

Abstract
Beta-N-methylamino-L-alanine (BMAA) has been demonstrated to contribute to the onset of the ALS/Parkinsonism-dementia complex (ALS/PDC) and is implicated in the progression of other neurodegenerative diseases. While the role of BMAA in these diseases is still debated, one of the suggested mechanisms involves the activation of excitatory glutamate receptors. In particular, the excitatory effects of BMAA are shown to be dependent on the presence of bicarbonate ions, which in turn forms carbamate adducts in physiological conditions. The formation of carbamate adducts from BMAA and bicarbonate is similar to the formation of carbamate adducts from non-proteinogenic amino acids. Structural, chemical, and biological information related to non-proteinogenic amino acids provide insight into the formation of and possible neurological action of BMAA. This article reviews the carbamate formation of BMAA in the presence of bicarbonate ions, with a particular focus on how the chemical equilibrium of BMAA carbamate adducts may affect the molecular mechanism of its function. Highlights of nuclear magnetic resonance (NMR)-based studies on the equilibrium process between free BMAA and its adducts are presented. The role of divalent metals on the equilibrium process is also explored. The formation and the equilibrium process of carbamate adducts of BMAA may answer questions on their neuroactive potency and provide strong motivation for further investigations into other toxic mechanisms.
Keywords: β-N-methylamino-L-alanine (BMAA), Carbamate, NMR, Chemical equilibrium
Introduction
The environmental neurotoxin β-N-methylamino-L-alanine (BMAA) was first isolated from cycad seeds in the late 1960s (Vega 1967). The non-proteinogenic amino acid, BMAA, has long been associated with an elevated incidence of amyotrophic lateral sclerosis/Parkinsonism-dementia complex (ALS/PDC) within the local Chamorro people of Guam (Murch et al. 2004a, b). The role of BMAA exposure has also been indicated in Alzheimer’s disease (AD) (Murch et al. 2004a, b; Pablo et al. 2009; Chiu et al. 2011). Numerous in vitro investigations reveal the detrimental effects of BMAA to neurons. The most commonly implicated mechanism is excitotoxicity-cell death due to activation of excitatory amino acid (EAA) receptors (notably glutamate receptors) (Choi 1988; Richter and Mena 1989; Brownson et al. 2002; Lobner et al. 2007; Chiu et al. 2012).
An EAA stimulates EAA receptors allowing them to modulate the efficacy of their synaptic transmission. However, overstimulation by EAA causes neuronal apoptosis (Monaghan et al. 1989). The main EAA receptors are subdivided into two categories: the ionotropic receptors and metabotropic glutamate receptors (mGluRs). Examples of ionotropic receptors include kainate, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and N-methyl-D-aspartate (NMDA). All ionotropic and metabotropic EAA receptors are activated by glutamate and structurally similar substances (Gahring and Rogers 2002; Catarzi et al. 2006; Xue and Field 2011). Dysfunction of glutamate receptors leads to excitotoxic cell death via prolonged depolarization of neurons and the activation of enzymatic and nuclear mechanisms of cell death-initiating neuronal apoptosis (Doble 1999). A noticeable increase in glutamate from the cerebrospinal fluid of ALS patients suggests excitotoxicity could be a significant contributor to the progression of neurodegenerative disease (Boillee et al. 2006a, b).
Though the excitatory action of neurotransmitters is necessary for the rapid transduction of signals throughout the nervous system, excessive stimulation of neurons by either environmental or biological factor can also trigger neuronal apoptosis, expediting the rate of neurodegeneration. The range of molecules able to depolarize the membrane is not solely limited to neurotransmitters but can encompass natural metabolites, such as amino acids, environmental triggers, pesticides, neurotoxins, and various other proteins. In particular, BMAA is a prevalent, non-amino acid and ever-growing evidence suggests that BMAA plays a role in the onset and progression of neurodegenerative diseases (Chiu et al. 2011, 2013; Faassen et al. 2013). In a seminal work, Weiss and Choi discovered that BMAA exhibited activity only in the presence of HCO3− ions in the culture media at physiological concentrations (10 mM and above). This discovery focused on the importance of not just BMAA but the interaction of BMAA with HCO3− ions and its critical role in the modality of BMAA’s excitotoxicity. Further studies (Richter and Mena 1989; Weiss et al. 1989; Myers and Nelson 1990) established the critical dependence of BMAA/HCO3− interaction on its function. The crux of these investigations suggested that the formation of a carbamate adduct of BMAA in the presence of HCO3− shares a high structural similarity with glutamic acid (Myers and Nelson 1990), leading to a possible explanation of the mechanism by which BMAA may bind to glutamate receptors.
Following the initial work by Nunn’s lab (Nunn and O’Brien 1989; Nunn et al. 1989, 1991; Davis et al. 1993b; Nunn 2009), we recently undertook a systematic evaluation of BMAA and HCO3− interaction using high-resolution NMR spectroscopy with a particular focus on the chemical equilibrium process of the carbamate formation (Zimmerman et al. 2016). In addition to confirming the earlier observations that BMAA/HCO3− interactions lead to the formation of both α-and β-carbamates, we observed that these adducts co-exist in the solution state at physiological conditions. The conformational preference of the carbamate adducts and the intermolecular dynamics may play a major role in how these adducts might function as a neurotransmitter to activate EAA receptors. Therefore, in this article, we review the fundamental idea of the carbamate formation of BMAA in the presence of bicarbonate ions with a particular focus on the chemical aspects and how the chemical equilibrium process of BMAA with its carbamate adducts may be essential towards an understanding of the molecular mechanism of its function.
Chemistry
Carbamylation
The formation of carbamic acid (NH2COOH) is known as carbamylation (or carbamoylation). Carbamate formation occurs when an uncharged amine interacts with carbon dioxide (CO2) or bicarbonate (HCO3−) to produce a carbamic acid functional group (NH2COOH) (Ewing et al. 1980). The spontaneous formation and decomposition of carbamates at physiological conditions (pH ~7.4) is an important biochemical control system (Lorimer 1983). At neutral pH (~7), the amines are predominantly in their protonated form (pKa > 8) and the carbon dioxide is in its hydrated form, HCO3− (H2CO3; pKa1 = 6.3). Though these conditions are unfavorable for carbamylation, the favorable ΔG can be attributed to the pKa values of the protonated amines lowered by their chemical environment and thus provide conditions suitable for carbamate formation (Lorimer 1983; Cleland et al. 1998). The dissociation of the hydrogen from the charged amine increases the nucleophilicity of the amine. The presence of an electron-deficient (electrophilic) CO2 or HCO3− allows the nucleophilic amine to attack the electrophilic carbon on either CO2 or HCO3−, leading to the carbamylation reaction, followed by stabilization of the carbamates by noncovalent intermolecular interactions (Lorimer 1983; Weiss and Choi 1988):
| (1) |
The Role of a Bicarbonate Buffer System
Physiologically, bicarbonate is the predominant buffering system that modulates the concentrations of both weak acids and bases in solution to maintain physiological pH. The rate of carbamate formation and decomposition in the presence of the bicarbonate buffering system may also be important in regulating adverse effects related to acidosis or alkalosis (Rossi-Bernardi and Roughton 1967). The regulation of physiological pH involves the formation of CO2 and H2O from H2CO3, catalyzed by the enzyme carbonic anhydrase. Dissociation of H2CO3 and the equilibrium between CO2, H2CO3, and HCO3− in the bicarbonate buffer system can be written as follows:
| (2) |
The presence of excess acid or base present within the system can disrupt the equilibrium between CO2, H2CO3, and HCO3− causing an overproduction of either CO2, H2CO3, and HCO3−, resulting in a reaction with amines (e.g., amine in the amino acids or protein side chains) leading to the formation of carbamic acid. The base-catalyzed reaction involving the formation of carbamates is further propagated by the presence of basic species. Amines (pKa 9–10), water (pKa 15), or carbonic acid (pKa 6.35, and 10.239) can facilitate the removal of an acidic hydrogen from the protonated amine allowing the compound to react with either CO2 or HCO3− resulting in the formation of a carbamate compound. The inter-linked reactions are sensitive to small changes in the chemical environment and highly relevant to carbamate formation in the case of BMAA.
Primary and Secondary Carbamates of BMAA
BMAA has a secondary amine in its side chain (Fig. 1). Typically, at neutral pH, the amino group of an amino acid is protonated, and the carboxyl group is deprotonated, resulting in a neutral charge. BMAA has two amino groups in its side chain: a primary (α) and a secondary (β) amino group. This is similar to the basic amino acid lysine, but different functionality. The pKa values of the carboxylic group (pK1), primary amine (pKα), and secondary amines (pKβ) were estimated to be 2.1, 6.5, and 9.8, respectively (Vega et al. 1968). This amine pKa values were further refined (Nunn and O’Brien 1989) to be 6.63 (pKα) and 9.76 (pKβ) and reproduced by the National Food Administration in Sweden (Sundh et al. 2007). The titration curve indicates well-separated pKa values for the primary and secondary amines with the isoelectric point (pI) at 8.09 (Fig. 2). Upon considering the charge state of each amine, Nunn and O’Brien determined that in solution BMAA was 86% α-deprotonated and 14% β-deprotonated (Nunn and O’Brien 1989). These results led to the fact that at neutral pH both α and β-amino groups are deprotonated at differing levels.
Fig. 1.
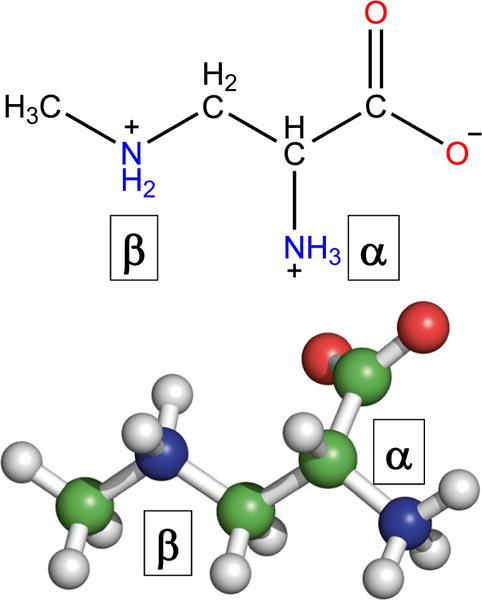
Chemical and three-dimensional structure of β-N-methylamino-L-alanine (BMAA)
Fig. 2.
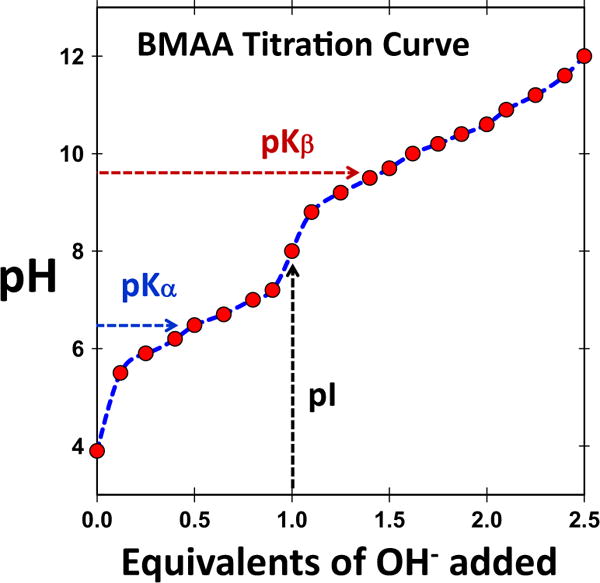
Titration curve of BMAA. Titration shows two pKa values, primary amine (pKα) and secondary amine (pKβ) and the pI (isoelectric point) is at 8.0. (Figure reproduced with permission from “Analysis, occurrence, and toxicity of ß-methylaminoalanine (BMAA),” published by Nordic Council of Ministers, Denmark (2007))
Considering the features mentioned above of BMAA and the carbamylation of amino acids presented earlier, several general features could be common between BMAA. However, there is a distinct difference between the carbamylation of non-proteinogenic amino acids and carbamylation of amino acids. The presence of the second amino group, however, alters the chemistry of carbamate formation of BMAA in a clear manner (Viso et al. 2005). In general, the formation of carbamates follows the schematic shown in Fig. 3. Physiologically, the CO2 released from cellular respiration reacts with water to produce carbonic acid, which dissociates to bicarbonate and subsequently converts back to CO2. The presence of HCO3− and CO2 can react with a deprotonated amine to produce a carbamate product (black arrows). Further reaction of HCO3− and CO2 with the primary or secondary amine lead to primary or secondary carbamylation of BMAA. The formation of primary carbamates of BMAA in the presence of bicarbonate ions was first suggested by Nunn and co-workers by characterizing these compounds using nuclear magnetic resonance (NMR) spectroscopy (Nunn and O’Brien 1989; Davis et al. 1993a, b). They first hypothesized that it was probably the formation of the second adduct of BMAA, but the second adduct could not be detected by NMR due to equipment capability (Fig. 2). A later study by Myers and Nelson (Myers and Nelson 1990) used 13C labeled bicarbonate and examined the formation of the BMAA/bicarbonate adducts using 13C experiments. Myers and Nelson observed the formation of two carbamate adducts and showed these products are formed when the amine from the BMAA binds to carbon dioxide, which is produced from bicarbonate. The equilibrium constant of both carbamates were calculated from this study based on the relative peak intensity of the α-carbamate (BMAA carbamate when the primary amine binds CO2), β-carbamate (BMAA carbamate when the secondary amine binds CO2), and the unbound CO2.
Fig. 3.
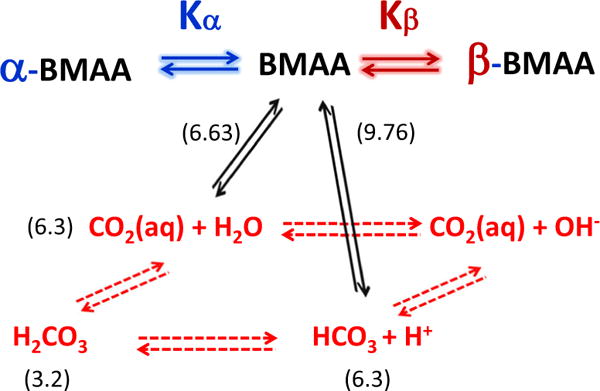
Kinetic equilibria of BMAA. The equilibria between CO2, H2CO3, and HCO3− are shown in red while the equilibrium between BMAA and its carbamates are shown in black. The pKa value of each reaction is given in parenthesis. The subscripts α and β represent the primary and secondary carbamates of BMAA
Experimentally, in an aqueous solution containing BMAA and bicarbonate, it was found that 14% of BMAAwas in its β-carbamate form while 86% was in the α-carbamate form (Myers and Nelson 1990). As discussed before, the predominance of the α-carbamate in comparison to the β-carbamate is due to the differences in pKa between the α-amine and the β-amine. In effect, the differential pKa values between the amines reduce the ability of the substituted secondary amine (β-amine) to be carbamylated in the presence of the primary amine (α-amine). The ability of the α-amine to be deprotonated rapidly at neutral pH allows for the nucleophilic attack of a CO2 and HCO3− leading to the formation of the α-carbamate as opposed to the formation of the β-carbamate from BMAA.
Structural Analogs of BMAA
It has been demonstrated that BMAA can be misincorporated in the place of L-serine biosynthetically (Rodgers and Shiozawa 2008), as well as in human proteins (Dunlop et al. 2013). The summary of molecules that resemble BMAA and their ability to form primary and secondary carbamates are shown in Fig. 4. In addition to glutamate (Fig. 4a), the other molecules included are DL-2,4-diaminobutyrate (2,4-DAB, Fig.4c), DL-2,3-diaminopropionate (2,3-DAP, Fig. 4d), and monoethanolamine (MEA, Fig.4e) along with BMAA (Fig. 4b) for comparison. The figure highlights the structural similarities between chemical structures by aligning the Cα and Cβ carbons for the α-carbamates (left) and β-carbamates (right), respectively. The figure also shows the pKα (pKa of primary amine) and pKβ (pKa of secondary amine) for each molecule.
Fig. 4.
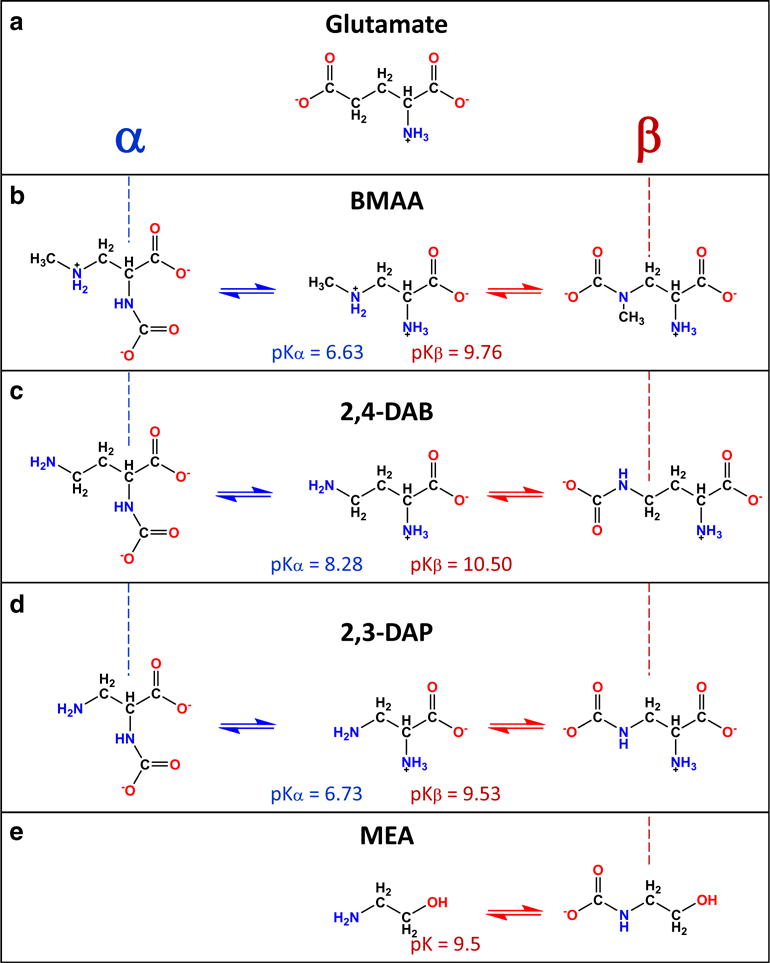
Structural similarities of non-proteinogenic amino acids and glutamate on BMAA. a Glutamate, b β-N-methylamino-L-alanine (BMAA), c DL-2,4-diaminobutyrate (2,4-DAB), d DL-2,3-diaminopropionate (2,3-DAP), and e monoethanolamine (MEA). The pKa values primary (pKα) and secondary (pKβ) are shown in parenthesis. The vertical dashed lines show the alignment to highlight the Cα (blue) and Cβ (red) carbons in the primary and secondary carbamates
Glutamate
Glutamate structurally resembles BMAA as it contains a α-amine and a α-carboxylic acid group. The resemblance of glutamate to the β-carbamate of BMAA (Fig. 4b, right) is structurally relevant due to the presence of the secondary carboxylic group. The difference between free BMAA and glutamate is the lack of the ω-moiety present on glutamate common to excitatory amino acids. Glutamate is one of the most prevalent and utilized excitatory amino acid found in the CNS. Glutamate is a neurotransmitter that activates both ionotropic and metabotropic receptors for the propagation of electrical signals within the CNS. The glutamate receptors, specifically the ionotropic receptors, have conservative regions within their binding pocket that interact with the α-carboxyl groups, α-amino, and ω-moiety groups of glutamate (Traynelis et al. 2010). Activation of the glutamate receptors is necessary for memory formation and learning in the hippocampus, as well as, motor coordination. Furthermore, excessive glutamate within the CNS is known to cause excessive depolarization of the neuronal membrane leading to neuronal apoptosis (Atlante et al. 2001). In the presence of bicarbonate, BMAA obtains the ω-electronegative moiety, increasing its functionality within the central nervous system (CNS) (Weiss et al. 1989). Therefore, the ability for carbamylated compounds to mimic the structure of glutamate indicates that they must also be activators at the various glutamate receptors present on the neuronal membrane.
DL-2,4-Diaminobutyrate
The non-protein amino acid 2,4-diaminobutyric (2,4-DAB, Fig. 4c) was isolated from the genus Lathyrus latifolus. Structurally, the compound contains a secondary amine, as seen in BMAA and DAP, but has an elongated methylene chain. The presence of two amino groups on 2,4-DAB allows the structure to undergo carbamylation at both amino sites (Fig. 4c). Due to its similarities to BMAA, 2,3-DAP, and other amino acids, it is presumed that 2,4-DAB can cross the blood-brain barrier using the amino acid transporter system. Once in the brain, 2,4-DAB may be able to react with either CO2 or HCO3− to form neuroactive carbamate products (Fig. 4c). As reported by Weiss et al., the neurotoxicity of 2,4-DAB was lower compared to 2,3-DAP (Weiss et al. 1989). The pKa values of the primary and secondary amines in 2,4-DAB were 8.28 (pKα) and 10.50 (pKβ), respectively. These pKa values are considerably higher than the corresponding pKa values in BMAA (pKα = 6.63 and pKβ = 9.76), in particular for the β-amine. This suggests that the neurotoxicity of 2,4-DAB can be related to the pKa of the β-amine, which could hinder the rapid formation of carbamate compounds and hence lower its potency (Friedman 1989). The increase in the pKβ of the β-amine, when compared to BMAA and DAP, is attributed to the inductive effect. The addition of an extra carbon to the carbon backbone increases the spatial distance between the electronegative groups, thus increasing the pKa and stabilizing the lone electron pair on the β-amine. Furthermore, the elongated carbon skeleton enhances the number of electron-donating groups within the proximity of the β-amine causing an increase in the electron density at the amine atom resulting in an increase in the basicity of the β-amine.
DL-2,3-Diaminopropionate
The non-protein amino acid oxalyldiaminopropionic acid (ODAP) was originally derived from the legume Lathyrus sativus, a known neurotoxic compound and the primary component in the neurological disease lathyrism (Nunn et al. 2010). Although ODAP does not form carbamates in the presence of CO2, or HCO3−, its structural similarities to glutamate causes ODAP to bind to ionotropic receptors and metabotropic receptors, causing cellular damage (Nunn et al. 2010). Dissociation of ODAP results in the metabolite 2,3-diaminopropionate (2,3-DAP, Fig.4d). For 2,3-DAP, the α-amine has a pKa of 6.73 while the pKa of the β-amine is 9.53 (Friedman 1989). These pKa values are similar to that of BMAA (6.63 and 9.76 for the α-amine and β-amine amines, respectively). By itself, 2,3-DAP is pharmacologically inactive in its normal state and upon reacting with CO2/HCO3−, it gains excitotoxic activity.
The slight reduction in the pKa of the β-amine, when compared to BMAA and DAB, is attributed to the decrease in the spatial distance between the electronegative groups. Reduction of the carbon backbone naturally decreases the distance between the electronegative groups, depleting the electron density of the carbon backbone and causing an overall negative moiety across the structure. The presence of electron-withdrawing groups shifts the electron density, making the molecule more electrophilic and unstable while increasing its reaction enthalpy to favor carbamate formation (Klebe 2015).
Ethanolamines
The ethanolamines are amino alcohols that can exist in three different forms: monoethanolamine, diethanolamine, and triethanolamine. Among the three, monoethanolamine (MEA) is a common intermediate derived from the amino acid serine. Decarboxylation of serine by pyridoxal phosphate is a crucial step in the formation of choline and subsequently acetylcholine (Scheiman 1962). Alternatively, monoethanolamine can be used to construct the cephalin found in the cell membranes of brain tissue. Pharmacologically, monoethanolamine is shown to inhibit cell growth, increase blood pressure (vasoconstriction), and convulsions. Given their importance, the formation of carbamate compounds from MEA was investigated (Fig. 4e) (Xie et al. 2015). Computational modeling of various substituted MEAs revealed characteristic thermodynamic and bonding properties involved in the formation of carbamates (Xie et al. 2015). These studies found that the addition of electronegative elements (N, O, or F) to the ethanolamine structure modulated the pKa of ethanolamine. These calculations further showed that the presence of electron-withdrawing groups decreases the pKa and raises the activation energy (Ea) of ethanolamine. The increase in reaction enthalpy of carbamate formation relates linearly with the basicity of the amine. The presence of electron-withdrawing groups creates transient changes in the charge distribution across the chemical structures that are inherently asymmetric. The changes in electron density influence the partial dipoles experienced by the chemical scaffold. Due to the negative moiety of CO2 (−0.2e–0.5e) and the positive moiety of the amine group (0.2e–0.5e), an electrostatic interaction arises between the CO2 and the amine group. Although BMAA contains two amines, α-and β-amine, the effect of electron-withdrawing and electron-donating groups on both amines are similar to that seen in monoethanolamines. Accordingly, due to the high pKa of the β-amine (9.8), it is presumed that the low activation energy (Ea) associated with this compound will favor the β-carbamate (Fig. 4e). However, due to the steric hindrance of the additional methyl group (Yamamoto et al. 2012), the β-amine is unfavorable; instead, the α-carbamate is favored due to the presence of the electron-withdrawing group (α-carboxylic group), promoting the formation of the C-N bond with either CO2 or HCO3−.
Equilibrium Kinetics of BMAA with Its Carbamates
Nunn and workers extensively studied the carbamate formation in BMAA and characterized its formation spectroscopically (Nunn and O’Brien 1989; Nunn et al. 1991; Davis et al. 1993a; Nunn 2009). Recently, we undertook a systematic evaluation of the BMAA and HCO3− interaction using high-resolution NMR spectroscopy with a particular focus on the chemical equilibrium process of the carbamate formation (Zimmerman et al. 2016). In addition to confirming the earlier observations that BMAA/HCO3− interactions lead to the formation of both α- and β-carbamates, we observed that these adducts co-exist in the solution state at physiological conditions. The conformational preference of the carbamate adducts and the intermolecular dynamics may play an important role in how these adducts might function as a neurotransmitter. More importantly, this study opens up the possibility of how one could control the activation by specific configurational changes of BMAA that might allow inhibition of the onset and progression of neurodegenerative diseases.
NMR Spectroscopy of BMAA and Its Carbamates
Figure 5 shows the 1H NMR spectra of BMAA and its carbamate adducts (1Hν = 400 MHz, in 100% D2O, pD of 7.6, and at 35 °C). The one-dimensional 1H NMR spectra of BMAA, in the presence of bicarbonate ions at a ratio of 1:20, show distinct spectral features from the free BMAA and the α- and β- carbamate adducts (Fig. 5a) as marked by B, α, and β respectively. All the resonances (1Hα, 1Hβ, and 1Hγ) from the BMAA, the α-carbamate, and the β-carbamate show the signs of conformational exchange among themselves. However, the methyl protons (1Hγ) are separated for free BMAA, α-carbamate, and β-carbamate at 2.72, 2.77, and 2.86 ppm, respectively. Two-dimensional exchange spectroscopy (EXSY) provides a powerful option to determine the equilibrium constants of nuclear spins undergoing conformation exchange (Jeener et al. 1979; Meier and Ernst 1979). The chemical exchange between the three species in the solution is shown in the two-dimensional EXSY experiment between the methyl protons (Fig. 5b). The chemical equilibrium between BMAA and its carbamate adducts may be considered as a pseudo-equilibrium process as the carbamate formation continue to change with an increase in the relative concentrations of the BMAA to HCO3− ions (Fig.5b).
Fig. 5.
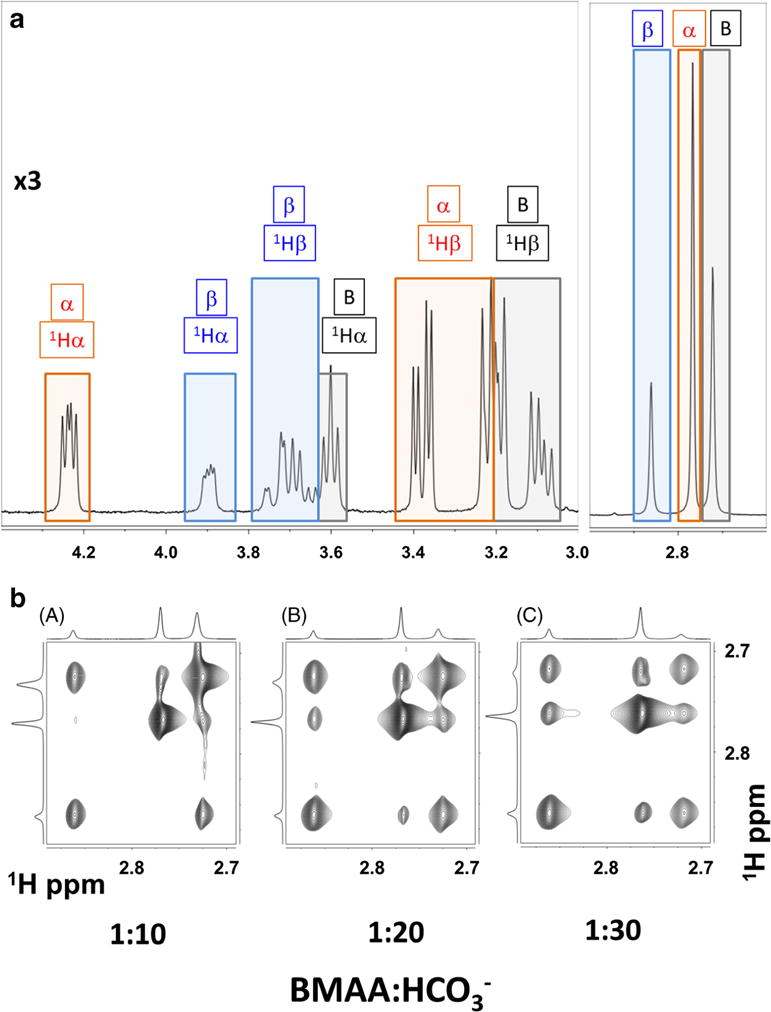
NMR spectral characterization of BMAA and its carbamate adducts. a 400 MHz 1H NMR spectrum of highlighting the coexistence of free BMAA (B: black boxes), α-carbamate adduct (α red boxes), and β-carbamate adduct (β: blue boxes). NMR spectra were recorded with 1:20 ratio of BMAA/HCO3− (10 mM/200 mM) in 100% D2O, pD of 7.6, and at 30 °C. b Effect of increasing bicarbonate concentration: Methyl region of the 400 MHz two-dimensional exchange spectroscopy (EXSY) spectra with increasing BMAA/HCO3− ratio: (A) 1:10, (B) 1:20, and (C) 1:30. The BMAA concentration was at 10 mM. EXSY spectrum is recorded at 30 °C and with a mixing time of 400 ms
The Equilibrium Between BMAA and Its Carbamates
Following the chemical exchange process for nuclear spin relaxation between the three spins (I = ½), the rate constants of magnetization exchange that are related to the chemical kinetics can be estimated using the well-established methods (McConnell 1958; Jeener et al. 1979; Meier and Ernst 1979). Using the total concentration of BMAA determined upon integrating the NMR signals from the 1D experiment (Fig. 5a) and estimations of the total concentration of carbon dioxide using the equilibration process of carbamate and carbamic acid (Fig. 3), the quasi-equilibrium kinetics of BMAA and its carbamates for a given sample conditions (BMAA/HCO3− concentration) can be estimated (Zimmerman et al. 2016). The concentrations of [BMAA] T, [α-carbamate], and [β-carbamate] were obtained from the integration of the 1D spectra (total BMAA concentration = [BMAA] T, or [α-carbamate] or [β-carbamate]). The concentration of BMAA (unprotonated) was calculated using the known pKa of amines (Arnold et al. 1969). Assuming that the pKa for primary and secondary amines are the same, temperature independent, and the total carbon dioxide concentration was estimated using the total NaHCO3 in the solution (pKa1 of H2CO3 = 6.34) (Dean 1992). At the neutral pH, the ratios [H2CO3]/[CO2] and [H2CO3]/[HCO3−] are small (< 0.005 or less) (Gibbons and Edsall 1963). Upon substituting these values and the corresponding rate constants determined from the EXSY spectrum, the equilibrium constant can be determined straightforwardly.
These results determine half-lives of the formation the α-and β-carbamate adducts (assuming a first order reaction) to be 4.18 ± 0.99 s (t½,1α) and 1.17 ± 0.10 s (t½,1β), while the cleavages are 3.08 ± 0.34 s (t½,2α) and 0.37 ± 0.01 s (t½,2β), respectively. The half-life of the α-carbamate formation decreases with increasing bicarbonate formation with increasing carbonate ion concentration, suggesting that the system is still in quasi-equilibrium condition. The equilibrium constant for the formation of α- and β-carbamate adducts (after accounting for the protonation) is estimated as 7.60 ± 1.75 × 106 and 3.11 ± 0.26 × 106, respectively. Assuming equilibrium conditions, the reaction Gibbs free energy (ΔGα*) for the α-carbamate formation was estimated to be −5.14 ± 1.18 kJ mol−1, and −2.89 ± 0.24 kJ mol−1 for the β-carbamate. The data does not show the presence of doubly carbamylated forms of BMAA, which follows a similar observation by Davis et al. (Davis et al. 1993a).
The quasi-equilibrium constant of the α-carbamate formation is larger than the β-carbamate with increasing concentration HCO3−. A linear trend between the equilibrium constants and the total concentration of NaHCO3 is observed where the α-carbamate equilibrium increased approximately 0.07 M−1 for an increase of 1 mM of HCO3− ion, while β-carbamate equilibrium increased approximately by 0.006 M−1 per 1 mM of HCO3−. Physiological concentrations of BMAA, as low as 10–30 μM have shown to cause neuronal injury (Rao et al. 2006; Lobner et al. 2007). Thus, with a physiological concentration of bicarbonate in the range of 20–25 mM, the HCO3− will be 1000 times higher than BMAA (BMAA/HCO3− = ~ 1:1000–1:2000). Based on the variation in the relative concentrations of the three species in solution, one may assume the concentration of BMAA and the β-carbamate will be the same (~ 25% each) while the α-carbamate will be twice that of the BMAA (or β-carbamate) with ~ 50% of the concentration.
Roles of Metal Ions on BMAA and Its Adducts—Zn2+
The presence of divalent metals (Mn2+, Zn2+, Mg2+, Ca2+, Cu2+) within the brain have various functions such as activation of glutamate receptors, influence synaptic transmission, cellular signaling, and as cofactors for different enzymes within the brain (Takeda 2003; Szewczyk 2013; Marchetti 2014). The most prevalent divalent metal within the brain is Zn2+. Zn2+ is abundantly present in its free ionic form and is a necessary cofactor for hundreds of enzymes (Szewczyk 2013).
BMAA is a known chelator of Cu2+, Zn2+, and Ni2+, suggesting the potential for BMAA to bind to these divalent metals (Nunn et al. 1989). Glover et al. examined the role of BMAA binding to zinc using mass spectrometry (Glover et al. 2012). Detailed mass spectroscopic investigation in combination with NMR and preliminary computational modeling suggest a possible distorted tetrahedral geometry of coordination; it was not possible to unambiguously determine the conformation of the Zn(BMAA)2 complex (Glover et al. 2012). Presumably, Zn2+ can coordinate with BMAA resulting in the formation of the Zn(BMAA)2 complex, similar to that of Cu(BMAA)2 to form a four-coordinate complex in which each amino acid binds through both N atoms (Nunn and O’Brien 1989). The mechanism of how BMAA can bind to Zn2+ is still unknown, but since BMAA and two carbamate adducts are present in the physiological condition it is possible that one of the BMAA carbamate adducts binds Zn2+ rather than the free BMAA. These experiments would provide insight on the ability of BMAA to chelate Zn2+, thus leading to a potential transport mechanism of BMAA and a second pathway for neurodegenerative effects (CuZnSOD disruption).
Biological Importance of Carbamate Formation
The biological importance of BMAA has been addressed extensively in the literature including many outstanding contributions in this particular volume of the journal by leading authorities in the area. Therefore, we briefly provide an overview of the potential implications of the carbamates of BMAA to function.
One of the outstanding questions that is yet to be addressed is how BMAA can bind to the GluR and if it does, how does this result in neuronal damage. Glutamate is a natural agonist for the GluR. Therefore, understanding how BMAA structurally compares to glutamate can provide insight on potential interactions at the receptor level. The first of three key characteristics for GluR binding is the molecule must have two carboxylate groups allowing for hydrogen bonding to occur between the residues of the ligand-binding domain and the agonist. Second, the two carboxylate groups must be located within 3.0 to 4.5 Å of one other. Moreover, the third feature is the main chain of the molecule should be between 4 to 5 carbons in length. BMAA lacks a second carboxylate group and would most likely not be able to hydrogen bond with the amino acid residues in the ligand-binding domain. The lack of hydrogen bonding could prevent free BMAA from binding to the GluR. However, BMAA in the presence of bicarbonate may bind to the GluR (Weiss et al. 1989).
The ability of BMAA to bind to the GluR in the presence of bicarbonate is cause for studying the carbamate adducts and their likelihood of forming under physiological conditions. Following the original experiments by Nunn and co-workers (Nunn and O’Brien 1989; Nunn 2009), our recent study shows that BMAA and its carbamates are all present in equilibrium at a pH of 7.6 (Zimmerman et al. 2016). The coexistence of the three molecules, BMAA adducts, and free BMAA is an important factor when discussing BMAA binding to the GluR. The newly formed carbamates have two carboxylate groups, whereas the free BMAA molecule only has one carboxylate group (Fig. 4b). Formation of the second carboxylate group on the α-carbamate of BMAA and β-carbamate of BMAA fulfill the first key characteristic of binding to the GluR. In addition to having the second carboxylate group from the carbamate formation, both the primary and secondary carbamate have the carboxylate groups located within 3.0 to 4.5 Å of each other, fulfilling the second key characteristic for possible GluR binding. The distance of 3.0 to 4.5 Å is not how close the carboxylate groups are located to each other on the backbone of the molecule. The distance of 3.0–4.5 Å is how far apart the carboxylate groups are when the molecule rotates to obtain the conformation needed to bind to the receptor. Both of the carbamate adducts have no double bonds or aromatic rings, allowing the molecules to be flexible and achieving the distance of 3.0–4.5 Å between carboxylate groups. The third key characteristic is fulfilled by both carbamate molecules. α-carbamate of BMAA and β-carbamate of BMAA have a main chain length between 4 and 5 carbons, which is small enough to fit into the ligand-binding domain of the receptor. The carbamate adducts both fulfill the characteristics for binding to the GluR, and there is a probability that α-carbamate of BMAA and β-carbamate of BMAA bind to the GluR. Not only are there two carbamates formed when BMAA/HCO3− interact, but the three molecules of the sample are in equilibrium with one another.
The HCO3− concentration in vivo has been estimated to be around 10 mM, which is far greater than the concentration of BMAA (Weiss et al. 1989). This means when BMAA is in the blood the majority of the BMAA can bind to CO2 and become α-carbamate of BMAA and β-carbamate of BMAA, with a small amount remaining as free BMAA. Since BMAA is in equilibrium with the carbamate adducts if β-carbamate of BMAA can bind to the GluR, the free BMAA and α-carbamate of BMAA would be converted to β-carbamate of BMAA to remain in equilibrium after β-carbamate of BMAA binds the receptor. The continued formation of β-carbamate of BMAA would allow for the possibility of more GluR binding by β-carbamate of BMAA resulting in overactivation and potential neuronal damage. Binding to the GluR is important, but in addition to just binding to the GluR, if a molecule has a higher binding affinity the receptor will be prone to overactivation.
The binding affinity of glutamate to the GluR allows glutamate to bind and activate the receptor while not binding so tightly to cause an irreversible reaction. Since the carbamate adducts are close to isosteric to glutamate with some slight structural difference, an extra methyl group, and an amine instead of carbon, there is a chance the adducts could have a higher binding affinity than glutamate. The additional amine located within BMAA could allow for additional hydrogen bonding to one of the residues located in the ligand-binding domain of the GluR. The methyl group of BMAA carbamate adducts could interact with the hydrophobic region of the ligand-binding domain. The possibility of increased interaction can produce a higher binding affinity for BMAA carbamates resulting in binding and possibly overactivation of the GluR. Furthermore, if the adducts are bound tightly to the receptor, there will be an excess of glutamate that is usually used for receptor binding. The excess glutamate can then bind to the GluR causing excitotoxicity. Understanding how and if BMAA/HCO3− interact to form two carbamate adducts could explain why the presence of bicarbonate is needed for BMAA to be neurotoxic.
BMAA’s neurotoxicity stems from the multiple modes of action it can take to cause neuronal damage. The first mode of action is the GluR binding and has been previously discussed. The second mode of action is BMAA being misincorporated into proteins causing misfolding. Misfolded proteins do not function properly, and could lead to excess proteins not capable of being metabolized, resulting in cell death. In addition to BMAA being misincorporated into proteins, it is thought BMAA can bind to metals causing enzyme malfunction. For example, the copper-zinc SOD enzyme has been hypothesized to be bound to BMAA affecting the metal active site in SOD, preventing the enzyme from correctly regulating superoxide anions. The excess of superoxide anions can damage cells and cause neuronal cell damage. Knowing that BMAA can form two carbamate adducts can provide insight on metal chelation and protein misincorporation. The carbamates of BMAA could be inserted into protein more readily than free BMAA, but more importantly, metal chelation of Zn2+ by the carbamate of BMAA could be causing damage to neurons.
Summary and Outlook
BMAA has been linked to ALS/PDC and has been hypothesized to contribute to the disease by binding to the GluR. NMR spectroscopy studies of the BMAA/HCO3− interaction validates the coexistence of three BMAA forms: α- and β-carbamate adducts of BMAA and free BMAA. The reaction between BMAA and CO2 produce the carbamate adducts, and the formation of the carbamates is a reversible reaction. Assuming a pseudo first-order reaction, the half-lives of the formation of α- and β-carbamate adducts are 4.18 ± 0.99 s and 1.17 ± 0.10 s, while the cleavages are 3.08 ± 0.34 s and 0.37 ± 0.01, respectively. Knowing the formation of the BMAA carbamate adducts provides a new set of experiments and hypothesis of how BMAA can be neurotoxic. The carbamate adducts could be responsible for the binding of the GluR resulting in overactivation. The binding to the GluR is more probable with the carbamates compared to free BMAA since the carbamates share a chemical structure more similar to glutamate. In addition to binding to the GluR, the carbamate adducts might be capable of readily binding to Zn2+ or other metals. If BMAA is determined to bind Zn2+, this could provide insight on how BMAA is transported throughout the body and possibly expose another route for BMAA neurotoxicity and metal chelation. Further studies on the carbamates of BMAA should be completed to determine how BMAA is causing neurodegenerative diseases.
Acknowledgments
JG and VVK thank Dr. Paul Cox at the Institute of Ethnomedicine for organizing the 2016 BMAA symposium where part of the work was presented. PD acknowledges the support by the Bridges to Doctorate Program (R25 GM115293). The authors thank David Zimmerman for part of the work related to NMR studies of carbamate formation. The authors thank C. Cortney for critical reading of the manuscript.
References
- Arnold H, Pahls K, Potsch D. Reaktion von N-(Chloräthyl)-2-oxazolidon mit primären Aminen. Tetrahedron Lett. 1969;10(3):137–139. [Google Scholar]
- Atlante A, Calissano P, Bobba A, Giannattasio S, Marra E, Passarella S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001;497(1):1–5. doi: 10.1016/s0014-5793(01)02437-1. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006a;312(5778):1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006b;52(1):39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Brownson DM, Mabry TJ, Leslie SW. The cycad neurotoxic amino acid, beta-N-methylamino-L-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J Ethnopharmacol. 2002;82(2–3):159–167. doi: 10.1016/s0378-8741(02)00170-8. [DOI] [PubMed] [Google Scholar]
- Catarzi D, Colotta V, Varano F. Competitive Gly/NMDA receptor antagonists. Curr Top Med Chem. 2006;6(8):809–821. doi: 10.2174/156802606777057544. [DOI] [PubMed] [Google Scholar]
- Chiu AS, Gehringer MM, Welch JH, Neilan BA. Does alpha-amino-beta-methylaminopropionic acid (BMAA) play a role in neurodegeneration? Int J Environ Res Public Health. 2011;8(9):3728–3746. doi: 10.3390/ijerph8093728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AS, Gehringer MM, Braidy N, Guillemin GJ, Welch JH, Neilan BA. Excitotoxic potential of the cyanotoxin beta-methyl-amino-L-alanine (BMAA) in primary human neurons. Toxicon. 2012;60(6):1159–1165. doi: 10.1016/j.toxicon.2012.07.169. [DOI] [PubMed] [Google Scholar]
- Chiu AS, Gehringer MM, Braidy N, Guillemin GJ, Welch JH, Neilan BA. Gliotoxicity of the cyanotoxin, beta-methyl-amino-L-alanine (BMAA) Sci Rep. 2013;3:1482. doi: 10.1038/srep01482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1(8):623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Cleland WW, Andrews TJ, Gutteridge S, Hartman FC, Lorimer GH. Mechanism of rubisco: the carbamate as general base. Chem Rev. 1998;98(2):549–562. doi: 10.1021/cr970010r. [DOI] [PubMed] [Google Scholar]
- Davis AJ, Hawkes GE, Haycock PR, O’Brien P, Kidd BL, Mapp PI, Naughton D, Grootveld M. Generation of substance P carbamate in neutral aqueous solution. Relevance to inflammatory joint diseases. FEBS Lett. 1993a;329(3):249–252. doi: 10.1016/0014-5793(93)80231-i. [DOI] [PubMed] [Google Scholar]
- Davis AJ, O’Brien P, Nunn PB. Studies of the stability of some amino acid carbamates in neutral aqueous solution. Bioorg Chem. 1993b;21(3):309–318. [Google Scholar]
- Dean JA. Lange’s handbook of chemistry. New York: McGraw-Hill Inc., USA; 1992. [Google Scholar]
- Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81(3):163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Dunlop RA, Cox PA, Banack SA, Rodgers KJ. The non-protein amino acid BMAA is misincorporated into human proteins in place of L-serine causing protein misfolding and aggregation. PLoS One. 2013;8(9):e75376. doi: 10.1371/journal.pone.0075376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing SP, Lockshon D, Jencks WP. Mechanism of cleavage of carbamate anions. J Am Chem Soc. 1980;102(9):3072–3084. [Google Scholar]
- Faassen EJ, Beekman W, Lurling M. Evaluation of a commercial enzyme linked immunosorbent assay (ELISA) for the determination of the neurotoxin BMAA in surface waters. PLoS One. 2013;8(6):e65260. doi: 10.1371/journal.pone.0065260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M. Absorption and utilization of amino acids. CRC Press; 1989. [Google Scholar]
- Gahring LC, Rogers SW. Autoimmunity to glutamate receptors in the central nervous system. Crit Rev Immunol. 2002;22(4):295–316. [PubMed] [Google Scholar]
- Gibbons BH, Edsall JT. Rate of hydration of carbon dioxide and dehydration of carbonic acid at 25 degrees. J Biol Chem. 1963;238:3502–3507. [PubMed] [Google Scholar]
- Glover WB, Liberto CM, McNeil WS, Banack SA, Shipley PR, Murch SJ. Reactivity of beta-methylamino-L-alanine in complex sample matrixes complicating detection and quantification by mass spectrometry. Anal Chem. 2012;84(18):7946–7953. doi: 10.1021/ac301691r. [DOI] [PubMed] [Google Scholar]
- Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. J Chem Phys. 1979;71(11):4546–4553. [Google Scholar]
- Klebe G. Applying thermodynamic profiling in lead finding and optimization. Nat Rev Drug Discov. 2015;14(2):95–110. doi: 10.1038/nrd4486. [DOI] [PubMed] [Google Scholar]
- Lobner D, Piana PM, Salous AK, Peoples RW. Beta-N-methylamino-L-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol Dis. 2007;25(2):360–366. doi: 10.1016/j.nbd.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorimer GH. Carbon dioxide and carbamate formation: the makings of a biochemical control system. Trends Biochem Sci. 1983;8(2):65–68. [Google Scholar]
- Marchetti C. Interaction of metal ions with neurotransmitter receptors and potential role in neurodiseases. Biometals. 2014;27(6):1097–1113. doi: 10.1007/s10534-014-9791-y. [DOI] [PubMed] [Google Scholar]
- McConnell HM. Reaction rates by nuclear magnetic resonance. J Chem Phys. 1958;28:430–431. [Google Scholar]
- Meier BH, Ernst RR. Elucidation of chemical exchange networks by two-dimensional NMR spectroscopy: the heptamethylbenzenonium ion. J Am Chem Soc. 1979;101(21):6441–6442. [Google Scholar]
- Monaghan DT, Bridges RJ, Cotman CW. The excitatory amino acid receptors: their classes, pharmacology, and distinct properties in the function of the central nervous system. Annu Rev Pharmacol Toxicol. 1989;29:365–402. doi: 10.1146/annurev.pa.29.040189.002053. [DOI] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA, Steele JC, Sacks OW. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol Scand. 2004a;110(4):267–269. doi: 10.1111/j.1600-0404.2004.00320.x. [DOI] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc Natl Acad Sci U S A. 2004b;101(33):12228–12231. doi: 10.1073/pnas.0404926101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers TG, Nelson SD. Neuroactive carbamate adducts of beta-N-methylamino-L-alanine and ethylenediamine. Detection and quantitation under physiological conditions by 13C NMR. J Biol Chem. 1990;265(18):10193–10195. [PubMed] [Google Scholar]
- Nunn PB. Three phases of research on beta-N-methylamino-L-alanine (BMAA)—a neurotoxic amino acid. Amyotroph Lateral Scler. 2009;10(Suppl 2):26–33. doi: 10.3109/17482960903272975. [DOI] [PubMed] [Google Scholar]
- Nunn PB, O’Brien P. The interaction of beta-N-methylamino-L-alanine with bicarbonate: an 1H-NMR study. FEBS Lett. 1989;251(1–2):31–35. doi: 10.1016/0014-5793(89)81423-1. [DOI] [PubMed] [Google Scholar]
- Nunn PB, O’Brien P, Pettit LD, Pyburn SI. Complexes of zinc, copper, and nickel with the nonprotein amino acid L-alpha-amino-beta-methylaminopropionic acid: a naturally occurring neurotoxin. J Inorg Biochem. 1989;37(2):175–183. doi: 10.1016/0162-0134(89)80040-6. [DOI] [PubMed] [Google Scholar]
- Nunn PB, Davis AJ, O’Brien P. Carbamate formation and the neurotoxicity of L-alpha amino acids. Science. 1991;251(5001):1619–1620. doi: 10.1126/science.1859531. [DOI] [PubMed] [Google Scholar]
- Nunn PB, Bell EA, Watson AA, Nash RJ. Toxicity of non-protein amino acids to humans and domestic animals. Nat Prod Commun. 2010;5(3):485–504. [PubMed] [Google Scholar]
- Pablo J, Banack SA, Cox PA, Johnson TE, Papapetropoulos S, Bradley WG, Buck A, Mash DC. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol Scand. 2009;120(4):216–225. doi: 10.1111/j.1600-0404.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- Rao SD, Banack SA, Cox PA, Weiss JH. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp Neurol. 2006;201(1):244–252. doi: 10.1016/j.expneurol.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Richter KE, Mena EE. L-beta-methylaminoalanine inhibits [3H]glutamate binding in the presence of bicarbonate ions. Brain Res. 1989;492(1–2):385–388. doi: 10.1016/0006-8993(89)90925-6. [DOI] [PubMed] [Google Scholar]
- Rodgers KJ, Shiozawa N. Misincorporation of amino acid analogues into proteins by biosynthesis. Int J Biochem Cell Biol. 2008;40(8):1452–1466. doi: 10.1016/j.biocel.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Rossi-Bernardi L, Roughton FJ. The specific influence of carbon dioxide and carbamate compounds on the buffer power and Bohr effects in human haemoglobin solutions. J Physiol. 1967;189(1):1–29. doi: 10.1113/jphysiol.1967.sp008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiman M. A review of monoethanolamine chemistry. DTIC Document 1962 [Google Scholar]
- Sundh UM, Andersoon C, Rosén J, Fonnum F, Knudsen I, Sippola S. Analysis, occurrence, and toxicity of ß-methylaminoalanine (BMAA) Denmark: Nordic Council of Ministers; 2007. [Google Scholar]
- Szewczyk B. Zinc homeostasis and neurodegenerative disorders. Front Aging Neurosci. 2013;5(33):1–12. doi: 10.3389/fnagi.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A. Manganese action in brain function. Brain Res Brain Res Rev. 2003;41(1):79–87. doi: 10.1016/s0165-0173(02)00234-5. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62(3):405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega A. α-amino-β-methylaminopropionic acid, a new amino acid from seeds of Cycas circinalis. Phytochemistry. 1967;6(5):759–762. [Google Scholar]
- Vega A, Bell EA, Nunn PB. The preparation of l- and d-α-amino-β-methylaminopropionic acids and the identification of the compound isolated from Cycas circinalis as the l-isomer. Phytochemistry. 1968;7(10):1885–1887. [Google Scholar]
- Viso A, Fernandez de la Pradilla R, Garcia A, Flores A. Alpha, beta-diamino acids: biological significance and synthetic approaches. Chem Rev. 2005;105(8):3167–3196. doi: 10.1021/cr0406561. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Choi DW. Beta-N-methylamino-L-alanine neurotoxicity: requirement for bicarbonate as a cofactor. Science. 1988;241(4868):973–975. doi: 10.1126/science.3136549. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Christine CW, Choi DW. Bicarbonate dependence of glutamate receptor activation by beta-N-methylamino-L-alanine: channel recording and study with related compounds. Neuron. 1989;3(3):321–326. doi: 10.1016/0896-6273(89)90256-0. [DOI] [PubMed] [Google Scholar]
- Xie H, Wang P, He N, Yang X, Chen J. Toward rational design of amines for CO2 capture: substituent effect on kinetic process for the reaction of monoethanolamine with CO2. J Environ Sci (China) 2015;37:75–82. doi: 10.1016/j.jes.2015.03.033. [DOI] [PubMed] [Google Scholar]
- Xue H, Field CJ. New role of glutamate as an immunoregulator via glutamate receptors and transporters. Front Biosci (Schol Ed) 2011;3:1007–1020. doi: 10.2741/205. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Hasegawa J, Ito Y. Kinetic investigation on carbamate formation from the reaction of carbon dioxide with amino acids in homogeneous aqueous solution. J Phys Org Chem. 2012;25(3):239–247. [Google Scholar]
- Zimmerman D, Goto JJ, Krishnan VV. Equilibrium dynamics of beta-N-methylamino-L-alanine (BMAA) and its carbamate adducts at physiological conditions. PLoS One. 2016;11(8):e0160491. doi: 10.1371/journal.pone.0160491. [DOI] [PMC free article] [PubMed] [Google Scholar]