic ventricular fibrillation is associated with early repolarization. J Am Coll Cardiol 2013;62:1015–1019. [DOI] [PubMed] [Google Scholar]
- 357. Nakagawa K, Nagase S, Morita H, Ito H. Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm 2013;11:314–317. [DOI] [PubMed] [Google Scholar]
- 358. Morita H, Zipes DP, Morita ST, Wu J. Temperature modulation of ventricular arrhythmogenicity in a canine tissue model of Brugada syndrome. Heart Rhythm 2007;4:188–197. [DOI] [PubMed] [Google Scholar]
- 359. Probst V, Veltmann C, Eckardt L et al. Long-term prognosis of aatients diagnosed with Brugada syndrome: results from the FINGER Brugada Syndrome Registry. Circulation 2010;121:635–643. [DOI] [PubMed] [Google Scholar]
- 360. Takagi M, Aonuma K, Sekiguchi Y, Yokoyama Y, Aihara N, Hiraoka M. The prognostic value of early repolarization (J wave) and ST-segment morphology after J wave in Brugada syndrome: multicenter study in Japan. Heart Rhythm 2013;10:533–539. [DOI] [PubMed] [Google Scholar]
- 361. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome. Pacing Clin Electrophysiol 2003;26:957. [DOI] [PubMed] [Google Scholar]
- 362. Huang Z, Patel C, Li W, Xie Q, Wu R, Zhang L, Tang R, Wan X, Ma Y, Zhen W, Gao L, Yan GX. Role of signal-averaged electrocardiograms in arrhythmic risk stratification of patients with Brugada syndrome: a prospective study. Heart Rhythm 2009;6:1156–1162. [DOI] [PubMed] [Google Scholar]
- 363. Priori SG. Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 2002;105:1342–1347. [DOI] [PubMed] [Google Scholar]
- 364. Brugada J, Brugada R, Brugada P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003;108:3092–3096. [DOI] [PubMed] [Google Scholar]
- 365. Cappato R, Furlanello F, Giovinazzo V et al. J wave, QRS slurring and ST elevation in athletes with cardiac arrest in the absence of heart disease: marker of risk, or innocent bystander? Circ Arrhythm Electrophysiol 2010;3:305–311. [DOI] [PubMed] [Google Scholar]
- 366. Perez-Riera AR, Abreu LC, Yanowitz F, Barros RB, Femenia F, McIntyre WF, Baranchuk A. “Benign” early repolarization versus malignant early abnormalities: clinical-electrocardiographic distinction and genetic basis. Cardiol J 2012;19:337–346. [DOI] [PubMed] [Google Scholar]