

Abstract
Immune checkpoint inhibitors have ushered in a new era in cancer therapy, although other therapies or combinations thereof are still needed for many patients for whom these drugs are ineffective. In this light, we have identified glypican‐3 an HLA‐24, HLA‐A2 restriction peptide with extreme cancer specificity. In this paper, we summarize results from a number of related clinical trials showing that glypican‐3 peptide vaccines induce specific CTLs in most patients (UMIN Clinical Trials Registry: UMIN000001395, UMIN000005093, UMIN000002614, UMN000003696, and UMIN000006357). We also describe the current state of personalized cancer immunotherapy based on neoantigens, and assess, based on our own research and experience, the potential of such therapy to elicit cancer regression. Finally, we discuss the future direction of cancer immunotherapy.
Keywords: cancer vaccine, CAR‐transduced T‐cell therapy, glypican‐3, neoantigen, TCR‐transduced T‐cell therapy
Abbreviations
- CAR
chimeric antigen receptor
- ELISpot
enzyme‐linked immunospot
- GPC3
glypican‐3
- HCC
hepatocellular carcinoma
- IFN‐γ
γ‐interferon
- iPS
induced pluripotent stem (cell)
- OCCC
ovarian clear cell carcinoma
- PD‐1
programmed cell death‐1
- TCR
T‐cell receptor
- TIL
tumor‐infiltrating lymphocyte
1. INTRODUCTION
In addition to surgery, radiation therapy, and chemotherapy, there are high hopes for a fourth alternative for cancer treatment: immunotherapy, which exploits the physiological immune system. For example, extremely high response rates have been reported for CAR‐transduced T‐cell therapy against CD19+ hematopoietic tumors,1 as well as for adoptive immunotherapy with TILs against malignant melanoma.2 In addition, emerging immunotherapies that block immune checkpoints, including antibodies to CTL‐associated protein‐4, PD‐1, PD‐ligand 1, and similar molecules, have been shown to have dramatic, long‐term antitumor effects.3, 4 The response rates for immune checkpoint blockers have been as high as 30% for melanoma excluding Hodgkin's lymphoma, with estimates for other cancers ranging from approximately 10% to 30%. However, the development of alternatives for the other 70%–80% of patients for whom this treatment is ineffective continues to be a challenge. To accelerate such development, it is essential to quantify tumor‐reactive T cells in an individual patient, and then to encourage such cells to infiltrate cancer tissues. Indeed, it may be necessary to expand the population of tumor‐reactive T cells in some patients by, for example, vaccination, adoptive immunotherapy, or similar strategies. Induction of inflammation by adjuvant therapy, chemotherapy, radiation therapy, and other approaches has been reported to promote T‐cell infiltration into cancer sites.5 For patients with numerous gene mutations, personalized cancer vaccines based on the variant antigens present (neoantigens) may be the only viable treatment. Indeed, clinical trials to assess such personalized cancer vaccines are already underway in Europe and North America, with results now starting to appear. In any case, it is clear that, even for patients with few gene mutations, immune reactions are difficult to trigger even by blocking natural immunosuppressive functions. Thus, it may not be possible to induce cancer regression in such cases,6 unless shared antigens like GPC3 are targeted in addition to neoantigens.
Despite initial impressions of ineffectiveness, our clinical trials have now shown that cancer peptide vaccines against GPC3 induce CTLs in vivo, but not autoimmune disease‐like and other significant adverse reactions. Alternatively, we speculate that even better antitumor effects can be achieved from adoptive immunotherapy with T cells transduced with TCRs obtained from CTLs generated by cancer peptide vaccines. In this paper, we summarize the results, mainly our own, of GPC3‐based cancer immunotherapy, and discuss future prospects. We also describe the current state and future direction of individualized immunotherapies, including cancer vaccines based on neoantigens and adoptive immunotherapy with engineered T cells. We note that, as of this writing, no official approval has been granted in Japan for any peptide vaccine that targets shared self‐antigens, further discouraging the development of such vaccines.
2. GLYPICAN‐3
We first identified GPC3 in August 2001 while searching for novel tumor‐associated antigens among several tens of thousands of genes in cDNA microarrays collected by Okabe et al7 against cancerous tissues, surrounding non‐cancerous tissues, and various normal organs. Glypican‐3 was found to be expressed in almost no other normal organs except the embryonal liver and kidney, the placenta in adults, and some renal tubes. However, expression in HCC arising from hepatitis B or C was in both cases 80% higher than in surrounding non‐cancerous tissues. Subsequently, we found that GPC3, a membrane protein, was also secreted, and was a specific and useful marker for HCC.8 In addition, we have also identified GPC3 peptides that induce specific CTLs, and that have now been tested clinically as cancer vaccines. In the last 15 years, we have yet to encounter a molecule with the same degree of cancer‐specific expression and ability to induce CTLs. Remarkably, Nakano et al also identified GPC3 at around the same time, despite the lack of interaction between our groups. They have also developed, and continue to develop, antibody therapies targeting GPC3.9
Glypican‐3 is encoded on the X chromosome, and is highly homologous between humans and mice. Just as knockout mice grow to an enormous size with accompanying deformities, patients with GPC3 deficiency due to mutation or deletion suffer from overgrowth and deformities characteristic of disorders such as Simpson–Golabi–Behmel syndrome (Figure 1a). Glypican‐3, a 65‐kDa protein of 580 amino acids, is a heparan sulfate proteoglycan anchored to the cell membrane by glycosylphosphatidylinositol (Figure 1b). Bound to the two heparan sulfate chains are various important proteins, including fibroblast growth factor and Wnt. The heparan chains are not yet fully defined in terms of function, but are believed to control the association of the bound proteins to the corresponding receptors. Glypican‐3 is expressed in HCC, OCCC, melanomas, lung squamous cell carcinomas, hepatoblastomas, nephroblastomas (Wilms’ tumors), and yolk sac tumors, as well as in certain stomach cancers, for example, gastric cancers that produce α‐fetoprotein. The function of secreted and membrane‐anchored GPC3 in these cancers is unknown, but it is almost certainly involved in neoplastic transformation in HCC.10 Strikingly, the protein is nearly absent in all other cancer forms. We note that in some GPC3‐positive HCC, a few surrounding normal cells also weakly express GPC3, and we believe that these cells may be involved in recurrence (Figure 1c).
Figure 1.
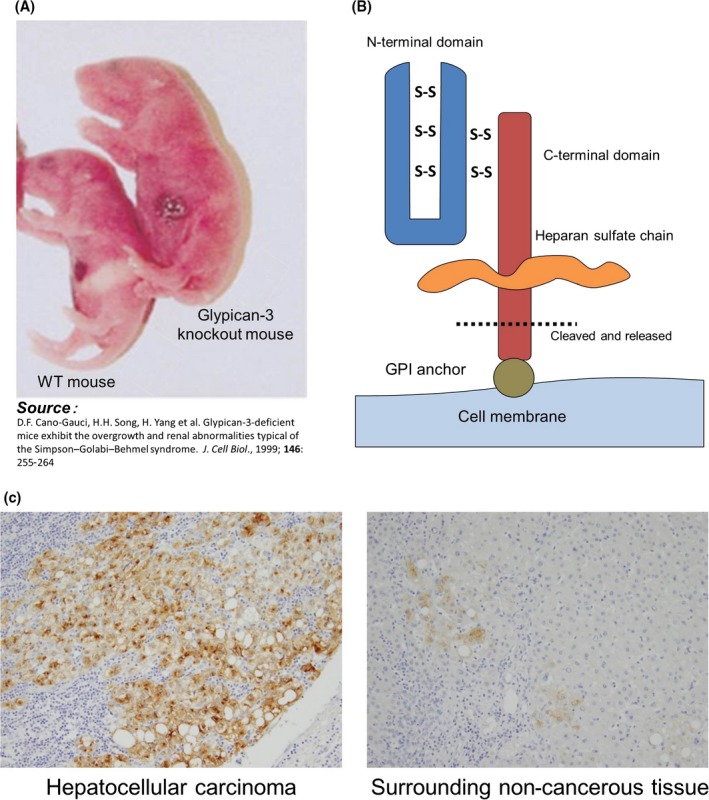
Characteristics of glypican‐3 (GPC3). A, A GPC3 knockout mouse of abnormally large size and with accompanying deformities. B, GPC3 expression and release. GPC3 is a membrane protein that may also be secreted, although the cleavage sites and underlying mechanisms are unknown. GPI, glycosylphosphatidylinositol. C, GPC3 is abundantly expressed in approximately 80% of hepatocellular carcinomas. In some cases, foci that weakly express GPC3 are observed in surrounding tissue
Based on these characteristics, GPC3 is an ideal target for cancer immunotherapy. Accordingly, we envision an array of GPC3‐based strategies, not only as cancer peptide vaccines, but also in antibody therapy, adoptive immunotherapy with TCR‐ or CAR‐transduced T cells, and others. We also anticipate that plasma GPC3 will be validated as a tumor marker for HCC.
3. PRECLINICAL STUDIES OF GPC3 PEPTIDE VACCINES
The GPC3 peptides in development as cancer vaccines are restricted to HLA‐A24 and HLA‐A2. Of note, the former is present in approximately 60% of Japanese, whereas the latter is present in 40% of Japanese and is also a major haplotype in Caucacians.11, 12 Mice immunized with such GPC3 peptides develop antigen‐specific CTLs and antitumor activity, but not autoimmune phenomena.11, 12 In support of subsequent clinical trials, we have completed dose‐ranging studies and investigated various adjuvants, including incomplete Freund's adjuvant, CpG, α‐GalCel, or aluminum.13 We found that, although the peptides by themselves are not immunogenic in mice, formulation with incomplete Freund's adjuvant elicited GPC3‐specific immune response.
4. PHASE I CLINICAL TRIAL AGAINST ADVANCED HCC
A phase I clinical trial of GPC3 peptide vaccines (UMIN Clinical Trials Registry: 000001395) was carried out from February 2007 to November 2009 at the National Cancer Center Hospital East (Kashiwa, Japan) among 33 cases of advanced HCC (Table 1).14, 15 The primary end‐points were the safety and immune response. Dose‐limiting toxicity was not observed, even for a single case, in a dose‐escalation study of 0.3, 1, 3, 10, and 30 mg. Thus, the maximum tolerated dose was difficult to set. This result also implied that GPC3 peptide vaccines are safe and GPC3 peptide‐specific CTL response was shown in 30 patients (90.1%) by IFN‐γ ELISpot assay. We reached primary end‐points in this study. However, partial clinical response in a patient dosed with 30 mg, as well as dose‐dependent immunological reactions, suggested that high doses were more effective. However, a 30‐mg dose would also require 6 mL vaccine and thus would entail additional issues in delivery and pain from reddening and duration at injection sites, even though these were generally of low grade. Based on such considerations, we recommended a 3‐mg dose as appropriate. In addition, some clinical response was noted, implying that the vaccines induced immune reactions (Figure 2a,b), including a decline in tumor markers and an increase of peptide‐specific CTLs in peripheral blood, as measured by IFN‐γ ELISpot assay. By collecting tumor biopsies before and after vaccination, we also found cases where infiltrating CD8+ CTLs were observed after, but not before vaccination, confirming the immunological effects of the vaccine (Figure 2c).14, 15
Table 1.
Summary of our clinical trials of GPC3‐derived peptide vaccine
| Trial | UMIN | Key inclusion criteria | Primary endpoint | Results |
|---|---|---|---|---|
| Phase I clinical study of GPC3 peptide vaccine in patients with advanced HCC | 000001395 | Advanced HCC patient |
|
GPC3 vaccination was well‐tolerated The vaccine induced a GPC3‐specific CTL response in 91% patients (30/33) |
| Clinical study evaluating immunological efficacy of GPC3 peptide vaccine in patients with advanced HCC | 000005093 | Advanced HCC patient | Increase of frequency of GPC3‐peptide specific CD8 positive T lymphocytes in the blood and into the tumor | After the vaccination, the number of GPC3 peptide‐specific CTLs in PBMC was found to have increased in 9 of 11 patients and tumor biopsy specimens after the vaccination ware obtained 3 patients, in which they found to infiltrate into the tumor |
| A Phase II study of GPC3 peptide vaccine as adjuvant treatment for HCC after surgical resection or Ragiofrequency ablation (RFA) | 000002614 |
|
The 1‐ and 2‐y recurrence rate | The 1‐ and 2‐y recurrence rates were 24.4% and 53.7%, respectively. The primary endpoint was not reached |
| Phase II study of GPC3 peptide vaccine as treatment for OCCC | 000003696 | Advanced OCCC patient | DCR at 6 mo | DCR at 6 mo was 9.4% (3/32) |
| A Phase I study of GPC3 peptide vaccine for pediatric patients with refractory tumors | 000006357 |
|
Incidence of DLT | No DLT or dose‐specific adverse events were observed |
GPC3, glypican‐3; HCC, Hepatocellular carcinoma; OCCC, ovarian clearcell carcinoma; PR, partial response; SD, stable disease; DCR, disease control rate; DLT, dose limiting toxicity; CTL, cytotoxic T lymphocyte.
Figure 2.
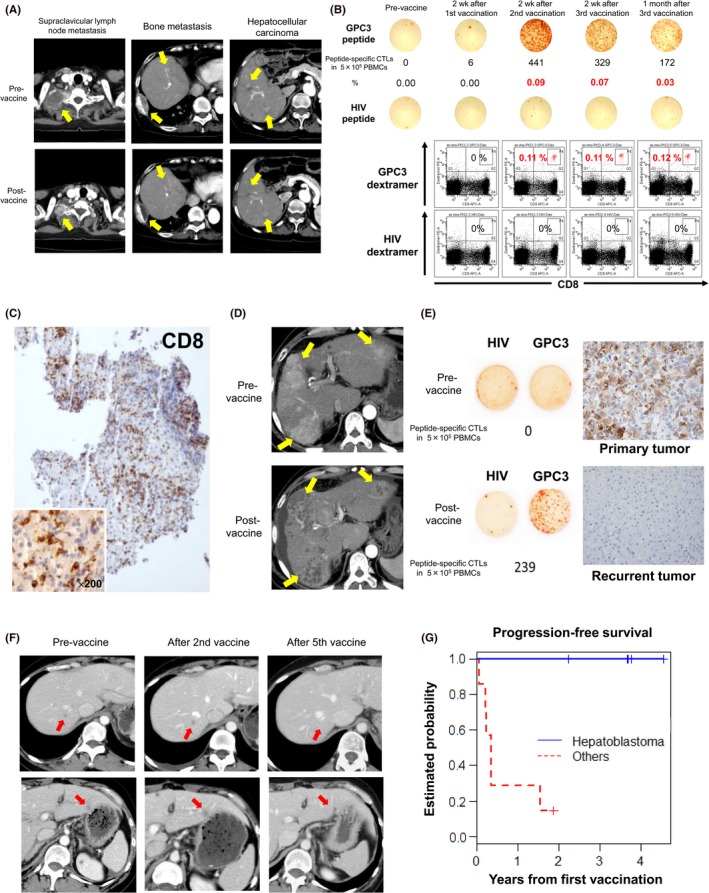
Results from clinical trials of glypican‐3 (GPC3) peptide vaccines. A, Computed tomography scans of a patient with partial response in a phase I trial show remarkable shrinkage of lymph node metastasis and disappearance of two hepatocellular carcinoma (HCC) nodules. Yellow arrows indicate those lesions. B, Peptide‐specific CTLs in peripheral blood, as measured by ex vivo γ‐interferon enzyme‐linked immunospot assay. Top panels indicate that the number of peptide‐specific CTLs in 0.5 million PBMCs increased from 0 to 441 in this patient. Bottom panels show a number of peptide‐specific CTLs after two vaccinations, as measured by flow cytometry of cells stained with GPC3 dextramer. C, Tumor biopsy of an HCC that did not change in size after vaccination, but became infiltrated with a large number of CD8+ killer T cells. D, Computed tomography scan of a case in which almost all HCCs became necrotic after two vaccinations. Yellow arrows indicate those lesions. E, In a clinical trial to prevent HCC recurrence after radical treatment, peptide‐specific CTLs were detected in peripheral blood after vaccination. However, the cancer relapsed thereafter, and was resected. GPC3 expression was observed in the primary tumor before vaccination, but not in the recurrent tumor. F, Computed tomography scans of a patient with ovarian clear cell carcinoma who responded to GPC3 peptide vaccines. After vaccination, all five metastasis sites became inflamed immediately, and disappeared after five vaccinations. Red arrows indicate those lesions. G, Phase I study in pediatric patients with refractory solid tumors. All five patients with hepatoblastoma in remission at enrollment survived and remained in remission by the end of the study
A subsequent phase I trial was initiated (UMIN Clinical Trials Registry: 000005093) to formally investigate, by means of tumor biopsies before and after vaccination, the extent of CTL infiltration into tumor tissues (Table 1).16 The primary end‐point was GPC3 peptide‐specific immune‐responses induced by GPC3 peptide vaccination. We note, however, that this trial was initiated after approval of sorafenib as chemotherapy, and patients with late‐stage cancer were recruited only after sorafenib was no longer effective. Most of these patients were nearly unresponsive to the vaccine and had progressive disease. Post‐vaccination biopsies also proved difficult, and were completed for just 11 cases. Nevertheless, the trial proved informative, with one valuable case in which HCC tissues became inflamed and then necrotic after two injections of the vaccine, despite ongoing liver dysfunction (Figure 2d).17 We showed that the present vaccine induced GPC3 peptide‐specific CTLs, which were found to infiltrate into the tumor. Moreover, we established GPC3 peptide‐specific CTL clones from a tumor biopsy specimen.16
5. PHASE II CLINICAL TRIAL TO INVESTIGATE RELAPSE PREVENTION FOLLOWING RADICAL TREATMENT OF HCC
We initiated a single‐arm phase II clinical trial (UMIN Clinical Trials Registry: 000002614) to evaluate 1‐ and 2‐year recurrence rates (primary end‐point) in 41 patients following radical treatment of HCC, using GPC3 peptide vaccine as adjuvant therapy (Table 1).18 Glypican‐3 peptide‐specific CTL responses were detected in 35 of the 41 patients (85.4%) after vaccination. However, the primary end‐point was not reached for 1‐ and 2‐year recurrence rates. As the absence of GPC3 expression correlates with good prognosis, we limited the control group in this trial strictly to GPC3‐negative cases, in order to isolate the effects of GPC3 vaccination post‐surgery. Notably, two cases of relapse were observed despite significant numbers of vaccine‐induced peptide‐specific CTLs in peripheral blood. In these cases, GPC3 was expressed in the primary tumor, but not in the recurrent tumor (Figure 2e).18 These results suggest that, although a peptide vaccine against a shared self‐antigen may eradicate tumor cells that express such antigen, cancer cells that do not express or lose the same antigen may then proliferate. In such cases, vaccines that target multiple shared antigens would be effective. One can also anticipate vaccine therapies that target multiple tumor‐associated antigens, as will be discussed below. In any case, we strongly expect that further clinical trials will validate the ability of GPC3 peptide vaccines to prevent recurrence after resection of GPC3‐positive HCC.
6. CLINICAL TRIALS AGAINST OCCC AND REFRACTORY PEDIATRIC CANCER
We also observed antitumor effects in a Nagoya University (Nagoya, Japan) clinical trial (UMIN Clinical Trials Registry: 000003696) of GPC3 peptide vaccines against OCCC (Figure 2f),19 highlighting the versatility of this vaccine (Table 1).20 The primary end‐point was disease control rate at 6 months. In the results, two cases showed partial response one case showed stable disease. The disease control rate at 6 months was 9.4% (3 of the 32 cases). Although response rates (partial response rates) tended to be higher than against HCC, this might be attributable only to the smaller volume of OCCC. We note that OCCCs are extremely recalcitrant to existing anticancer drugs, and effective immunotherapies are eagerly awaited.
As GPC3 expression was also detected in some pediatric cancers, we initiated a clinical trial against GPC3‐positive refractory pediatric cancers at the National Cancer Center (Tokyo and Kashiwa, Japan), St. Luke's International Hospital (Tokyo, Japan), Osaka City General Hospital, (Osaka, Japan) and Kyushu University Hospital (Fukuoka, Japan) (UMIN Clinical Trials Registry: 000006357) (Table 1).21 Eighteen patients with pediatric solid tumors expressing GPC3 underwent GPC3 peptide vaccination. The primary end‐point was the incidence of dose‐limiting toxicity. In the results, no dose‐limiting toxicity was observed. The GPC3 peptide vaccine induced a GPC3‐specific CTL response in 7 of the 18 patients (39%), and almost all of the patients showing increased GPC3‐specific CTL frequency were in remission and had diagnosed hepatoblastoma. By contrast, GPC3‐specific CTL frequency never increased in the refractory advanced progression group. Results from this trial suggest that the vaccine may suppress hepatoblastoma after the second relapse, which is considered to be unavoidable (Figure 2g). Accordingly, we will continue to aggressively pursue potentially effective therapies and approaches against hepatoblastoma relapse.
7. NOVEL APPROACHES TO BOOST THE EFFECTS OF CANCER VACCINES
At present, cancer peptide therapies based on shared self‐antigens and that target advanced cancers may have limited efficacy. Indeed, even if a superior peptide vaccine was developed to abundantly induce peptide‐specific CTLs, the effects of such a vaccine would still depend critically on the amount of target peptide presented to HLA class I molecules on cancer cell surfaces. Hence, we initiated preclinical research on intratumor injection of peptide vaccines,22 and on concurrent therapy with antibodies to PD‐1,23 CD4,24 and other immunomodulators. These studies indicate that such approaches generally enhance the efficacy of peptide vaccines.
8. THERAPY WITH GPC3‐SPECIFIC CTL CLONES ESTABLISHED FROM VACCINATED PATIENTS
We have successfully established multiple peptide‐specific CTL clones from peripheral blood and cancer tissues of patients immunized with GPC3 peptide vaccines during clinical trials (Figure 3).14, 16, 25 Some of these clones efficiently kill cancer cells that present GPC3 peptides in vitro. In collaboration with Kaneko et al, Kyoto University (Kyoto, Japan), and a private company, we are now developing adoptive immunotherapies based on T cells transduced with TCRs obtained from the best CTL clones. The engineered T cells themselves are artificially differentiated from iPS cells (Figure 4). We anticipate the advantages of ensuring quality, shortening treatment periods, and suppressing costs by using iPS cell‐derived T cells instead of autologous T cells. We note that adoptive immunotherapies based on T cells engineered in this manner are generally more effective than peptide vaccines, and that their application against advanced cancers is hotly anticipated.
Figure 3.
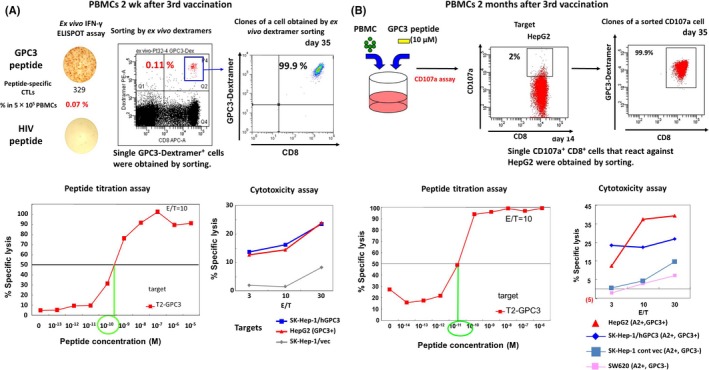
Establishment and analysis of glypican‐3 (GPC3)‐specific CTL clones. Peptide‐specific CTL clones were established from PBMCs from a patient with HLA‐A * 02:01 and immunized with 30 mg peptide vaccine (A), and from a patient with HLA‐A * 02:01 and immunized with 3 mg peptide vaccine (B). These clones react even to low concentrations of the peptide, and are toxic to cancer cell lines that express GPC3. The T‐cell receptors in these clones can then be transduced to other T cells for use in therapy. cont, control; E/T, effector/target; vec, vector
Figure 4.
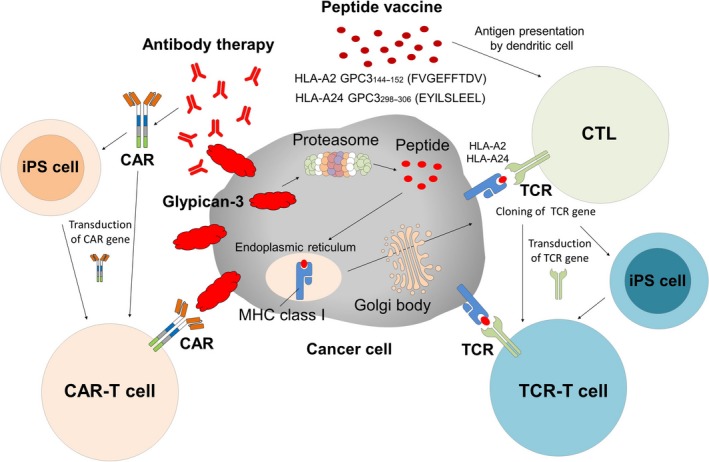
Cancer immunotherapy against glypican‐3 (GPC3). GPC3 is strongly cancer‐specific and extremely promising as a therapeutic target. Existing GPC3 therapies include peptide vaccine therapy and therapy with T cells transduced with a suitable T‐cell receptor (TCR). We are presently engaged in developing such T cells from induced pluripotent stem (iPS) cells. Therapies that target membrane‐bound GPC3 include antibody therapy and chimeric antigen receptor (CAR)‐transduced T‐cell therapies, which we are also striving to develop using iPS cells or by other approaches
9. ANTIBODY THERAPY TARGETING MEMBRANE GPC3
GC33, a humanized mAb to GPC3, induces antibody‐dependent cell‐mediated cytotoxicity against GPC3‐positive HCC, as reported in patient‐derived xenografted tumors.26 In a first‐in‐human phase I trial in the USA among patients with advanced HCC, GC33 was well tolerated, and antitumor effects were observed in some tumors that abundantly expressed GPC3.27 A similar phase I trial in Japan confirmed the tolerability of GC33, although complete or partial response was not observed. Nevertheless, stable disease was noted in seven of 13 patients, of whom three did not present disease progression beyond 3 months.28 Currently, GPC3 stratification is determined by immunohistochemistry, and an international phase II trial with placebo is underway. Results from this trial are eagerly awaited. In 2017, the results of fundamental research about ERY974, an anti‐GPC3/CD3 bispecific T cell‐redirecting antibody, were reported.29 We would like to expect future clinical applications of this novel drug.
10. CHIMERIC ANTIGEN RECEPTOR‐TRANSDUCED T‐CELL THERAPY AGAINST MEMBRANE GPC3
Although CAR‐transduced T‐cell therapy elicits remarkable response rates exceeding 80% against blood tumors, its effectiveness has not been established to date against solid carcinomas. Intriguingly, a clinical trial of CAR‐transduced T‐cell therapy based on GC33 is already underway in China.30 In a joint project with Tamada et al, Yamaguchi university (Ube, Japan), we are also developing next‐generation CAR‐transduced T‐cell therapy against solid carcinomas based on novel GPC3 antibodies. In contrast to traditional modes, we combine cancer‐specific antibodies conjugated to FITC with CAR‐transduced T cells that react to FITC.31 In this approach, therapeutic effects could be tightly controlled by the dose of FITC‐conjugated antibody, which also maintaining the survival of CAR‐transduced T cells. Additionally, in collaboration with Kaneko et al, we are now developing iPS cell‐derived GPC3 CAR‐T cell therapy (Figure 4).
11. DEVELOPMENT OF BIOMARKERS BASED ON SERUM FULL‐LENGTH GPC3
Glypican‐3 is subjected to shedding into serum in HCC patients. Serum GPC3 could be a biomarker for early diagnosis, prediction of recurrence, and assessment of response to anti‐GPC3 therapy against HCC. However, such soluble GPC3 protein might block the anti‐GPC3 antibody and CAR‐T cells. Serum GPC3 could be a treatment effect predictive biomarker or patient eligibility criteria of the anti‐GPC3 antibody or anti‐GPC3 CAR‐T cell therapy. In partnership with a private company, we have developed an assay to quantify serum full‐length GPC3, which we have shown to be predictive of HCC recurrence after surgery.32 In any case, we continue to investigate the value of this assay in early diagnosis, as well as in the prediction and assessment of response to anti‐GPC3 therapy.
12. NEOANTIGENS
Mutant antigens in individual tumors, or neoantigens, are of major interest. For example, UV‐induced melanoma and smoking‐induced lung cancer harbor multiple gene mutations,33 which are then presented as MHC peptides to which T cells might react. From the 1990s into the early 2000s, patients who were effectively treated by adoptive immunotherapy with TILs were also found to have accumulated gene mutations, and had therefore developed strong T‐cell immunity against neoantigens.34, 35, 36, 37, 38 Unfortunately, these mutations were in many cases unique to individual patients, and thus were considered inappropriate as universal targets for cancer vaccine therapy. Accordingly, neoantigens did not progress significantly from academic laboratories to the clinic. Today, however, mutations throughout the entire genome of individual cancer patients can now be catalogued, thanks to next‐generation sequencing and bioinformatics. Intriguingly, cancers that recur after therapy with immune checkpoint inhibitors were reported to be genomically unstable, and to accumulate numerous mutations.39, 40, 41, 42 Consequently, neoantigens are once again back in the spotlight as immunotherapy targets.
13. ADOPTIVE IMMUNOTHERAPY AND NEOANTIGENS
Rosenberg et al at the NCI (USA) have been developing various adoptive immunotherapeutics. For example, they showed that TILs are effective in adoptive immunotherapy against advanced melanoma, with outstanding response rates of over 70%. In this approach, T cells isolated from the tumor are cultivated in vitro, and returned to patients from whom lymphocytes had been depleted. With more than 1000 melanoma patients having already been treated in this fashion,4 the effectiveness of this approach, as well as that of immune checkpoint inhibitors, is thought to be due to the expression of numerous tissue‐specific antigens probably loaded with mutations, as well as to the abundance of CTLs in the tumor that recognize such mutated antigens.43 Indeed, multiple neoantigens recognized by T cells have been identified from several patients who responded to treatment.44, 45, 46
14. SOMATIC MUTATIONS AND IMMUNE CHECKPOINT INHIBITORS
A major paradigm shift has occurred in cancer therapy due to the effectiveness of immune checkpoint inhibitors. Indeed, cancer regression was repeatedly reported to be achievable by harnessing the immune system, not only in mouse models but also in multiple clinical cases. An urgent issue now for stand‐alone or combination therapies with immune checkpoint inhibitors is predicting treatment efficacy and side‐effects, one predictive marker being accumulation of gene mutations.41, 42, 43, 44, 45, 46, 47 Indeed, whole‐genome analyses of cancer tissues indicate that many more mutations than expected are accumulated in multiple cancer types. The mutated gene products are themselves potential therapeutic targets, especially if such gene products contain “driver mutations” that promote carcinogenesis, such as those in epidermal growth factor receptor, p53, and Ras. In addition, even “passenger mutations” that at first sight seem unrelated to carcinogenesis, may affect immunogenicity and are thus valid targets for immunotherapy. Indeed, non‐small‐cell lung cancers with large numbers of gene mutations and neoantigens are as responsive to anti‐PD‐141 as melanoma.43 Similarly, anti‐PD‐1 was initially considered ineffective against colorectal cancer until remarkable extension of survival was reported for nine of 10 patients with highly unstable genomes.48
15. VACCINE THERAPY AGAINST NEOANTIGENS
As noted, next‐generation sequencing and other technologies have ushered a new era in immunogenomics. Accordingly, numerous papers have described the identification of mutated peptides in murine cancer cells by next‐generation sequencing, the use of vaccines against these antigens, and the resulting antitumor effects.49, 50, 51, 52, 53 On the basis of these lines of evidence, clinical trials of personalized cancer vaccines have been initiated in Europe, North America, and China.54, 55, 56, 57, 58 More elaborate approaches have also been attempted, such as first predicting mutated peptides that will bind to a patient's MHC, and then treating the same patient with vaccines containing fusions of such peptides or with dendritic cells loaded with such fusions.59 In recent issues of Nature, clinical trials were described to prevent post‐resection recurrence of advanced melanoma using cancer vaccines targeting neoantigens. One report was of a vaccine using a long peptide,60 and another was of an RNA vaccine.61 In both trials, T‐cell activation was consistently reported, along with relapse prevention. In cases of relapse, anti‐PD‐1 proved effective. Similarly, we have carried out exome sequencing and HLA‐binding prediction for lung, hepatic–biliary–pancreatic, and gynecologic cancers, and are currently developing a cancer vaccine based on predicted, personalized neoantigenic peptides. We expect that studies of personalized cancer vaccines will be launched in Japan in the future.
16. ADOPTIVE IMMUNOTHERAPY AGAINST NEOANTIGENS
Rosenberg et al have also identified TCRs that recognize neoantigens, and are now striving to develop personalized cancer immunotherapy using T cells transduced with such TCRs, although it is still not clear whether T cells that recognize neoantigens or shared antigens accumulate in tumors. Under current investigation is whether adoptive immunotherapy with TILs, found to be effective against melanoma, is also effective against other solid carcinomas. Moreover, we are currently developing a high‐throughput system to isolate multiple tumor‐reactive populations from TILs in individual patients, as it is essential to identify T cells and TCRs that effectively eliminate carcinoma as speedily as possible.
We seek to develop adoptive immunotherapy with such T cells, but transduced with individual TCRs identified from TILs in patients. To this end, we are identifying antigens that react with individual TCRs, concurrent with our efforts to develop cancer vaccines that target neoantigens. In addition, we are isolating T cells that recognize shared antigens such as GPC3 or neoantigens unique to an individual, and that are also dominant in the tumor tissue (Figure 5).
Figure 5.
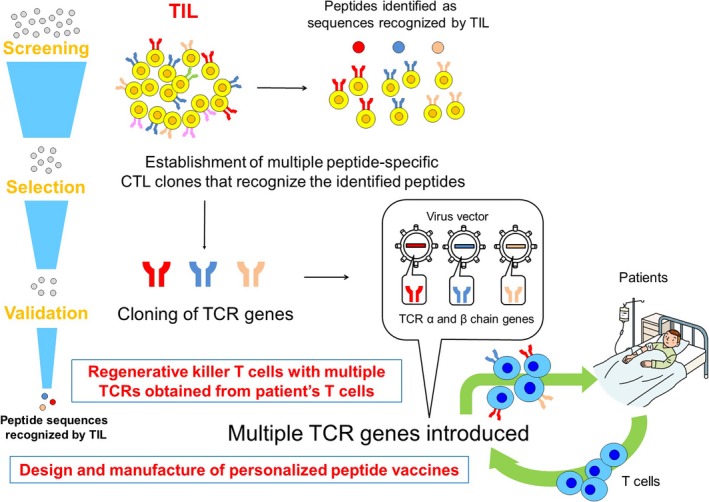
Production of individualized T cells transduced with a suitable T‐cell receptor (TCR) from autologous T cells. TIL, tumor‐infiltrating lymphocyte
17. FUTURE PERSPECTIVES
Cancer immunotherapy is critically dependent on T cells, even those therapies based on immune checkpoint inhibitors, which have claimed much of the spotlight in recent years. To recognize a cancer cell, T cells must first interact with surface complexes of tumor‐associated peptides and MHC. Since tumor‐associated peptides were first identified by Boon et al, we—like numerous other researchers—have continuously strived to identify suitable tumor‐associated antigens and the corresponding epitopes that can be exploited. Indeed, target identification is a key step forward in the development of cancer immunotherapy, regardless of technique. Thus, we now look forward to future developments on neoantigens or on shared antigens such as GPC3, developments that may show which of these can best serve as immunotherapeutic targets.
CONFLICT OF INTEREST
T. N. is a scientific advisor for Ono Pharmaceutical Co., Ltd. T. N., T. S., and T. Y. are supported by fundamental research funding from Takeda Pharmaceutical Co., Ltd., and BrightPath Biotherapeutics Co., Ltd. T. N. and T. S. are supported by fundamental research funding from Ono Pharmaceutical Co., Ltd., T. N. is supported by fundamental research funding from Noile‐Immune Biotech Inc. and Sysmex Co., Ltd. The other authors have no conflict of interest.
ACKNOWLEDGMENTS
This study was supported in part by the National Cancer Center Research and Development Fund (25‐A‐7) and (28‐A‐8), as well as Health and Labor Science Research Grants for Clinical Research, Japan and joint research funding from Takeda Pharmaceutical Co, Ltd., Noile‐Immune Biotech Inc., Ono Pharmaceutical Co., Ltd., BrightPath Biotherapeutics Co., Ltd., and Sysmex Co., Ltd. This study was performed as a part of a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P‐Direct), Ministry of Education, Culture, Sports, Science and Technology of Japan and supported by AMED under Grant Number JP15cm0106115h0002 and JP17ck0106204h0002
Shimizu Y, Suzuki T, Yoshikawa T, et al. Cancer immunotherapy‐targeted glypican‐3 or neoantigens. Cancer Sci. 2018;109:531–541. https://doi.org/10.1111/cas.13485
Funding information
National Cancer Center; Health and Labor Science Research Grants for Clinical Research, Japan; Takeda Pharmaceutical Co., Ltd.; Noile‐Immune Biotech Inc.; Ono Pharmaceutical Co., Ltd.; BrightPath Biotherapeutics Co., Ltd.; Sysmex Co., Ltd.
REFERENCES
- 1. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gattinoni L, Powell DJ Jr, Rosenberg SA, et al. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N Engl J Med. 2013;369:134‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Callahan MK, Postow MA, Wolchok JD. CTLA‐4and PD‐1 pathway blockade: combinations in the clinic. Front Oncol. 2015;4:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll‐like receptor 4‐dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050‐1059. [DOI] [PubMed] [Google Scholar]
- 6. Toh JW, de Souza P, Lim SH, et al. The potential value of immunotherapy in colorectal cancers: review of the evidence for programmed death‐1 inhibitor therapy. Clin Colorectal Cancer. 2016;15:285‐291. [DOI] [PubMed] [Google Scholar]
- 7. Okabe H, Satoh S, Kato T, et al. Genome‐wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001; 61: 2129‐2137. [PubMed] [Google Scholar]
- 8. Nakatsura T, Yoshitake Y, Senju S, et al. Glypican‐3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. 2003;306:16‐25. [DOI] [PubMed] [Google Scholar]
- 9. Nakano K, Orita T, Nezu J, et al. Anti‐glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2009; 378: 279‐284. [DOI] [PubMed] [Google Scholar]
- 10. Shirakawa H, Suzuki H, Shimomura M, et al. Glypican‐3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009;100:1403‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakatsura T, Komori H, Kubo T, et al. Mouse homologue of a novel human oncofetal antigen, glypican‐3, evokes T‐cell‐mediated tumor rejection without autoimmune reactions in mice. Clin Cancer Res. 2004;10:8630‐8640. [DOI] [PubMed] [Google Scholar]
- 12. Komori H, Nakatsura T, Senju S, et al. Identification of HLA‐A2‐ or HLA‐A24‐restricted CTL epitopes possibly useful for glypican‐3‐specific immunotherapy of hepatocellular carcinoma. Clin Cancer Res. 2006;12:2689‐2697. [DOI] [PubMed] [Google Scholar]
- 13. Motomura Y, Ikuta Y, Kuronuma T, et al. HLA‐A2 and ‐A24‐restricted glypican‐3‐derived peptide vaccine induces specific CTLs: preclinical study using mice. Int J Oncol. 2008;32:985‐990. [PubMed] [Google Scholar]
- 14. Yoshikawa T, Nakatsugawa M, Suzuki S, et al. HLA‐A2‐restricted glypican‐3 peptide‐specific CTL clones induced by peptide vaccine show high avidity and antigen‐specific killing activity against tumor cells. Cancer Sci. 2011;102:918‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I trial of a glypican‐3‐derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686‐3696. [DOI] [PubMed] [Google Scholar]
- 16. Tsuchiya N, Yoshikawa T, Fujinami N, et al. Immunological efficacy of glypican‐3 peptide vaccine in patients with advanced hepatocellular carcinoma. Oncoimmunology. 2017;6:e1346764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sawada Y, Yoshikawa T, Fujii S, et al. Remarkable tumor lysis in a hepatocellular carcinoma patient immediately following glypican‐3‐derived peptide vaccination: an autopsy case. Hum Vaccin Immunother. 2013;9:1228‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sawada Y, Yoshikawa T, Ofuji K, et al. Phase II study of the GPC3‐derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. 2016;5:e1129483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki S, Shibata K, Kikkawa F, et al. Significant clinical response of progressive recurrent ovarian clear cell carcinoma to glypican‐3‐derived peptide vaccine therapy: two case reports. Hum Vaccin Immunother. 2014;10:338‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki S, Sakata J, Utsumi F, et al. Efficacy of glypican‐3‐derived peptide vaccine therapy on the survival of patients with refractory ovarian clear cell carcinoma. Oncoimmunology. 2016;5:e1238542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsuchiya N, Hosono A, Yoshikawa T, et al. Phase I study of glypican‐3‐derived peptide vaccine therapy for patients with refractory pediatric solid tumors. OncoImmunology. 2017;6:e1377872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nobuoka D, Yoshikawa T, Takahashi M, et al. Intratumoral peptide injection enhances tumor cell antigenicity recognized by cytotoxic T lymphocytes: a potential option for improvement in antigen‐specific cancer immunotherapy. Cancer Immunol Immunother. 2013;62:639‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sawada Y, Yoshikawa T, Shimomura M, et al. Programmed death‐1 blockade enhances the antitumor effects of peptide vaccine‐induced peptide‐specific cytotoxic T lymphocytes. Int J Oncol. 2015;46:28‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujinami N, Yoshikawa T, Sawada Y, et al. Enhancement of antitumor effect by peptide vaccine therapy in combination with anti‐CD4 antibody: study in a murine model. Biochem Biophys Rep. 2016;5:482‐491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tada Y, Yoshikawa T, Shimomura M, et al. Analysis of cytotoxic T lymphocytes from a patient with hepatocellular carcinoma who showed a clinical response to vaccination with a glypican‐3‐derived peptide. Int J Oncol. 2013;43:1019‐1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishiguro T, Sugimoto M, Kinoshita Y, et al. Anti‐glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 2008;68:9832‐9838. [DOI] [PubMed] [Google Scholar]
- 27. Zhu AX, Gold PJ, El‐Khoueiry AB, et al. First‐in‐man phase I study of GC33, a novel recombinant humanized antibody against glypican‐3, in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2013;19:920‐928. [DOI] [PubMed] [Google Scholar]
- 28. Ikeda M, Ohkawa S, Okusaka T, et al. Japanese phase I study of GC33, a humanized antibody against glypican‐3 for advanced hepatocellular carcinoma. Cancer Sci. 2014;105:455‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishiguro T, Sano Y, Komatsu SI, et al. An anti‐glypican 3/CD3 bispecific T cell‐redirecting antibody for treatment of solid tumors. Sci Transl Med. 2017;9:eaal4291. [DOI] [PubMed] [Google Scholar]
- 30. Jiang Z, Jiang X, Chen S, et al. Anti‐GPC3‐CAR T cells suppress the growth of tumor cells in patient‐derived xenografts of hepatocellular carcinoma. Front Immunol. 2017;7:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tamada K, Geng D, Sakoda Y, et al. Redirecting gene‐modified T cells toward various cancer types using tagged antibodies. Clin Cancer Res. 2012;18:6436‐6445. [DOI] [PubMed] [Google Scholar]
- 32. Ofuji K, Saito K, Suzuki S, et al. Perioperative plasma glypican‐3 level may enable prediction of the risk of recurrence after surgery in patients with stage I hepatocellular carcinoma. Oncotarget. 2017;8:37835‐37844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alexandrov LB, Nil‐Zainai S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wölfel T, Hauer M, Schneider J, et al. A p16INK4a‐insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281‐1284. [DOI] [PubMed] [Google Scholar]
- 35. Robbins PF, El‐Gamil M, Li YF, et al. A mutated beta‐catenin gene encodes a melanoma‐specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. 1996;183:1185‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mandruzzato S, Brasseur F, Andry G, et al. A CASP‐8 mutation recognized by cytolytic T lymphocytes on a human head and neck carcinoma. J Exp Med. 1997;186:785‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saeterdal I, Bjørheim J, Lislerud K, et al. Frameshift‐mutation‐derived peptides as tumor‐specific antigens in inherited and spontaneous colorectal cancer. Proc Natl Acad Sci U S A. 2001;98:13255‐13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Linard B, Bézieau S, Benlalam H, et al. A ras‐mutated peptide targeted by CTL infiltrating a human melanoma lesion. J Immunol. 2002;168:4802‐4808. [DOI] [PubMed] [Google Scholar]
- 39. van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen‐specific T‐cell reactivity in an ipilimumab‐responsive melanoma. J Clin Oncol. 2013;31:e439‐e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371:2189‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515:577‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gros A, Parkhurst MR, Tran E, et al. Prospective identification of neoantigen‐specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22:433‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robbins PF, Lu YC, El‐Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor‐reactive T cells. Nat Med. 2013;19:747‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lu YC, Yao X, Li YF, et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J Immunol. 2013;190:6034‐6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu YC, Yao X, Crystal JS, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20:3401‐3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081‐1091. [DOI] [PubMed] [Google Scholar]
- 50. Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324‐327. [DOI] [PubMed] [Google Scholar]
- 51. Kreiter S, Vormehr M, van de Roemer N, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yadav M, Jhunjhunwala S, Phung QT, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515:572‐576. [DOI] [PubMed] [Google Scholar]
- 53. Duan F, Duitama J, Al Seesi S, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. 2014;211:2231‐2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69‐74. [DOI] [PubMed] [Google Scholar]
- 55. Boisguérin V, Castle JC, Loewer M, et al. Translation of genomics‐guided RNA‐based personalised cancer vaccines: towards the bedside. Br J Cancer. 2014;111:1469‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fritsch EF, Hacohen N, Wu CJ. Personal neoantigen cancer vaccines: the momentum builds. Oncoimmunology. 2014;3:e29311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vonderheide RH, Nathanson KL. Immunotherapy at large: the road to personalized cancer vaccines. Nat Med. 2013;19:1098‐1100. [DOI] [PubMed] [Google Scholar]
- 58. Rammensee HG, Singh‐Jasuja H. HLA ligandome tumor antigen discovery for personalized vaccine approach. Expert Rev Vaccines. 2013;12:1211‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kranz LM, Diken M, Haas H, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016;534:396‐401. [DOI] [PubMed] [Google Scholar]
- 60. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly‐specific therapeutic immunity against cancer. Nature. 2017;547:222‐226. [DOI] [PubMed] [Google Scholar]