

Abstract
Background: It is widely accepted that cleft lip with or without cleft palate (CL/P) results from the complex interplay between multiple genetic and environmental factors. However, a robust investigation of these gene-environment (GxE) interactions at a genome-wide level is still lacking for isolated CL/P.
Materials and Methods: We used our R-package Haplin to perform a genome-wide search for GxE effects in isolated CL/P. From a previously published GWAS, genotypes and information on maternal periconceptional cigarette smoking, alcohol intake, and vitamin use were available on 1908 isolated CL/P triads of predominantly European or Asian ancestry. A GxE effect is present if the relative risk estimates for gene-effects in the offspring are different across exposure strata. We tested this using the relative risk ratio (RRR). Besides analyzing all ethnicities combined (“pooled analysis”), separate analyses were conducted on Europeans and Asians to investigate ethnicity-specific effects. To control for multiple testing, q-values were calculated from the p-values.
Results: We identified significant GxVitamin interactions with three SNPs in “Estrogen-related receptor gamma” (ESRRG) in the pooled analysis. The RRRs (95% confidence intervals) were 0.56 (0.45–0.69) with rs1339221 (q = 0.011), 0.57 (0.46–0.70) with rs11117745 (q = 0.011), and 0.62 (0.50–0.76) with rs2099557 (q = 0.037). The associations were stronger when these SNPs were analyzed as haplotypes composed of two-SNP and three-SNP combinations. The strongest effect was with the “t-t-t” haplotype of the rs1339221-rs11117745-rs2099557 combination [RRR = 0.50 (0.40–0.64)], suggesting that the effects observed with the other SNP combinations, including those in the single-SNP analyses, were mainly driven by this haplotype. Although there were potential GxVitamin effects with rs17734557 and rs1316471 and GxAlcohol effects with rs9653456 and rs921876 in the European sample, respectively, none of the SNPs was located in or near genes with strong links to orofacial clefts. GxAlcohol and GxSmoke effects were not assessed in the Asian sample because of a lack of observations for these exposures.
Discussion/Conclusion: We identified significant interactions between vitamin use and variants in ESRRG in the pooled analysis. These GxE effects are novel and warrant further investigations to elucidate their roles in orofacial clefting. If validated, they could provide prospects for exploring the impact of estrogens and vitamins on clefting, with potential translational applications.
Keywords: gene-environment interaction, GWAS, case-parent triad, orofacial cleft, cleft lip with or without cleft palate, birth defects, genetic epidemiology, Haplin
Introduction
With a prevalence of 3.4–22.9 per 10,000 live births (Mossey and Castilla, 2003), cleft lip with or without cleft palate (CL/P) ranks among the most common birth defects in humans. It is widely accepted that CL/P results from the complex interplay between multiple genetic and environmental factors (GxE), but only recently have data and practical approaches become available to enable an investigation of these effects at a genome-wide level. Identifying GxE interactions may not only provide important insights into the etiology of orofacial clefts, but may also be important from a public health perspective because of the potential for interventions on environmental risk factors alone, particularly in genetically more susceptible subgroups of the population. This rationale has long been demonstrated in animal models (Millicovsky and Johnston, 1981a,b; Juriloff, 2002), but the evidence in humans is less conclusive. Just as some murine strains are more susceptible to external teratogens, human fetuses harboring high-risk alleles may also be more sensitive to particular environmental risk factors, and identifying those that interact with genetic risk variants may lead to important inroads in our understanding of the causes of CL/P.
Over the years, a growing list of maternal exposures has been reported to influence the risk of isolated CL/P. In particular, cigarette smoking (Zeiger and Beaty, 2002; Little et al., 2004; Lie et al., 2008), alcohol consumption (DeRoo et al., 2008), folic acid and other B-complex vitamin supplementation (Hayes, 2002; Munger, 2002; Munger et al., 2004; Wilcox et al., 2007), and anti-folate medication (Hernández-Díaz et al., 2000; Holmes et al., 2001) have been among the most widely studied exposures. Associations with other environmental risk factors have also been reported (reviewed in Dixon et al., 2011), but these have been less consistent across studies and no consensus has yet emerged on the harmful effects of these exposures.
A wide variety of study designs have been used to study GxE effects in orofacial clefts, one of which involves treating a case-parent triad as the unit of analysis. This family-based “triad design” handles bias due to population stratification by using non-transmitted parental alleles as controls, to be compared with the alleles transmitted to the case child (Gjessing and Lie, 2006). We and others have used the triad design to investigate GxE effects in orofacial clefts (Jugessur et al., 2003; Shi et al., 2007; Wu et al., 2010). In terms of statistical power, a unit comprising one case and one control provides approximately the same power as a complete triad when studying single-SNP associations (Schaid, 2002). For haplotype reconstruction, however, the triad design offers the additional advantage of allowing haplotypes to be deduced from the family structure (Gjessing and Lie, 2006).
A handful of genome-wide association studies (GWAS) have now been published on orofacial clefts (reviewed in Dixon et al., 2011; Mangold et al., 2011; Rahimov et al., 2012; Beaty et al., 2016), offering unprecedented opportunities for investigating GxE effects at a genome-wide level. Using data on case-parent triads from a previously published GWAS (Beaty et al., 2010), Wu et al. (2014) and Beaty et al. (2011) screened for GxE effects in one category of orofacial clefts: isolated cleft palate only (CPO). Here, we used the same GWAS dataset to search for GxE effects in the larger category of isolated CL/P.
Materials and methods
Study participants
Participants in this study stem from an international cleft collaboration involving seven Asian and six European/US populations. Characteristics of the study populations and details of the GWAS have been provided in Beaty et al. (2010). Briefly, genotyping was performed on an Illumina Human610-Quad® platform and genotypes for 589,945 SNPS were deposited in the Database of Genotypes and Phenotypes (dbGaP; http://www.ncbi.nlm.nih.gov/gap) under study accession ID phs000094.v1.p1. Besides genotypes, information was also available on maternal cigarette smoking, alcohol intake, and vitamin use in the periconceptional period (i.e., 3 months prior to conception through the first trimester of pregnancy). Interviews and questionnaires were used to assess maternal exposures and the data were coded as simple yes/no responses for cigarette smoking, any reported alcohol consumption, and any use of vitamin supplements. For a detailed description of these maternal exposures, see the recent work on isolated CPO by Wu et al. (2014) and the study outline at dbGAP under study accession ID phs000094.v1.p1. Quality control was performed using the approach outlined in Haaland et al. (2017). The number of SNPs before and after the pruning process is provided in Table 1. Figure 1 shows the distribution of triads by ethnicity and environmental exposure after quality control was performed. Note that we did not analyze GxSmoke or GxAlcohol in the Asian sample because very few of the mothers reported smoking cigarettes or drinking alcohol in the periconceptional period. The final version of the dataset included genotypes for 1,908 nuclear families; 825 were of European ancestry, 1,024 of Asian descent, and 59 of other ethnicities. Table 2 provides an overview of triad completeness and ethnicity. Among the 1,908 nuclear families, 1,594 were complete mother-father-child triads and 314 were parent–child dyads only.
Table 1.
Number of SNPs before and after pruning.
| Criteria | Number |
|---|---|
| Total no. of SNPs before pruning | 569,244 |
| Failed HWE test | 173,955 |
| Failed missingness test | 1,934 |
| Failed SNP frequency test | 61,167 |
| Mendelian errors detected | 349 |
| Remaining SNPs after pruninga | 341,191 |
Remaining SNPs refer to those without deviations from Hardy-Weinberg equilibrium (HWE) (p < 0.001), and those having less than 5% missed calls, minor allele frequencies >5%, and Mendelian errors <1%. Note that because some SNPs may fail several criteria, the number of remaining SNPs is not equal to the total number of SNPs minus those removed in the pruning process.
Figure 1.
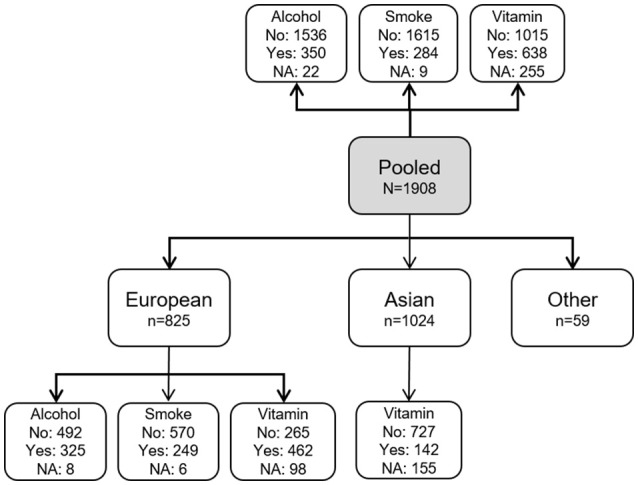
Distribution of families according to ethnicity and maternal exposure status. The gray box shows the pooled sample consisting of all ethnicities (N = 1,908). White boxes show the number of families according to ethnicity and exposure (yes: exposed; no: unexposed; NA: data not available). Note that we did not consider GxSmoke or GxAlcohol analyses in the Asian sample because of a very low number of observations for these exposures.
Table 2.
Triad completeness by ethnicitya.
| Ethnicity | Complete triadsc | Incomplete triadsd | Total | |||
|---|---|---|---|---|---|---|
| Individuals | Families | Individuals | Families | Individuals | Families | |
| European | 2,024 | 670 | 310 | 155 | 2,334 | 825 |
| Asian | 2,670 | 890 | 268 | 134 | 2,938 | 1,024 |
| Otherb | 102 | 34 | 50 | 25 | 152 | 59 |
| Pooled | 4,796 | 1,594 | 628 | 314 | 5,424 | 1,908 |
In total, 317 individuals had genotype call rates <10% and were removed from the analyses. Columns show the number of remaining families and individuals.
Analyses were not conducted for this group because of small numbers.
Some families had more than one offspring.
These are parent-offspring dyads.
Statistical analysis
We are continuously extending a comprehensive R-package, Haplin, for analyzing different constellations of family-based data (Gjessing and Lie, 2006). Haplin uses maximum likelihood to estimate the relative risk (RR), p-value, and 95% confidence intervals for a given risk allele or haplotype. To identify GxE effects, a Wald test is used to test whether RR estimates for gene-effects in the offspring are significantly different across strata of exposed and unexposed mothers, as described in our previous works (Skare et al., 2012; Gjerdevik et al., 2017). A GxE effect is assessed as the ratio of relative risks (RRR). That is, RRR = RR(1)/RR(0), where RR(1) and RR(0) are the relative risks for the offspring of exposed and unexposed mothers, respectively. If RRR ≠ 1, the RR among the exposed is different from the RR among the unexposed, which would indicate a GxE effect. See Haaland et al. (2017) for more details on the calculation of RRR.
Since its inception, Haplin has incorporated haplotype estimation for triads. By using the expectation-maximization (EM) algorithm (Dempster et al., 1977), Haplin reconstructs haplotypes from the multi-SNP data even though phase is not known from the observed markers alone. The EM-algorithm also enables Haplin to account for missing parental genotypes, thus allowing the 314 parent–child dyads in the current dataset to be included in the analyses.
If an offspring was homozygous for the risk allele at a locus, we assumed a multiplicative dose-response model. In other words, the relative risk with two copies of the risk allele was assumed to be simply RR × RR. For each maternal exposure (smoking, alcohol use, vitamin use), we performed separate analyses on Asians (n = 1,024), Europeans (n = 825), and all participants combined irrespective of their ethnicity (“pooled sample”; N = 1,908). The different sets of analyses are illustrated in Figure 1.
Further, we conducted haplotype analyses for SNPs that had very low q-values. This was done using the haplinSlide function in Haplin, as described in our previous works on candidate genes for clefts (Jugessur et al., 2008, 2009b, 2012a,b).
Next, we performed statistical power calculations using the hapPowerAsymp function in Haplin. It is based on the asymptotic variance-covariance structure of the Wald test, as described in our recent work (Gjerdevik et al., 2017). The sample sizes used in these calculations reflect those that were available in the current GWAS dataset (Figure 1).
To facilitate the interpretation of the large number of statistical tests being performed simultaneously, we generated Quantile-Quantile (QQ) plots (Wilk and Gnanadesikan, 1968) using the pQQ function in Haplin to screen for significant p-values. This was done separately for the analysis of each exposure. To control the false discovery rate, we used the method described by Storey and Tibshirani (2003) where q-values are computed from the p-values. The function qvalue from the R-package “q-value” in the Bioconductor repository [https://www.bioconductor.org/, Gentleman et al., 2004] was used to generate the q-values. A low q-value indicates a low likelihood that a statistically significant association is a false positive. A q-value of 0.1, for instance, corresponds to a false discovery rate of 10%.
A regional plot was generated to map the chromosomal area flanking the most significant SNP (“lead SNP”). This plot provides a visual display of linkage disequilibrium between the lead SNP and other SNPs, along with their positions. It also provides the position of the closest genes and the recombination rates in the region. We generated the plot by modifying an R-script available at http://www.broadinstitute.org/files/shared/diabetes/scandinavs/assocplot.R (Pruim et al., 2010).
The plethora of publicly available data makes it feasible to explore the connections between different entities, e.g., genes and vitamins. Hetionet integrates different sources of information from actively maintained meta-databases and allows the visualization of possible paths between various diseases, genes and compounds (Himmelstein et al., 2017, https://neo4j.het.io/). For example, to explore possible links between a given disease, compounds and gene nodes, Hetionet uses the following databases: Disease Ontology (Kibbe et al., 2015), Drug Bank (Law et al., 2014), and Entrez Gene (Maglott et al., 2005). Furthermore, Hetionet can also integrate information from the Human Interactome Database (Rolland et al., 2014), CMap (https://clue.io/) or Bgee (Bastian et al., 2008) to identify potential gene-gene interactions, compound-gene interactions and gene expression in specific tissue, respectively. We queried Hetionet to visualize the relationship between genes and maternal exposures for the cleft lip phenotype. The complete query and output codes are provided in the online Supplementary Text 1.
Ethics approvals
Ethics approvals for the International Cleft Consortium were obtained from the respective institutional review boards of the participating sites. The consortium was formed in 2007 and each participating institution approved research protocols for the recruitment of case-parent triads from 13 individual sites. All participants have granted their written informed consents. The participating sites included institutions in the US (Johns Hopkins University; University of Iowa; Utah State University; National Institute of Environmental Health Sciences (NIEHS); University of Pittsburgh), Denmark (University of Southern Denmark), Norway (University of Bergen), China (Peking University Health Science Center; Wuhan University; Peking Union Medical College; West China School of Stomatology, Sichuan University; School of Stomatology, Beijing University), Korea (Yonsei University), Taiwan (Chang Gung Memorial Hospital), and Singapore (KK Women's & Children's Hospital; National University of Singapore). For more details on the recruitment sites, the research approvals and protocols, see the online “Supplementary Note” of the original publication (Beaty et al., 2010), as well as the study outline at dbGAP (https://www.ncbi.nlm.nih.gov/gap) under study accession number phs000094.v1.p1.
Results
Tables 3–5 provide the relative risk ratios (RRRs) and 95% confidence intervals (CIs) for the top 20 SNPs in each set of analyses depicted in Figure 1. These SNPs were ranked by p-value, and their corresponding q-values are also provided in these tables. RRRs were calculated as previously described by Haaland et al. (2017). Figures 2–4 show the QQ-plots for each maternal exposure. The corresponding Manhattan plots are provided in Figures 5–7. For SNPs that are not associated with isolated CL/P, –log10 (p-values) in the QQ-plots are expected to fall along the straight diagonal line representing the null distribution, within the 95% confidence band (gray area). Conversely, –log10 (p-values) would fall above this line, outside the confidence band and in the upper right-hand corner of the plot. Several SNPs were significantly associated with isolated CL/P in the GxVitamin analyses of the pooled sample (Figure 2). In the other analyses, none of the SNPs were outside the confidence bands, suggesting that the distribution of p-values is as expected if there are no associations. Still, in each of the GxVitamin and GxAlcohol analyses of the European sample (Tables 3, 5), two SNPs had markedly lower p-values than expected (see also Figures 5, 7). However, none of these SNPs was located in or near genes with obvious links to orofacial clefts. In the remainder of this section, therefore, we focus mainly on the results of the GxVitamin analysis of the pooled sample.
Table 3.
Relative risk ratio (RRR) and 95% confidence interval (CI) for the top 20 SNPs in the GxVitamin analyses.
| Analysis | Top 20 SNPsa | Chromosomal band locationb | Location of SNP in or near a given gene(s)b | p-value | q-value | RRR (95% CI) |
|---|---|---|---|---|---|---|
| Pooled | rs1339221 | 1q41 | In ESRRG | 5.9e-08 | 0.011 | 0.56 (0.45–0.69) |
| rs11117745 | 1q41 | In ESRRG | 6.8e-08 | 0.011 | 0.57 (0.46–0.70) | |
| rs13027140 | 2p16 | Between FLJ30838 and LOC101927285 | 1.0e-06 | 0.117 | 1.70 (1.38–2.11) | |
| rs4233974 | 2p16 | Between FLJ30838 and LOC101927285 | 2.2e-06 | 0.15 | 1.67 (1.35–2.06) | |
| rs1316471 | 7p12 | Nearest gene is COBL | 2.2e-06 | 0.15 | 2.36 (1.66–3.38) | |
| rs16970288 | 19q13.1 | Between pseudogene EEF1A1P7a and LINC01531 | 6.2e-06 | 0.337 | 1.69 (1.35–2.13) | |
| rs2099557 | 1q41 | In ESRRG | 7.0e-06 | 0.337 | 0.62 (0.50–0.76) | |
| rs13022580 | 2q21.1 | Between pseudogenes RPL19P4 and TEKT4P3 | 8.4e-06 | 0.337 | 1.60 (1.30–1.96) | |
| rs5762534 | 22q12.1 | In TTC28 | 9.0e-06 | 0.337 | 1.90 (1.43–2.51) | |
| rs938868 | 2q21.1 | Between pseudogenes MTND5P29 and RPL19P4 | 1.02e-05 | 0.343 | 1.59 (1.29–1.95) | |
| rs10992247 | 9q22.2 | Between pseudogenes IL6RP1 and OR7E31P | 1.19e-05 | 0.367 | 0.42 (0.28–0.62) | |
| rs130413 | 22q12.1 | In TTC28 | 1.61e-05 | 0.453 | 1.81 (1.38–2.37) | |
| rs10072077 | 5q14 | Near LOC101929423 | 1.81e-05 | 0.468 | 2.35 (1.59–3.48) | |
| rs949771 | 2q21.1 | In pseudogene MTND5P29 | 1.94e-05 | 0.468 | 1.56 (1.27–1.92) | |
| rs6696232 | 1p22.2 | Between ZNF644 and HFM1 | 2.12e-05 | 0.477 | 0.60 (0.47–0.76) | |
| rs247405 | 16q23 | Nearest gene is HSBP1 | 2.59e-05 | 0.5 | 0.48 (0.34–0.67) | |
| rs621188 | 11q23.3 | In DSCAML1 | 2.63e-05 | 0.5 | 1.59 (1.28–1.97) | |
| rs6431784 | 2p25.3 | Nearest gene is SOX11 | 2.66e-05 | 0.5 | 1.74 (1.34–2.26) | |
| rs2042743 | 18p11.2 | In LDLRAD4 | 3.04e-05 | 0.539 | 1.55 (1.26–1.91) | |
| rs11676593 | 2q21.1 | Between pseudogenes RPL19P4 and TEKT4P3 | 3.18e-05 | 0.539 | 1.56 (1.26–1.92) | |
| European | rs17734557 | 7p12 | Nearest gene is COBL | 5e-07 | 0.163 | 3.22 (2.04–5.09) |
| rs1316471 | 7p12 | Nearest gene is COBL | 1e-06 | 0.168 | 3.36 (2.07–5.46) | |
| rs10875883 | 12q13.1 | Nearest gene is CCDC65 | 1.17e-05 | 0.607 | 0.49 (0.36–0.68) | |
| rs2838965 | 21q22.3 | Nearest gene is SLC19A1 | 1.29e-05 | 0.607 | 0.49 (0.35–0.67) | |
| rs7741153 | 6p23 | Nearest gene is JARID2 | 1.5e-05 | 0.607 | 0.45 (0.31–0.64) | |
| rs247405 | 16q23 | Nearest gene is HSBP1 | 1.52e-05 | 0.607 | 0.23 (0.12–0.45) | |
| rs9544295 | 13q22 | Nearest gene is KCTD12 | 1.63e-05 | 0.607 | 2.77 (1.74–4.41) | |
| rs13027140 | 2p16 | Between FLJ30838 and LOC101927285 | 1.83e-05 | 0.607 | 2.02 (1.46–2.78) | |
| rs2159963 | 12p13.3 | Between A2ML1 and PHC1 | 1.9e-05 | 0.607 | 2.35 (1.59–3.48) | |
| rs10746803 | 9q21.3 | Between C9orf170 and DAPK1 | 2.23e-05 | 0.607 | 0.34 (0.21–0.56) | |
| rs2575625 | 4q25 | Between OSTC and ETNPPL | 2.35e-05 | 0.607 | 1.96 (1.44–2.68) | |
| rs3792390 | 3q21 | In PDIA5 | 2.46e-05 | 0.607 | 0.44 (0.30–0.65) | |
| rs17651808 | 15q21.2 | Between ARPP19 and FAM214A | 2.49e-05 | 0.607 | 0.28 (0.15–0.50) | |
| rs4932844 | 19p12 | In pseudogene ZNF724 | 2.53e-05 | 0.607 | 0.51 (0.37–0.70) | |
| rs295367 | 19p12 | In pseudogene LOC100132815 | 3.21e-05 | 0.719 | 0.51 (0.38–0.70) | |
| rs9821623 | 3p22 | In ULK4 | 3.66e-05 | 0.768 | 2.19 (1.51–3.18) | |
| rs2362803 | 19p12 | Nearest gene is ZNF724P | 4.16e-05 | 0.783 | 0.52 (0.38–0.71) | |
| rs260818 | 11q22.3 | In PDGFD | 4.2e-05 | 0.783 | 1.94 (1.41–2.67) | |
| rs10743549 | 12p13.3 | Between A2ML1 and PHC1 | 4.45e-05 | 0.786 | 2.33 (1.55–3.51) | |
| rs642307 | 1p32.1 | In C1orf87 | 6.4e-05 | 0.905 | 1.89 (1.38–2.59) | |
| Asian | rs13261120 | 8p22 | In SGCZ | 3.4e-06 | 0.73 | 2.45 (1.68–3.57) |
| rs8055365 | 16q24 | Between TLDC1 and COTL1 | 6.2e-06 | 0.73 | 0.39 (0.26–0.59) | |
| rs12546303 | 8p22 | In SGCZ | 8.4e-06 | 0.73 | 2.35 (1.62–3.43) | |
| rs4773818 | 13q32 | In LOC101927284 | 9.9e-06 | 0.73 | 3.16 (1.90–5.25) | |
| rs247832 | 16q24 | Between TLDC1 and COTL1 | 1.29e-05 | 0.73 | 0.42 (0.29–0.62) | |
| rs792306 | 13q32 | In LOC101927284 | 1.33e-05 | 0.73 | 3.10 (1.86–5.17) | |
| rs12374531 | 5q32 | Between PPP2R2B and STK32A | 1.53e-05 | 0.73 | 3.22 (1.90–5.47) | |
| rs4695853 | 4q34 | Nearest gene is HAND2-AS1 | 2.34e-05 | 0.897 | 3.12 (1.84–5.29) | |
| rs6860289 | 5p15.3 | Nearest gene is MTRR | 2.65e-05 | 0.897 | 2.35 (1.58–3.50) | |
| rs1615355 | 12q24.33 | In LOC100190940 | 2.98e-05 | 0.897 | 2.29 (1.55–3.38) | |
| rs13418410 | 2p22 | Between RASGRP3 and FAM98A | 3.24e-05 | 0.897 | 2.35 (1.57–3.52) | |
| rs423909 | 1p36.2 | Nearest gene is SLC45A1 | 3.52e-05 | 0.897 | 2.18 (1.51–3.16) | |
| rs4670209 | 2p22 | Between RASGRP3 and FAM98A | 3.59e-05 | 0.897 | 2.25 (1.53–3.31) | |
| rs1448561 | 2p22 | Nearest gene is FAM98A | 4.12e-05 | 0.897 | 2.32 (1.55–3.46) | |
| rs9286945 | 1p35 | Nearest gene is PTPRU | 4.49e-05 | 0.897 | 0.45 (0.30–0.66) | |
| rs2002022 | 13q21.3 | Between pseudogenes NPM1P22 and OR7E111P | 4.6e-05 | 0.897 | 2.35 (1.56–3.54) | |
| rs17138910 | 10p13 | In RSU1 | 5.05e-05 | 0.897 | 2.22 (1.51–3.26) | |
| rs4290780 | 3q13.2 | In CCDC80 | 5.19e-05 | 0.897 | 3.85 (2.01–7.41) | |
| rs10503525 | 8p22 | In SGCZ | 5.96e-05 | 0.897 | 2.39 (1.56–3.65) | |
| rs13388224 | 2p22 | Between RASGRP3 and FAM98A | 6.04e-05 | 0.897 | 2.27 (1.52–3.39) |
SNPs with q-values <0.2 are shown in bold.
Location of SNP was determined using the 1,000 Genomes browser at https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/.
Table 5.
Relative risk ratio (RRR) and 95% confidence interval (CI) for the top 20 SNPs in the GxAlcohol analyses.
| Analysisa | Top 20 SNPsb | Chromosomal band locationc | Location of SNP in or near a given gene(s)c | p-value | q-value | RRR (95% CI) |
|---|---|---|---|---|---|---|
| Pooled | rs17023089 | 3p24.1 | Nearest gene is RBMS3 | 7.4e-06 | 0.858 | 0.32 (0.19–0.53) |
| rs7164773 | 15q22.2 | In RORA | 9.8e-06 | 0.858 | 1.73 (1.35–2.20) | |
| rs6748903 | 2q14.3 | Near LOC101927881 | 1.65e-05 | 0.858 | 0.51 (0.38–0.69) | |
| rs11691558 | 2q21.1 | In LOC101927881 | 1.72e-05 | 0.858 | 0.51 (0.38–0.69) | |
| rs4132008 | 1q32.1 | In PLEKHA6 | 1.94e-05 | 0.858 | 1.75 (1.35–2.27) | |
| rs2118769 | 2q22.3 | Nearest gene is ACVR2A | 2.04e-05 | 0.858 | 0.58 (0.46–0.75) | |
| rs1335594 | 13q33 | In ITGBL1 | 2.3e-05 | 0.858 | 1.74 (1.35–2.25) | |
| rs3805025 | 3p26 | In ITPR1 | 2.77e-05 | 0.858 | 1.96 (1.43–2.69) | |
| rs30306 | 5q32 | Nearest gene is ADBR2 | 3.58e-05 | 0.858 | 1.66 (1.30–2.11) | |
| rs6685648 | 1p36.2 | In CASP9 | 3.89e-05 | 0.858 | 0.59 (0.46–0.76) | |
| rs2064317 | 6p21.3 | In TULP1 | 4.45e-05 | 0.858 | 1.65 (1.30–2.11) | |
| rs686805 | 11q13.4 | In SHANK2 | 4.66e-05 | 0.858 | 2.24 (1.52–3.30) | |
| rs9653456 | 2q12 | Nearest gene is EDAR | 4.76e-05 | 0.858 | 1.85 (1.37–2.49) | |
| rs2163752 | 5q32 | Between HTR4 and ADRB2 | 4.77e-05 | 0.858 | 1.64 (1.29–2.09) | |
| rs13086826 | 3q22 | In CPNE4 | 4.94e-05 | 0.858 | 0.59 (0.45–0.76) | |
| rs4810165 | 20q13.3 | Between EDN3 and peudogene PIEZO1P1 | 5.01e-05 | 0.858 | 1.75 (1.34–2.30) | |
| rs4859190 | 3q27 | Nearest gene is MCF2L2 | 5.12e-05 | 0.858 | 0.58 (0.45–0.76) | |
| rs2740882 | 8p23.3 | In CSMD1 | 5.15e-05 | 0.858 | 0.48 (0.33–0.68) | |
| rs13077768 | 3q22 | In CPNE4 | 5.29e-05 | 0.858 | 0.59 (0.45–0.76) | |
| rs11076508 | 16q12.1 | Between ZNF423 and pseudogene RPL34P29 | 5.56e-05 | 0.858 | 0.61 (0.48–0.78) | |
| European | rs9653456 | 2q12 | Between EDAR and SH3RF3 | 4e-07 | 0.148 | 2.40 (1.71–3.37) |
| rs921876 | 2q12 | Nearest gene is EDAR | 9e-07 | 0.153 | 2.09 (1.56–2.81) | |
| rs11076508 | 16q12.1 | Between ZNF423 and pseudogene RPL34P29 | 2.25e-05 | 0.892 | 0.54 (0.40–0.72) | |
| rs7713774 | 5q14 | In ACOT12 | 3.05e-05 | 0.892 | 0.51 (0.37–0.70) | |
| rs10784765 | 12q15 | Between CPM and CPSF6 | 4.76e-05 | 0.892 | 0.41 (0.27–0.63) | |
| rs893912 | 15q22.3 | Between LCTL and SMAD6 | 5.04e-05 | 0.892 | 2.28 (1.53–3.39) | |
| rs17199679 | 9q31 | Between pseudogene RPL36AP35 and ACTL7B | 5.1e-05 | 0.892 | 2.86 (1.72–4.76) | |
| rs3020054 | 11q21 | Nearest gene is CCDC67 | 6.7e-05 | 0.892 | 0.52 (0.38–0.72) | |
| rs2192926 | 2p12 | Nearest gene is TACR1 | 7.24e-05 | 0.892 | 0.50 (0.36–0.71) | |
| rs7862797 | 9q31 | Between pseudogene RPL36AP35 and ACTL7B | 7.49e-05 | 0.892 | 2.77 (1.67–4.58) | |
| rs851674 | 7q35 | In CNTNAP2 | 7.85e-05 | 0.892 | 1.87 (1.37–2.56) | |
| rs6581864 | 12q15 | Between CPM and CPSF6 | 8.72e-05 | 0.892 | 1.81 (1.34–2.43) | |
| rs6495940 | 15q14 | Between TMCO5A and SPRED1 | 8.82e-05 | 0.892 | 0.41 (0.26–0.64) | |
| rs6822683 | 4q34 | Nearest gene is GALNT7 | 9.58e-05 | 0.892 | 1.82 (1.35–2.46) | |
| rs2276049 | 11q24 | In VWA5A | 9.78e-05 | 0.892 | 0.48 (0.33–0.69) | |
| rs2417976 | 9q31 | Between TMEM245 and FRRS1L | 9.82e-05 | 0.892 | 2.04 (1.42–2.91) | |
| rs10759878 | 9q33 | In ASTN2 | 9.89e-05 | 0.892 | 1.93 (1.39–2.69) | |
| rs4778036 | 15q26.1 | Between SLCO3A1 and pseudogene DUXAP6 | 0.0001033 | 0.892 | 1.80 (1.34–2.43) | |
| rs17104078 | 10q23.1 | Nearest gene is CCSER2 | 0.0001042 | 0.892 | 2.48 (1.57–3.93) | |
| rs12540601 | 7p15.3 | Between TRA2A and pseudogene LOC442517 | 0.0001044 | 0.892 | 0.56 (0.42–0.75) |
We were unable to assess GxAlcohol effects in the Asian sample because very few of the mothers reported drinking alcohol in the periconceptional period.
SNPs with q-values <0.2 are shown in bold.
Location of SNP was determined using the 1,000 Genomes browser at https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/.
Figure 2.

QQ-plots for the GxVitamin analyses in the pooled, European and Asian samples.
Figure 4.

QQ-plots for the GxAlcohol analyses in the pooled and European samples.
Figure 5.

Manhattan plots for GxVitamin effects in the pooled, Europeans-only and Asians-only analyses. SNPs with p-values below 10−6 are shown in blue.
Figure 7.

Manhattan plots for GxAlcohol effects in the pooled and Europeans-only analyses. SNPs with p-values below 10−6 are shown in blue.
Table 4.
Relative risk ratio (RRR) and 95% confidence interval (CI) for the top 20 SNPs in the GxSmoke analyses.
| Analysisa | Top 20 SNPs | Chromosomal band locationb | Location of SNP in or near a given gene(s)b | p-value | q-value | RRR (95% CI) |
|---|---|---|---|---|---|---|
| Pooled | rs820930 | 7q31.1 | Nearest locus is C7orf66 | 1.06e-05 | 0.994 | 1.93 (1.44–2.58) |
| rs210625 | 6q22.2 | In DCBLD1 | 1.36e-05 | 0.994 | 1.79 (1.38–2.33) | |
| rs7258736 | 19q12 | In LOC101927210 | 1.56e-05 | 0.994 | 2.99 (1.82–4.91) | |
| rs7247342 | 19q12 | In LOC101927210 | 2.59e-05 | 0.994 | 2.89 (1.76–4.73) | |
| rs2024140 | 19q12 | In LOC101927210 | 2.91e-05 | 0.994 | 2.97 (1.78–4.94) | |
| rs6810129 | 3q26.1 | Nearest gene is CT64 | 2.94e-05 | 0.994 | 0.57 (0.44–0.74) | |
| rs1182865 | 6p21.2 | Between KCNK5 and KCNK17 | 3.43e-05 | 0.994 | 0.51 (0.37–0.70) | |
| rs1013592 | 19q12 | In LOC101927210 | 3.47e-05 | 0.994 | 2.81 (1.72–4.57) | |
| rs10085496 | 7p21 | Between pseudogenes GAPDHP68 and PER4 | 3.85e-05 | 0.994 | 0.58 (0.45–0.75) | |
| rs4805661 | 19q12 | Nearest gene is TSHZ3 | 4.05e-05 | 0.994 | 0.57 (0.43–0.74) | |
| rs2543146 | 8p22 | In TUSC3 | 4.19e-05 | 0.994 | 1.73 (1.33–2.24) | |
| rs4804843 | 19q12 | In LOC101927210 | 4.41e-05 | 0.994 | 2.86 (1.73–4.73) | |
| rs10417454 | 19q12 | In LOC101927210 | 4.63e-05 | 0.994 | 2.84 (1.72–4.69) | |
| rs244226 | 21q21 | Between pseudogenes MAPK6PS2 and ZNF299P | 4.88e-05 | 0.994 | 1.72 (1.32–2.23) | |
| rs352806 | 8p22 | In TUSC3 | 5.21e-05 | 0.994 | 0.53 (0.38–0.72) | |
| rs2191018 | 19q12 | In LOC101927210 | 5.4e-05 | 0.994 | 2.82 (1.71–4.67) | |
| rs6806286 | 3q22 | In CPNE4 | 6.37e-05 | 0.994 | 1.72 (1.32–2.25) | |
| rs2834061 | 21q22.1 | Nearest gene is OLIG2 | 6.37e-05 | 0.994 | 1.80 (1.35–2.41) | |
| rs12204228 | 6q22.1 | Between RFX6 and VGLL2 | 6.44e-05 | 0.994 | 0.40 (0.25–0.63) | |
| rs11014205 | 10p12.2 | In ARHGAP21 | 8.06e-05 | 0.994 | 0.55 (0.41–0.74) | |
| European | rs10106898 | 8q22.1 | Between pseudogenes GAPDHP30 and TUBBP7 | 4.1e-06 | 0.684 | 0.29 (0.18–0.50) |
| rs10095562 | 8q22.1 | Between pseudogenes GAPDHP30 and TUBBP7 | 1.4e-05 | 0.684 | 0.30 (0.17–0.52) | |
| rs10282921 | 8q22.1 | Between pseudogenes GAPDHP30 and TUBBP7 | 1.43e-05 | 0.684 | 0.30 (0.18–0.52) | |
| rs820930 | 7q31.1 | Between LOC646614 and LOC100421901 | 1.45e-05 | 0.684 | 2.11 (1.51–2.97) | |
| rs266642 | 5q23.2 | Nearest gene is GRAMD3 | 1.55e-05 | 0.684 | 2.69 (1.72–4.22) | |
| rs601904 | 11q13.4 | Between peudogene CYCSP27 and LIPT2 | 1.59e-05 | 0.684 | 0.49 (0.35–0.68) | |
| rs17608059 | 17p12 | Nearest gene is COX10-AS1 | 1.68e-05 | 0.684 | 1.97 (1.44–2.67) | |
| rs657339 | 11q13.4 | In LOC100287896 | 1.85e-05 | 0.684 | 0.49 (0.35–0.68) | |
| rs617989 | 11q13.4 | In LIPT2 | 1.9e-05 | 0.684 | 0.49 (0.36–0.68) | |
| rs649460 | 11q13.4 | Nearest gene is LIPT2 | 2.28e-05 | 0.684 | 0.49 (0.36–0.68) | |
| rs2509563 | 11q13.4 | Nearest gene is LIPT2 | 2.29e-05 | 0.684 | 0.50 (0.36–0.69) | |
| rs1013592 | 19q12 | In LOC101927210 | 2.45e-05 | 0.684 | 3.84 (2.05–7.17) | |
| rs1783196 | 11q13.4 | Nearest gene is LIPT2 | 2.85e-05 | 0.684 | 0.50 (0.36–0.69) | |
| rs648677 | 11q13.4 | Between LIPT2 and POLD3 | 2.85e-05 | 0.684 | 0.50 (0.36–0.69) | |
| rs10028174 | 4q22 | In CCSER1 | 3.65e-05 | 0.796 | 0.40 (0.26–0.62) | |
| rs1655475 | 11q13.4 | Between LIPT2 and POLD3 | 3.79e-05 | 0.796 | 0.51 (0.37–0.70) | |
| rs2089253 | 4q21.1 | In USO1 | 4.34e-05 | 0.858 | 0.28 (0.16–0.52) | |
| rs3317 | 5q22 | In REEP5 and SRP19 | 5.55e-05 | 0.89 | 0.53 (0.39–0.72) | |
| rs11692230 | 2q12 | In IL1RL2 | 5.67e-05 | 0.89 | 1.89 (1.39–2.58) | |
| rs4805661 | 19q12 | Nearest gene is TSHZ3 | 6.19e-05 | 0.89 | 0.53 (0.38–0.72) |
We were unable to assess GxSmoke effects in the Asian sample because very few of the mothers reported smoking cigarettes in the periconceptional period.
Location of SNP was determined using the 1,000 Genomes browser at https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/.
Figure 3.

QQ-plots for the GxSmoke analyses in the pooled and European samples.
Figure 6.

Manhattan plots for GxSmoke effects in the pooled and Europeans-only analyses.
All the top 20 SNPs in the GxVitamin analysis of the pooled sample had q-values below 0.54 (Table 3). The two top SNPs among these, rs1339221 and rs11117745, both had a q-value of 0.01 and are located in the gene for “Estrogen-related receptor gamma” (ESRRG). These SNPs are in strong linkage disequilibrium (LD) with one another (R2 = 0.77 and D' = 1.00). A third SNP, rs2099557 (q = 0.037), is also located in ESRRG but is in weaker LD with the above two SNPs (R2 = 0.33 and D' = 0.57 with rs1339221; R2 = 0.28 and D' = 0.61 with rs11117745). A regional plot centered around the lead SNP in ESRRG (rs1339221) was generated to visualize the strength of the association signals and regional information around that SNP (Figure 8). Next, we performed stratified analyses of the SNPs in ESRRG by first testing for an overall child effect in the unstratified sample, followed by the effects among children who were exposed and unexposed to maternal vitamin use, respectively (Table 6). All three SNPs exhibited a so-called “qualitative interaction” (Clayton, 2009), in that the effect of the SNP-allele among mothers taking vitamins was in the opposite direction of that among mothers not taking vitamins. Specifically, RR>1 among non-takers, whereas RR < 1 among takers, and none of the 95% CIs included 1 (Table 6). There were no statistically significant overall effects of the child's allele alone for any of the SNPs; all the 95% CIs included RR = 1. As mentioned earlier, the total GxE effect was measured as the ratio of the RRs in each stratum of vitamin use (i.e., RRVitamins/RRNo vitamins). RRRs and 95% CIs were 0.56 (0.45–0.69) with the variant allele at rs1339221 (q = 0.011), 0.57 (0.46–0.70) with the variant allele at rs11117745 (q = 0.011), and 0.62 (0.50–0.76) with the variant allele at rs2099557 (q = 0.037) (Table 6).
Figure 8.

Regional association plot to assess the strength of association and regional information flanking the lead SNP in ESRRG, rs1339221, shown here in blue alongside its p-value.
Table 6.
Stratified analyses of the top three SNPs in ESRRG.
| SNP namea | Stratum | RR (95% CI)b | p-value | Frequency |
|---|---|---|---|---|
| rs1339221 | All (child effect) | 0.94 (0.84–1.04) | 0.22 | 0.40 |
| No vitamin use | 1.17 (1.03–1.33) | 0.016 | 0.38 | |
| Vitamin use | 0.66 (0.56–0.78) | 5 × 10−7 | 0.43 | |
| Across strata | 0.56 (0.45–0.69) | 6 × 10−8 | – | |
| rs11117745 | All (child effect) | 0.96 (0.87–1.06) | 0.41 | 0.46 |
| No vitamin use | 1.19 (1.05–1.35) | 0.0064 | 0.46 | |
| Vitamin use | 0.68 (0.58–0.80) | 2 × 10−6 | 0.46 | |
| Across strata | 0.57 (0.46–0.70) | 7 × 10−8 | – | |
| rs2099557 | All (child effect) | 0.96 (0.87–1.06) | 0.40 | 0.40 |
| No vitamin use | 1.15 (1.01–1.31) | 0.031 | 0.37 | |
| Vitamin use | 0.71 (0.61–0.84) | 5 × 10−5 | 0.44 | |
| Across strata | 0.62 (0.50–0.76) | 5 × 10−6 | – |
All three SNPs are located within chromosomal region 1q41 according to the 1000 Genomes browser (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/).
Relative risk (RR) for the overall child effect, and effects within each stratum of vitamin use. Across strata, an effect is measured as the ratio of the RRs (i.e., RRR) for vitamin use and no vitamin use.
Next, we considered haplotypes of the top three SNPs in ESRRG (Table 7) and analyzed the following two-SNP and three-SNP combinations: (i) rs2099557-rs1339221, (ii) rs1339221-rs11117745, and (iii) rs2099557-rs1339221-rs11117745. The RRRs and 95% CIs were 0.52 (0.41-0.66) for SNP combination (i), 0.55 (0.44–0.68) for SNP combination (ii), and 0.50 (0.40–0.64) for SNP combination (iii) (Table 7). The pattern of RRRs was similar to that in the single-SNP analyses. Provided that vitamin use itself is not harmful, the haplotypes with the lowest p-values appeared to be protective among vitamin-takers, but detrimental among non-takers (Table 7). The RRs and RRRs were even further away from 1, and the p-values were lower than in the single-SNP analyses. The most prominent effect was with the “t-t-t” haplotype in the analysis of SNP combination (iii) above, suggesting that the observed effects with SNP combinations (i) and (ii), as well as those observed in the single-SNP analyses, are likely to be driven by this haplotype.
Table 7.
Haplotype analysis for the top three SNPs in ESRRG.
| SNP combinationa | Haplotype | Topb | Ref.c | Frequency (top vs. ref) | Stratum | RR (95% CI)d | p-value |
|---|---|---|---|---|---|---|---|
| rs2099557-rs1339221 | t/C × t/C | t-t | C-C | 0.30 vs. 0.50 | All (child effect) | 0.93 (0.83–1.04) | 0.20 |
| 0.27 vs. 0.51 | No vitamin use | 1.21 (1.04–1.40) | 0.013 | ||||
| 0.35 vs. 0.48 | Vitamin use | 0.63 (0.53–0.75) | 6 × 10−7 | ||||
| – | Across strata | 0.52 (0.41–0.66) | 6 × 10−8 | ||||
| rs1339221-rs11117745 | t/C × t/G | t-t | C-G | 0.40 vs. 0.54 | All (child effect) | 0.94 (0.85–1.04) | 0.25 |
| 0.39 vs. 0.54 | No vitamin use | 1.19 (1.04–1.36) | 0.0091 | ||||
| 0.43 vs. 0.54 | Vitamin use | 0.65 (0.55–0.77) | 6 × 10−7 | ||||
| – | Across strata | 0.55 (0.44–0.68) | 4 × 10−8 | ||||
| rs2099557-rs1339221-rs11117745 | t/C × t/C × t/G | t-t-t | C-C-G | 0.30 vs. 0.46 | All (child effect) | 0.94 (0.83–1.05) | 0.27 |
| 0.27 vs. 0.45 | No vitamin use | 1.24 (1.07–1.45) | 0.0061 | ||||
| 0.35 vs. 0.46 | Vitamin use | 0.63 (0.52–0.75) | 7 × 10−7 | ||||
| – | Across strata | 0.50 (0.40–0.64) | 2 × 10−8 |
All three SNPs are located within the chromosomal band 1q41 according to the 1,000 Genomes browser (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/).
“Top” refers to the haplotype with the lowest p-value in the analysis.
The most frequent haplotype was used as reference.
Relative risk (RR) for the overall child effect, and effects within each stratum of vitamin use. Across strata, an effect is measured as the ratio of the RRs (i.e., RRR) for vitamin use and no vitamin use.
Our in silico analyses using Hetionet revealed that the cleft lip phenotype is influenced by vitamins A and D through a network of genes connected to ESRRG (Figure 9). Except for “Platelet derived growth factor subunit A” (PDGFA), few of the genes in the network have previously been linked to orofacial clefts. Figure 9 shows that ESRRG is expressed in three major tissues/organs and that the cleft lip phenotype is localized to the embryo, head, and telencephalon (the most highly developed and anterior part of the forebrain). ESRRG regulates or interacts with various genes that are regulated by the levels of vitamin A and D. Note that several genes are up- or down-regulated in the telencephalon (marked by blue arrows in Figure 9) and they also interact directly with ESRRG.
Figure 9.

Relationships between cleft lip, vitamins and ESRRG, visualized as a heterogenic network, where each relationship evidenced in public databases is depicted as an arrow connecting two nodes. Different types of nodes (e.g., genes, diseases, etc.) are connected by different types of relationships (e.g., regulation, interaction, etc.). The nodes and arrows are colored according to the legend at the top of the figure. The thickness of the “DOWNREGULATES_AdG” arrow is less than that of “UPREGULATES_AuG,” and similarly for “DOWNREGULATES_CdG” and “UPREGULATES_CuG.” The numbers on the two latter arrows show the Z-score, which is a measure for the strength of the influence. The figure was generated using the Hetionet network (see the Materials and Methods section for details).
Discussion
The current genome-wide search for GxE effects in isolated CL/P is based on the largest available GWAS dataset on orofacial cleft triads (Beaty et al., 2010). We identified a statistically significant GxE effect between maternal periconceptional vitamin use and genetic variants in ESRRG in the pooled analysis. We also identified potential GxVitamin and GxAlcohol effects in the European sample (Tables 3, 5), but none of the SNPs were outside the 95% confidence bands in the QQ plots (Figures 2, 4), nor were they located in or near genes with obvious connections to orofacial clefts. Because of a lack of observations, we were unable to perform GxSmoke and GxAlcohol analyses in the Asian sample. The low level of alcohol intake and cigarette smoking appears to be a general trend among Asian women, and is likely to be even lower among those who are pregnant or planning pregnancy (Yang et al., 2010; Ng et al., 2014; Haaland et al., 2017). Although GxVitamin analysis of the Asian sample was not hampered by a lack of observations, vitamin intake was nevertheless markedly lower in this group compared to European women (14 vs. 56%) (Figure 1).
The current genome-wide scan for GxE effects in isolated CL/P was motivated by a number of observations. First, several studies have reported associations between orofacial clefts and periconceptional maternal cigarette smoking, alcohol intake and vitamin use (reviewed in Jugessur et al., 2009a; Dixon et al., 2011; Marazita, 2012; Rahimov et al., 2012; Leslie and Marazita, 2013; Beaty et al., 2016), but our comprehensive analysis of 334 autosomal cleft candidate genes showed little evidence of interaction with these maternal exposures despite being the largest GxE study of clefts at the time (Skare et al., 2012). Second, the lack of GxE effects may be due to a combination of limited statistical power to detect nothing but the largest GxE effects and the a priori small environmental contributions to CL/P (~9%) and CPO (~10%) (Grosen et al., 2011). Because most previous GxE studies have used a candidate-gene approach and are based on relatively small sample sizes, the small environmental contributions are likely to have further reduced the power to detect a GxE effect. Third, only two genome-wide studies of GxE effects have so far been published in orofacial clefts—in isolated CPO (Beaty et al., 2011; Wu et al., 2014). We thus focused on the larger sample of isolated CL/P and screened for GxE effects using the same GWAS dataset as in Wu et al. (2014).
The connection between vitamins and variants in ESRRG is novel in the context of orofacial clefts. To shed more light on how vitamins, ESRRG and clefting might relate to one another, we performed in silico analyses using the Hetionet database (Figure 9). By harnessing data from several publicly available meta-databases, Hetionet generates a detailed overview of the relationships between a given disorder/disease, genes and compounds that might easily be overlooked if the focus were solely on specific aspects of disease-gene associations (Greene et al., 2015; Himmelstein et al., 2017). Our analyses revealed a rich network of genes connecting cleft lip to ESRRG and to two vitamins in particular—vitamins A and D. These genes are significantly influenced by the levels of these two vitamins, which might partially explain why the extra vitamin intake by pregnant mothers appears to protect the fetus against clefts. There is some evidence in the literature linking vitamin A itself and genes related to vitamin A, e.g., retinoic acid receptor alpha, RARA, with the risk of orofacial clefts (Rothman et al., 1995; Mitchell et al., 2003; Bille et al., 2007; Johansen et al., 2008; Boyles et al., 2009; Skare et al., 2012). For instance, early studies in mice showed that the timing of exposure to retinoic acid (a metabolite of vitamin A) is important in the disruption of the expression patterns of key growth factors, resulting in abnormally small palatal shelves that cannot fuse (Abbott et al., 1989, 2005; Abbott and Birnbaum, 1990). Compared to vitamin A or vitamin B complex, few studies have examined the effects of high or low dose of vitamin D on clefting risk. Unfortunately, the cleft consortium (Beaty et al., 2010) did not include detailed information on the use of different types of vitamins, which would have allowed a more targeted analysis of vitamins A and D in relation to the ESRRG variants.
The impact of specific variants in ESRRG on the risk of orofacial clefts is also uncharted territory, but several lines of evidence point to a biologically plausible link between estrogens, ESRRG and craniofacial malformations. First, estrogens are a group of steroid-based sex hormones that are involved in several important developmental and physiological processes, including cartilage proliferation and growth, and formation of the craniofacial complex (Ahi, 2016). Second, sex hormones are involved in several traits associated with sexual dimorphism (Callewaert et al., 2010; Randall et al., 2013; Sanger et al., 2014). Given the consistently observed higher male-to-female ratio of isolated CL/P (~2:1 in Caucasians), it is plausible that the skewed sex prevalence is a manifestation of opposing sex steroid actions. Third, exposure to very high or very low doses of estrogens during embryonic development results in craniofacial skeletal malformations in various animal models (Fushimi et al., 2009; Cohen et al., 2014; Morthorst et al., 2016). For instance, when zebrafish are exposed to bisphenol-A (an endocrine-disrupting chemical that mimics estrogen), they develop craniofacial malformations (Kramer et al., 1990). Signaling and dosage regulation of estrogens are finely orchestrated by estrogen receptors (ERs) and estrogen related receptors (ESRRs). These two closely-related families of receptors share target genes, co-regulators and promoters (Maglott et al., 2005). ESRRG, the gene identified in our GxVitamin analyses, encodes the orphan nuclear receptor “Estrogen-related receptor γ” (ERRγ). ERRγ itself does not appear to be important for skeletal development, but it is a sex-dependent negative regulator of postnatal bone formation (Cardelli and Aubin, 2014).
The majority of the genes in Figure 9 have not previously been linked to orofacial clefts, except perhaps for PDGFA. This gene belongs to the PDGF family of genes that play important roles in the PDGF receptor-alpha (PDGFR-α) signaling pathway. Compared to PDGFA, PDGFC has a well-substantiated role in palatogenesis (Eberhart et al., 2008). Mice with the Pdgfc gene knocked out (Pdgfc−/−) exhibit a complete cleft of the secondary palate (Ding et al., 2004). The phenotype is less severe in Pdgfa−/− mice. The ligands PDGFA and PDGFC share a common pathway with the PDGFR-α receptor in regulating the development of craniofacial structures. Targeted deletion of murine Pdgfra causes neural tube closure defects, including midfacial and palatal clefts (Soriano, 1997). Retinoic acid, on the other hand, inhibits proliferation of embryonic palatal shelves in mice by downregulating Pdgfc activity (Han et al., 2006). Pedigo et al. (2007) later reported the location of a complex retinoic acid response element in a region upstream of the transcription start site of PDGFA that was previously shown to harbor basal and vitamin D-inducible enhancer activity, which lends support to the connections seen between these entities in Figure 9.
In addition to the novel insights from the biological findings, we aimed at developing a robust method for genome-wide screening of GxE effects in GWAS data. Using a log-linear model with multiplicative dose-response is a very efficient statistical approach, and case-parent triads provide sufficient information to reconstruct haplotypes with high precision. Yet, our study may still lack power to detect effects that are small. Figure 10 depicts power simulations for different sample sizes and different proportions of exposed to unexposed mothers, reflecting those that were available in the current GWAS dataset. The figure shows that there is acceptable power at a nominal significance level of 5% to detect a single-SNP GxE effect with relative risk ratio (RRR) of 1.4 or higher in a sample consisting of 2,000 triads, and an effect with RRR of 1.6 or higher with 1,000 triads. The proportion of exposed to unexposed cases does not have a major impact on power as long as it does not deviate substantially from 1-to-1. In a GWAS setting, there is of course the extra burden of extensive multiple testing. Controlling the FDR in our study for each exposure is less taxing on statistical power and better suited for discovery than, for instance, a strict Bonferroni correction. However, it increases the need for independent verification of top hits.
Figure 10.
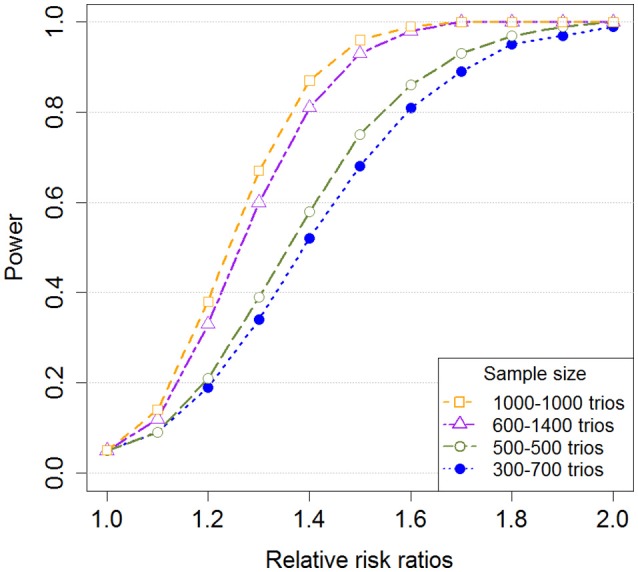
Single-SNP power for different sample sizes and proportions of exposed to unexposed mothers. The figure shows the power for detecting a GxE effect with increasing relative risk ratios (RRR). The relative risk in the smallest exposure group was increased, but the relative risk in the largest exposure group was set equal to 1. Additionally, we used a minor allele frequency (MAF) of 0.2, a nominal significance level of 5%, and a total sample size of 1,000 triads and 2,000 triads with different proportions of exposed/unexposed mothers. Note that the minor allele was used as the risk allele.
Our study has limitations. Even though it was based on the largest collection of isolated CL/P triads to date, it still had limited statistical power in the smallest group of exposed mothers. This was especially true for the GxSmoke and GxAlcohol analyses in the Asian sample, where the low level of exposure prevented any meaningful GxE analysis. The lack of a replication cohort was also a major shortcoming. Our study is the first to investigate the risk of isolated CL/P using GWAS data in two major ethnicities, and there are currently no other similar studies on isolated CL/P that can be used to confirm our findings. Further, our assumption of a detectable RRR of 1.4 with a sample size of 2,000 triads and an RRR of 1.6 with 1,000 triads may be optimistic in the context of birth defects and complex traits in general. The current scan for GxE effects in isolated CL/P should therefore be considered exploratory and hypothesis-generating at this stage, assuming that other researchers who in the future have access to comparable cleft data and more detailed information on different types of vitamins (including vitamins A and D) would be interested in replicating these findings.
To summarize, we identified significant interactions between variants in ESRRG and vitamin use in the pooled analysis. Our in silico analyses revealed an intricate network of genes linking cleft lip, ESRRG and two vitamins in particular: vitamins A and D. These GxE effects are novel and warrant further investigations to unravel the potential interplay between vitamins and ESRRG variants. If confirmed in other cleft samples, they could provide prospects for exploring the impact of estrogens and vitamins on orofacial clefts, with potential translational applications.
Author contributions
AJ, HG, RL, ØH, MG, and JR: Conception of the work, study design, data analysis and interpretation, manuscript preparation and final manuscript approval; HG, MG, JR, and ØH: Statistical modeling and software design; AJ, HG, and RL: Funding acquisition.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to the families who contributed to this study, and the orofacial cleft consortium as a whole. We also sincerely thank everyone involved in the recruitment process and the genotyping of DNA from the families.
Footnotes
Funding. This research was supported by the Bergen Medical Research Foundation (BMFS) grant 807191, by the Research Council of Norway (RCN) through its Centres of Excellence funding scheme, grant 262700, and by the Biobank Norway II grant 245464/F50 from the RCN.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00060/full#supplementary-material
References
- Abbott B. D., Best D. S., Narotsky M. G. (2005). Teratogenic effects of retinoic acid are modulated in mice lacking expression of epidermal growth factor and transforming growth factor-α. Birth Defects Res. Part A Clin. Mol. Teratol. 73, 204–217. 10.1002/bdra.20117 [DOI] [PubMed] [Google Scholar]
- Abbott B. D., Birnbaum L. S. (1990). TCDD-induced altered expression of growth factors may have a role in producing cleft palate and enhancing the incidence of clefts after coadministration of retinoic acid and TCDD. Toxicol. Appl. Pharmacol. 106, 418–432. 10.1016/0041-008X(90)90337-T [DOI] [PubMed] [Google Scholar]
- Abbott B. D., Harris M. W., Birnbaum L. S. (1989). Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 40, 533–553. 10.1002/tera.1420400602 [DOI] [PubMed] [Google Scholar]
- Ahi E. P. (2016). Signalling pathways in trophic skeletal development and morphogenesis: insights from studies on teleost fish. Dev. Biol. 420, 11–31. 10.1016/j.ydbio.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Bastian F., Parmentier G., Roux J., Moretti S., Laudet V., Robinson-Rechavi M. (2008). Bgee: integrating and comparing heterogeneous transcriptome data among species, in Data Integration in the Life Sciences: 5th International Workshop, DILS 2008, Evry, France, June 25-27, (2008). eds Bairoch A., Cohen-Boulakia S., Froidevaux C. (Berlin; Heidelberg: Springer; ), 124–131. [Google Scholar]
- Beaty T. H., Marazita M. L., Leslie E. J. (2016). Genetic factors influencing risk to orofacial clefts: today's challenges and tomorrow's opportunities. F1000Res 5:2800. 10.12688/f1000research.9503.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty T. H., Murray J. C., Marazita M. L., Munger R. G., Ruczinski I., Hetmanski J. B., et al. (2010). A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat. Genet. 42, 525–529. 10.1038/ng.580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaty T. H., Ruczinski I., Murray J. C., Marazita M. L., Munger R. G., Hetmanski J. B., et al. (2011). Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate. Genet. Epidemiol. 35, 469–478. 10.1002/gepi.20595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bille C., Olsen J., Vach W., Knudsen V. K., Olsen S. F., Rasmussen K., et al. (2007). Oral clefts and life style factors–a case-cohort study based on prospective Danish data. Eur. J. Epidemiol. 22, 173–181. 10.1007/s10654-006-9099-5 [DOI] [PubMed] [Google Scholar]
- Boyles A. L., Wilcox A. J., Taylor J. A., Shi M., Weinberg C. R., Meyer K., et al. (2009). Oral facial clefts and gene polymorphisms in metabolism of folate/one-carbon and vitamin A: a pathway-wide association study. Genet. Epidemiol. 33, 247–255. 10.1002/gepi.20376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert F., Sinnesael M., Gielen E., Boonen S., Vanderschueren D. (2010). Skeletal sexual dimorphism: relative contribution of sex steroids, GH-IGF1, and mechanical loading. J. Endocrinol. 207, 127–134. 10.1677/JOE-10-0209 [DOI] [PubMed] [Google Scholar]
- Cardelli M., Aubin J. E. (2014). ERRgamma is not required for skeletal development but is a RUNX2-dependent negative regulator of postnatal bone formation in male mice. PLoS ONE 9:e109592 10.1371/journal.pone.0109592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D. G. (2009). Prediction and interaction in complex disease genetics: experience in type 1 diabetes. PLoS Genet. 5:e1000540. 10.1371/journal.pgen.1000540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. P., LaChappelle A. R., Walker B. S., Lassiter C. S. (2014). Modulation of estrogen causes disruption of craniofacial chondrogenesis in Danio rerio. Aquat. Toxicol. 152, 113–120. 10.1016/j.aquatox.2014.03.028 [DOI] [PubMed] [Google Scholar]
- Dempster A. P., Laird N. M., Rubin D. B. (1977). Maximum likelihood from incomplete Data via the EM algorithm. J. R. Stat. Soc. Ser. B 39, 1–38. [Google Scholar]
- DeRoo L. A., Wilcox A. J., Drevon C. A., Lie R. T. (2008). First-trimester maternal alcohol consumption and the risk of infant oral clefts in Norway: a population-based case-control study. Am. J. Epidemiol. 168, 638–646. 10.1093/aje/kwn186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H., Wu X., Boström H., Kim I., Wong N., Tsoi B., et al. (2004). A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat. Genet. 36, 1111–1116. 10.1038/ng1415 [DOI] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H., Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167–178. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart J. K., He X., Swartz M. E., Yan Y. L., Song H., Boling T. C., et al. (2008). MicroRNA Mirn140 modulates Pdgf signaling during palatogenesis. Nat. Genet. 40, 290–298. 10.1038/ng.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi S., Wada N., Nohno T., Tomita M., Saijoh K., Sunami S., et al. (2009). 17β-Estradiol inhibits chondrogenesis in the skull development of zebrafish embryos. Aquat. Toxicol. 95, 292–298. 10.1016/j.aquatox.2009.03.004 [DOI] [PubMed] [Google Scholar]
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerdevik M., Haaland O. A., Romanowska J., Lie R. T., Jugessur A., Gjessing H. K. (2017). Parent-of-origin-environment interactions in case-parent triads with or without independent controls. Ann. Hum. Genet. 82, 60–73. 10.1111/ahg.12224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjessing H. K., Lie R. T. (2006). Case-parent triads: estimating single- and double-dose effects of fetal and maternal disease gene haplotypes. Ann. Hum. Genet. 70(Pt 3), 382–396. 10.1111/j.1529-8817.2005.00218.x [DOI] [PubMed] [Google Scholar]
- Greene C. S., Krishnan A., Wong A. K., Ricciotti E., Zelaya R. A., Himmelstein D. S., et al. (2015). Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576. 10.1038/ng.3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosen D., Bille C., Petersen I., Skytthe A., Hjelmborg J. B., Pedersen J. K., et al. (2011). Risk of oral clefts in twins. Epidemiology 22, 313–319. 10.1097/EDE.0b013e3182125f9c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaland Ø. A., Jugessur A., Gjerdevik M., Romanowska J., Shi M., Beaty T. H., et al. (2017). Genome-wide analysis of parent-of-origin interaction effects with environmental exposure (PoOxE): an application to European and Asian cleft palate trios. PLoS ONE 12:e0184358. 10.1371/journal.pone.0184358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Xiao Y., Lin J., Li Y. (2006). PDGF-C controls proliferation and is down-regulated by retinoic acid in mouse embryonic palatal mesenchymal cells. Birth Defects Res. B Dev. Reprod. Toxicol. 77, 438–444. 10.1002/bdrb.20094 [DOI] [PubMed] [Google Scholar]
- Hayes C. (2002). Environmental risk factors and oral clefts, in Cleft Lip and Palate: From Origin to Treatment, ed Wyszynski D. F. (New York, NY: Oxford University Press; ), 159–169. [Google Scholar]
- Hernández-Díaz S., Werler M. M., Walker A. M., Mitchell A. A. (2000). Folic acid antagonists during pregnancy and the risk of birth defects. N. Engl. J. Med. 343, 1608–1614. 10.1056/NEJM200011303432204 [DOI] [PubMed] [Google Scholar]
- Himmelstein D. S., Lizee A., Hessler C., Brueggeman L., Chen S. L., Hadley D., et al. (2017). Systematic integration of biomedical knowledge prioritizes drugs for repurposing. Elife 6:e26726. 10.7554/eLife.26726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes L. B., Harvey E. A., Coull B. A., Huntington K. B., Khoshbin S., Hayes A. M., et al. (2001). The teratogenicity of anticonvulsant drugs. N. Engl. J. Med. 344, 1132–1138. 10.1056/NEJM200104123441504 [DOI] [PubMed] [Google Scholar]
- Johansen A. M., Lie R. T., Wilcox A. J., Andersen L. F., Drevon C. A. (2008). Maternal dietary intake of vitamin A and risk of orofacial clefts: a population-based case-control study in Norway. Am. J. Epidemiol. 167, 1164–1170. 10.1093/aje/kwn035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A., Farlie P. G., Kilpatrick N. (2009a). The genetics of isolated orofacial clefts: from genotypes to subphenotypes. Oral Dis. 15, 437–453. 10.1111/j.1601-0825.2009.01577.x [DOI] [PubMed] [Google Scholar]
- Jugessur A., Lie R. T., Wilcox A. J., Murray J. C., Taylor J. A., Saugstad O. D., et al. (2003). Variants of developmental genes (TGFA, TGFB3, and MSX1) and their associations with orofacial clefts: a case-parent triad analysis. Genet. Epidemiol. 24, 230–239. 10.1002/gepi.10223 [DOI] [PubMed] [Google Scholar]
- Jugessur A., Rahimov F., Lie R. T., Wilcox A. J., Gjessing H. K., Nilsen R. M., et al. (2008). Genetic variants in IRF6 and the risk of facial clefts: single-marker and haplotype-based analyses in a population-based case-control study of facial clefts in Norway. Genet. Epidemiol. 32, 413–424. 10.1002/gepi.20314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A., Shi M., Gjessing H. K., Lie R. T., Wilcox A. J., Weinberg C. R., et al. (2009b). Genetic determinants of facial clefting: analysis of 357 candidate genes using two national cleft studies from Scandinavia. PLoS ONE 4:e5385. 10.1371/journal.pone.0005385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugessur A., Skare Ø., Harris J. R., Lie R. T., Gjessing H. K. (2012a). Using offspring-parent triads to study complex traits: a tutorial based on orofacial clefts. Norweg. J. Epidemiol. 21, 251–267. 10.5324/nje.v21i2.1503 [DOI] [Google Scholar]
- Jugessur A., Skare Ø., Lie R. T., Wilcox A. J., Christensen K., Christiansen L., et al. (2012b). X-linked genes and risk of orofacial clefts: evidence from two population-based studies in Scandinavia. PLoS ONE 7:e39240. 10.1371/journal.pone.0039240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff D. M. (2002). Mapping studies in animal models, in Cleft Lip and Palate: From Origin to Treatment, ed Wyszynski D. F. (New York, NY: Oxford University Press; ), 265–282. [Google Scholar]
- Kibbe W. A., Arze C., Felix V., Mitraka E., Bolton E., Fu G., et al. (2015). Disease ontology 2015 update: an expanded and updated database of human diseases for linking biomedical knowledge through disease data. Nucleic Acids Res. 43, D1071–D1078. 10.1093/nar/gku1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer P. L., de Leon D., Ozelius L., Risch N., Bressman S. B., Brin M. F., et al. (1990). Dystonia gene in Ashkenazi Jewish population is located on chromosome 9q32-34. Ann. Neurol. 27, 114–120. 10.1002/ana.410270203 [DOI] [PubMed] [Google Scholar]
- Law V., Knox C., Djoumbou Y., Jewison T., Guo A. C., Liu Y., et al. (2014). DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097. 10.1093/nar/gkt1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie E. J., Marazita M. L. (2013). Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin. Med. Genet. 163C, 246–258. 10.1002/ajmg.c.31381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie R. T., Wilcox A. J., Taylor J., Gjessing H. K., Saugstad O. D., Aabyholm F., et al. (2008). Maternal smoking and oral clefts: the role of detoxification pathway genes. Epidemiology 19, 606–615. 10.1097/EDE.0b013e3181690731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J., Cardy A., Munger R. G. (2004). Tobacco smoking and oral clefts: a meta-analysis. Bull. World Health Organ. 82, 213–218. [PMC free article] [PubMed] [Google Scholar]
- Maglott D., Ostell J., Pruitt K. D., Tatusova T. (2005). Entrez gene: gene-centered information at NCBI. Nucleic Acids Res. 33, D54–D58. 10.1093/nar/gki031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangold E., Ludwig K. U., Nöthen M. M. (2011). Breakthroughs in the genetics of orofacial clefting. Trends Mol. Med. 17, 725–733. 10.1016/j.molmed.2011.07.007 [DOI] [PubMed] [Google Scholar]
- Marazita M. L. (2012). The evolution of human genetic studies of cleft lip and cleft palate. Annu. Rev. Genomics Hum. Genet. 13, 263–283. 10.1146/annurev-genom-090711-163729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millicovsky G., Johnston M. C. (1981a). Hyperoxia and hypoxia in pregnancy: simple experimental manipulation alters the incidence of cleft lip and palate in CL/Fr mice. Proc. Natl. Acad. Sci. U.S.A. 78, 5722–5723. 10.1073/pnas.78.9.5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millicovsky G., Johnston M. C. (1981b). Maternal hyperoxia greatly reduces the incidence of phenytoin-induced cleft lip and palate in A/J mice. Science 212, 671–672. 10.1126/science.7221553 [DOI] [PubMed] [Google Scholar]
- Mitchell L. E., Murray J. C., O'Brien S., Christensen K. (2003). Retinoic acid receptor alpha gene variants, multivitamin use, and liver intake as risk factors for oral clefts: a population-based case-control study in Denmark, 1991-1994. Am. J. Epidemiol. 158, 69–76. 10.1093/aje/kwg102 [DOI] [PubMed] [Google Scholar]
- Morthorst J. E., Korsgaard B., Bjerregaard P. (2016). Severe malformations of eelpout (Zoarces viviparus) fry are induced by maternal estrogenic exposure during early embryogenesis. Mar. Environ. Res. 113, 80–87. 10.1016/j.marenvres.2015.11.007 [DOI] [PubMed] [Google Scholar]
- Mossey P., Castilla E. E. (2003). Global Registry and Database on Craniofacial Anomalies. Geneva: World Health Organization. [Google Scholar]
- Munger R. G. (2002). Maternal nutrition and oral clefts, in Cleft Lip and Palate: From Origin to Treatment, ed Wyszynski D. F. (New York, NY: Oxford University Press; ), 170–192. [Google Scholar]
- Munger R. G., Sauberlich H. E., Corcoran C., Nepomuceno B., Daack-Hirsch S., Solon F. S. (2004). Maternal vitamin B-6 and folate status and risk of oral cleft birth defects in the Philippines. Birth Defects Res. Part A Clin. Mol. Teratol. 70, 464–471. 10.1002/bdra.20037 [DOI] [PubMed] [Google Scholar]
- Ng M., Freeman M. K., Fleming T. D., Robinson M., Dwyer-Lindgren L., Thomson B., et al. (2014). Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. JAMA 311, 183–192. 10.1001/jama.2013.284692 [DOI] [PubMed] [Google Scholar]
- Pedigo N. G., Zhang H., Mishra A., McCorkle J. R., Ormerod A. K., Kaetzel D. M. (2007). Retinoic acid inducibility of the human PDGF-a gene is mediated by 5′-distal DNA motifs that overlap with basal enhancer and vitamin D response elements. Gene Expr. 14, 1–12. 10.3727/000000007783991763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim R. J., Welch R. P., Sanna S., Teslovich T. M., Chines P. S., Gliedt T. P., et al. (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337. 10.1093/bioinformatics/btq419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F., Jugessur A., Murray J. C. (2012). Genetics of nonsyndromic orofacial clefts. Cleft Palate Craniofac. J. 49, 73–91. 10.1597/10-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall J. C., Winkler T. W., Kutalik Z., Berndt S. I., Jackson A. U., Monda K. L., et al. (2013). Sex-stratified genome-wide association studies including 270,000 individuals show sexual dimorphism in genetic loci for anthropometric traits. PLoS Genet. 9:e1003500. 10.1371/journal.pgen.1003500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T., Taşan M., Charloteaux B., Pevzner S. J., Zhong Q., Sahni N., et al. (2014). A proteome-scale map of the human interactome network. Cell 159, 1212–1226. 10.1016/j.cell.2014.10.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman K. J., Moore L. L., Singer M. R., Nguyen U. S., Mannino S., Milunsky A. (1995). Teratogenicity of high vitamin A intake. N. Engl. J. Med. 333, 1369–1373. 10.1056/NEJM199511233332101 [DOI] [PubMed] [Google Scholar]
- Sanger T. J., Seav S. M., Tokita M., Langerhans R. B., Ross L. M., Losos J. B., et al. (2014). The oestrogen pathway underlies the evolution of exaggerated male cranial shapes in Anolis lizards. Proc. Biol. Sci. 281:20140329. 10.1098/rspb.2014.0329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaid D. J. (2002). Disease-marker association, in Biostatistical Genetics and Genetic Epidemiology, eds Elston R. C., Olson J. M., Palmer L. (Chichester: John Wiley & Sons Ltd; ), 206–217. [Google Scholar]
- Shi M., Christensen K., Weinberg C. R., Romitti P., Bathum L., Lozada A., et al. (2007). Orofacial cleft risk is increased with maternal smoking and specific detoxification-gene variants. Am. J. Hum. Genet. 80, 76–90. 10.1086/510518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skare O., Jugessur A., Lie R. T., Wilcox A. J., Murray J. C., Lunde A., et al. (2012). Application of a novel hybrid study design to explore gene-environment interactions in orofacial clefts. Ann. Hum. Genet. 76, 221–236. 10.1111/j.1469-1809.2012.00707.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1997). The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development 124, 2691–2700. [DOI] [PubMed] [Google Scholar]
- Storey J. D., Tibshirani R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445. 10.1073/pnas.1530509100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox A. J., Lie R. T., Solvoll K., Taylor J., McConnaughey D. R., Abyholm F., et al. (2007). Folic acid supplements and risk of facial clefts: national population based case-control study. BMJ 334:464. 10.1136/bmj.39079.618287.0B [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk M. B., Gnanadesikan R. (1968). Probability plotting methods for the analysis of data. Biometrika 55, 1–17. 10.2307/2334448 [DOI] [PubMed] [Google Scholar]
- Wu T., Liang K. Y., Hetmanski J. B., Ruczinski I., Fallin M. D., Ingersoll R. G., et al. (2010). Evidence of gene-environment interaction for the IRF6 gene and maternal multivitamin supplementation in controlling the risk of cleft lip with/without cleft palate. Hum. Genet. 128, 401–410. 10.1007/s00439-010-0863-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T., Schwender H., Ruczinski I., Murray J. C., Marazita M. L., Munger R. G., et al. (2014). Evidence of gene-environment interaction for two genes on chromosome 4 and environmental tobacco smoke in controlling the risk of nonsyndromic cleft palate. PLoS ONE 9:e88088. 10.1371/journal.pone.0088088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W., Lu J., Weng J., Jia W., Ji L., Xiao J., et al. (2010). Prevalence of diabetes among men and women in China. N. Engl. J. Med. 362, 1090–1101. 10.1056/NEJMoa0908292 [DOI] [PubMed] [Google Scholar]
- Zeiger J. S., Beaty T. H. (2002). Gene-environment interaction and risk to oral clefts, in Cleft Lip and Palate: From Origin to Treatment, ed Wyszynski D. F. E. (New York, NY: Oxford University Press; ), 283–289. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.