

Abstract
In recent years, cancers once viewed as relatively homogeneous in terms of organ location and treatment strategy are now better understood to be increasingly heterogeneous across biomarker and genetically defined patient subgroups. This has produced a shift toward development of biomarker-targeted agents during a time when funding for cancer research has been limited; as a result, the need for improved operational efficiency in studying many agent-and-target combinations in parallel has emerged. Platform trials, basket trials, and umbrella trials are new approaches to clinical research driven by this need for enhanced efficiency in the modern era of increasingly specific cancer subpopulations and decreased resources to study treatments for individual cancer subtypes in a traditional way. In this review, we provide an overview of these new types of clinical trial designs, including discussions of motivation for their use, recommended terminology, examples, and challenges encountered in their application.
Keywords: basket trial, biomarker-based trial, master protocol, platform trial, precision medicine, umbrella trial
Introduction
A new cancer treatment paradigm
In recent years, substantial progress in the areas of genomics technology, tumor biology, computational analysis, and drug discovery have motivated exciting advances in clinical and translational cancer research. In particular, rapid development, decreased cost, and increased availability of next-generation genomic sequencing and other methods for molecular classification of tumors has produced a new paradigm for understanding and treating cancer. In some cases, this new molecular viewpoint of ‘precision oncology’ has led to therapeutic discoveries where the targeted treatment paradigm actually worked; for most other hypotheses, however, the significance of molecular classification remains unclear.
The advantages of targeting oncogenic pathways believed to be responsible for the growth and metastasis of tumors is now well established in a variety of cancer settings, including BRAF-mutant melanoma [1], HER2-positive breast cancer [2, 3], KRAS wild-type colorectal cancer [4, 5], and EGFR or ALK-mutated lung cancer [6–8], among others. The success of most of these discoveries hinged upon identification of a fairly prevalent biomarker which happened to be an actionable mutation responsible for driving the clinical behavior of a fairly common tumor; where low prevalence mutations exist, particularly in rare diseases, less progress has been made to date. Additionally, immunotherapies including nivolumab [9, 10] and pembrolizumab [11] have been approved for treatment of advanced squamous cell and non-squamous cell non-small-cell lung cancer (NSCLC) and advanced melanoma, respectively.
Development of novel therapeutics is challenged by the heterogeneity that exists not only among patients within any given tumor type (inter-patient heterogeneity), but also the heterogeneity within an individual (intra-patient heterogeneity) as demonstrated by molecular evolution of a tumor through time (through sequences of therapy) and space (primary tumor to metastasis) [12]. The respective concepts of ‘oncogenic driver’ and ‘oncogene addiction’ have thus shifted the course of drug development in oncology [13–15]. In parallel, increased understanding of the complex structural paradigm of genetic alterations activating intracellular proteins along multiple pathways, as well as several years of early clinical experience with success of (and subsequent resistance to) single-target therapies, has led to the widely accepted hypothesis that multiple targeted therapeutics will likely be required to overcome tumor resistance and yield sustained clinical benefit for patients [16–20].
Clearly, we have entered an era where a patient's tumor and its treatment will no longer be viewed primarily in terms of fixed organ location and pathology, but also (or instead) will be viewed in terms of potentially dynamic genomic, proteomic, transcriptomic, and immunologic abnormalities and features that may be specifically targeted with novel agents.
Challenges of traditional clinical trial designs in the era of new treatment strategies
Despite remarkable advances in the development of molecularly targeted agents, progress on the clinical front has outpaced modernization of the clinical trial designs used to study novel therapies within possibly heterogeneous—and potentially rare—patient subgroups [21]. Cancer statistician Dr Don Berry has called clinical trials the ‘weakest links in the chain of knowledge for determining therapeutic advances’, further stating that ‘it is ironic that we take the same clinical trial approach to evaluate all manner of potentially amazing transformative experimental therapies and yet we don't experiment with the design of the clinical trial itself’ [22]. Limitations inherent to both the existing phase I–II–III clinical development paradigm and the types of designs typically employed within single-phase cancer trials are evident when clinical cancer ‘success’ is viewed on a broader scale. Currently, the time from initial drug discovery to clinical testing and regulatory review can take up to 15 years [23]. While preclinical evaluations are currently underway for thousands of compounds intended for the treatment of cancer [24] and 771 new cancer medicines and vaccines are currently in development or awaiting regulatory review [25], it remains the case that only ∼13% of cancer drugs initiating phase I studies ultimately achieve final market approval [26]. Of those compounds that make it to confirmatory phase III trials, only 34% may be expected to achieve a ‘statistically significant’ result [27, 28].
A 2010 report by the Institute of Medicine echoed these concerns, highlighting the challenges faced by cooperative oncology groups and calling for restructuring of the entire clinical trials system to increase efficiency and avoid redundancy at a time where novel compounds waiting for evaluation are many and resources are limited [29]. In the same year, the National Cancer Institute's Investigational Drug Steering Committee convened The Clinical Trial Design Task Force, the members of which endorsed several types of clinical trial design modifications to meet these objectives, including sequential learning for early termination (for efficacy or futility), possible trial extensions to establish or identify predictive subgroups, multi-stage (e.g. phase II/III) designs to enable seamless transitions to confirmatory studies, and ‘platform’ designs that allow for mid-trial adding or dropping of new experimental treatment arms [30, 31].
Concerns regarding simple application of ‘off-the-shelf’ designs within a traditional sequence of phase I, II, and III trials (often utilizing different end points) are especially well founded in the context of targeted therapies studied within ever-shrinking ‘targeted’ patient populations [22]. Specifically, when molecularly targeted treatments are studied in early phase trials, it must be decided whether to allow all eligible patients with a given disease to enroll, whether to restrict enrollment to those patients hypothesized to experience the greatest benefit from targeted therapy, or whether to do some combination of the two through mid-trial adaptive measures [32, 33]. In the case of a standard ‘all-comers’ design, the targeted agent may only benefit a selected group of patients and thus a strong subgroup effect may instead appear as a weak overall effect, though randomization of marker-positive and marker-negative patients to targeted versus non-targeted treatment can play an important role of validating the treatment-by-marker interaction hypothesis. On the other hand, so-called ‘enriched’ clinical trials enrolling only those patients hypothesized to benefit (such as those whose tumor DNA harbors a particular mutation) may demonstrate a large effect in theory, but such a trial may be challenging to conduct in practice, particularly within low-prevalence populations where a high screen failure rate dampens enthusiasm for the trial. Choice of an unselected versus enriched design for a single marker-therapy combination should be made based on the existing level of evidence for the putative predictive biomarker [34].
As many have noted, performing clinical development of targeted therapies according to a traditionally isolated one-agent-at-a-time fashion lacks efficiency and may be prone to high overhead and low feasibility, particularly in low-prevalence subgroups of a given disease [16, 17, 22, 35]. While nearly 1.7 million adults are diagnosed with cancer in the United States each year [36], only 3%–5% of these patients enroll in clinical trials, further contributing to underpowered studies and early trial discontinuation [37].
This changing cancer paradigm and ongoing segmentation of broadly recognized cancers into comparatively rare subcancers have produced an urgent need to streamline the development and approval of new compounds [16, 19].
A movement toward increased efficiency in clinical research
In response to the aforementioned challenges and pressures of conducting single biomarker-based trials in this new era of clinical cancer research, a trend has emerged toward investigating multiple target–treatment pairs in parallel, either within or across recognized tumor types. These so-called ‘master protocols’ may utilize a centralized screening platform and common protocol format for each of several biomarker-driven substudies, with benefits including enhanced patient participation due to increased likelihood of eligibility for at least one of the accruing cohorts. In the sections that follow, we review different types of master protocols including ‘basket’ and ‘umbrella’ trials that operationalize these advantages. We further clarify terminology related to these types of trials, describe features they have in common, provide examples of recently completed or ongoing master protocols, and discuss challenges and important considerations in their implementation.
Overview of master protocols
Master protocols: motivation and common features
The term ‘master protocol’ refers generally to a framework in which multiple parallel drug studies are operated under one overarching (‘master’) protocol, wherein the parallel studies are differentiated by the marker–treatment combinations under investigation (Figure 1) [16]. Master protocols require endorsement by a broad consortium of academic and industry partners, pharmaceutical companies, and government agencies. The main goals of constructing a master protocol in place of several truly independent trials in biomarker-defined cohorts include increased genomic screening efficiency, accelerated and streamlined clinical development timeline, and enhanced enthusiasm for patient accrual due to inclusion of a broad range of molecular subtypes.
Figure 1.

General schema of a master protocol.
increased efficiency
Master protocols increase genomic screening efficiency in several ways, including: use of a common platform or set of assays capable of detecting abnormalities in multiple potential targets; immediate assignment of patients to an appropriate substudy on the basis of screening results; strict guidelines ensuring that sufficient tumor is evaluated for simultaneous assessment of multiple markers or abnormalities, and a structural framework for allowing substudies of new agents and biomarkers to be added or dropped in a preplanned, expeditious fashion. Although not always the case, substudies of master protocols are often designed to detect only large efficacy signals (in single-arm cohorts) or large treatment effects (in randomized cohorts), with efficiency resulting from lower sample size requirements [16, 19, 22]. Additionally, master protocols often utilize a master budget with shared costs for a common infrastructure across substudies, resulting in financial efficiency.
accelerated clinical development
Master protocols often include enhanced regulatory input to support accelerated clinical development, as agencies usually review the high-level study design of a master protocol or platform trial up front, with lesser requirements for subsequent regulatory review when modifications to treatments or cohorts are made later in time [16, 38–40]. In this case, the protocol serves as an operational structure allowing for new therapies to enter and subsequently exit a standing trial (for efficacy or futility) while the trial is underway, without the need for overall protocol modifications and associated administrative delays. The use of a master protocol with a standardized screening platform may also facilitate US Food and Drug Administration (FDA) approval of new therapies, making them available to marker-defined subgroups of patients more quickly than may have been possible otherwise.
stakeholder enthusiasm
Inclusion of a potentially large number of marker–treatment combinations within the same study has the potential to bolster the enthusiasm of study stakeholders, including treating physicians, participating institutions, sponsors, and patients themselves. In particular, where an additional cohort is included to capture patients negative on all markers (and where such patients are assigned to an experimental therapy or standard of care), an initially eligible patient may enter the trial with the knowledge that he or she will almost certainly be able to receive a potentially promising experimental treatment regardless of the outcome of the screening process [16, 35].
flexible objectives
Substudies based on biomarkers or genomic groups of interest may share common design features (e.g. power, sample size, type I error), or different study designs may be used across the master protocol, reflecting differences in study objectives among the protocol cohorts. A master protocol may contain substudies with discovery-based or confirmatory objectives, or in some cases, sequential objectives are addressed by multi-phase designs within patient cohorts [16, 21, 38, 39]. An exploratory master protocol typically comprises single-arm studies within targeted cohorts where patients received a matched experimental (targeted) drug; a confirmatory master protocol randomizes patients to targeted versus non-targeted or standard of care therapy within targeted cohorts in order to confirm the predictive nature of the biomarker for regulatory purposes [21].
Baskets, umbrellas, and platforms: definitions and terminology
Rapid introduction of master protocols and the subsequent frenzy of scientific interest have motivated widespread use (and misuse) of related terms in the literature such as ‘basket trial’, ‘umbrella trial’, and ‘platform trial’, among others. A number of authors have noted confusion regarding the definitions of these terms [16, 19, 22, 39, 41, 42]; we attempt to harmonize some definitions here.
A basket trial is a master protocol for which patient eligibility is defined by the presence of a particular biomarker or molecular alteration rather than a particular cancer type. Basket trials are predicted on the hypothesis that the molecular characterization of a particular tumor predicts response to a matched (targeted) treatment to a greater extent or independent of tumor histology [16, 18–20, 21, 22, 39, 41, 42]. Basket trials are generally tumor-agnostic to some degree; for example, enrollment to a basket trial may be restricted to patients with solid tumors, while the molecularly defined subtrials (or ‘baskets’) may comprise patients with many different types of solid tumors. For this reason, randomization to a common control arm within molecular cohorts is uncommon, due to differences in standard of care across tumor types. A general schema for a basket trial is shown in Figure 2.
Figure 2.
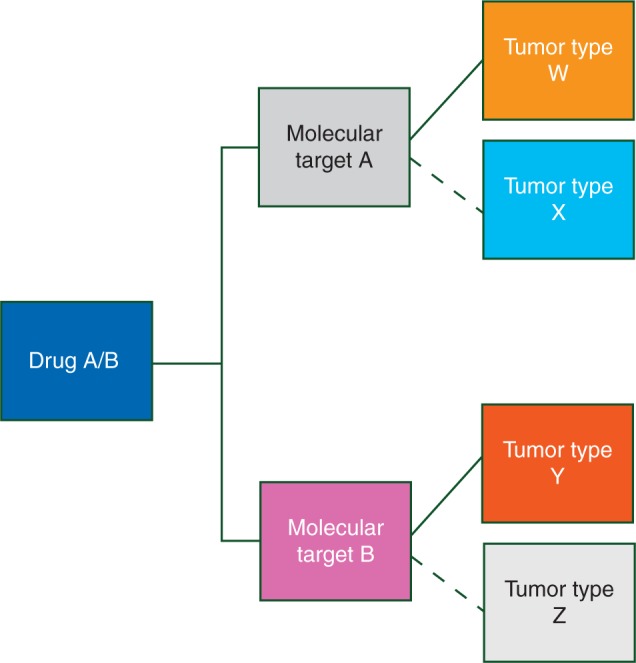
General schema of a basket trial (one ‘basket’ of potentially many baskets shown).
Unlike basket trials, where drug–mutation pairs are tested on a variety of tumor types, an umbrella trial generally restricts enrollment to a single type or class of cancers. In an umbrella trial, patients with tumors from the specified cancer type are centrally screened and assigned to one of several molecularly defined subtrials where they receive (or perhaps are randomized to) a matched targeted treatment [16, 18–21, 39, 42]. In such trials, the relevant markers are regarded as refinements of (rather than replacements of) tumor type. Common features of umbrella trials include: an enrollment and screening process that occurs before an actionable biomarker has been identified, and central profiling using a standardized platform or set of assays. While basket studies are generally limited to single-arm substudies with discovery objectives, umbrella trials may include single-arm or randomized subtrials, where the latter includes confirmatory objectives. An example schema for an umbrella trial is shown in Figure 3. Hyman et al. [39] refer to umbrella trials as ‘molecular allocation studies’.
Figure 3.
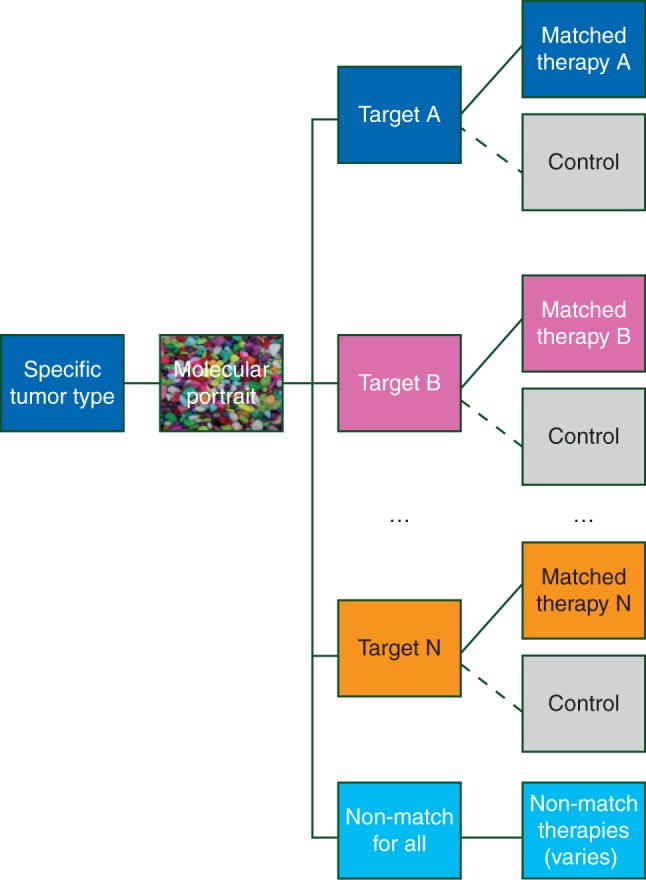
General schema of an umbrella trial.
Another type of master protocol described in the literature is the platform trial (or ‘standing trial’), a generic term for a randomized design with a common control arm and many different experimental arms that enter and exit the trial as futility or efficacy are demonstrated, often according to Bayesian decision rules [22, 38, 40]. The trial itself then comprises a platform or standing infrastructure to which novel therapies may be added or from which they may be dropped. While biomarker cohorts in a platform trial may not be explicitly separate, the treatment effects of various experimental treatments are usually modeled as independent parameters across molecularly defined subtypes, often according to a Bayesian hierarchical model. Adaptive randomization, i.e. mid-trial shifts in the randomization ratios for patients with a given biomarker signature to favor the treatment showing the most promise in that signature, may also be present. Berry [22], Saville and Berry [38], and Hobbs et al. [40] have described platform trials in the context of randomization to a single control arm versus many experimental arms (which may enter and exit the trial seamlessly), in order to reduce overall sample size and improve efficiency when compared with an equivalent set of two-arm trials using a common control arm.
In the sections that follow, we describe basket trials, umbrella trials, and other master protocol designs in more detail, including advantages, limitations, and examples.
Basket trials: marker specific, histology-independent cohorts
Advantages of basket trials
Basket trials have several advantages. First, they can provide access to molecularly targeted agents for patients across a broad range of tumor types, potentially including those not otherwise studied in clinical trials of targeted therapies [20]. Secondly, in many cases, molecular testing is carried out locally and confirmation by a central assay is not required before patient enrollment [20, 39], though tumor and plasma are often banked for subsequent companion diagnostic testing and validation. This feature reduces the time between initial diagnosis and/or eligibility confirmation and later cohort assignment and initiation of treatment. Thirdly, cohorts within basket trials are often small and utilize single-stage or two-stage designs, which yield quick results, given sufficient accrual.
Limitations of basket trials
One major limitation of basket trials is the assumption that molecular profiling may be sufficient to replace histological tumor typing, as, in some cases, histological tumor type has been found to predict response to treatment more strongly than the biomarkers or mutations comprising the studied cohorts (see, e.g. [39]). Even outside the context of a basket trial, it was recognized that V600E BRAF-mutant melanoma or hairy cell leukemia are responsive to BRAF inhibition, while colon tumors with the same BRAF mutation are not [18, 20, 43–46]. This issue may be anticipated, as it is well accepted that the environment and location in which a tumor develops may impact its mutational profile as well as differentially predict treatment response across similar profiles. To this end, many have noted that current clinical evidence is insufficient to conclude that molecular descriptors should replace histological tumor typing [18, 22, 41, 47], and it has been suggested that future studies integrate anatomic with mutational and functional molecular profiling through the use of proteomic technologies [47] and explore multi-gene signatures with combination therapies [48].
Basket trial examples
NCI MATCH
Activated in August 2015, NCI MATCH (Molecular Analysis for Therapy CHoice) is an ongoing basket trial that was initially intended to screen ∼3000 patients with advanced solid tumors or lymphoma who have progressed on at least one therapy with the goal of assigning 800–1000 patients to 25 single-arm substudies based on disease classification by next-generation sequencing [20, 22, 49, 50]. In MATCH, it is hypothesized that the overall response rate will be improved from 5% to 25% in one or more cohorts through the use of matched targeted therapies, and this hypothesis will be tested in independent analyses of 30 patients with heterogeneous tumor types per substudy. Each arm will enroll at least 25% of patients from rare tumor types and repeat biopsies over time may also occur, such that patients may be reclassified to other arms of the study if they remain eligible. In the event that NCI-MATCH investigators detect a promising efficacy signal within one of the ‘baskets’ that is mostly or completely attributable to a particular tumor type, a location or histology-dependent expansion or separate phase II or III study could be launched to confirm the finding [50]. Patients are not eligible for participation in NCI MATCH if they have a tumor type and mutation for which a targeted agent has been FDA approved. Additional details of biopsy processing, molecular classification, and required levels of evidence for NCI MATCH are described by Moore and Mannel [20].
While some discussions of NCI MATCH have been mostly positive [20, 50], others have expressed strong concerns [22, 50]. Importantly, all patients are regarded to be exchangeable across tumor types in terms of expected response rate, even when patients with different tumor types (e.g. breast versus colon) are understood to have very different prognoses. Currently, NCI MATCH has temporarily suspended accrual after experiencing a lower-than-anticipated match rate among eligible patients [51, 52]. At this time, feasibility is being assessed, along with the possible addition of molecular cohorts to the trial to enhance the overall match rate. Despite these challenges, NCI MATCH is one of the first basket trials to be activated and will serve as a foundation for subsequent learning, both in terms of cancer biology and targeted trial design.
SIGNATURE
Another ongoing basket trial is SIGNATURE, a basket trial sponsored by Novartis that is currently enrolling patients harboring any solid tumor or lymphoma refractory to standard therapies (excluding those where molecular agents have already proved effective). SIGNATURE comprises several independent biomarker-driven single-arm trials with matched targeted treatments, and to date, three arms are open to enrollment [20, 53].
AcSé
AcSé is a large-scale multi-center phase II trial from the French program UNICANCER assessing the efficacy and safety of crizotinib as monotherapy in 23 cohorts of patients harboring at least one mutation among ALK, MET, RON, or ROS1 across a variety of solid tumors (gastrointestinal, breast, kidney, ovarian, thyroid, and sarcomas, among others). While AcSé primarily resembles a basket trial in that it enrolls patients from many tumor types, within AcSé, cohorts are defined both by alteration and histopathology (e.g. gastric cancer with MET amplification), with each cohort following a single-arm, two-stage design. This study is currently accruing patients [19, 54].
Umbrella trials: one tumor type, many marker cohorts
Advantages of umbrella trials
One immediate advantage of umbrella trials (relative to basket trials) is the ability to draw meaningful conclusions that are specific to a tumor type and therefore less prone to chance tumor heterogeneity present within a given trial cohort. Furthermore, when randomization to targeted versus non-targeted treatments within cohorts takes place (and particularly when a marker-negative cohort is included), the drug's purported mechanism of action can be more thoroughly evaluated, and prognostic versus predictive marker effects can be empirically distinguished. Such trial designs may lend stronger evidence to support the activity of a new drug with a readily describable population and indication.
Limitations of umbrella trials
A direct consequence of the greatest strength of umbrella trials (conclusions applicable to a single tumor type) is one great disadvantage: feasibility. Particularly within rare diseases, further subclassification of an already-rare tumor type by molecular alterations may lead to poor accrual within cohorts, and slow progress of the trial overall. An already lengthy trial may be further exacerbated by inclusion of randomization (generally requiring larger cohort-specific sample sizes) and changes in the treatment landscape of the tumor type under study, e.g. introduction of new standard of care regimen to the market while the umbrella trial is underway [38, 40].
Umbrella trial examples
FOCUS4
One ongoing umbrella trial is FOCUS4, which enrolls previously untreated metastatic colorectal cancer patients and assigns them to one of four biomarker-enriched cohorts following 16 weeks of standard front-line chemotherapy. Within each cohort, patients are randomized to an experimental targeted agent versus placebo [55, 56]. FOCUS4 further contains a cohort for all-wild-type patients where patients may be randomized to treatments showing promise in the marker-positive cohorts, thereby enhancing participation as potential access to experimental treatment is ensured for all eligible patients. FOCUS4 opened to enrollment in 2014 and plans to follow patients for up to 5 years.
ALCHEMIST
An umbrella trial similar to FOCUS4 in its design is ALCHEMIST (Adjuvant Lung Cancer Enrichment Marker Identification and Sequencing Trial), a study of targeted therapy in patients with resectable adenocarcinoma of the lung harboring EGFR or ALK mutations [16, 34, 57, 58]. In ALCHEMIST, following initial treatment with standard non-targeted therapy, patients are randomized to experimental targeted agents versus placebo within respective target-enriched cohorts. Because the prevalence of the mutations under study is quite low (10% combined), ALCHEMIST further enrolls all-wild-type patients whose tumors undergo whole exome sequencing at registration and at relapse following standard care, so that the natural course of disease may be studied in this group.
LUNG-MAP
LUNG-MAP [16, 22, 59, 60] is a phase II/III study of targeted therapies in patients with previously treated advanced squamous NSCLC led by the Southwest Oncology Group (SWOG) in cooperation with NCI. In LUNG-MAP, four randomized phase II trials of targeted therapy versus standard of care are conducted in parallel within mutation-enriched cohorts, with graduation of a cohort to a phase III registration study if progression-free survival crosses an efficacy boundary during phase II (phase II patients are included in the phase III analysis, contributing critical additional length of follow-up). Once eligibility is determined, patients in LUNG-MAP are screened for mutations and amplifications of interest by the research version of the Foundation One panel of Foundation Medicine, Inc., which improves screening efficiency in a disease where available tissue may be limited. LUNG-MAP was additionally designed as a platform trial allowing for seamless integration of new cohorts via nomination by its Drug Selection committee. No cross-cohort statistical analyses are planned, and patients without a mutation of interest were initially randomized to receive an anti-PD-L1 therapy versus standard of care within a ‘non-match’ study. Patients with tumors harboring multiple markers are randomized in a weighted fashion to qualifying cohorts with priority given to those cohorts with lower prevalence markers.
Despite its sound design, regulatory efficiency, and ability to assign each eligible patient to a promising experimental therapy, LUNG-MAP has faced challenges since its initiation. In March 2015, shortly after the trial began, the FDA approved the immunotherapy drug nivolumab for the same population based on a trial where it had shown superiority over docetaxel, the standard of care drug used in the control arms of the LUNG-MAP cohorts. Also, one cohort of the study (for c-MET-positive patients) closed early for toxicity concerns when it became known that one of the study drugs had caused harm in patients with gastric cancer. Following temporary suspension to respond to these events [16], LUNG-MAP is currently recruiting patients to some of the study cohorts with treatment modifications [59].
Beyond baskets and umbrellas: platform trials and other designs
Here, we provide several examples of master protocols that do not fit neatly in the categories of basket or umbrella trials, including strategy trials and Bayesian adaptive platform designs.
SHIVA
In the multi-histology SHIVA trial, 195 patients with solid tumors refractory to standard treatments were randomized to targeted treatment according to molecular characteristics versus non-targeted treatment with standard therapy (physician's choice), with crossover to targeted therapy upon progression allowed for patients randomized to the control arm [61]. SHIVA is considered a ‘strategy’ trial, as the strategy of each patient's treatment (biomarker-based versus standard) was the differentiating feature at randomization, rather than comparison of specific treatments. Unfortunately, the experimental targeted strategy arm of SHIVA failed to show improved progression-free survival compared with standard of care [62].
NCI-MPACT
Similar to the SHIVA trial in its objectives, NCI-MPACT (Molecular Profiling-Based Assignment of Cancer Therapy) is an ongoing pilot strategy trial for patients with advanced solid tumors harboring a mutation in one of three specific genetic pathways. In MPACT, patients are randomized in a 2:1 ratio to receive targeted therapy for their identified pathway mutation versus a treatment not known to be pathway-specific, with crossover to targeted therapy allowed upon progression in the control arm [63, 64]. Currently, MPACT is studying treatments for the DNA repair, PI3K, and RAS/RAF/MEK pathways, with a total of four experimental treatment arms (two experimental arms are included for the DNA repair pathway). It is anticipated that 700 patients will be screened to enroll 180 patients to the initial 4 cohorts of the trial; additional pathway/treatment cohorts may be added at a later date. Dual end points of this trial include overall response rate and 4-month progression-free survival.
BATTLE trials
BATTLE was a Bayesian adaptive trial in advanced NSCLC and one of the first clinical trials specifically designed to investigate differential biomarker-driven treatment effects [22, 65, 66]. BATTLE employed a master protocol and individual protocols for each of four treatment arms, among which response-adaptive randomization was used to modify the randomization probabilities within each of five biomarker-based subgroups based on observed 8-week disease control rates within each marker–treatment combination. The final results of the BATTLE trial and its inherent challenge were detailed in several manuscripts [66–70]. Kim et al. [66] stated that in a follow-up trial, BATTLE-2 [67], a prospectively defined learning period would occur, from which only biomarkers showing sufficiently strong predictive ability would be subsequently used to guide patient assignments. BATTLE-2 is currently ongoing.
I-SPY2
Another biomarker-based Bayesian adaptive design is I-SPY2, a phase II trial of neoadjuvant treatment of women with locally advanced breast cancer [22, 71]. In I-SPY2, patients are biopsied at baseline and directly assigned to one of many biomarker signature cohorts, wherein patients are subsequently randomized to one of several experimental treatments. The primary end point for each cohort is pathologic complete response supported by longitudinal MRI measurements. A drug performing well within a specific marker signature (in terms of Bayesian predictive probability) triggers adaptive randomization at higher probabilities for subsequent patients enrolled within the same signature, and definitively successful drugs are ‘graduated’ to a phase III study within the signature. Meanwhile, treatments not showing promise within a signature are randomized at lower probabilities and are ultimately removed from consideration, and drugs reaching futility across all signatures are dropped from the trial. This platform design framework allows novel targeted agents of interest to continually enter and exit the trial protocol in an operationally seamless manner, taking advantage of established infrastructure and site participation. To date, at least four cohorts have graduated to the phase III setting [22, 72]. As noted by several authors [65–69], the utility of Bayesian adaptive randomization depends on quick marker assessment, a relatively quick end point to inform the randomization algorithm, and a slow-to-moderate accrual rate to ensure that early adaptations may benefit subsequent patients.
CUSTOM
The Molecular Profiling and Targeted Therapies in Advanced Thoracic Malignancies (CUSTOM) study is a master protocol that simultaneously evaluated 5 targeted therapies in 15 different patient cohorts defined by 5 groupings of molecular features crossed with three different tumor histologies. One trial arm studying erlotinib in NSCLC patients with EGFR mutations demonstrated an overall response rate of 60%; in combination with other published reports of erlotinib's efficacy in this setting, this arm of CUSTOM was terminated for early efficacy. Studies of targeted therapies in other arms are ongoing [18, 73].
CREATE
The cross-tumoral phase II study with Crizotinib (CREATE) study [19, 74] is evaluating crizotinib's efficacy in patients with advanced disease and ALK and/or MET mutations in one of six heterogeneous tumor types. In CREATE, each tumor type constitutes a subtrial, and each subtrial contains two subcohorts to enroll patients with ALK/MET+ versus ALK/MET– tumors. CREATE is hoping to recruit up to 420 patients from several countries, and enrollment is ongoing.
Discussion: considerations and challenges in master protocols
New challenges and considerations often affect the conduct and feasibility of trials with a master protocol design. Here, we discuss several in more detail. A detailed review of biomarker-based trial designs from a statistical design and analysis perspective was recently given by Renfro et al. [33].
New collaborative paradigm
To be successful, a master protocol requires a new collaborative paradigm defined by the close collaboration of multiple industry, academic, regulatory, and community oncology stakeholders, often including participation by multiple pharmaceutical companies providing drugs to the same trial [38]. Experts from many disparate areas are often involved, including cooperative group and local hospital leadership, and specialists in oncology, pathology, molecular medicine, computational biology, clinical and translational research, pharmacology, biostatistics, and patient advocates. This is of clear long-term benefit to the research enterprise, but is a short-term challenge as new relationships and trust are established.
Inclusion of marker-negative patients
As described earlier, inclusion of a treatment cohort for all-negative or all-wild-type patients within a master protocol design may enhance enthusiasm for the trial, as all otherwise eligible patients will be offered access to a potentially beneficial experimental treatment regardless of screening results. In some settings, however, inclusion of a marker-negative cohort may not be feasible or reasonable, or its patient population may shift due to mid-trial addition or cancellation of marker-positive arms. In the latter case, potential regulatory approval of new therapies on the basis of a master protocol's marker-negative substudy would be challenged by a poorly defined patient population for labeling purposes.
Classification of patients with multiple genetic mutations
In many of the master protocols described, patients may potentially qualify for more than one targeted cohort on the basis of multiple positive biomarkers. Assignment of such patients to just one qualifying matched cohort must be decided. If prevalence of a mutation or biomarker group is not too low and accrual is not a concern, patients may be assigned to one of the cohorts at random. In other cases, particularly where one of the mutations is rare and feasibility needs to be optimized, the patient may be directly assigned to the rarest mutation or the cohort with the lowest patient accrual to date. In all of these cases, it is also possible to reassess patients for mutations or markers upon initial disease progression and, on the basis of those results, assign such patients to second matched cohorts within in the same trial (e.g. NCI MATCH).
Effect size versus sample size
The sample size versus effect size trade-off is often an issue when defining targeted cohort-specific objectives within basket, umbrella, or platform designs. To keep the sample size small within cohorts and maintain overall trial feasibility, it is often necessary to target a large effect size (versus a randomized comparator arm or historical control), often with lower power and higher type I error than traditional phase II or III trials. Several authors such as Menis et al. [19] and Burock et al. [75] have stated that the goal of cancer clinical trials in this era of precision medicine should be to conduct ‘trials designed to learn’ which lead to ‘trials designed to conclude’, which begins with identifying large and meaningful differences within small, molecularly enriched groups of patients, often referred to as ‘home runs’ [76].
Challenges in assay evaluation, validation, and implementation
Validation of biomarkers for use in patient screening and clinical trials remains fraught with challenges, including a multitude of available assessment methods (e.g. immunohistochemistry, fluorescence in situ hybridization, and next-generation sequencing), variable reliability (sensitivity and specificity), reproducibility, feasibility in terms of tissue requirements and turn-around time, and related costs [19]. Several authors have noted that an optimal drug/device co-development program should include simultaneous evaluation of assay methodology and companion drugs from prospective or retrospective analysis of clinical trials [35, 77].
Future directions of master protocols in oncology
As next-generation sequencing continues to develop, master protocols including basket and umbrella trials are likely to see increased use with more nuanced assignment of patients to matched treatments, e.g. to combination treatments according to multiple driving mutations, biomarkers, or pathways [18]. In the future, molecular evaluation of patients who show remarkable improvement in their disease following treatment with cancer drugs that have otherwise shown low activity in other patients (so-called ‘exceptional responders’ as defined by the NCI) may motivate future master protocol designs within specific disease types [78]. The FDA has also demonstrated a willingness to approve agents that have been evaluated in only a small number of patients, even based on single-arm trials, as traditionally large registration trials may never be feasible or ethically appropriate within some cancer subtypes [79]. How these large shifts in the standard paradigm of cancer clinical research will ultimately improve success rates for new therapies in oncology remains to be seen, but we are confident that if we continue to learn and refine, the net impact will be better treatment of cancer patients.
Funding
This work was supported by the National Institutes of Health [CTSA Grant Number KL2 TR000136 from the National Center for Advancing Translational Science (NCATS) to LAR]; and the National Cancer Institute at the National Institutes of Health (R01 CA174779 to DJS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosure
The authors have declared no conflicts of interest.
References
- 1. Chapman PB, Hauschild A, Robert C et al. . Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slamon DJ, Leyland-Jones B, Shak S et al. . Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792. [DOI] [PubMed] [Google Scholar]
- 3. Swain SM. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 2015; 372: 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Armado RG, Wolf M, Peeters M et al. . Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 1626–1634. [DOI] [PubMed] [Google Scholar]
- 5. Jonker DJ, O'Callaghan CJ, Karapetis CS et al. . Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357: 2040–2048. [DOI] [PubMed] [Google Scholar]
- 6. Zhou C, Wu YL, Chen G et al. . Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2011; 12: 735–742. [DOI] [PubMed] [Google Scholar]
- 7. Shaw AT, Kim DW, Nakagawa K et al. . Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368: 2385–2394. [DOI] [PubMed] [Google Scholar]
- 8. Shaw AT, Ou SH, Bang YJ et al. . Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 2014; 371: 1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brahmer J, Reckamp KL, Baas P et al. . Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med 2015; 373: 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. FDA Food and Drug Administration. FDA expands approved use of Opdivo in advanced lung cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm466413.htm
- 11. Robert C, Schachter J, Long GV et al. . Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015; 372: 2521–2532. [DOI] [PubMed] [Google Scholar]
- 12. Catennaci DVT. Next generation clinical trials: novel strategies to address the challenge of tumor molecular heterogeneity. Mol Oncol 2015; 9: 967–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008; 68: 3077–3080. [DOI] [PubMed] [Google Scholar]
- 14. Weinstein IB. Cancer. Addiction to oncogenes: the Achilles heal of cancer. Science 2002; 297: 63–64. [DOI] [PubMed] [Google Scholar]
- 15. Vogelstein B, Papadopoulos N, Velculescu VE et al. . Cancer genome landscapes. Science 2013; 339: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Redman MW, Allegra CJ. The master protocol concept. Semin Oncol 2015; 42: 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Galbraith S. The changing world of oncology drug development—a global pharmaceutical company's perspective. Chin Clin Oncol 2014; 3: 20. [DOI] [PubMed] [Google Scholar]
- 18. Redig AJ, Jänne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol 2015; 33: 975–977. [DOI] [PubMed] [Google Scholar]
- 19. Menis J, Hasan B, Besse B. New clinical research strategies in thoracic oncology: clinical trial design, adaptive, basket and umbrella trials, new end-points and new evaluations of response. Eur Respir Rev 2014; 23: 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moore KN, Mannel RS. Is the NCI MATCH trial a match for gynecologic oncology? Gynecol Oncol 2016; 140: 161–166. [DOI] [PubMed] [Google Scholar]
- 21. Billingham L, Malottki K, Steven N. Research methods to change clinical practice for patients with rare cancers. Lancet Oncol 2016; 17: e70–e80. [DOI] [PubMed] [Google Scholar]
- 22. Berry DA. The Brave New World of clinical cancer research: adaptive biomarker-driven trials integrating clinical practice with clinical research. Mol Oncol 2015; 9: 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dilts DM, Cheng SK, Crites JS et al. . Phase III clinical trial development: a process of chutes and ladders. Clin Cancer Res 2010; 16: 5381–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pharmaceutical Research and Manufacturers of America. Drug discovery and development. http://www.phrma.org/sites/default/files/pdf/rdbrochure022307.pdf
- 25. Pharmaceutical Research and Manufacturers of America. 771 medicines in development for cancer. http://www.phrma.org/research/cancer
- 26. DiMasi JA, Reichert JM, Feldman L et al. . Clinical approval success rates for investigational cancer drugs. Clin Pharmacol Ther 2013; 94: 329–335. [DOI] [PubMed] [Google Scholar]
- 27. Sutter S, Lamotta L. Cancer drugs have the worst phase III track record. Internal Medicine News Digital Network 2011. [Google Scholar]
- 28. Amiri-Kordestani L, Fojo T. Why do phase III trials in oncology fail so often? J Natl Cancer Inst 2012; 104: 568–569. [DOI] [PubMed] [Google Scholar]
- 29. Nass SJ, Moses HL, Mendelsohn J. Institute of Medicine: A National Cancer Clinical Trials System for the 21st Century: Reinvigorating the NCI Cooperative Group Program. Washington, DC: National Academies Press, 2010. [PubMed] [Google Scholar]
- 30. Seymour L, Ivy SP, Sargent DJ et al. . The design of phase II clinical trials testing cancer therapeutics: consensus recommendations from the clinical trial design task force of the National Cancer Institute investigational drug steering committee. Clin Cancer Res 2010; 16: 1764–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med 2015; 372: 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Renfro LA, Mallick H, An MW, Sargent DJ, Mandrekar SJ. Clinical trial designs incorporating predictive biomarkers. Cancer Treat Rev 2016; 43: 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Renfro LA, An MW, Mandrekar SJ. Precision oncology: a new era of cancer clinical trials. Cancer Lett 2016; epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freidlin B, Korn EL. Biomarker enrichment strategies: matching trial design to biomarker credentials. Nat Rev Clin Oncol 2014; 11: 81–90. [DOI] [PubMed] [Google Scholar]
- 35. Malik SM, Pazdur R, Abrams JS et al. . Consensus report of a joint NCI thoracic malignancy steering committee: FDA workshop on strategies for integrating biomarkers into clinical development of new therapies for lung cancer leading to the inception of ‘master protocols’ in lung cancer. J Thorac Oncol 2014; 9: 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. SEER stat facts sheet, cancer of any site. National Cancer Institute Survey, Epidemiology, and End Results Program; http://seer.cancer.gov/statfacts/html/all.html [Google Scholar]
- 37. A simple act: increasing clinical trial participation. American Association for Cancer Research; http://blog.aacr.org/a-simple-act-increasing-clinical-trial-participation/ (25 March 2015, date last accessed) [Google Scholar]
- 38. Saville BR, Berry SM. Efficiencies of platform clinical trials: a vision of the future. Clin Trials 2016; 13: 358–66. [DOI] [PubMed] [Google Scholar]
- 39. Hyman DM, Solit DB, Arcila ME et al. . Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today 2015; 20: 1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hobbs BP, Chen N, Lee JJ. Controlled multi-arm platform design using predictive probability. Stat Methods Med Res 2016; epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simon R, Geyer S, Subramanian J, Roychowdhury S. The Bayesian basket design for genomic variant-driven phase II trials. Semin Oncol 2016; 43: 13–18. [DOI] [PubMed] [Google Scholar]
- 42. Mandrekar SJ, Dahlberg SE, Simon R. Improving clinical trial efficiency: thinking outside the box. Am Soc Clin Oncol Educ Book 2015; e141–e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hyman DM, Puzanov I, Subbiah V et al. . Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015; 373: 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Flaherty KT, Puzanov I, Kim KB et al. . Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363: 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tiacci E, Trifonov V, Schiavoni G et al. . BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364: 2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prahallad A, Sun C, Huang S et al. . Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483: 100–103. [DOI] [PubMed] [Google Scholar]
- 47. Klauschen F, Andreeff M, Keilholz U et al. . The combinatorial complexity of cancer precision medicine. Oncoscience 2014; 1: 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stenzinger A, Weichert W, Lennerz JK, Klauschen F. Basket trials: just the end of the first quarter. J Clin Oncol 2015; 33: 2823–2824. [DOI] [PubMed] [Google Scholar]
- 49. National Cancer Institute. NCI-Molecular Analysis for Therapy Choice (NCI-MATCH) Trial. http://www.cancer.gov/about-cancer/treatment/clinical-trials/nci-supported/nci-match
- 50. Mullard A. NCI-MATCH trial pushes cancer umbrella trial paradigm. Nat Rev Drug Discov 2015; 14: 513–515. [DOI] [PubMed] [Google Scholar]
- 51. National Cancer Institute. NCI-Match: status report and future directions. http://www.cancer.gov/news-events/cancer-currents-blog/2016/nci-match-update
- 52. Genomeweb. NCI-Match sees lots of enthusiasm in initial months; not many matches. https://www.genomeweb.com/molecular-diagnostics/nci-match-sees-lots-enthusiasm-initial-months-not-many-matches
- 53. Signature: bring the protocol to the patient. https://www.signaturetrial.com/en/hcp?remember_choice=true#all
- 54. Clinicaltrials.gov. Phase 2 study assessing efficacy and safety of crizotinib in patients harboring an alteration on ALK, MET or ROS1 (AcSe). https://clinicaltrials.gov/ct2/show/NCT02304809
- 55. Kaplan R, Maughan T, Crook A et al. . Evaluating many treatments and biomarkers in oncology: a new design. J Clin Oncol 2013; 36: 4562–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Medical Research Council (MRC) Clinical Trials Unit. FOCUS4: molecular selection of therapy in metastatic colorectal cancer: a molecularly stratified randomized controlled trial programme. http://www.focus4trial.org
- 57. Gerber DE, Oxnard GR, Govindan R. ALCHEMIST: bringing genomic discovery and targeted therapies to early-stage lung cancer. Clin Pharmacol Ther 2015; 97: 447–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Colwell J. Trial offers new model for drug development. Cancer Discov 2014; 4: 266–267. [DOI] [PubMed] [Google Scholar]
- 59. LUNG-MAP. A groundbreaking collaborative approach to clinical trials. http://www.lung-map.org
- 60. Ferrarotto R, Redman MW, Gandara DR et al. . Lung-MAP—framework, overview, and design principles. Chin Clin Oncol 2015; 4: 36–41. [DOI] [PubMed] [Google Scholar]
- 61. Le Tourneau C, Paoletti X, Servant N et al. . Randomised proof-of-concept phase II trial comparing targeted therapy based on tumour molecular profiling vs. conventional therapy in patients with refractory cancer: results of the feasibility part of the SHIVA trial. Br J Cancer 2014; 111: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Le Tourneau C, Delord J-P, Gonçalves A et al. . Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomized, controlled phase 2 trial. Lancet Oncol 2015; 16: 1324–1334. [DOI] [PubMed] [Google Scholar]
- 63. NCI-MPACT: molecular profiling-based assignment of cancer therapy for patients with advanced solid tumors. https://clinicaltrials.gov/ct2/show/NCT01827384
- 64. Do K, O'Sullivan Coyne G, Chen AP. An overview of the NCI precision medicine trials NCI MATCH and MPACT. Chin Clin Oncol 2015; 4: 31. [DOI] [PubMed] [Google Scholar]
- 65. Zhou X, Liu S, Kim ES et al. . Bayesian adaptive design for targeted therapy development in cancer—a step toward personalized medicine. Clin Trials 2008; 5: 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim ES, Herbst RS, Wistuba II et al. . The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov 2011; 1: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu S, Lee JJ. An overview of the design and conduct of the BATTLE trials. Chin Clin Oncol 2015; 4: 33. [DOI] [PubMed] [Google Scholar]
- 68. Berry DA, Herbst RS, Rubin EH. Reports from 2010 Clinical and Translational Cancer Research Think Tank Meeting: design strategies for personalized therapy trials. Clin Cancer Res 2012; 18: 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Marchenko O, Fedorov V, Lee JJ et al. . Adaptive clinical trials: overview of early-phase designs and challenges. Ther Innov Regul Sci 2014; 48: 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lara PN Jr, Redman MW, Kelly K et al. . Disease control rate at 8 weeks predicts clinical benefit in advanced non-small cell lung cancer: results from Southwest Oncology Group randomized trials. J Clin Oncol 2008; 26: 463–467. [DOI] [PubMed] [Google Scholar]
- 71. Barker AD, Sigman CC, Kelloff GJ et al. . I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy. Clin Pharmacol Ther 2009; 86: 97–100. [DOI] [PubMed] [Google Scholar]
- 72. Park JW, Liu MC, Yee D et al. . Adaptive randomization of neratinib in early breast cancer. N Engl J Med 2016; 375: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lopez-Chavez A, Thomas A, Rajan A et al. . Molecular profiling and targeted therapy for advanced thoracic malignancies: a biomarker-derived, multiarm, multihistology phase II basket trial. J Clin Oncol 2015; 33: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Clinicaltrials.gov. CREATE: cross-tumoral phase 2 with crizotinib. NCT01524926 http://clinicaltrials.gov/ct2/show/NCT01524926?term5NCT01524926&rank51
- 75. Burock S, Mounier F, Lacombe D. How can innovative forms of clinical research contribute to deliver affordable cancer care in an evolving health care environment? Eur J Cancer 2013; 49: 2777–2783. [DOI] [PubMed] [Google Scholar]
- 76. Sleiffer S, Bogaerts J, Siu LL et al. . Designing transformative clinical trials in the cancer genome era. J Clin Oncol 2013; 31: 1834–1841. [DOI] [PubMed] [Google Scholar]
- 77. Polley MY, Freidlin B, Korn EL et al. . Statistical and practical considerations for clinical evaluation of predictive biomarkers. J Natl Cancer Inst 2013; 105: 1677–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. National Cancer Institute. Exceptional responders initiative: questions and answers. http://www.cancer.gov/news-events/press-releases/2014/ExceptionalRespondersQandA
- 79. Willyard C. Basket studies will hold intricate data for cancer drug approvals. Nat Med 2013; 19: 655. [DOI] [PubMed] [Google Scholar]