

Abstract
Cholangiopathies are a diverse group of progressive diseases whose primary cell targets are cholangiocytes. To identify shared pathogenesis and molecular connectivity among the three main human cholangiopathies (biliary atresia [BA], primary biliary cholangitis [PBC] and primary sclerosing cholangitis [PSC]), we built a comprehensive platform of published data on gene variants, gene expression and functional studies, and applied network-based analytics in search for shared molecular circuits. Mining the data platform with largest connected component and interactome analyses, we validated previously reported associations and identified essential- and hub-genes. In addition to disease-specific modules, we found a substantial overlap of disease neighborhoods, and uncovered a group of 34 core genes that are enriched for immune processes and abnormal intestine/hepatobiliary mouse phenotypes. Within this core, we identified a gene subcore containing STAT3, IL6, TNF and FOXP3 prominently placed in a regulatory connectome of genes related to cellular immunity and fibrosis. We also found substantial gene enrichment in the AGE-RAGE pathway, and showed that RAGE activation induced cholangiocyte proliferation.
Conclusion
Human cholangiopathies share pathways enriched by immunity genes and a molecular connectome that links different pathogenic features of BA, PBC and PSC.
Keywords: biliary atresia, primary biliary cholangitis, primary sclerosing cholangitis, pathogenesis, liver
Introduction
Although cholangiocytes are the primary target of injury in biliary syndromes, the location of injured cholangiocytes along the anatomical domains is one of the distinguishing features among the three primary cholangiopathies. The anatomical sites of injury obey a reproducible pattern focused predominantly either on intrahepatic bile ducts (as seen in primary biliary cholangitis [PBC]), extrahepatic ducts and intrahepatic bile ducts (as in primary sclerosing cholangitis [PSC]), or extrahepatic bile ducts with secondary involvement of intrahepatic cholangiocytes (as in biliary atresia [BA]). The anatomical continuity and functional integrity of intra- and extra-hepatic bile ducts are key to the homeostasis of bile production, modification and excretion (1, 2). If any of these functions are disrupted by genetic defects or injuries to cholangiocytes, bile flow is impaired and associated with disease phenotypes (3-5). Clinically, they manifest as diverse cholangiopathies that often progress to cirrhosis, and account for 16% of all liver transplants performed in the United States (6).
BA is a severe neonatal cholangiopathy characterized by a progressive fibro-inflammatory obstruction of extrahepatic bile ducts in children with estimated frequency of 1 in 15,000 live births in USA (7). In contrast, PBC has a higher frequency of nearly 4 per 10,000 people of which >90% are women, with a mean age of 55 years at diagnosis (6, 8). Different than the other two cholangiopathies, PSC affects children and adults with a frequency as high as 1.62 in 10,000 people, a male predominance, and a median age of 30 to 40 years at diagnosis (6, 8). Despite these age- and anatomy-related features and heterogeneity in clinical presentation, growing evidence points to a potential commonality in pathogenic mechanisms that involve the innate and adaptive immune systems, cholangiocyte damage and proliferation, accumulation of potentially toxic bile acids, and the recruitment of neighboring cells in the reparative response (3-5).
Recent advances from patient-based studies examining the genome and liver transcriptome identified several genes, gene groups, and pathways linked to the pathogenesis of individual cholangiopathies (9-11). For example, genome-wide association studies (GWAS) reported genetic associations of ADD3 with BA, the IL-12/STAT4 pathway with PBC, and the CD28/IL-2 pathway with PSC (11-13). Based on the common cellular target and the continuity of the biliary epithelium within and outside the liver, we hypothesized that these cholangiopathies have common genes and gene groups that relate, at least in part, to shared pathogenic mechanisms of disease. To test this hypothesis, we applied comprehensive bioinformatics algorithms to published databases on gene sequence variants and tissue expression on BA, PBC, and PSC. We found that despite the signatures in each disease, they share gene groups and pathways highly enriched by immune response processes assembled into a biliary connectome containing a high degree of interaction networks. Within the networks, STAT3, IL6, TNF, and FOXP3 have greater connectivity and, when combined with a high enrichment in the AGE-RAGE pathway, they emerge as shared regulators of BA, PBC and PSC.
Methods
Database for cholangiopathy-associated genes
To collect information on genes associated with BA, PBC, and PSC, we curated genes from PubMed and DisGeNET (v4.0) databases (14) using the keywords “biliary atresia,” “primary biliary cholangitis,” or “primary biliary cirrhosis” (for PBC), and “primary sclerosing cholangitis” (for PSC). A gene was considered as associated with a cholangiopathy if it met at least one of the following criteria: 1) genetic association or mutation in one of the diseases; 2) functional association obtained from studies using experimental models; 3) differential expression in patients compared to controls; and 4) function as auto-antigen by the encoded protein. These criteria identified disease-causing genes and genetic association with cholangiopathies, or captured genes that are involved in late-stage disease. We mapped the genes to human Entrez gene symbols and identification numbers. For GWAS studies, we set threshold of P value <5×10-8 (standard threshold) for PBC and PSC, and P value <1×10-5 (suggestive threshold) for BA because of relatively small sample size of BA cohort in published studies. Genes associated with IHC (intrahepatic cholestasis) and PLD (polycystic liver disease) were downloaded from DisGeNET (v4.0) databases. Detailed lists of disease-associated genes and overlapping genes are summarized in Supplemental Tables 1 and 2.
Mining tools and databases
We compiled multiple types of data including protein interactions, essential genes, and binding information for transcription factors and miRNAs from literature and databases. Details for data mining methods and analytical approaches are provided in the Supporting Methods.
Dysregulated miRNAs in cholangiopathies
To collect a set of common dysregulated miRNAs in cholangiopathies, we conducted literature search for studies that assessed miRNAs dysregulation in cholangiopathies. Dysregulated miRNAs are defined as those that show differential expression between patients and healthy controls in published studies. A list of dysregulated miRNAs is summarized in Supplemental Table 7.
Biological processes, mouse phenotype enrichment and KEGG pathway analyses
Biological processes, mouse phenotype enrichment and KEGG pathway analyses were performed using ToppFun tool in ToppGene Suite (https://toppgene.cchmc.org/) with core genes as input (15). We used terms with Bonferroni corrected P value <0.05 for 2-way CIMminer clustering with defaults settings.
Statistical analyses
The statistical significance of overlap between two gene lists in comparison with a genomic background was tested by Fisher's exact test using GeneOverlap (version 1.10.0) R package. We performed 1,000 random permutations and calculated z-scores to test the significance of disease-associated gene localization and network-based disease separation S using methods/Python scripts from Menche et al (7) and TF-miRNA feed forward loops using in-house Python scripts (https://github.com/Zhenhualuo/TFs-miRNAs-Feed-Forward-Loop). Details for the calculation are provided in the Supporting Methods.
Data visualization
Venn diagrams were generated by Venny 2.1.0 (http://bioinfogp.cnb.csic.es/tools/venny/). Networks were visualized using Cytoscape (version 3.4.0) and bar graphs were created using GraphPad Prism 6.
Scoring and ranking of the gene subcore
To provide a hierarchical ranking among the subcore genes, we applied a binary functional system that scores gene associations based on biological processes, involvement in abnormal liver and biliary function, report in studies of gene expression or sequence variants by GWAS, evaluation in experimental models, and relationship to clinical parameters. We defined “1” for association and “0” for no association. We summed all scores to generate a functional score for each gene. Then, connectivity scores were calculated by averaging the number of connections for each subcore genes from two connectomes. The overall scores were the sum of functional and connectivity scores.
Cell proliferation assay and real-time PCR
Human immortalized nonmalignant cholangiocyte H69 cells were cultured in 96-well plates (5,000 cells/well) (16). After 24 hours, cells were cultured with DMEM/F-12 medium/0.5% serum ± pre-treatment with 5 ug/mL recombinant human RAGE-Fc chimera (R&D, Minneapolis, MN) for 1 hour ± 100 ng/mL recombinant human HMGB1 (R&D) for 48 hours. Cellular proliferation was quantified with a colorimetric assay (CellTiter 96Aqueous; Promega). RNA was isolated from H69 cells in different wells using RNeasy Mini Kit (Qiagen, Valencia, CA, USA) and subjected to real-time PCR with the Brilliant III SYBR Green QPCR Master Mix Gene Expression Assay Kit and the Mx3005p system (Stratagene), normalized with GAPDH. Primer sequences are listed in Supplemental Table 8.
Results
Building of a database of genes associated with human cholangiopathies
To explore the existence of molecular pathways that are shared among the human cholangiopathies, we developed a systems biology analytic pipeline that started with manually curated lists of genes associated with BA, PBC, and PSC from PubMed and DisGeNET databases (Figure 1). DisGeNET is a comprehensive platform integrating information on human disease-associated genes and gene variants using the selection criteria described above (14). After removal of duplication from different sources, we identified 276, 387 and 166 human genes associated with BA, PBC and PSC, respectively (Supplemental Table 1).
Figure 1. An outline of the systems biology analytical pipeline used in this study.

Cholangiopathy-associated genes form disease modules and encode hub proteins
Previous studies of the human interactome suggest that disease-associated proteins interact with each other and tend to cluster in the same neighborhood, forming a connected subgraph often referred to as a “disease module” (17); the disruption of a disease module results in a particular disease phenotype. To investigate whether the proteins encoded by the genes reported in studies of BA, PBC and PSC form disease modules, we measured the size of the Largest Connected Component (LCC) to quantify the degree to which the proteins tend to agglomerate (17). Examining the significance of protein agglomeration, we also compared the observed LCC size with randomly permutated LCC sizes by computing z-scores, in which a threshold z-score ≥1.65 is larger than expected by chance at a P value <0.05. We found that the gene lists for each of the three diseases formed significantly connected disease modules, with observed LCC size z-scores of 10.6 for BA, 8.5 for PBC, and 19.1 for PSC (Figure 2A-C).
Figure 2. Topological characteristics of cholangiopathy-associated genes.
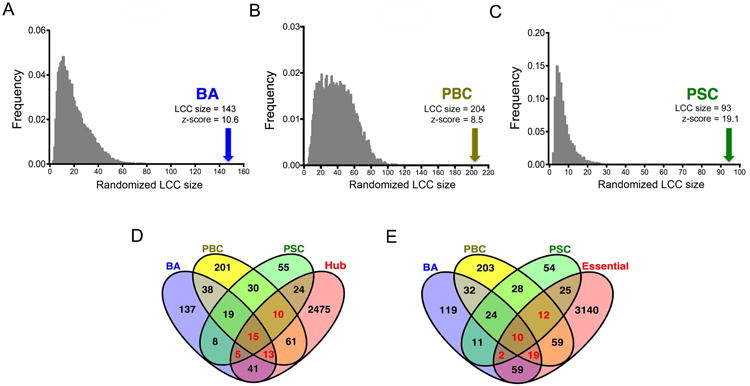
(A-C) Distribution of the size of largest connected component (LCC) obtained from 1,000 random permutations. Arrow indicates the observed LCC size and the corresponding z-score. (D) Venn diagram showing overlapping of disease genes and hub genes in the human interactome. (E) Venn diagram showing overlapping of disease genes and essential genes.
To further analyze the functional relevance of genes in each disease, we applied predictive algorithms to determine whether they encode essential proteins and form hubs, as done previously in human interactome studies (18, 19). Defining hubs by genes placed at the top 20% of nodes ranked by degrees in the human interactome and using an updated list of 3,326 essential genes (19, 20), we found that each disease associated gene list significantly overlaps with hub genes or essential genes (all P value < 0.05, Supplemental Table 9). Importantly, we found 43 genes (highlighted in red in Figure 2D) that overlap or are shared by at least 2 diseases that were also classified as hub genes (Figure 2D), and 43 genes (highlighted in red in Figure 2E) classified as essential genes. These data suggested that the changes in sequence or expression of the selected genes are predicted to disrupt important biological processes and may relate to disease phenotypes.
Cholangiopathies have overlapping disease neighborhoods
Another method to evaluate clinical and pathobiological similarities between two diseases is the network-based distance that is shared by the diseases, which is typically quantified as network-based Separation (or “S” = the mean shortest distances dAA and dBB within the respective cholangiopathies with the mean shortest distance dAB between their proteins) (17). In this approach, S>0 indicates that the two disease modules are topologically separated and are pathobiologically distinct, with S<0 indicating similarities (17). Using this approach, we found that each cholangiopathy pair has a negative S and z-score < -1.65, indicating that they significantly overlap disease neighborhoods (Figure 3A-C), thus supporting the premise that cholangiopathies share pathobiological and clinical similarities at the network level.
Figure 3. Gene-sharing and molecular overlap among BA, PBC and PSC.
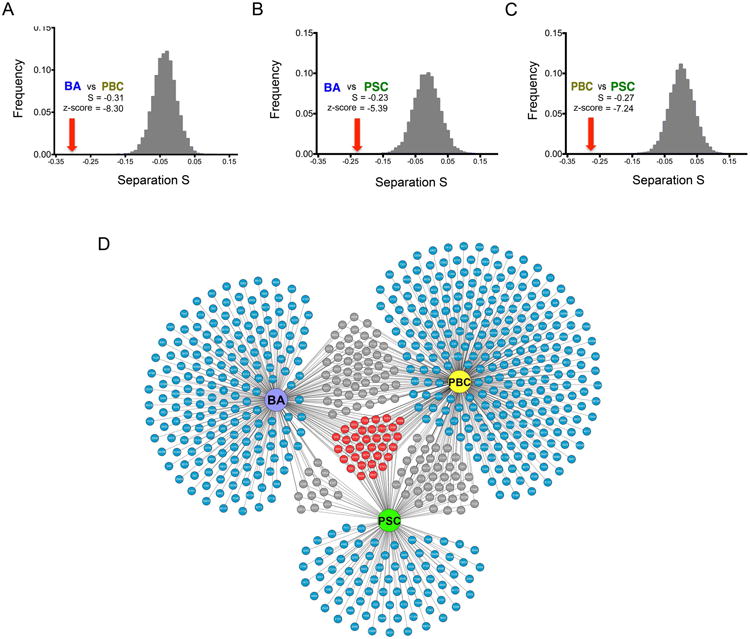
(A-C) Distribution of the network-based separation S values obtained from 1,000 random permutations for the cholangiopathies pairs. Red arrow indicates the observed separation S value and the corresponding z-score. (D) Network of cholangiopathy-associated genes, where blue depict genes specific to individual diseases, grey for genes shared by two diseases, and red containing genes shared by the three diseases.
Identification of core genes that are shared by BA, PBC, and PSC
In order to identify genes that are shared among the primary cholangiopathies, we subjected the disease-associated genes listed in Supplemental Table 1 (above) to gene overlapping analysis using Fisher's exact test (Figure 3D and Supplemental Table 9). We found that several genes segregate with individual diseases or are shared by pairs of diseases, with a distinct group of 34 genes that are shared by all three cholangiopathies (Figure 3D and Supplemental Table 2), which we refer to as “core genes” (Red colors in Figure 3D). These findings suggested that the three cholangiopathies share functionally related genes that may be linked to pathogenic mechanisms of disease.
Cholangiopathy core genes are predominantly related to immunity
To explore whether the core genes may identify molecular pathways involved in pathogenic mechanisms of diseases, we studied biological and phenotypic properties of all 34 core genes. Enrichment analyses using a right-tailed Fisher's exact test with Bonferroni correction of P<0.05 revealed that the core genes were linked to 576 biological processes and 298 mouse phenotypes. Among these, responses to biotic stimuli, immune-related processes and abnormal hepatobiliary/intestinal phenotypes comprised the top 10 highest levels of significance (Figure 4A-B), with additional enrichments for inflammation/immunity, and metabolism. Applying 2-way CIMminer clustering analyses to investigate the functional relatedness of the core genes, we next analyzed the genes based on “biological processes” and uncovered a cluster of genes encoding the cytokines IL10, TLR4, TNFα, IL6, TGFB1, and IL2 and the transcription factor FOXP3 (Figure 4C). Analyzing the same group of core genes according to their relationships to “mouse phenotypes,” we found a second cluster of genes encoding the same cytokines (IL10, TLR4, TNFα, IL6, TGFB1, and IL2) in addition to IL17A and PTGS2, and a new group with the transcription factors NFKB1, STAT3, TP53, SMAD3, and HIF1A (Figure 4D). These analyses pointed to a predominance of inflammation/immunity genes in the main core genes assigned to the cholangiopathies, and identified a highly connected immunity gene subcore containing 14 genes (TNF, IL10, FOXP3, TGFB1, IL2, IL17A, NFKB1, STAT3, TP53, SMAD3, HIF1A, PTGS2, IL6 and TLR4).
Figure 4. Cluster analysis of core genes based on biological processes and experimental phenotypes.
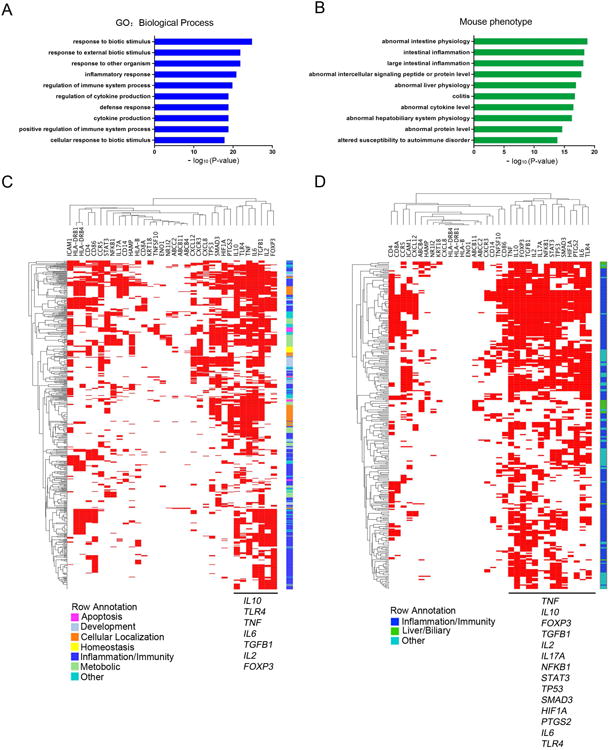
(A) Top 10 enriched biological processes for core genes for BA, PBC, and PSC. Enrichment scores are represented as – log10(P value), with a threshold of 1.3 as the cut-off for significance (P < 0.05). (B) Top 10 enriched mouse phenotypes for core genes are shown. Enrichment scores are represents as – log10 (P value), with a threshold of 1.3 as the cut-off for significance (P < 0.05). (C) Functional enrichment analysis by CIMminer classifies 34 genes into 7 functional categories (shown in colors). (D) A mouse phenotype enrichment analysis by CIMminer classifies 34 genes into 3 main phenotype categories. The red areas in the heatmap indicate closely related biological processes/mouse phenotypes that are shared by the subgroup of genes shown on the horizontal axis.
Co-regulatory network for TF-miRNA subcore genes
As an initial strategy to investigate how the immunity gene subcore may relate to shared pathogenic mechanisms of the three diseases, we searched for evidence of higher orders of regulation by miRNAs and transcription factors (TF). There is increasing evidence that changes in the expression of TFs and miRNAs may lead to dysregulation of genes and encoded proteins involved in the pathogenesis of cholangiopathies (21-25). In a first approach, we examined Feed Forward Loop (FFL), an important element in regulatory networks (26) that can be classified into three types according to the master regulators: transcription factors FFL (TF-FFL), miRNAs FFL and transcription factors-miRNAs composite FFL (TF-miRNA composite FFL) (27). In the co-regulatory network, there were 5,575 TF-FFLs, 1,385 miRNA-FFLs and 332 TF-miRNA composite FFLs (Supplemental Table 6). We then tested the significance of FFLs by comparing the observed number of FFLs with random permutated number of FFLs. Notably, all z-scores were larger than 1.65 (Figure 5A-C), suggesting that the observed FFLs in the set of transcription factors, miRNAs and the subcore genes were significantly enriched. These data support a strong relatedness among the components of the immune subcore genes.
Figure 5. Co-regulatory network of transcription factor-miRNA subcore genes.
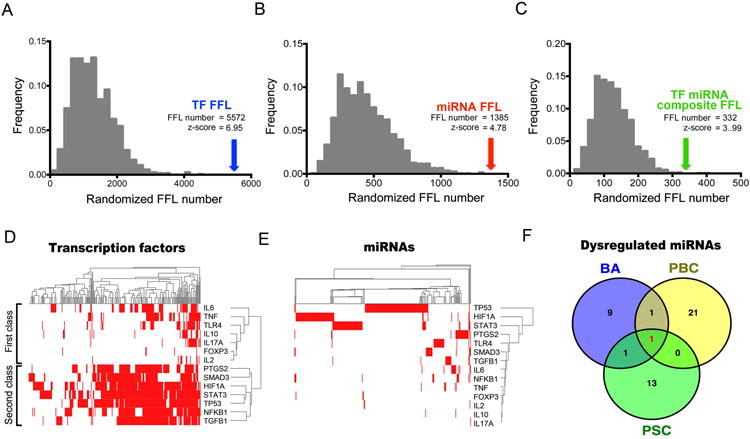
(A-C) Distribution of the number of feed forward loops (FFLs) obtained from 1,000 random permutations. Arrow indicates the observed FFL number and the corresponding z-score. (D-E) Clustering of transcription factor-miRNA (horizontal axis) and subcore genes (vertical axis) based on regulation events (red). (F) Venn diagram showing overlapping of BA, PBC and PSC associated dysregulated miRNAs.
In the second approach, we used 2-way CIMminer cluster analysis and found that the 14 genes can be grouped into two major gene classes based on transcription factor regulation (Figure 5D). The first class comprised all cytokines including IL6, TNF, IL10, IL17A and IL2, while the second gene class contains mostly transcription factors such as NFKB1 and STAT3 (Figure 5D), indicating these genes can function as master regulators by integrating different signals coming from diverse pathways. However, we did not observe a similar regulatory pattern in the analysis of miRNAs (Figure 5E).
To further explore the potential regulation of miRNA-subcore genes in cholangiopathies, we curated a list of dysregulated miRNAs shared by the 3 diseases (Figure 5F and Supplemental Table 7). We found that only the miRNA hsa-miR-122-5p is shared by BA, PBC and PSC (Figure 5F, highlighted in red). All these data suggest that transcription factors are the master regulators in shared mechanisms of cholangiopathies, with a less prominent presence of distinct miRNA profiles.
Building a connectome for the immune subcore genes
As a strategy to investigate how the immune subcore genes relate to each other, we next searched for an integrated model for the genes taking into account their biological roles (transcription factors or cytokines/chemokines) and their functional relatedness. Using transcription factor-subcore regulatory information, we generated a 3-disease immune subcore connectome that showed FOXP3, SMAD3, and NFKB1 in the periphery, with minimal relationship with the cytokines (Figure 6A). Most notably, STAT3, TP53 and HIF1A were centrally linked to all genes, among which STAT3 formed a node with the highest connectivity with a first level link to all genes except for TGFB1. Using the STRING database-v10 to generate a second immune subcore connectome, we found a higher frequency of level-1 connectivity for STAT3, FOXP3, TP53 and IL6 (Supplemental Figure 1).
Figure 6. Connectome for the cholangiopathy subcore genes and STAT3, IL6, TNF and FOXP3 centered network.
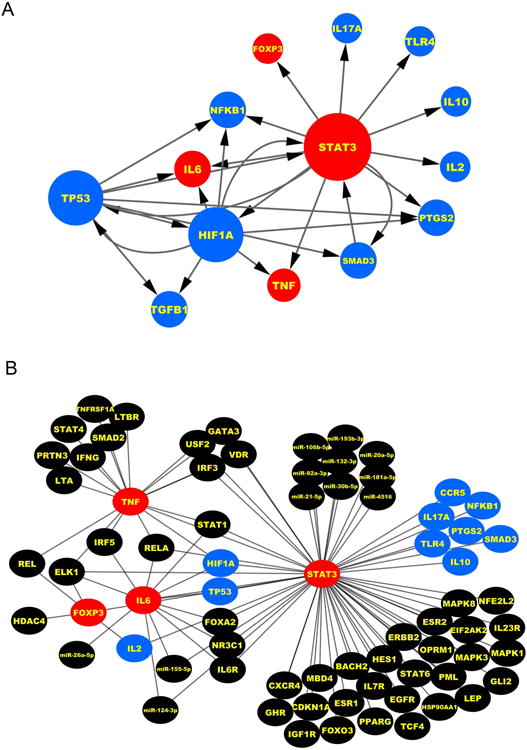
(A) Regulatory network of subcore genes and grey edges indicating regulatory interaction obtain from transcription factor-miRNA co-regulatory network. The size of the nodes is proportional to the number of edges connected to the node. Red represents the key genes; Blue represents the genes most shared among the cholangiopathies. (B) Network linking STAT3, IL6, TNF and FOXP3 with cholangiopathy-associated genes and miRNAs. Red nodes: key genes; blue nodes: genes shared by three diseases; black nodes: genes/miRNAs for individual disease or shared by two diseases.
Next, to identify which genes of the immune subcore have the highest level of shared association among the BA, PBC and PSC, we performed an initial ranking of genes based on their combined involvement in several parameters, including biological and disease processes, involvement in abnormal liver and biliary processes, studies of gene expression or GWAS, evaluation in experimental models, and link to clinical parameters (Supplemental Table 10). The genes with the highest level of shared association were TNF, IL2, IL6, TGFB1, FOXP3 and IL10 (functional scores, from Table 1). Then, to develop a final rank of shared genes, we developed an overall score based on first-level connectivity (connectivity score) (from Figures 6A and Supplemental Figure 1) and functional studies (functional score, from Supplemental Table 10). As also shown in Supplemental Table 10 (overall scores), STAT3, IL6, TNF and FOXP3 are among the top 6 genes encoding proteins related to inflammation/immunity. Analyzing these genes against all cholangiopathy-associated genes (from Supplemental Table 1) and miRNAs (from Supplemental Table 6), we found their relationships with genes that are shared by BA, PBC, and PSC, as well as with each disease separately, and a highest connectivity for STAT3 (Figure 6B).
Table 1.
Summary of clinical features, primary or secondary anatomical sites of biliary injury and pathogenic mechanisms of tissue injury proposed for biliary atresia (BA), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC).
| Age | 1° site | 2° site | IL6-STAT3 | TNF | FOXP3 | |
|---|---|---|---|---|---|---|
| BA | Children | EHBD | IHBD | Epithelial repair: Cholangiocyte proliferation | Epithelial injury: TNFa-TNFR2 | Suppression of Treg cells, Epithelial injury |
| PBC | Adults | IHBD | None | Injury:exacerbates cholangitis via TGFβ signaling | Increased expression in affected livers | Increase in Treg cells, Epithelial injury |
| PSC | Children & adults | EHBD | IHBD | Injury and fibrosis in Mdr2-/- mice | TNFA polymorphisms and dysregulation of T and NK lymphocytes | Suppression of Treg cells, Epithelial injury |
Last, to test the specificity and of the connectome to the three cholangiopathies, we examined whether the 34 core genes overlapped with the genes for the two disease controls IHC and PLD. Only ABCB11, ABCB4, HLA-DRB1, ABCC2, and NR1I2 genes were shared with IHC, and none with PLD (Supplemental Figure 2A); these 5 genes are involved in bile acid metabolism and transport systems (Supplemental Figure 2B-C). Notably, the genes STAT3, IL6, TNF and FOXP3 were not shared by IHC or PLD. These findings support the involvement of metabolism and transport systems as a shared process among all 5 cholestatic and biliary diseases analyzed. They also support the specificity of the connectome to the key biological processes of inflammation and fibrosis that are shared by BA, PBC and PSC.
Identification of RAGE in human cholangiopathies
To generate insight into potential molecular mechanisms not previously linked to BA, PBC or PSC, we subjected the connectome genes to KEGG pathway analysis. Six of 14 subcore genes (TGFB1, SMAD3, NFKB1, IL6, TNF, and STAT3) resided in the AGE-RAGE pathway, a less-characterized circuit that activates inflammatory and pro-fibrogenic signaling (Figure 7A). The AGE-RAGE pathway was also enriched in genes signatures for each disease individually, to any combination of two of these diseases, and to TFs that regulate the 14 subcore genes, but not in genes associated with IHC and PLD (Figure 7B).
Figure 7. Activation of the AGE-RAGE signaling enhances cholangiocyte proliferation and expression of IL6 and TGFB1.
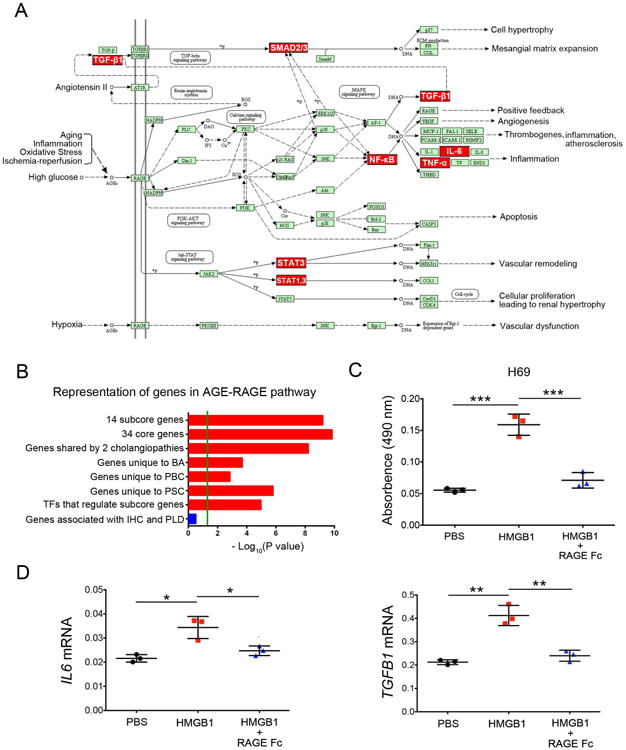
(A) Representation of subcore genes (red) in the AGE-RAGE pathway (downloaded from KEGG pathway database). (B) Graph showing that genes in the AGE-RAGE pathway are enrichment in individual diseases, but not in intrahepatic cholestasis (IHC) or polycystic liver disease (PLD). Enrichment scores are represented as – log10, with a threshold of 1.3 as the cut-off for significance (P < 0.05; green line). (C) H69 cholangiocyte proliferation by the MTS assay after 48 hours of culture with PBS control, HMGB1 or HMGB1 plus RAGE Fc protein. (D) IL6 and TGFB1 mRNA expression in H69 cells. Expression was normalized against GAPDH. Values as mean ± SD, N=3; *P < 0.05, **P < 0.01, ***P < 0.001
Exploring the potential biological relevance for the pathway, we tested whether the activation of AGE-RACE influences cholangiocyte proliferation, a hallmark of cholangiopathies (5). Culture of human cholangiocyte H69 cells with HMGB1 (a ligand for RAGE) for 48 hours increased cell proliferation by 3 fold, an effect that was abolished by the addition of recombinant human RAGE-Fc chimera protein to block the receptor (Figure 7C). Interestingly, the activation of this pathway also upregulated the mRNA expression for the biliary mitogen IL6 and pro-fibrogenic factor TGFB1, but not other genes (Figure 7D and Supplemental Figure 3).
Discussion
Applying systems biology algorithms to a comprehensive data platform on gene variant and expression from patients with BA, PBC and PSC, we found gene groups that form modules for each disease independently and that encode hub proteins. Among the genes, 34 formed a core shared by the three diseases and related to immune-regulated processes, abnormal hepatobiliary and intestinal function, and abnormal mouse phenotypes. Within this core, 14 genes were functionally connected and formed a subcore encoding proteins regulating immune processes and fibrosis, with two major functional classes of cytokines and transcription factors, and high level of connectivity based on relationships to transcription factors. Ranking the genes in this subcore according to composite scores for functional relatedness and links to the disease phenotypes, we generated a connectome shared by BA, PBC, and PSC, which contained STAT3, IL6, FOXP3 and TNF as key genes. Further, pathway analyses identified the AGE-RAGE pathway in the cholangiopathies, with experimental evidence that activation of RAGE induces cholangiocyte proliferation and the expression of IL6 and TGFB1.These data demonstrate that BA, PBC and PSC share common molecular pathways predominantly linked to immunity and are linked to the AGE-RAGE pathway and other genes related to fibrosis.
The prominence of STAT3 and secondarily of IL6, FOXP3 and TNF in every analytical strategy applied to the data platform suggests essential roles of pro- and anti-inflammatory circuits in biliary damage and repair. Both IL-6 and TNFα are expressed by cholangiocytes in response to an insult or injury (28), and may function in an autocrine and paracrine fashion to amplify the damage and or trigger repair signals (28). IL-6 binding to its receptor on cholangiocyte activates STAT3 and MAPK signaling to promote cholangiocyte proliferation (29). In parallel or independently, TNFα increases the expression of many pro-inflammatory cytokines including IL-6 and MHC epitopes (29, 30). If expressed concomitantly with IFNγ, TNFα induces cholangiocyte apoptosis (31). Moreover, TNFα and IL-6 regulate the immune response by modulating FOXP3 expression levels in T cells (32-36). Combined, these studies support an inter-relatedness between STAT3, IL6, FOXP3 and TNF in cholangiocyte biology and pathogenesis of biliary injury in BA, PBC, and PSC.
Evidence from human data and mouse models of cholangiopathies suggests these four genes play a role in mechanisms of tissue injury and clinical phenotype. For example, the activation of the IL6-STAT3 pathway has been demonstrated in patients with BA, PBC and PSC (10, 37, 38). This pathway has been studied in experimental model of cholangiopathies. In BA, increased IL-6 levels are associated with cholangiocyte proliferation (38). In PBC, the targeted disruption of the IL6 gene exacerbates cholangitis in transforming growth factor beta (TGFβ) receptor II dominant-negative (dnTGFbetaRII) mouse model (39). In PSC, the loss of functional Stat3 protects the liver from injury and fibrosis in the Mdr2 knockout mouse model (40). In contrast to IL6-STAT3 pathway, the role of TNFα in cholangiopathies is less explored and understood. TNFα is upregulated in liver of experimental mouse model of BA (41), but an earlier study showed that the constitutive inactivation of the TNFR1 gene, one of TNFα receptors, failed to decrease the severity of experimental BA (41). More recently, we reported that a preferential use of the TNFR2 by TNFα may be an important circuit that is more relevant to pathogenesis of BA (42). In PBC, increased TNFα levels were found in liver samples of patients (37), while TNFA polymorphisms were associated with the risk of PSC (43). TNFα has been shown to impair the function of liver derived T cells and NK cells in PSC patients (44).
Regulatory T cells and their marker FOXP3 have emerged as key regulatory components in BA, PBC and PSC. FOXP3 expression levels were higher in livers of BA and PBC patients compared to normal controls, while decreased levels were observed in peripheral blood mononuclear cells from PBC patients (34-36). In experimental BA, Foxp3+ regulatory T cells inhibit Th1 cell-mediated injury and control DC dependent NK cell activation and CD8 T cell response (34, 45, 46). And in PSC, CD25highFOXP3+CD127low Treg cells were reduced in peripheral blood and livers of PSC patients and associated with IL2RA polymorphisms (47). These data suggest that the dysregulation of the four key genes and their related cell types play important roles in the immune mechanisms of cholangiopathies. Interestingly, the similar behavior of FOXP3 and Treg cells in patients and experimental models of BA and PSC suggests a closer link of pathogenic mechanisms between these two diseases (Table 1).
The expression of RAGE was has been reported as higher in cholangiocytes when compared to other cell types in cholestatic livers (48) and in proliferating bile ducts after bile duct ligation (49), where the administration of anti-RAGE antibodies attenuated bile duct proliferation and liver fibrosis (50). This is particularly relevant to our findings of a connectome gene enrichment in the AGE-RAGE pathway. Directly testing this pathway in cholangiocytes, we showed that HMGB1, a damage-associated molecular pattern released from injured cells (5), induces cholangiocyte proliferation and the expression of IL6 and TGFB1 mRNAs through RAGE. These data raise several experimental opportunities to modulate the pathway in animal models of BA, PBC and PSC in future pre-clinical studies to explore its potential as a therapeutic target for the cholangiopathies.
Among the limitations of our study, we recognize that there is an intrinsic bias towards well-studied genes that have been well annotated and extensively investigated, and that components of the gene signatures for individual diseases or the connectome may overlap with other unrelated diseases or be part of a stereotypical tissue (or cellular) response to an injury. In addition, the measures of significance vary among the studies, either focusing on differences in the frequency of gene variants or in the levels of expression in individual cholangiopathies, with limited information on how studies controlled for racial and sex differences. Last, the co-regulation of the core immune genes by transcription factors and miRNAs are cell type- and content-dependent. Experimental validation is needed to determine their relevance to cholangiopathies. These limitations notwithstanding, our report of a novel connectome that generates a molecular link among the three human cholangiopathies of BA, PBC and PSC underscores the existence of important roles and molecular crosstalks involving multiple immune signaling modules in pathogenesis of the diseases. The connectome and the network of transcription factors-miRNAs-core immune genes can serve as a valuable tool for hypothesis generation, and enhance our understanding of regulatory mechanism of cholangiopathies.
Supplementary Material
Acknowledgments
We thank Dr. Pranavkumar Shivakumar and members of the Bezerra laboratory for insightful discussions and suggestions for this study.
Financial Support: Supported by the NIH grants DK-64008 and DK-83781 to JAB and by the Gene Analysis Cores of the Digestive Health Center (DK-78392). The studies used the infrastructure for discoveries and innovations that catalyzes patient-based research in the Center for Autoimmune Liver Disease of Cincinnati Children's Hospital Medical Center. The work received generous support from the Junior Co-Operative Society of Cincinnati Children's Hospital Medical Center.
List of Abbreviations
- BA
biliary atresia
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- GWAS
genome-wide association studies
- FFL
feed forward loop
- LCC
largest connected component
- TF
transcription factor
- miRNA
microRNA
- AGE-RAGE
advanced glycation endproduct - receptor for advanced glycation endproducts
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- IHC
intrahepatic cholestasis
- PLD
polycystic liver disease
Footnotes
Author names in bold designate shared co-first authorship.
Contributor Information
Zhenhua Luo, Email: zhenhua.luo@cchmc.org.
Anil G Jegga, Email: anil.jegga@cchmc.org.
Jorge A Bezerra, Email: jorge.bezerra@cchmc.org.
References
- 1.Tietz PS, Larusso NF. Cholangiocyte biology. Curr Opin Gastroenterol. 2006;22:279–287. doi: 10.1097/01.mog.0000218965.78558.bc. [DOI] [PubMed] [Google Scholar]
- 2.Marzioni M, Glaser SS, Francis H, Phinizy JL, LeSage G, Alpini G. Functional heterogeneity of cholangiocytes. Semin Liver Dis. 2002;22:227–240. doi: 10.1055/s-2002-34501. [DOI] [PubMed] [Google Scholar]
- 3.Syal G, Fausther M, Dranoff JA. Advances in cholangiocyte immunobiology. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1077–1086. doi: 10.1152/ajpgi.00227.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol. 2015;62:934–945. doi: 10.1016/j.jhep.2014.11.027. [DOI] [PubMed] [Google Scholar]
- 5.O'Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58:575–582. doi: 10.1016/j.jhep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lazaridis KN, LaRusso NF. The Cholangiopathies. Mayo Clin Proc. 2015;90:791–800. doi: 10.1016/j.mayocp.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol. 2015;12:342–352. doi: 10.1038/nrgastro.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56:1181–1188. doi: 10.1016/j.jhep.2011.10.025. [DOI] [PubMed] [Google Scholar]
- 9.Bezerra JA, Tiao G, Ryckman FC, Alonso M, Sabla GE, Shneider B, Sokol RJ, et al. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653–1659. doi: 10.1016/S0140-6736(02)11603-5. [DOI] [PubMed] [Google Scholar]
- 10.Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW. Identification of novel molecules and pathogenic pathways in primary biliary cirrhosis: cDNA array analysis of intrahepatic differential gene expression. Gut. 2001;49:565–576. doi: 10.1136/gut.49.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webb GJ, Hirschfield GM. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J Autoimmun. 2016;66:25–39. doi: 10.1016/j.jaut.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Tsai EA, Grochowski CM, Loomes KM, Bessho K, Hakonarson H, Bezerra JA, Russo PA, et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum Genet. 2014;133:235–243. doi: 10.1007/s00439-013-1368-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, Andreassen OA, et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet. 2013;45:670–675. doi: 10.1038/ng.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinero J, Queralt-Rosinach N, Bravo A, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, et al. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. doi: 10.1093/nar/gkp427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grubman SA, Perrone RD, Lee DW, Murray SL, Rogers LC, Wolkoff LI, Mulberg AE, et al. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol. 1994;266:G1060–1070. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 17.Menche J, Sharma A, Kitsak M, Ghiassian SD, Vidal M, Loscalzo J, Barabasi AL. Disease networks Uncovering disease-disease relationships through the incomplete interactome. Science. 2015;347:1257601. doi: 10.1126/science.1257601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang M, Zhu C, Jacomy A, Lu LJ, Jegga AG. The orphan disease networks. Am J Hum Genet. 2011;88:755–766. doi: 10.1016/j.ajhg.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dickinson ME, Flenniken AM, Ji X, Teboul L, Wong MD, White JK, Meehan TF, et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fida S, Myers MA, Whittingham S, Rowley MJ, Ozaki S, Mackay IR. Autoantibodies to the transcriptional factor SOX13 in primary biliary cirrhosis compared with other diseases. J Autoimmun. 2002;19:251–257. doi: 10.1006/jaut.2002.0622. [DOI] [PubMed] [Google Scholar]
- 22.Ellinghaus D, Folseraas T, Holm K, Ellinghaus E, Melum E, Balschun T, Laerdahl JK, et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology. 2013;58:1074–1083. doi: 10.1002/hep.25977. [DOI] [PubMed] [Google Scholar]
- 23.Pisarello MJ, Loarca L, Ivanics T, Morton L, LaRusso N. MicroRNAs in the Cholangiopathies: Pathogenesis, Diagnosis, and Treatment. J Clin Med. 2015;4:1688–1712. doi: 10.3390/jcm4091688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munoz-Garrido P, Garcia-Fernandez de Barrena M, Hijona E, Carracedo M, Marin JJ, Bujanda L, Banales JM. MicroRNAs in biliary diseases. World J Gastroenterol. 2012;18:6189–6196. doi: 10.3748/wjg.v18.i43.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bessho K, Shanmukhappa K, Sheridan R, Shivakumar P, Mourya R, Walters S, Kaimal V, et al. Integrative genomics identifies candidate microRNAs for pathogenesis of experimental biliary atresia. BMC Syst Biol. 2013;7:104. doi: 10.1186/1752-0509-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 27.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yasoshima M, Kono N, Sugawara H, Katayanagi K, Harada K, Nakanuma Y. Increased expression of interleukin-6 and tumor necrosis factor-alpha in pathologic biliary epithelial cells: in situ and culture study. Lab Invest. 1998;78:89–100. [PubMed] [Google Scholar]
- 29.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–1043. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]
- 30.Ayres RC, Neuberger JM, Shaw J, Joplin R, Adams DH. Intercellular adhesion molecule-1 and MHC antigens on human intrahepatic bile duct cells: effect of pro-inflammatory cytokines. Gut. 1993;34:1245–1249. doi: 10.1136/gut.34.9.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erickson N, Mohanty SK, Shivakumar P, Sabla G, Chakraborty R, Bezerra JA. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology. 2008;47:1567–1577. doi: 10.1002/hep.22229. [DOI] [PubMed] [Google Scholar]
- 32.Gao Y, Tang J, Chen W, Li Q, Nie J, Lin F, Wu Q, et al. Inflammation negatively regulates FOXP3 and regulatory T-cell function via DBC1. Proc Natl Acad Sci U S A. 2015;112:E3246–3254. doi: 10.1073/pnas.1421463112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liberal R, Grant CR, Longhi MS, Mieli-Vergani G, Vergani D. Regulatory T cells: Mechanisms of suppression and impairment in autoimmune liver disease. IUBMB Life. 2015;67:88–97. doi: 10.1002/iub.1349. [DOI] [PubMed] [Google Scholar]
- 34.Miethke AG, Saxena V, Shivakumar P, Sabla GE, Simmons J, Chougnet CA. Post-natal paucity of regulatory T cells and control of NK cell activation in experimental biliary atresia. J Hepatol. 2010;52:718–726. doi: 10.1016/j.jhep.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki M, Ikeda H, Sawada S, Sato Y, Nakanuma Y. Naturally-occurring regulatory T cells are increased in inflamed portal tracts with cholangiopathy in primary biliary cirrhosis. J Clin Pathol. 2007;60:1102–1107. doi: 10.1136/jcp.2006.044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rong G, Zhou Y, Xiong Y, Zhou L, Geng H, Jiang T, Zhu Y, et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol. 2009;156:217–225. doi: 10.1111/j.1365-2249.2009.03898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, Matsumura S, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. 1999;19:422–427. doi: 10.1023/a:1020511002025. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Y, Wang J, Yan W, Zhou Y, Chen Y, Zhou K, Wen J, et al. Dysregulated miR-124 and miR-200 expression contribute to cholangiocyte proliferation in the cholestatic liver by targeting IL-6/STAT3 signalling. J Hepatol. 2015;62:889–896. doi: 10.1016/j.jhep.2014.10.033. [DOI] [PubMed] [Google Scholar]
- 39.Zhang W, Tsuda M, Yang GX, Tsuneyama K, Rong G, Ridgway WM, Ansari AA, et al. Deletion of interleukin-6 in mice with the dominant negative form of transforming growth factor beta receptor II improves colitis but exacerbates autoimmune cholangitis. Hepatology. 2010;52:215–222. doi: 10.1002/hep.23664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mair M, Zollner G, Schneller D, Musteanu M, Fickert P, Gumhold J, Schuster C, et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology. 2010;138:2499–2508. doi: 10.1053/j.gastro.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 41.Tucker RM, Hendrickson RJ, Mukaida N, Gill RG, Mack CL. Progressive biliary destruction is independent of a functional tumor necrosis factor-alpha pathway in a rhesus rotavirus-induced murine model of biliary atresia. Viral Immunol. 2007;20:34–43. doi: 10.1089/vim.2006.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shivakumar PMT, Mourya R, Gutta S, Yang L, Luo Z, Bezerra JA. Preferential TNFα signaling via TNFR2 regulates epithelial injury and duct obstruction in experimental biliary atresia. JCI Insight. 2017 doi: 10.1172/jci.insight.88747. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li S, Huang X, Zhong H, Chen Z, Peng Q, Deng Y, Qin X. Tumour necrosis factor alpha (TNF-alpha) genetic polymorphisms and the risk of autoimmune liver disease: a meta-analysis. J Genet. 2013;92:617–628. [PubMed] [Google Scholar]
- 44.Bo X, Broome U, Remberger M, Sumitran-Holgersson S. Tumour necrosis factor alpha impairs function of liver derived T lymphocytes and natural killer cells in patients with primary sclerosing cholangitis. Gut. 2001;49:131–141. doi: 10.1136/gut.49.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tucker RM, Feldman AG, Fenner EK, Mack CL. Regulatory T cells inhibit Th1 cell-mediated bile duct injury in murine biliary atresia. J Hepatol. 2013;59:790–796. doi: 10.1016/j.jhep.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lages CS, Simmons J, Chougnet CA, Miethke AG. Regulatory T cells control the CD8 adaptive immune response at the time of ductal obstruction in experimental biliary atresia. Hepatology. 2012;56:219–227. doi: 10.1002/hep.25662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sebode M, Peiseler M, Franke B, Schwinge D, Schoknecht T, Wortmann F, Quaas A, et al. Reduced FOXP3(+) regulatory T cells in patients with primary sclerosing cholangitis are associated with IL2RA gene polymorphisms. J Hepatol. 2014;60:1010–1016. doi: 10.1016/j.jhep.2013.12.027. [DOI] [PubMed] [Google Scholar]
- 48.Butscheid M, Hauptvogel P, Fritz P, Klotz U, Alscher DM. Hepatic expression of galectin-3 and receptor for advanced glycation end products in patients with liver disease. J Clin Pathol. 2007;60:415–418. doi: 10.1136/jcp.2005.032391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lohwasser C, Neureiter D, Popov Y, Bauer M, Schuppan D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J Gastroenterol. 2009;15:5789–5798. doi: 10.3748/wjg.15.5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia P, Deng Q, Gao J, Yu X, Zhang Y, Li J, Guan W, et al. Therapeutic effects of antigen affinity-purified polyclonal anti-receptor of advanced glycation end-product (RAGE) antibodies on cholestasis-induced liver injury in rats. Eur J Pharmacol. 2016;779:102–110. doi: 10.1016/j.ejphar.2016.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.