

Abstract
Animals that wield toxins face self-intoxication. Poison frogs have a diverse arsenal of defensive alkaloids that target the nervous system. Among them is epibatidine, a nicotinic acetylcholine receptor (nAChR) agonist that is lethal at microgram doses. Epibatidine shares a highly conserved binding site with acetylcholine, making it difficult to evolve resistance yet maintain nAChR function. Electrophysiological assays of human and frog nAChR revealed that one amino acid replacement, which evolved three times in poison frogs, decreased epibatidine sensitivity but at a cost of acetylcholine sensitivity. However, receptor functionality was rescued by additional amino acid replacements that differed among poison frog lineages. Our results demonstrate how resistance to agonist toxins can evolve and that such genetic changes propel organisms towards an adaptive peak of chemical defense.
Acquiring chemicals from the environment and recycling them for anti-predator defense is a survival strategy that has evolved in nearly every major branch of life (1). Exposure to toxic chemicals may have high physiological costs, but it can also be an opportunity for organisms to capitalize on these substances as new resources. Organisms that accumulate these chemicals risk self-intoxication unless they can resist their own defenses through compartmentalization, metabolic detoxification, or target-site insensitivity, i.e., changes in the molecular target of the toxin that affect its ability to bind (2). Many toxins target evolutionarily conserved proteins such as ion channels, which govern key nervous system functions. Thus, revealing the mechanistic basis of toxin resistance deepens our understanding of protein function and provides insights into nervous system evolution (3, 4). Moreover, the physiology of toxin resistance is a crucial aspect of chemical defense and characterizing the evolution of resistance might elucidate how and why organisms acquire toxic defenses (5).
Neotropical poison frogs (Dendrobatidae) have independently evolved chemical defenses at least four times (6). The origins of chemical defense are usually accompanied by shifts towards bright coloration, resulting in a complex phenotype or syndrome known as aposematism (6). Theoretically, aposematic and non-aposematic poison frogs represent alternative peaks on an adaptive landscape that arose as a result of disruptive selection that favored more extreme phenotypes over intermediate ones (e.g., conspicuous but not well defended, or defended but not aposematic) (7). The multiple origins of aposematism within dendrobatids suggest that the switch from non-aposematic to aposematic phenotypes is easily attained within this group. Characterizing the evolution of toxin resistance, a key step in this phenotypic transition, may reveal pathways between these adaptive peaks in which toxin resistance facilitates origins of toxin sequestration.
Chemically defended dendrobatids take up from their diet over 800 types of lipophilic alkaloids (8), many of which modulate nervous system function (9). Their effects vary from benign to lethal (10), but most are bitter-tasting and thus generally aversive to predators (11). Epibatidine, one of the best known of these alkaloids, was first isolated from the phantasmal poison frog Epipedobates anthonyi in 1974 (12). Epibatidine has an analgesic effect 200 times that of morphine, yet it targets a specific subset of nicotinic acetylcholine receptors (nAChRs) rather than opioid receptors (13). Because of these qualities, epibatidine has inspired pharmacological innovations, although its toxicity has prohibited its successful development as a pharmaceutical (14).
Toxic animals, including poison frogs, often evolve resistance to their toxins via amino acid (AA) replacements in toxin-binding sites (target-site insensitivity; 15, 16). The location of these replacements is constrained by protein function, leading to predictable and convergent mechanisms of resistance (17). For example, resistance to tetrodotoxin (TTX), a NaV1 voltage-gated sodium channel blocker, evolved many times in toxic pufferfish, newts, and snakes that feed on newts via various AA replacements at residues in NaV1 proteins that interact with TTX (17–19). Similarly, resistance to cardiac glycosides, which inhibit the sodium-potassium pump, has evolved at least fourteen times in toxic insects and amphibians as well as their predators via AA replacements in the cardiac-glycoside binding site (4, 20).
Evolving epibatidine resistance involves different strategies at the molecular level, as epibatidine is an agonist that shares a binding site with ACh, the endogenous ligand of nAChRs, while TTX and cardiac glycosides act on receptors that are not ligand-gated (21, 22). Resistance to epibatidine thus requires decreased sensitivity to epibatidine while preserving sensitivity to the endogenous agonist ACh that interacts with many of the same AAs, all without disrupting the normal receptor function.
Phylogenetic identification of AA replacements in the poison frog nAChR
Based on what is known about the toxin and ligand, we hypothesized that epibatidine-bearing frogs would have nAChRs that resist epibatidine yet display normal ACh sensitivity, and that the basis of resistance would involve genetic changes in the ligand-binding site. To test this hypothesis, we sequenced genes in poison frogs encoding the primary molecular target of epibatidine in the brain, the α4β2 nAChR (chrna4 and chrnb2) (23). Epibatidine has been detected in two distinct lineages of dendrobatids, Epipedobates and Ameerega (12), so we predicted two origins of resistance. Consequently, we sequenced 9 species of these genera as well as 19 other species of poison frogs, including 8 species of Dendrobatinae (Dendrobates+Phyllobates), a clade of chemically defended poison frogs lacking epibatidine, and 11 non-defended species (table S1, 24).
Four sites in the β2 subunit (F106, S108, A110, and I118, numeration of the mature human protein) have unique AA replacements in the alkaloid-sequestering dendrobatids Epipedobates, Ameerega, and Dendrobates (subgenus Oophaga sensu 25; Fig. 1A), the last of which is not known to have epibatidine defenses. These replacements are near the epibatidine-binding site in the α+–β− interface: between loops A and E, and in loop E (Fig. 1B–E) (21, 22). Each of these replacements involves a single nucleotide change in the first or second codon position (table S2, 24), suggesting non-neutral evolution. Five additional sites in α4 were found to have AA replacements unique to these poison frogs, but only one of these (D176N) was near the epibatidine-binding site (table S3 and fig. S1, 24).
Fig. 1. AA replacements in β2 associated with alkaloid-defended poison frogs.
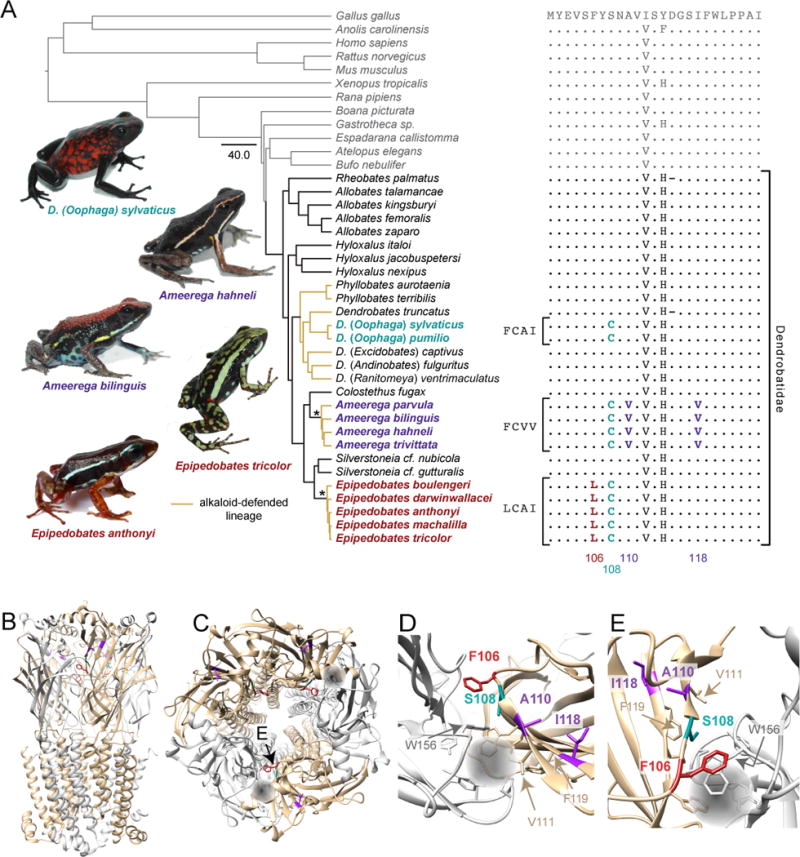
(A)
Alignment of dendrobatid (black), non-dendrobatid (grey), and outgroup (grey) β2 sequences (table S4). Yellow branches in the phylogeny (adapted from 36) indicate alkaloid-defended lineages; asterisks indicate clades in which epibatidine has been detected; the unit of the scale bar is number of expected substitutions per site. Focal species names are in bold and colored by their AA replacement pattern. The AA replaced only in Epipedobates poison frogs is in red (F106L); AAs replaced only in Ameerega are in purple (A110I and I118V); the convergently evolved replacement is in cyan (S108). Genotypes of clades with replacements are indicated to the left of the alignment (see Table 1). Structure of the human (α4)2(β2)3 nAChR (22) (B) from the side and (C) from extracellular space. α4 subunits are in light grey, β2 subunits are in gold, and the ligand-binding sites are indicated by grey spheres. AA residues identified with grey and gold arrows are known to be involved in ACh and/or epibatidine binding (21, 28, 29). Closer view of the binding site from (D) extracellular space and (E) viewpoint indicated by labeled arrow in (C).
Electrophysiology of AA replacements in the poison frog nAChR
The α4β2 nAChR is a pentameric protein that exhibits two different stoichiometries: a high-ACh sensitivity conformation (HS), (α4)2(β2)3, and a low-ACh sensitivity conformation (LS), (α4)3(β2)2 (26, 27). To determine experimentally whether the identified AA replacements provide resistance to epibatidine, we used site-directed mutagenesis to introduce poison frog AA replacements into human nAChRs. We then co-expressed the wild-type and mutated β2 subunits with human α4 nAChR subunits in Xenopus laevis oocytes and measured acetylcholine and epibatidine concentration-response curves (CRCs) through two-electrode voltage clamp (parameters, results, and statistical analyses from all CRCs are shown in tables S5–S16, 24). For each subunit combination, we injected different ratios of α4 and β2 transcripts to favor the formation of either HS or LS conformations (24). For brevity, we describe only HS nAChRs in the main text, as we found the same general pattern of channel sensitivity to ACh and epibatidine in both stoichiometries. For LS nAChR results see fig. S2 and tables S5–S7 (24). We also performed electrophysiology experiments to test whether the one replacement in the α4 subunit near the ligand-binding site (D176N) affected LS nAChR function, but we found no evidence for an effect (24; fig. S1 and S4, table S8).
For clarity, we denote all nAChR genotypes with four letters indicating the AA residue at each of the four sites of interest (106, 108, 110, and 118, see Table 1). Bold letters in each genotype indicate AA replacements introduced into a transcript via site-directed mutagenesis.
Table 1. Effects of AA replacements on ligand responses in human and Epipedobates high-ACh-sensitivity (HS) nAChRs.
Relative fold change in sensitivity induced by AA replacements was calculated as [mutant EC50]/[FSAI EC50] for each genetic background (i.e., [mutant]/[reference]; see tables S5−S7; 24). Values greater than 1 indicate that relatively more ligand is required to elicit the same response; thus, higher values indicate lower sensitivity. ACh assessments for biphasic curves (>1) are qualitative, but both cases result in lowered sensitivity (see tables S5−S6).
| Human genetic background | Epipedobates genetic background | ||||||
|---|---|---|---|---|---|---|---|
| Genotype | AA replacement(s) | Fold change in EC50 ACh | Fold change in EC50 epibatidine | Genotype | AA replacement(s) | Fold change in EC50 ACh | Fold change in EC50 epibatidine |
| FSAI | (wild-type) | reference | reference | FSAI | L106F C108S |
reference | reference |
| LCAI | F106L S108C |
1 | 17** | LCAI | (wild-type) | 1 | 44** |
| LSAI | F106L | 1 | 1 | LSAI | C108S | 1 | 1 |
| FCAI | S108C | >1 | 49** | FCAI | L106F | 2* | 138** |
| FCVV | S108C A110V I118V |
1 | 6** | – | – | – | – |
| FSVI | A110V | 1 | 1 | – | – | – | – |
| FSAV | I118V | >1 | 75** | – | – | – | – |
, p < 0.01;
, p < 0.001 (two-way ANOVA, corrected for multiple comparisons using Tukey’s test; see tables S9, S11, S13, and S14).
Human-to-frog mutants
The Epipedobates and Ameerega replacement patterns (LCAI and FCVV genotypes) produced by mutagenesis showed ACh concentration-response curves (CRCs) identical to that of wild-type human FSAI genotype, while the subgenus Oophaga replacement pattern (FCAI) showed a decrease in sensitivity to ACh (Fig. 2A). All three nAChRs with poison frog AA replacement patterns (LCAI, FCVV, FCAI) were less sensitive to epibatidine than the wild-type receptor (Fig. 2B), indicating that these replacement patterns are sufficient to produce epibatidine-resistant phenotypes (Tables 1, S5−S7; 24). Interestingly, the ACh CRC is biphasic for the Oophaga replacement pattern, suggesting that in the human genetic background the S108C replacement may induce assembly of low-sensitivity (LS) nAChRs. As the LS stoichiometry possess two kinds of binding sites, application of increasing concentration of ACh results in a biphasic curve that reflects activation of the two HS binding sites at low ACh concentrations and of the single LS binding site at high ACh concentrations (24). Thus, resistance to epibatidine conferred by the S108C replacement incurs a cost of ACh sensitivity in the human β2 subunit.
Fig. 2. ACh and epibatidine concentration-response curves in high-sensitivity α4β2 nAChRs.

Left panels show responses to ACh and right panels show responses to epibatidine. (A, B) Human α4β2 nAChRs: wild-type genotype (FSAI) and receptors containing the AA patterns identified in Epipedobates, Ameerega, and Dendrobates (Oophaga) poison frogs (LCAI, FCVV, and FCAI genotypes, respectively). (C, D) Human α4β2 nAChRs: wild-type (FSAI) and Ameerega genotypes (FCVV, FCAI, FSVI and FSAV). (E, F) Human α4β2 nAChRs: wild-type (FSAI) and Epipedobates genotypes (LCAI, LSAI and FCAI). (G, H) Epipedobates α4β2 nAChRs: wild-type (LCAI genotype) and human genotypes (FSAI, FCAI and LSAI). Dotted lines (⋯) correspond to human FSAI and LCAI curves from C and D panels. Error bars smaller than the symbols are not visible. Data were fitted to either mono- (−) or biphasic (–) curves. Inset: schematic of HS α4β2 nAChR stoichiometry; ligand-binding sites indicated by arrows.
We then characterized the physiological effect of each individual replacement in poison frogs by generating human α4β2 nAChR transcripts with single amino acid replacements (LSAI, FSVI, and FSAV). As with the S108C replacement, human transcripts with the I118V replacement (FSAV, derived in Ameerega) provided moderate resistance to epibatidine at a cost of ACh sensitivity, possibly because this AA replacement also induced assembly of LS nAChRs (Fig. 2C and D, Table 1). In contrast, human receptors with either F106L (Epipedobates) or A110V (Ameerega) displayed no change in ACh and epibatidine sensitivity, indicating that these replacements probably do not contribute to epibatidine resistance (Fig. 2E to H, Table 1). Instead, these replacements appear to compensate for the decrease in ACh sensitivity incurred by the replacements that provided resistance (Table 1), as human receptors with the LCAI genotype (Epipedobates) or the FCVV genotype (Ameerega) both showed normal ACh response (Fig. 2A and B; Table 1; 24).
Epipedobates-to-human mutants
We synthesized and expressed the wild-type Epipedobates α4β2 nAChR (LCAI genotype) and a double mutant replicating the plesiomorphic human genotype (FSAI) in Xenopus laevis oocytes, and performed electrophysiology assays. The Epipedobates-to-human mutant (FSAI) showed greatly increased sensitivity to epibatidine but no change in sensitivity to ACh (Table 1 and S7; Fig. 2G and H) indicating that the replacements in Epipedobates were necessary for resistance.
To understand the contributions of each replacement when it occurs in the poison frog genetic background, we expressed the single mutant genotypes FCAI and LSAI in the Epipedobates β2 subunit. While S108C incurred a drastic cost in ACh sensitivity in the human genetic background (Fig. 2A, compare FSAI and FCAI), the Epipedobates-to-human FCAI mutant demonstrated only a minor (but significant) decrease in sensitivity to ACh (compared to FSAI), suggesting that some other aspects of the poison frog genetic background ameliorate the large cost of this replacement in the human FCAI genotype (Table 1). This difference may be explained by the observation that the S108C replacement in human receptors appeared to induce formation of LS nAChRs (Fig. 2A), which are less sensitive to ACh than HS nAChRs. However, the Epipedobates nAChR never appeared to form the LS stoichiometry, even when the injected cRNA subunit ratio favored its formation (compare Fig. 2G and H to fig. S2G and H, 24). Little is known about the poison frog α4β2 nAChR, but the apparent absence of the LS stoichiometry in Epipedobates (evidenced by the lack of a biphasic, right-shifted curve) lessens the cost of the S108C replacement, and might be related to epibatidine exposure and resistance.
As predicted, the Epipedobates FCAI receptor displayed a decrease in sensitivity to epibatidine compared with FSAI (Tables 1 and S7; 24), confirming the role of S108C in epibatidine resistance (Fig. 2G and H). As with the human-to-frog LSAI receptor, the Epipedobates-to-human LSAI receptor affected neither ACh nor epibatidine sensitivities compared to FSAI (Table 1). The LCAI genotype (wild-type in Epipedobates) displayed normal responses to ACh and decreased sensitivity to epibatidine (Table 1). Thus, as in the human receptor, C108 provides epibatidine resistance and L106 appears to compensate by normalizing α4β2 receptor function in Epipedobates poison frogs.
AA replacements in poison frog nAChR are proximal to the epibatidine binding site
We found that AA replacements in the poison frog β2 subunit (Fig. 1) alter α4β2 nAChR sensitivity to epibatidine (Fig. 2B). We propose that this is in part due to the proximity of the AA replacements to the epibatidine binding site. Namely, the β2C108 residue directly contacts the side chain of α4W156, one of the main determinants in stabilizing epibatidine binding (Fig. 1D and E) (21, 28). The sulfur-containing side chain of C108 is bulkier than that of serine, and it could modify the epibatidine-W156 interaction. The I118V replacement in Ameerega, which also contributes to epibatidine resistance (Fig. 1D and E), is next to F119, a residue that interacts with the epibatidine chloropyridine ring and stabilizes the epibatidine chlorine atom through its backbone carboxyl group. Moreover, the A110V replacement is next to V111, another AA residue that interacts with epibatidine via van der Waals forces (21, 28, 29). These replacements are located in β-sheets that are involved in epibatidine binding, but are less involved in ACh binding (21, 28, 30). The β2− side of the binding pocket is further from ACh than is the α4+ side, and thus forms looser interactions with ACh, such that AA replacements in the β− region that allow changes in epibatidine binding may be less likely to affect ACh sensitivity. This structure-function problem was apparently solved via an identical genetic change three times within poison frogs and refined via different genetic changes at least twice in these lineages.
Evolutionary pathways towards epibatidine resistance
Toxin resistance often evolves in response to recurrent exposure to toxins (2, 4, 31, 32); thus patterns of resistance should reflect the evolutionary history of toxin exposure. The evolutionary patterns of AA replacements in the poison frog β2 nAChR subunit suggest that in each of the Epipedobates, Ameerega, and Dendrobates (Oophaga) clades (Fig. 1A), an ancestral species was likely exposed to epibatidine, resulting in selection for and evolution of epibatidine resistance approximately 5, 10, and 8MYA, respectively (25). Although no clade of poison frogs that has epibatidine defense lacks AA replacements in the β2 nAChR, epibatidine has only been detected in two of three sampled species of Epipedobates, in two of twelve sampled species of Ameerega, and in none of nine sampled species of Dendrobates (Oophaga) (9, 12). It is possible that some populations with epibatidine defense are extinct or have not been detected, or that the dietary source of epibatidine, presumed to be an arthropod, is not as available as it was long ago (12). While epibatidine resistance may have arisen as a side effect of some other change to the protein, mutations in the ligand-binding domain are uncommon (Fig. 1A) and presumably evolve under strong selective pressures. Regardless of the apparent rarity of epibatidine in poison frogs, the epibatidine-resistant phenotype (determined by electrophysiology) does not appear to have been lost in any resistant lineages (Oophaga, Epipedobates, or Ameerega), suggesting that lack of resistance has a high cost, that reversion to a non-resistant phenotype is physiologically difficult, or that maintenance of epibatidine resistance is not costly.
The evolutionary patterns underlying origins of epibatidine resistance in poison frogs reflect an adaptive landscape with two peaks that maximize fitness of alternative phenotypes: toxin-resistant and defended or toxin-sensitive and undefended. Given that S108C provides significant epibatidine resistance and that it is found in all three resistant clades, we argue that it provides a substantial selective advantage. We suggest two possible evolutionary pathways for acquisition of toxin resistance. In the first, initial replacements may provide a small selective benefit of resistance yet carry some physiological cost in receptor function. For example, the S108C replacement arose independently in all three lineages and is sufficient to produce an epibatidine-resistance phenotype. However, it also incurs decreased sensitivity to ACh in both the human and the Epipedobates backgrounds (Table 1), and the fitness cost of this replacement in living organisms is not clear. We speculate that yet unidentified mutants in the poison frog nAChR sustained receptor functionality, i.e., by inducing nAChR expression changes, until other replacements such as F106L evolved to rescue receptor sensitivity to ACh. Disruptive selection on populations with both genotypes may have propelled the populations with S108C toward a new adaptive peak.
In the second possible trajectory, certain mutants already present in the gene pool provide a genetic background in which resistance arises without cost (e.g., F106). For example, the artificial genotype LSAI (F106L) shows no reduction in either ACh or epibatidine sensitivity (Table 1). Thus, a frog species with F106L has evolved a novel genotype (LSAI), intermediate between FSAI (plesiomorphic) and LCAI in Epipedobates, without incurring a cost, which subsequently allows the C108 replacement to also evolve without cost. However, the LSAI genotype does not exist in any taxa we sampled. It is not present in Silverstoneia, the sister-group of Epipedobates (two of eight species sampled), nor in the closely related taxa Ameerega and Colostethus (Fig. 1A). Thus, this second pathway, in which a novel genotype evolves without apparent cost, is not found in poison frogs. However, this pathway is known in the brown plant-hopper (Nilaparvata lugens) (33), in which two AA replacements confer resistance to fipronil, a noncompetitive antagonist of GABA receptors. This occurs in an apparently sequential process where the second AA change provides high resistance yet has a high fitness cost and never occurs without the first (33). It is unclear how common such pre-existing compensatory mutations are, although it appears that mutations providing incremental increases in resistance are quite common. In Danainae butterflies, newts, garter snakes, and poison frogs, toxin resistance tends to increase over evolutionary time via additional AA replacements that occur in parallel with increased concentrations of chemical defenses (15, 16, 34, 35). It is possible that pre-adaptive mutations that allow resistance to evolve with little cost are present in these organisms and simply have not been identified. The presence of such pre-adaptive mechanisms would imply a shallow, “neutral” valley on the adaptive landscape that facilitates the movement from one adaptive peak to another.
The Epipedobates, Ameerega, and Dendrobates (Oophaga) clades, which are evolutionarily young (6, 36), are an example of rapid and ongoing diversification possibly driven by the evolution of resistance to anti-predator toxins (15). We demonstrate that resistance to epibatidine involves finely tuning a highly conserved binding site without disrupting receptor function, providing insights into evolutionary pathways culminating in chemical defenses. Thus, evolution, with millions of years and subjects, can solve complex problems in systems biology that may otherwise seem impossible.
Supplementary Material
One Sentence Summary.
Mutations combine to confer epibatidine resistance without altering acetylcholine response in a poison frog nicotinic acetylcholine receptor.
Acknowledgments
Data are available in the Supplementary Materials; sequences are archived in GenBank under Accession Numbers MF598757–MF598762, MF580080–MF580125, and MF619959–MF619960. RDT is indebted to the many herpetological societies that funded this project: American Society of Ichthyologists and Herpetologists, Herpetologists’ League, Texas Herpetological Society, Minnesota Herpetological Society, Society for the Study of Amphibians and Reptiles, Chicago Herpetological Society, and North Carolina Herpetological Society, in addition to the National Geographic Society 9468-14, the Society for the Study of Evolution, the Society of Systematic Biologists, and NSF DEB-1404409. RDT was supported by a Graduate School Continuing Fellowship at UT Austin and an NSF Graduate Research Fellowship. JCS was supported by start-up funds at St. John’s University and by the NSERC-CREATE Training Program in Biodiversity Research at UBC. JCS also thanks J. W. Sites, Jr. for his support as a postdoctoral advisor at BYU with grants NSF EF-1241885 and EF-1241848. WS thanks Prof. Dr. D. R. Dietrich of U. Konstanz for his support as a thesis advisor. LAO was supported by a Bauer Fellowship at Harvard University, a William F. Milton grant from Harvard Medical School, and a L’Oreal For Women in Science Fellowship. DCC was supported by NSF DEB-1556967. HHZ was supported by NSF IOS-1557857 and PLR-1443637. CMB and RAH were supported by the UT Waggoner Center for Alcohol and Addiction Research and NIH R01-AA006399. We thank the two anonymous reviewers and P. Andolfatto for comments that greatly improved the manuscript. We also thank L. Coloma, A. Amézquita, M. Betancourth, S. Ron, and T. LaDuc in facilitating specimen collection and access to museum tissues. All specimens were collected under approved protocols (Harvard University IACUC # 12-10 and UT Austin IACUC # 2012-00032 and 2015-00205) and valid permits (001-13 IC-FAU-DNB/MA and 001-11 IC-FAU-DNB/MA [Ecuador], IBD0359 – Res 1177-2014 [Colombia], and SPR-1097-912 [USA]).
References and Notes
- 1.Brodie EDI. Toxins and venoms. Curr Biol. 2009;19:931–935. doi: 10.1016/j.cub.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Després L, David JP, Gallet C. The evolutionary ecology of insect resistance to plant chemicals. Trends Ecol Evol. 2007;22:298–307. doi: 10.1016/j.tree.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 3.McGlothlin JW, et al. Historical contingency in a multigene family facilitates adaptive evolution of toxin resistance. Curr Biol. 2016;26:1616–1621. doi: 10.1016/j.cub.2016.04.056. [DOI] [PubMed] [Google Scholar]
- 4.Ujvari B, et al. Widespread convergence in toxin resistance by predictable molecular evolution. Proc Natl Acad Sci U S A. 2015;112:11911–11916. doi: 10.1073/pnas.1511706112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobler S. Evolutionary aspects of defense by recycled plant compounds in herbivorous insects. Basic Appl Ecol. 2001;2:15–26. [Google Scholar]
- 6.Santos JC, et al. Aposematism increases acoustic diversification and speciation in poison frogs. Proc R Soc B Biol Sci. 2014;281:20141761. doi: 10.1098/rspb.2014.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright S. The roles of mutation, inbreeding, crossbreeding, and selection in evolution. Proc Sixth Int Congr Genet. 1932;1:356–366. [Google Scholar]
- 8.Daly JW, Spande TF, Garraffo HM. Alkaloids from amphibian skin: A tabulation of over eight-hundred compounds. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 9.Santos JC, Tarvin RD, O’Connell LA. In: Chemical Signals in Vertebrates 13. Schulte BA, Goodwin TE, Ferkin MH, editors. Springer Science + Business Media; New York: 2016. pp. 305–337. [Google Scholar]
- 10.Roberts MF, Wink M. Alkaloids: biochemistry, ecology and medicinal applications. Springer Science + Business Media, LLC; New York: 1998. [Google Scholar]
- 11.Darst CR, Cummings ME. Predator learning favours mimicry of a less-toxic model in poison frogs. Nature. 2006;440:208–211. doi: 10.1038/nature04297. [DOI] [PubMed] [Google Scholar]
- 12.Daly JW, Garraffo HM, Spande TF. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 13. Pergamon; New York: 1999. pp. 1–161. [Google Scholar]
- 13.Spande TF, et al. Epibatidine: a novel (chloropyridyl)azabicycloheptane with potent analgesic activity from an Ecuadoran poison frog. J Am Chem Soc. 1992;114:3475–3478. [Google Scholar]
- 14.Dukat M, Glennon RA. Epibatidine: impact on nicotinic receptor research. Cell Mol Neurobiol. 2003;23:365–378. doi: 10.1023/A:1023692705700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tarvin RD, Santos JC, O’Connell LA, Zakon HH, Cannatella DC. Convergent substitutions in a sodium channel suggest multiple origins of toxin resistance in poison frogs. Mol Biol Evol. 2016;33:1068–1081. doi: 10.1093/molbev/msv350. [DOI] [PubMed] [Google Scholar]
- 16.Hanifin CT, Gilly WF. Evolutionary history of a complex adaptation: Tetrodotoxin resistance in salamanders. Evolution (N Y) 2015;69:232–244. doi: 10.1111/evo.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldman CR, Brodie EDJ, Brodie EDI, Pfrender ME. Constraint shapes convergence in tetrodotoxin-resistant sodium channels of snakes. Proc Natl Acad Sci U S A. 2012;109:4556–4561. doi: 10.1073/pnas.1113468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jost MC, et al. Toxin-resistant sodium channels: parallel adaptive evolution across a complete gene family. Mol Biol Evol. 2008;25:1016–1024. doi: 10.1093/molbev/msn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGlothlin JW, et al. Parallel evolution of tetrodotoxin resistance in three voltage-gated sodium channel genes in the garter snake. Thamnophis sirtalis Mol Biol Evol. 2014;31:2836–2846. doi: 10.1093/molbev/msu237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobler S, Dalla S, Wagschal V, Agrawal AA. Community-wide convergent evolution in insect adaptation to toxic cardenolides by substitutions in the Na,K-ATPase. Proc Natl Acad Sci U S A. 2012;109:13040–13045. doi: 10.1073/pnas.1202111109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kouvatsos N, Giastas P, Chroni-Tzartou D, Poulopoulou C, Tzartos SJ. Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: ligand-bound α2 homopentamer. Proc Natl Acad Sci U S A. 2016;113:9635–40. doi: 10.1073/pnas.1602619113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales-Perez CL, Noviello CM, Hibbs RE. X-ray structure of the human α4β2 nicotinic receptor. Nature. 2016;538:411–415. doi: 10.1038/nature19785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan JP, et al. (±)-Epibatidine elicits a diversity of in vitro and in vivo effects mediated by nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 1994;271:624–631. [PubMed] [Google Scholar]
- 24.See the Supplementary Materials.
- 25.Santos JC, et al. Amazonian amphibian diversity is primarily derived from late Miocene Andean lineages. PLOS Biol. 2009;7:0448–0461. doi: 10.1371/journal.pbio.1000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.B I, Moroni M, Zwart R, Sher E, Cassels BK. α4β2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol Pharmacol. 2006;70:755–768. doi: 10.1124/mol.106.023044. [DOI] [PubMed] [Google Scholar]
- 27.DeDominicis KE, et al. The (α4)3(β2)2 stoichiometry of the nicotinic acetylcholine receptor predominates in the rat motor cortex. Mol Pharmacol. 2017;92:327–337. doi: 10.1124/mol.116.106880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li SX, et al. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat Neurosci. 2011;14:1253–1259. doi: 10.1038/nn.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamouda AK, et al. Photoaffinity labeling the agonist binding domain of α4β4 and α4β2 neuronal nicotinic acetylcholine receptors with [125I]epibatidine and 5[125I]A-85380. Biochim Biophys Acta. 2009;1788:1987–1995. doi: 10.1016/j.bbamem.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen JA, Balle T, Gajhede M, Ahring PK, Kastrup JS. Molecular recognition of the neurotransmitter acetylcholine by an acetylcholine binding protein reveals determinants of binding to nicotinic acetylcholine receptors. PLOS One. 2014;9:e91232. doi: 10.1371/journal.pone.0091232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang X, Lonsdale DJ, Gobler CJ. Rapid gain and loss of evolutionary resistance to the harmful dinoflagellate Cochlodinium polykrikoides in the copepod. Acartia tonsa Limnol Oceanogr. 2011;56:947–954. [Google Scholar]
- 32.Feldman CR, Brodie EDJ, Brodie EDI, Pfrender ME. The evolutionary origins of beneficial alleles during the repeated adaptation of garter snakes to deadly prey. Proc Natl Acad Sci U S A. 2009;106:13415–13420. doi: 10.1073/pnas.0901224106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, et al. Synergistic and compensatory effects of two point mutations conferring target-site resistance to fipronil in the insect GABA receptor RDL. Sci Rep. 2016;6:32335. doi: 10.1038/srep32335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petschenka G, et al. Stepwise evolution of resistance to toxic cardenolides via genetic substitutions in the Na+/K+-ATPase of milkweed butterflies (Lepidoptera: Danaini) Evolution (N Y) 2013;67:2753–2761. doi: 10.1111/evo.12152. [DOI] [PubMed] [Google Scholar]
- 35.Geffeney SL, Fujimoto E, Brodie EDI, Brodie EDJ, Ruben PC. Evolutionary diversification of TTX-resistant sodium channels in a predator–prey interaction. Nature. 2005;434:759–763. doi: 10.1038/nature03444. [DOI] [PubMed] [Google Scholar]
- 36.Tarvin RD, Powell EA, Santos JC, Ron SR, Cannatella DC. The birth of aposematism: high phenotypic divergence and low genetic diversity in a young clade of poison frogs. Mol Phylogenet Evol. 2017;109:283–295. doi: 10.1016/j.ympev.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 37.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 38.Lassmann T, Hayashizaki Y, Daub CO. TagDust – A program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dlugosch K, Lai Z, Bonin A, Hierro J, Rieseberg L. Allele identification for transcriptome-based population genomics in the invasive plant Centaurea solstitialis. G3 Genes|Genomes|Genetics. 2013;3:359–367. doi: 10.1534/g3.112.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic Local Alignment Search Tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 43.Bairoch A, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–9. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hahn C, Bachmann L, Chevreux B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Res. 2013;41:e129. doi: 10.1093/nar/gkt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harpsøe K, et al. Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors. J Neurosci. 2011;31:10759–10766. doi: 10.1523/JNEUROSCI.1509-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou Y, et al. Human α4β2 acetylcholine receptors formed from linked subunits. J Neurosci. 2003;23:9004–9015. doi: 10.1523/JNEUROSCI.23-27-09004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lucero LM, et al. Differential α4(+)/(−)β2 agonist-binding site contributions to α4β2 nicotinic acetylcholine receptor function within and between isoforms. J Biol Chem. 2016;291:2444–2459. doi: 10.1074/jbc.M115.684373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kreienkamp HJ, Maeda RK, Sine SM, Taylor P. Intersubunit contacts governing assembly of the mammalian nicotinic acetylcholine receptor. Neuron. 1995;14:635–644. doi: 10.1016/0896-6273(95)90320-8. [DOI] [PubMed] [Google Scholar]
- 50.Nichols WA, et al. Mutation linked to autosomal dominant nocturnal frontal lobe epilepsy reduces low-sensitivity α4β2, and increases α5α4β2, nicotinic receptor surface expression. PLOS One. 2016;11:e0158032. doi: 10.1371/journal.pone.0158032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weltzin MM, Lindstrom JM, Lukas RJ, Whiteaker P. Distinctive effects of nicotinic receptor intracellular-loop mutations associated with nocturnal frontal lobe epilepsy. Neuropharmacology. 2016;102:158–173. doi: 10.1016/j.neuropharm.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.