

Abstract
Angioimmunoblastic T‐cell lymphoma (AITL) is an age‐related malignant lymphoma, characterized by immune system‐dysregulated symptoms. Recent sequencing studies have clarified the recurrent mutations in ras homology family member A (RHOA) and in genes encoding epigenetic regulators, tet methyl cytosine dioxygenase 2 (TET2), DNA methyl transferase 3 alpha (DNMT3A) and isocitrate dehydrogenase 2, mitochondrial (IDH2), as well as those related to the T‐cell receptor signaling pathway in AITL. In this review, we focus on how this genetic information has changed the understanding of the developmental process of AITL and will in future lead to individualized therapies for AITL patients.
Keywords: angioimmunoblastic T‐cell lymphoma, epigenetic regulator, multistep and multilineage tumorigenesis, ras homology family member A, T‐cell receptor‐signaling
1. INTRODUCTION
Recent progress in next‐generation sequencing has provided emerging evidence of characteristic genetic abnormalities in angioimmunoblastic T‐cell lymphoma (AITL). In this review, we provide insight into how the biologic and clinical aspects of AITL are linked to its genetic features.
1.1. Angioimmunoblastic T‐cell lymphoma belongs to a nodal T‐cell lymphoma with T follicular helper phenotype
Angioimmunoblastic T‐cell lymphoma (AITL) is a subtype of malignant lymphoma. Together with nodal peripheral T‐cell lymphomas (PTCL) with T follicular helper (TFH) phenotype and follicular T‐cell lymphoma (FTCL), AITL belongs to nodal T‐cell lymphoma with TFH phenotype, a newly proposed entity in the 2016 revised WHO classification.1 Follicular helper T cells, a subset of helper T cells, reside mainly in the follicles to support B‐cell survival, proliferation, maturation and migration.2 The TFH phenotype is determined by expression of 2 or 3 markers that are expressed both in normal follicular helper T cells and in tumor cells: CD279/programmed death‐1 (PD1) and inducible T‐cell costimulator (ICOS), T‐cell coinhibitory and costimulatory molecules; CD10, a membrane metalloendopeptidase; B‐cell lymphoma 6 protein (BCL6), a key transcription factor for TFH development; C‐X‐C motif chemokine ligand 13 (CXCL13) and c‐x‐c motif chemokine receptor 5 (CXCR5), a chemokine and chemokine receptor; and signaling lymphocyte activation molecule (SLAM)‐associated protein (SAP), an adaptor protein for SLAM family receptors.1 Some gene mutations are commonly found in diseases categorized into nodal T‐cell lymphomas with TFH phenotype, and AITL‐specific mutations have been identified. (Note: See the Section below, “1.4.”)
1.2. Angioimmunoblastic T‐cell lymphoma is an age‐related lymphoma, presenting with symptoms of immune system dysregulation
The incidence of AITL increases with age, with the median age at onset reported to be 59‐65 years.3 The prevalence of AITL in elderly individuals may be tightly linked to the age‐related premalignant mutations in AITL. (Note: See the Section, “1.9.”) AITL patients display generalized lymphadenopathy, a characteristic symptom of malignant lymphomas. Furthermore, the symptoms suggestive of immunologic hyperactivation are also present in AITL: fever, rash, Coombs test‐positive hemolytic anemia and polyarthritis.3 Again, these immune system‐related symptoms may be attributable to genetic events involving multiple components of T‐cell receptor (TCR) signaling pathways. (Note: See the Section below, “1.4.”)
1.3. Massive infiltration of accessory cells occurs in angioimmunoblastic T‐cell lymphoma
Various immune cells, including nontumor reactive T cells, B cells (some of which are infected by Epstein–Barr virus [EBV]), eosinophils and macrophages, invade AITL tissues.3 Moreover, the blood vessels are markedly increased and often surrounded by AITL tumor cells. In addition, follicular dendritic cells (FDC) are also prominently present near the tumor cells and blood vessels.3 As mentioned above, AITL tumor cells resemble cytokine‐producing and chemokine‐producing TFH cells.4 Cytokines and chemokines released from TFH‐like tumor cells may recruit immune cells, blood vessels and FDC into AITL tissues, and activate them to further produce cytokines and chemokines. This positive circuit of cytokines and chemokines may exacerbate the trafficking of these cells into AITL tissues. For instance, the CXCL13 and its receptor CXCR5 network is thought to promote recruitment of B cells and FDC as well as tumor cells into AITL tissues.3 Vascular endothelial growth factor (VEGF), a cytokine that promotes angiogenesis, is expressed in both tumor and vascular endothelial cells.5 Cytokine‐producing helper T17 (Th17) cells as well as CD8‐positive T cells are also enriched in AITL tissues.6 Mast cells in AITL tissues function as producers of VEGF, to recruit endothelial cells,7 and of interleukin‐6 (IL‐6), to proliferate Th17 cells.8
Although the cytokine and chemokine circuit originating from TFH‐like tumor cells may account for the massive infiltration of immune cells into AITL tissues, novel genetic evidence indicated that tumor‐infiltrating cells may not be entirely attributable to the reactive process.9 The infiltrating B cells in AITL tissues had gene mutations distinct from those found in tumor cells.9 The genetic events in tumor‐infiltrating cells may synergize with the cytokine‐and chemokine‐mediated reactions to produce the pathologic features of AITL. (Note: See the Section below, “1.10.”)
1.4. Ras homology family member A, epigenetic regulators, and T‐cell signaling molecules are the main players in the genetic abnormalities of angioimmunoblastic T‐cell lymphoma
Recent genetic studies identified recurrent mutations in ras homolog family member A (RHOA) (50%‐70%)10, 11, 12 and in genes encoding the epigenetic regulators, tet methyl cytosine dioxygenase 2 (TET2) (47%‐83%),10, 13 DNA methyltransferase 3 alpha (DNMT3A) (20%‐30%)10, 11, 14 and isocitrate dehydrogenase 2, mitochondrial (IDH2) (20%‐45%),10, 15 as well as the components of the TCR signaling pathways, phospholipase C gamma 1 (PLC γ) (14%),16 CD28 (9%‐11%),16, 17 FYN protooncogene, Src family tyrosine kinase (FYN) (3%‐4%)11, 16 and vav guanine nucleotide exchange factor 1 (VAV1) (5%)16 in AITL (Table 1).
Table 1.
Recurrent gene mutations in AITL
| Frequencies (%) | References | |
|---|---|---|
| RAS superfamily | ||
| RHOA | 50‐70 | 10, 11, 12 |
| Epigenetic regulators | ||
| TET2 | 47‐83 | 10, 13 |
| DNMT3A | 20‐30 | 10, 11, 14 |
| IDH2 | 20‐45 | 10, 15 |
| TCR signaling pathway | ||
| PLCγ | 14 | 16 |
| CD28 | 9‐11 | 16, 17 |
| FYN | 3‐4 | 11, 16 |
| VAV1 | 5 | 16 |
AITL, angioimmunoblastic T‐cell lymphoma; DNMT3A, DNA methyltransferase 3 alpha; FYN, FYN protooncogene, Src family tyrosine kinase; IDH2, isocitrate dehydrogenase 2, mitochondrial; PLCγ, phospholipase C gamma 1; RHOA, ras homolog gene family, member A; TCR, T‐cell receptor; TET2, tet methylcytosine dioxygenase 2; VAV1, vav guanine nucleotide exchange factor 1.
Almost all the RHOA mutations found in AITL were p.G17V (G17V RHOA mutations).10, 11, 12 G17V RHOA mutations were commonly observed in the other nodal T‐cell lymphomas with TFH phenotype: 57%‐62% of nodal PTCL with TFH phenotype10, 14 and 60% of FTCL,14 while they were quite rare in other diseases. In contrast, TET2 and DNMT3A mutations were found in a broad range of hematologic malignancies,18 and even in healthy elderly individuals.19, 20 (Note: See the Section below, “1.9.”) Among T‐cell lymphomas, TET2 mutations were more prevalent in nodal T‐cell lymphomas with TFH phenotype than those without TFH phenotype (nodal PTCL with TFH phenotype vs FTCL vs PTCL without the TFH phenotype: 64% vs 75% vs 17%).14 IDH2 mutations were also found in myeloid malignancies. However, IDH2 mutations were not detectable in the other T‐cell lymphomas,10, 15 even those with the TFH phenotype,14 suggesting that IDH2 mutations may provide AITL with its specific pathologic features. The mutations involving components of the TCR signaling pathways were commonly observed in nodal PTCL with TFH phenotype,16 although CD28 mutations were specific to AITL.17 Notably, the AITL genome exhibited a specific combination of these mutations: the RHOA‐mutated samples also had TET2 mutations, while a part of the RHOA and TET2‐mutated samples had IDH2 mutations.10 These combinations may have a synergistic effect on oncogenesis.
1.5. Diagnostic impact of G17V ras homology family member A mutations
As mentioned above, G17V RHOA mutations were commonly identified in nodal T‐cell lymphomas with the TFH phenotype,10, 14 although they were also observed in a few cases of adult T‐cell leukemia/lymphoma (ATLL),21 which can be distinguished by its integration of human T‐lymphotropic virus (HTLV)‐1. Therefore, G17V RHOA mutations serve as a genetic indicator to detect nodal T‐cell lymphomas with the TFH phenotype. The tumor ratio is sometimes low because of the prominent reactive cells, which makes it difficult to detect G17V RHOA mutations by direct sequencing. It is reported that the allele‐specific PCR (AS‐PCR) assay is an easy‐to‐use method to detect G17V RHOA mutations.22 The positive and negative concordance rates between AS‐PCR and amplicon‐based deep sequencing were as high as 95%.22 G17 RHOA mutations were also detectable in cell‐free DNA, enabling their application in noninvasive diagnostic testing of AITL.23
1.6. Oncogenic roles of ras homology family member A mutations are under investigation
RHOA is a small guanine nucleotide triphosphate (GTP)‐binding protein. RHOA mediates fundamental biologic processes, including cell mortality, adhesion, the cell cycle and cytokinesis. While the functions of RHOA in peripheral T cells have not been fully elucidated, the conditional deletion of the RhoA gene under the control of the CD2 or Lck promoters resulted in severe defects in thymocyte development in mice.24 RHOA carries out a switch‐like function by making a round trip between the guanine nucleotide diphosphate (GDP)‐bound inactive state and the guanine nucleotide triphosphate (GTP)‐bound active state.25 The 17th glycine of RHOA is located at a position essential for binding to GTP.10 In a Rhotekin pull‐down assay to detect GTP‐bound RHOA, the G17V RHOA mutant was not bound to GTP,10, 11, 12 indicating that the G17V RHOA mutant does not mediate classical RHOA signaling. Curiously, the p.K18N mutant existing in a few AITL samples had higher GTP‐binding capacity.16 Therefore, the oncogenic roles of the G17V RHOA mutant in AITL development may not be due to the disruption of classical RHOA signaling. Rather, the existence of mutations at a single amino acid strongly suggests that the G17V mutant acquires a specific oncogenic role. Recently it was reported that the G17V mutant activated TCR pathway through direct binding to VAV1, an essential mediator of the TCR pathway.26 Together with the frequent mutations in TCR pathway, aberrant activation of TCR pathway by the G17V RHOA mutant may be a clue for AITL development. (See the Section, “1.8.”)
Other RHOA mutations are reported in diffuse‐type gastric carcinoma,27 Burkitt lymphoma28 and ATLL.21 The most frequent RHOA mutations in gastric carcinoma and Burkitt lymphoma were the p.Y42C and p.R5Q mutations,27, 28 while the p.C16R mutations were the most frequent in ATLL.21 Whether these various RHOA mutations share common downstream molecules essential for oncogenesis remains to be elucidated.
1.7. Mutations in epigenetic regulators
TET2 encodes a methylcytosine, dioxygenase, to convert methylation cytosine (mC) to hydroxymethylcytosine (hmC), formylcytosine (fC) and carboxylcytosine (CaC).29 These modified cytosines function as intermediates of the passive or active demethylating process, and as epigenetic marks.29 Nonsense and frameshift mutations were distributed throughout the entire TET2 protein, while missense mutations almost always existed at the C‐terminal catalytic domain in AITL, as in myeloid malignancies.10, 13 This distribution of mutations indicates that TET2 mutations are loss‐of‐function mutations. DNMT3A encodes a DNA methyltransferase, which methylates nonmethylated CpG. DNMT3A mutations were distributed across the entire protein. Hotspot p.R882 mutations accounted for approximately 15% of DNMT3A mutations in AITL,10 while they accounted for more than half of the mutations in myeloid malignancies.30 The p.R882H DNMT3A mutant was shown to have reduced methyltransferase activity and also to dominant‐negatively inhibit wildtype DNMT3A by interference with homotetramer formation.31 DNMT3A and TET2 mutations were sometimes seen together in AITL32 as well as in myeloid malignancies, although the epigenetic effects were opposite. The synergistic effects of TET2 and DNMT3A loss on AITL development were shown using a mouse model33 (Note: See the Section below, “1.11.”) In AITL, IDH2 mutations were exclusively present at the p.R172 position,10, 15 while both p.R140 and p.R172 IDH2 mutations and p.R132 IDH1 mutations were seen in myeloid malignancies.34 Under physiologic conditions, IDH enzymes convert isocitric acid to α‐ketoglutaric acid (α‐KG) in an NADP+‐dependent manner. α‐KG functions as an intermediate metabolite of the tricarboxylic acid (TCA) cycle and also as a substrate in enzymes that are not included in the TCA cycle. IDH mutants have been shown to aberrantly produce D‐2‐hydroxyglutarate (D‐2‐HG), a so‐called oncometabolite. D‐2‐HG inhibits the α‐KG‐dependent enzymatic activity of dioxygenases, including the TET family of proteins and Jumonji‐C histone demethylases.34 As mentioned above, IDH2 and TET2 mutations coexist in AITL samples, suggesting that TET proteins other than TET2 or Jumonji‐C histone demethylases may be the main targets of the oncometabolite. In fact, it was shown that both DNA methylation and histone H3K27 methylation were more prominent in AITL samples with TET2 and IDH2 mutations than in those with TET2/without IDH2 mutations.35
Hypomethylating reagents are currently in clinical use for myelodysplastic syndrome. These reagents tend to be more effective in TET2‐mutated cases than in TET2 wildtype cases. Highly prevalent TET2 mutations in AITL suggest that AITL may also respond to these hypomethylating reagents. In fact, several AITL cases effectively treated with azacytidine have been reported.36, 37
1.8. T‐cell receptor‐related mutations
Upon TCR stimulation, CD28 functions as a costimulatory molecule to support full and sustained T‐cell activation. Subsequently, FYN, a Src kinase, is activated, resulting in further phosphorylation of downstream molecules (ie PLCγ and VAV1). VAV1, known as a GEF protein, also functions as an adaptor to facilitate and activate the TCR proximal signaling complex, involving PLCγ and SLP‐76. PLCγ catalyzes phosphatidylinositol 4, 5‐bisphosphate (PI(4,5)P2) into inositol‐1, 4, 5‐trisphosphate (IP3) and diacylglycerol (DAG), leading to intracellular signal transduction through calcium mobilization and activation of protein kinase C (PKC). Activating mutations were observed in these players participating in TCR signaling.
CD28 mutations were accumulated at 2 hotspots, p.D124 and p.T195.16, 17 The p.D124 mutant was shown to have higher affinity for the ligands CD80 and CD86,17 while the p.T195 mutant had higher affinity for the intracellular adaptor proteins GADS/GRAP2 and GRB2.17, 38 The CTLA4‐CD28 39 and ICOS‐CD28 fusion genes17 have also been described. FYN mutations found in the SH2 domain and C terminus were activating mutations, presumably through the disruption of the intramolecular inhibitory interaction between the SH2 domain and the C terminus.11 PLCγ mutations were found in several functional motifs, including the PI‐PLC, SH2, SH3 and C2 domain. PLCγ mutations were also shown to be activating mutations,16 although the biologic consequence of these mutations has yet to be clarified. VAV1 mutations were found at several hotspots,16 although the oncogenic mechanisms of these mutations remain unclear. In addition, the C‐terminal portion of VAV1, participating in intramolecular inhibition, was recurrently deleted by 2 distinct mechanisms: an alternative splicing mechanism resulting from in‐flame deletion of the N‐terminal site of the CSH3 domain40 and formation of fusion genes with several distinct partners.40, 41
Although the genetic evidence suggests that activation of TCR signaling by gene mutations may play a role in the symptoms and progression of AITL, it has not been exactly proven by in vivo experiments. Cyclosporin A, a calcineurin inhibitor that blocks TCR signaling, is widely used for the treatment of immune system‐mediated diseases.42 Cyclosporin A as a single reagent43 or with other immunosuppressive reagents44 was shown to effectively ameliorate the progression and symptoms of AITL. The effectiveness of cyclosporine A supports the hypothesis that activation of TCR signaling may actually contribute to AITL progression and that it can be a candidate pathway in targeted therapies.
1.9. Age‐related mutations may precede angioimmunoblastic T‐cell lymphoma
Hematologic malignancies are classified according to their normal counterparts; that is, normal cells sharing the characteristics of tumor cells. Furthermore, they had previously been thought to originate from their normal counterparts; for example, AITL had been thought to originate from its normal counterpart, TFH cells. However, we now believe that at least some hematologic malignancies including AITL may originate from immature blood cells.18 The TET2 and DNMT3A mutations detected in tumor cells were also recognized in the tumor‐free peripheral blood cells,45 bone marrow cells10, 32, 45 and hematopoietic progenitors32, 45 of AITL patients. Some patients simultaneously or serially developed both AITL and myeloid malignancies. Identical TET2 and DNMT3A mutations were reported to be present in both diseases, suggesting that both diseases originate from premalignant cells harboring these mutations.32, 37 When a nationwide survey was conducted to examine the cooccurrence of myeloid and lymphoid malignancies, 72 cases were identified: 45 cases having the diseases simultaneously and 27 cases sequentially.46 Whether the multiple diseases in these cases actually had common ancestors remains to be elucidated.
Finally, somatic mutations were also detected even in healthy individuals.19, 20 Mutation frequencies were reportedly increased with age: by 5% for those in their 60s, by 10% to 15% for those in their 70s, and by 10% to 25% for those in their 80s.19, 20 Somatic mutations were also detected in 95% of individuals aged 50‐60 years when the detection sensitivity was set at 0.0003 variant allele frequencies (VAF).47 The most frequently mutated genes in the healthy individuals were DNMT3A, TET2 and ASXL1.19, 20 Although these mutations were first found in hematologic malignancies, they may be defined as age‐related mutations. The status of having somatic mutations without any evidence of hematologic diseases is called clonal hematopoiesis of indeterminate potential (CHIP).48 CHIP was related to a high incidence of blood cancers and inferior overall survival.19, 20 The presence of premalignant mutations in AITL patients suggests that CHIP may precede AITL in most cases. However, the actual incidence rate of AITL caused by CHIP has not been determined. Indeed, because of its rarity, it would be tough to determine the incidence rate.
TET2 and DNMT3A mutations themselves in premalignant cells may not be sufficient to induce AITL development. Multiple TET2 mutations were frequently observed in AITL tissues,10 while TET2 mutations were heterozygous in CHIP as well as in myeloid malignancies. When the distribution of TET2 mutations were examined in 19 AITL/PTCL samples using laser microdissection followed by targeted sequencing, 10 samples had 2 distinct TET2 mutations, while 6 had one TET2 mutation.9 Although both mutations were determined as premalignant mutations in 5 of the samples, the 5 samples had 1 mutation as a premalignant mutation and the other as a tumor‐specific mutation.9 These observations suggest that the profound defect in TET2 function may skew premalignant cells into tumor cells. In addition, RHOA and IDH2 mutations were detected in tumor cells,9 suggesting that acquisition of these mutations together with preexisting TET2 and DNMTA mutations leads to AITL development.
1.10. Clonal evolution in tumor‐infiltrating B cells
As mentioned above, the massive infiltration of immune cells into AITL tissues is partly due to the cytokine and chemokine storm, beginning from the cytokine and chemokine production from tumor cells and being amplified by the tumor‐infiltrating inflammatory cells. At the same time, it is well known that rearrangement of immunoglobulin (Ig) genes in addition to that of TCR genes is found in 0% to 40% of AITL samples,3 suggesting that B cells as well as T‐lineage tumor cells proliferate clonally in AITL tissues. EBV infection observed in 66% to 86% of cases49, 50, 51 may partly explain the clonal expansion of B cells. Notably, AITL and B‐cell lymphomas simultaneously cooccur as composite lymphomas, or serially during the disease course in up to 20% of AITL patients.52, 53 Although EBV may account for oncogenic mechanisms of EBV‐positive B‐cell lymphomas, EBV is negative in a substantial proportion of B‐cell lymphomas.52, 53, 54
TET2‐and DNMT3A‐mutated premalignant cells may be differentiated into tumor‐infiltrating B cells as well as tumor cells. In fact, when the distribution of these mutations was examined in AITL tissues using laser microdissection followed by targeted sequencing, TET2 mutations were detected even in B cells as well as in tumor cells in 15 of the 16 cases, and DNMT3A mutations were also found in both B cells and tumor cells in 4 of the 7 cases (Figures 1 and 2).9 Remarkably, B‐cell specific mutations were also identified.9 In particular, all 3 NOTCH1 mutations exhibited B‐cell‐specific distribution (Figures 1 and 2). These observations suggest that B cells residing in AITL tissues may have undergone clonal selection.
Figure 1.
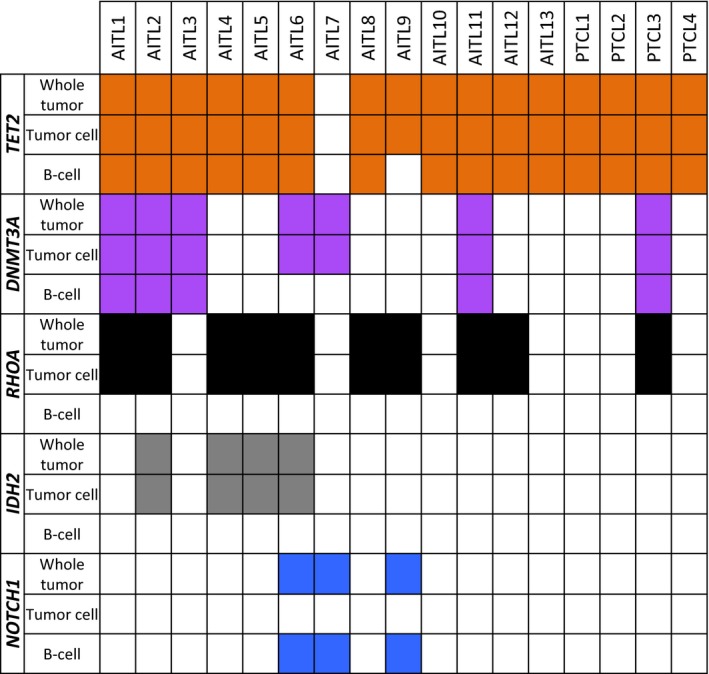
Distribution of common gene mutations in angioimmunoblastic T‐cell lymphoma (AITL). The orange boxes show TET2 mutations; the purple boxes, DNMT3A mutations; the black boxes, RHOA mutations; the gray boxes, IDH2 mutations; the blue boxes, NOTCH1 mutations; and the white boxes, no mutation
Figure 2.
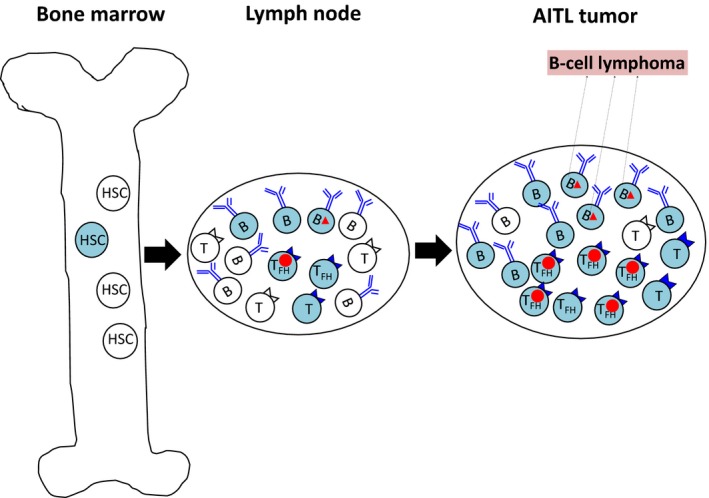
Multistep and multilineage tumorigenesis in angioimmunoblastic T‐cell lymphoma (AITL). The blue cells show cells that acquired TET2/DNMT3A mutations. The circles indicate RHOA/IDH2 mutations, and the triangles, NOTCH1 mutations. Hematopoietic stem/progenitor cells (HSC/HSPC) acquire TET2/DNMT3A mutations and become premalignant cells. These cells can be differentiated into both T and B cells. Acquisition of RHOA/IDH2 mutations in T cells leads the cells to transform into AITL tumor cells. In contrast, acquisition of NOTCH1 mutations in B cells may lead the cells to transform into B‐lymphoma cells
1.11. Angioimmunoblastic T‐cell lymphoma mouse model mimicking the human angioimmunoblastic T‐cell lymphoma genome
The impact of genetic events on AITL development can be examined using mouse models. As mentioned above, loss‐of‐function TET2 mutations were highly frequent in AITL.10 It was reported that TFH cells were gradually increased and finally T‐cell lymphomas with the TFH phenotype developed at long latencies in Tet2 gene‐trap mice.55 The lymphoma cells exhibited increased methylation at the transcriptional start site (TSS) regions, gene bodies and CpG islands, and decreased hydroxymethylation at the TSS regions.55 In particular, the negative regulatory region of BCL6 encoding a fate‐determinant of TFH cells was highly methylated in lymphoma cells.55 Decitabine treatment results in demethylation of the loci, accompanying downregulation of Bcl6 expression.55 Furthermore, human PTCL samples also had hypermethylation of the corresponding loci, especially when they had TET2 mutations.56 The impaired TET2 function may induce BCL6 upregulation, resulting in the skewed differentiation toward TFH cells in both humans and mice. The synergistic effect of TET2 and DNMT3A mutations on AITL development was proven using mice transplanted with Tet2‐null hematopoietic stem/progenitor cells expressing genes transduced with R882H DNMT3A mutant cDNA.33 The synergistic effect of TET2 and G17V RHOA mutations, the most frequent combinations in human AITL, was also shown by mice transplanted with Tet2‐null T cells expressing genes transduced with G17V RHOA mutant cDNA.57
2. CONCLUSION
The biology of AITL has become gradually understood as a result of the multistep and multilineage tumorigenesis concept: premalignant cells having epigenetic mutations evolve into tumor and tumor‐infiltrating cells through clonal selection of the mutated cells. The multistep and multilineage acquisition of mutations may contribute to the formation of the striking pathologic features of AITL. Concurrently, these characteristic gene mutations have begun to change the clinical approach to AITL. G17V RHOA mutations will be used in a clinical setting to assist diagnosis of AITL. This genetic information may lead to individualized therapies for AITL patients in future.
CONFLICT OF INTEREST
S.C. received research funding from the following companies: Kyowa Hakko Kirin, Shionogi, Takeda Pharmaceutical, Chugai Pharmaceutical and Bristol‐Myers Squibb. The other authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
We thank Dr Flaminia Miyamasu for helping to improve the grammar in the present paper. This work was supported by Grants‐in‐Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (JP16K15497 to M.S.‐Y).
Fukumoto K, Nguyen TB, Chiba S, Sakata‐Yanagimoto M. Review of the biologic and clinical significance of genetic mutations in angioimmunoblastic T‐cell lymphoma. Cancer Sci. 2018;109:490–496. https://doi.org/10.1111/cas.13393
REFERENCES
- 1. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Leval L, Gisselbrecht C, Gaulard P. Advances in the understanding and management of angioimmunoblastic T‐cell lymphoma. Br J Haematol. 2010;148:673‐689. [DOI] [PubMed] [Google Scholar]
- 4. de Leval L, Rickman DS, Thielen C, et al. The gene expression profile of nodal peripheral T‐cell lymphoma demonstrates a molecular link between angioimmunoblastic T‐cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952‐4963. [DOI] [PubMed] [Google Scholar]
- 5. Zhao WL, Mourah S, Mounier N, et al. Vascular endothelial growth factor‐A is expressed both on lymphoma cells and endothelial cells in angioimmunoblastic T‐cell lymphoma and related to lymphoma progression. Lab Invest. 2004;84:1512‐1519. [DOI] [PubMed] [Google Scholar]
- 6. Gaulard P, de Leval L. The microenvironment in T‐cell lymphomas: emerging themes. Semin Cancer Biol. 2014;24:49‐60. [DOI] [PubMed] [Google Scholar]
- 7. Fukushima N, Satoh T, Sano M, Tokunaga O. Angiogenesis and mast cells in non‐Hodgkin's lymphoma: a strong correlation in angioimmunoblastic T‐cell lymphoma. Leuk Lymphoma. 2001;42:709‐720. [DOI] [PubMed] [Google Scholar]
- 8. Tripodo C, Gri G, Piccaluga PP, et al. Mast cells and Th17 cells contribute to the lymphoma‐associated pro‐inflammatory microenvironment of angioimmunoblastic T‐cell lymphoma. Am J Pathol. 2010;177:792‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nguyen TB, Sakata‐Yanagimoto M, Asabe Y, et al. Identification of cell‐type‐specific mutations in nodal T‐cell lymphomas. Blood Cancer J. 2017;7:e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakata‐Yanagimoto M, Enami T, Yoshida K, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171‐175. [DOI] [PubMed] [Google Scholar]
- 11. Palomero T, Couronne L, Khiabanian H, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014;46:166‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoo HY, Sung MK, Lee SH, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:371‐375. [DOI] [PubMed] [Google Scholar]
- 13. Lemonnier F, Couronne L, Parrens M, et al. Recurrent TET2 mutations in peripheral T‐cell lymphomas correlate with TFH‐like features and adverse clinical parameters. Blood. 2012;120:1466‐1469. [DOI] [PubMed] [Google Scholar]
- 14. Dobay MP, Lemonnier F, Missiaglia E, et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T‐cell lymphoma and other nodal lymphomas of follicular helper T‐cell origin. Haematologica. 2017;102:e148‐e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cairns RA, Iqbal J, Lemonnier F, et al. IDH2 mutations are frequent in angioimmunoblastic T‐cell lymphoma. Blood. 2012;119:1901‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vallois D, Dobay MP, Morin RD, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T‐cell‐derived lymphomas. Blood. 2016;128:1490‐1502. [DOI] [PubMed] [Google Scholar]
- 17. Rohr J, Guo S, Huo J, et al. Recurrent activating mutations of CD28 in peripheral T‐cell lymphomas. Leukemia. 2016;30:1062‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sakata‐Yanagimoto M. Multistep tumorigenesis in peripheral T cell lymphoma. Int J Hematol. 2015;102:523‐527. [DOI] [PubMed] [Google Scholar]
- 19. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jaiswal S, Fontanillas P, Flannick J, et al. Age‐related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagata Y, Kontani K, Enami T, et al. Variegated RHOA mutations in adult T‐cell leukemia/lymphoma. Blood. 2015;127:596‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakamoto‐Matsubara R, Sakata‐Yanagimoto M, Enami T, et al. Detection of the G17V RHOA mutation in angioimmunoblastic T‐cell lymphoma and related lymphomas using quantitative allele‐specific PCR. PLoS One. 2014;9:e109714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sakata‐Yanagimoto M, Nakamoto‐Matsubara R, Komori D, et al. Detection of the circulating tumor DNAs in angioimmunoblastic T‐cell lymphoma. Ann Hematol. 2017;96:1471‐1475. [DOI] [PubMed] [Google Scholar]
- 24. Zhang S, Konstantinidis DG, Yang JQ, et al. Gene targeting RhoA reveals its essential role in coordinating mitochondrial function and thymocyte development. J Immunol. 2014;193:5973‐5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Etienne‐Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629‐635. [DOI] [PubMed] [Google Scholar]
- 26. Fujisawa M, Sakata‐Yanagimoto M, Nishizawa S, et al. Activation of RHOA‐VAV1 signaling in angioimmunoblastic T‐cell lymphoma. Leukemia. 2017. https://doi.org/10.1038/leu.2017.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kakiuchi M, Nishizawa T, Ueda H, et al. Recurrent gain‐of‐function mutations of RHOA in diffuse‐type gastric carcinoma. Nat Genet. 2014;46:583‐587. [DOI] [PubMed] [Google Scholar]
- 28. Rohde M, Richter J, Schlesner M, et al. Recurrent RHOA mutations in pediatric Burkitt lymphoma treated according to the NHL‐BFM protocols. Genes Chromosom Cancer. 2014;53:911‐916. [DOI] [PubMed] [Google Scholar]
- 29. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Russler‐Germain DA, Spencer DH, Young MA, et al. The R882H DNMT3A mutation associated with AML dominantly inhibits wild‐type DNMT3A by blocking its ability to form active tetramers. Cancer Cell. 2014;25:442‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25‐38. [DOI] [PubMed] [Google Scholar]
- 33. Scourzic L, Couronne L, Pedersen MT, et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia. 2016;30:1388‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730‐741. [DOI] [PubMed] [Google Scholar]
- 35. Wang C, McKeithan TW, Gong Q, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T‐cell lymphoma. Blood. 2015;126:1741‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheminant M, Bruneau J, Kosmider O, et al. Efficacy of 5‐Azacytidine in a TET2 mutated angioimmunoblastic T cell lymphoma. Br J Haematol. 2015;168:913‐916. [DOI] [PubMed] [Google Scholar]
- 37. Saillard C, Guermouche H, Derrieux C, et al. Response to 5‐azacytidine in a patient with TET2‐mutated angioimmunoblastic T‐cell lymphoma and chronic myelomonocytic leukaemia preceded by an EBV‐positive large B‐cell lymphoma. Hematol Oncol. 2016. https://doi.org/10.1002/hon.2319. [DOI] [PubMed] [Google Scholar]
- 38. Lee SH, Kim JS, Kim J, et al. A highly recurrent novel missense mutation in CD28 among angioimmunoblastic T‐cell lymphoma patients. Haematologica. 2015;100:e505‐e507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yoo HY, Kim P, Kim WS, et al. Frequent CTLA4‐CD28 gene fusion in diverse types of T‐cell lymphoma. Haematologica. 2016;101:757‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abate F, da Silva‐Almeida AC, Zairis S, et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T‐cell lymphomas. Proc Natl Acad Sci USA. 2017;114:764‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boddicker RL, Razidlo GL, Dasari S, et al. Integrated mate‐pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T‐cell lymphoma. Blood. 2016;128:1234‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wiederrecht G, Lam E, Hung S, Martin M, Sigal N. The mechanism of action of FK‐506 and cyclosporin A. Ann N Y Acad Sci. 1993;696:9‐19. [DOI] [PubMed] [Google Scholar]
- 43. Advani R, Horwitz S, Zelenetz A, Horning SJ. Angioimmunoblastic T cell lymphoma: treatment experience with cyclosporine. Leuk Lymphoma. 2007;48:521‐525. [DOI] [PubMed] [Google Scholar]
- 44. Chen XG, Huang H, Tian YL, et al. Cyclosporine, prednisone, and high‐dose immunoglobulin treatment of angioimmunoblastic T‐cell lymphoma refractory to prior CHOP or CHOP‐like regimen. Chin J Cancer. 2011;30:731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nguyen TB, Sakata‐Yanagimoto M, Nakamoto‐Matsubara R, et al. Double somatic mosaic mutations in TET2 and DNMT3A – origin of peripheral T cell lymphoma in a case. Ann Hematol. 2015;94:1221‐1223. [DOI] [PubMed] [Google Scholar]
- 46. Sakata‐Yanagimoto M, Yokoyama Y, Muto H, et al. A nationwide survey of co‐occurrence of malignant lymphomas and myelodysplastic syndromes/myeloproliferative neoplasms. Ann Hematol. 2016;95:829‐830. [DOI] [PubMed] [Google Scholar]
- 47. Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML‐associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T‐cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837‐2843. [DOI] [PubMed] [Google Scholar]
- 50. Zhou Y, Attygalle AD, Chuang SS, et al. Angioimmunoblastic T‐cell lymphoma: histological progression associates with EBV and HHV6B viral load. Br J Haematol. 2007;138:44‐53. [DOI] [PubMed] [Google Scholar]
- 51. Mourad N, Mounier N, Briere JA, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T‐cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte (GELA) trials. Blood. 2008;111:4463‐4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Attygalle AD, Kyriakou C, Dupuis J, et al. Histologic evolution of angioimmunoblastic T‐cell lymphoma in consecutive biopsies: clinical correlation and insights into natural history and disease progression. Am J Surg Pathol. 2007;31:1077‐1088. [DOI] [PubMed] [Google Scholar]
- 53. Willenbrock K, Brauninger A, Hansmann ML. Frequent occurrence of B‐cell lymphomas in angioimmunoblastic T‐cell lymphoma and proliferation of Epstein‐Barr virus‐infected cells in early cases. Br J Haematol. 2007;138:733‐739. [DOI] [PubMed] [Google Scholar]
- 54. Suefuji N, Niino D, Arakawa F, et al. Clinicopathological analysis of a composite lymphoma containing both T‐ and B‐cell lymphomas. Pathol Int. 2012;62:690‐698. [DOI] [PubMed] [Google Scholar]
- 55. Muto H, Sakata‐Yanagimoto M, Nagae G, et al. Reduced TET2 function leads to T‐cell lymphoma with follicular helper T‐cell‐like features in mice. Blood Cancer J. 2014;4:e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nishizawa S, Sakata‐Yanagimoto M, Hattori K, et al. BCL6 locus is hypermethylated in angioimmunoblastic T‐cell lymphoma. Int J Hematol. 2016;105:465‐469. [DOI] [PubMed] [Google Scholar]
- 57. Zang S, Li J, Yang H, et al. Mutations in 5‐methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J Clin Invest. 2017;127:2998‐3012. [DOI] [PMC free article] [PubMed] [Google Scholar]