

Abstract
Hypoxia‐inducible factor 1 (HIF‐1) is a transcriptional activator of various genes related to cellular adaptive responses to hypoxia. Dysfunctions in the regulatory systems of HIF‐1 activity have been implicated in the pathogenesis of various diseases including malignant tumors and, thus, elucidating the molecular mechanisms underlying the activation of HIF‐1 is eagerly desired for the development of novel anti‐cancer strategies. The importance of oxygen‐dependent and ubiquitin‐mediated proteolysis of the regulatory subunit of HIF‐1 (HIF‐1α) was first reported in 1997. Since then, accumulating evidence has shown that HIF‐1α may become stable and active even under normoxic conditions; for example, when disease‐associated genetic and functional alterations in some genes trigger the aberrant activation of HIF‐1 regardless of oxygen conditions. We herein review the last two decades of knowledge, since 1997, on the regulatory mechanisms of HIF‐1 activity from conventional oxygen‐ and proteolysis‐dependent mechanisms to up‐to‐the‐minute information on cancer‐associated genetic and functional alteration‐mediated mechanisms.
Keywords: gene expression, hypoxia‐inducible factor 1 (HIF‐1), molecular mechanism, tumor hypoxia
Abbreviations
- AMPK
AMP‐activated protein kinase
- ATR
ataxia telangiectasia and Rad3‐related protein
- CBP
CREB‐binding protein
- CK1δ
casein kinase 1δ
- D‐2HG
D‐2‐hydroxyglutarate
- FH
fumarate hydratase
- FIH‐1
factor‐inhibiting HIF‐1
- GSK‐3
glycogen synthase kinase 3
- HDAC
histone deacetylase
- HIF‐1
hypoxia‐inducible factor 1
- HIF‐1α
hypoxia‐inducible factor 1α
- HIF‐1β
hypoxia‐inducible factor 1β
- HRE
hypoxia‐response element
- HSP
heat‐shock protein
- IDH
isocitrate dehydrogenase
- IRES
internal ribosomal entry site
- IRF9
interferon regulatory factor 9
- ISGF3
interferon‐stimulated gene factor 3
- L‐2HG
L‐2‐hydroxyglutarate
- LSD1
lysine‐specific demethylase‐1
- LY6E
lymphocyte antigen 6 complex locus E
- MEF
mouse embryonic fibroblast
- ND6
NADH dehydrogenase subunit 6
- NF‐κΒ
nuclear factor kappa B
- NLS
nuclear localization signal
- NRF‐1
NF‐E2‐related factor 1
- NQO1
NAD(P)H:quinone oxidoreductase 1
- PAN2
poly(A) nuclease 2
- PCAF
p300/CBP‐associated factor
- PHD
prolyl‐4‐hydroxylase
- PI3K
phosphoinositide 3‐kinase
- PKC
protein kinase C
- PLK3
polo‐like kinase 3
- pVHL
von Hippel‐Lindau protein
- RACK1
receptor for activated C kinase
- RSUME
RWD‐containing SUMOylation enhancer
- SDH
succinate dehydrogenase
- SENP
sentrin/SUMO‐specific protease
- SIRT
sirtuin
- STAT
signal transducer and activator of transcription
- TCA
tricarboxylic acid
- USP52
ubiquitin‐specific protease 52
- VDU
pVHL‐interacting deubiquitinating enzyme
- VEGF
vascular endothelial growth factor
- VHL
von Hippel‐Lindau
- YB‐1
Y‐box‐binding protein 1
- 2HG
2‐hydroxyglutarate
- 2OG
2‐oxoglutarate
1. INTRODUCTION
The existence of a hypoxia‐inducible transcriptional factor was first predicted in 1991 through an investigation on the cis‐acting regulatory elements of the human erythropoietin gene (Epo), the expression of which is known to be induced under hypoxic conditions1, 2 (Figure 1). Wang and Semenza subsequently succeeded in purifying the responsible factor in 1995, identified it as a heterodimer composed of approximately 120‐kDa and 90‐kDa proteins, and designated them as HIF‐1α and HIF‐1β, respectively (Figure 1)3. Since then, HIF‐1 has been shown to play important roles in the growth and malignant progression of tumors by inducing the transcription of hundreds of genes that function in improving oxygen availability in tumor tissues (angiogenesis/neovascularization4, 5, 6, 7), the escape of cancer cells from tumor hypoxia (invasion and metastasis of cancer cells5, 8, 9), and the adaption of cancer cells to a hypoxic microenvironment (metabolic reprogramming7, 10, 11, 12, 13, 14). As a result of these diverse functions of its downstream genes, HIF‐1 has been widely recognized as a rational target for cancer therapy5, 15, 16 and, thus, marked efforts have been devoted to elucidating the molecular mechanisms underlying its activation.
Figure 1.
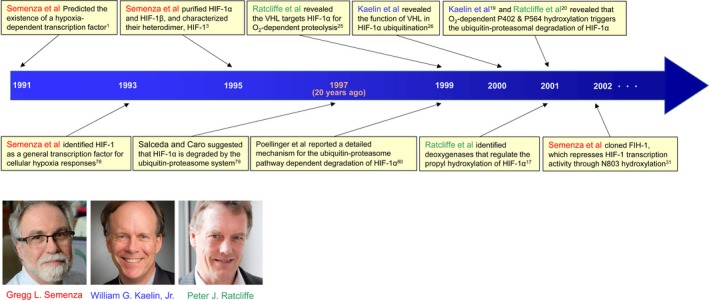
History of hypoxia‐inducible factor 1 (HIF‐1) biology in the first era. Gregg L. Semenza, William G. Kaelin, Jr, and Peter J. Ratcliffe have contributed to elucidating the molecular mechanism responsible for HIF‐1‐mediated adaptive responses to hypoxia in the first era of HIF‐1 biology. The importance of the oxygen‐dependent and ubiquitin‐mediated proteolysis of hypoxia‐inducible factor 1α (HIF‐1α) was first reported by Salceda and Caro 20 years ago in 1997.79 VHL, von Hippel‐Lindau. Images courtesy of the Albert and Mary Lasker Foundation
Regulatory mechanisms, which have the biggest impact on HIF‐1 activity, are regulated through post‐translational modifications to the HIF‐1α protein (Figure 2). The hydroxylation of HIF‐1α by oxygen‐, iron‐, and 2‐oxoglutarate (2OG)‐dependent dioxygenase enzymes at two specific proline residues (P402 and P564) and one specific asparagine residue (N803) leads to proteolysis and a decrease in the transactivation activity of HIF‐1α, respectively, such that HIF‐1 may express its transcriptional activity under hypoxic conditions.17, 18, 19, 20 Consequently, the expression of HIF‐1α itself and its downstream genes are detected in hypoxic regions, which are located 70‐100 μm from functional blood vessels in malignant solid tumors.21 However, the expression of HIF‐1α is often observed regardless of the distance from functional tumor blood vessels (and potentially regardless of oxygen availability) in immunohistochemical analyses of clinical tumor samples.22 These findings suggest that there are mechanisms other than the above‐described hypoxia‐dependent mechanism. In the last decade, HIF‐1 activity has consistently been reported to be regulated not only at the post‐translational level in an oxygen‐dependent way, but also at other levels, such as transcription initiation, translation initiation, heterodimer formation, translocation into the nucleus, and transactivation, regardless of the surrounding oxygen conditions.11 Moreover, prolyl‐ and asparagine‐hydroxylation‐mediated decreases in HIF‐1 activity were shown to be abrogated, even in the presence of oxygen, in some cases.7, 23, 24 We herein review the last two decades of knowledge on the regulatory mechanisms of HIF‐1 activity including not only well‐established oxygen‐ and hydroxylation‐dependent mechanisms, but also up‐to‐the‐minute information on cancer‐associated genetic and functional alteration‐mediated mechanisms.
Figure 2.
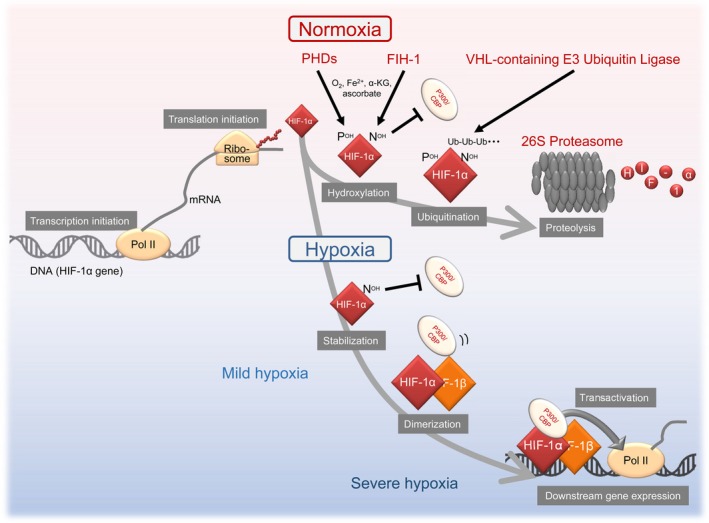
Conventional oxygen‐ and dioxygenase‐dependent regulatory mechanisms for hypoxia‐inducible factor 1 (HIF‐1) activity. FIH‐1, factor‐inhibiting HIF‐1; HIF‐1α, hypoxia‐inducible factor 1α; HIF‐1β, hypoxia‐inducible factor 1β; PHD, prolyl‐4‐hydroxylase; VHL, von Hippel‐Lindau
2. CONVENTIONAL DOGMA REGARDING DIOXYGENASE‐DEPENDENT REGULATION OF HIF‐1 ACTIVITY
After HIF‐1 was reported to be a heterodimer,3 HIF‐1α was recognized as the main regulatory subunit for HIF‐1 activity, whereas HIF‐1β was a constitutively expressed subunit. Although the expression levels of HIF‐1α are now known to be regulated at multiple levels, the initially elucidated and most influential molecular mechanism on HIF‐1 activity is that mediated by the dioxygenase‐dependent and ubiquitin‐proteasome pathway‐mediated system (Figures 1 and 2). Two proline residues positioned at amino acids (a.a.) 402 and 564 of HIF‐1α are hydroxylated by dioxygenases, called PHD (including PHD1‐3), in an oxygen‐dependent way (Figures 2 and 3).17, 19, 20 Prolyl hydroxylation triggers a ubiquitination reaction by pVHL‐containing E3 ubiquitin ligase, leading to the proteolysis of HIF‐1α through the ubiquitin‐proteasome system with a half‐life of 6‐8 min (Figure 2).25, 26, 27 As HIF‐1 promotes angiogenesis by inducing the expression of proangiogenic factors, such as VEGF, this mechanism appears to be reasonable from the viewpoint of a characteristic feature of VHL disease whereby a deficiency in functional VHL as a result of a mutation in chromosome 3p25.3 causes vascular‐rich hemangioblastomas, renal cell carcinomas (mainly clear cell carcinomas), and pheochromocytomas.28, 29
Figure 3.
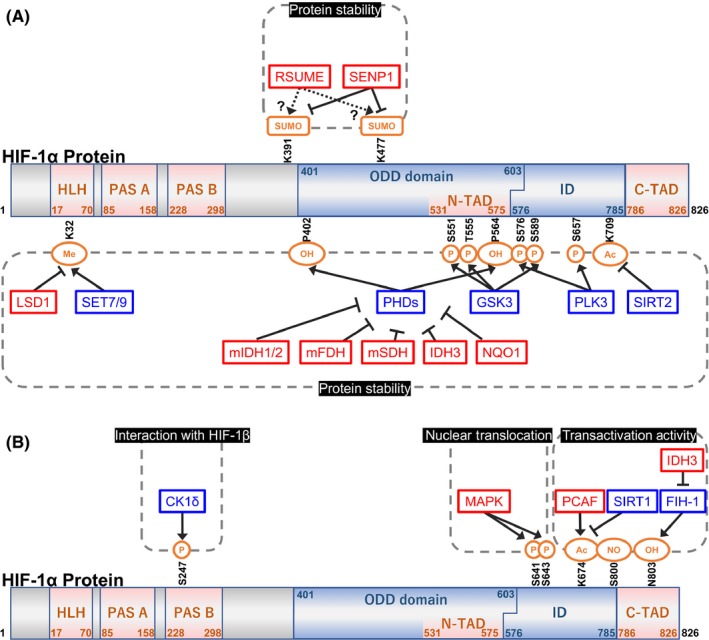
Schematic diagram showing the primary structure and hydroxylated (OH), SUMOylated (Sumo), phosphorylated (P), acetylated (Ac), nitrosylated (NO), and methylated (Me) amino acid residues of the hypoxia‐inducible factor 1α (HIF‐1α) protein. Gene symbols represented in red and blue indicate positive and negative regulators of hypoxia‐inducible factor 1 (HIF‐1), respectively. Factors are categorized into 4 groups by dashed squares according to the levels to which HIF‐1 activity is regulated; at the level of protein stability (A), interactions with hypoxia‐inducible factor 1β (HIF‐1β) (B), nuclear translocation (B), and transactivation activity (B). CK1δ, casein kinase 1δ; C‐TAD, C‐terminal transactivation domain; FIH‐1, factor‐inhibiting HIF‐1; GSK‐3, glycogen synthase kinase 3; HLH, helix‐loop‐helix; ID, inhibitory domain; IDH, isocitrate dehydrogenase; LSD1, lysine‐specific demethylase‐1; N‐TAD, N‐terminal transactivation domain; NQO1, NAD(P)H:quinone oxidoreductase 1; ODD, oxygen‐dependent degradation; PAS, Per‐Arnt‐Sim; PCAF, p300/CBP‐associated factor; PHD, prolyl‐4‐hydroxylase; PLK3, polo‐like kinase 3; RSUME, RWD‐containing SUMOylation enhancer; SDH, succinate dehydrogenase; SENP, sentrin/SUMO‐specific protease; SIRT, sirtuin
In addition to the proteolytic mechanism triggered by PHD, another dioxygenase, FIH‐1, was found to play a key role in the inactivation of HIF‐1 under normoxic conditions (Figures 2 and 3). FIH‐1 hydroxylates the asparagine residue positioned at a.a. 803 of HIF‐1α in an oxygen‐dependent way, and suppresses recruitment of the transcriptional co‐activator complex, p300/CBP acetyltransferases, to the HIF‐1α protein.30, 31 Thus, asparagine hydroxylation results in the inactivation of the transactivation activity of HIF‐1α under normoxic conditions.
Under hypoxic conditions, as a result of decreases in the hydroxylation activity of prolyl and asparagine dioxygenases, HIF‐1α becomes stable and acquires transactivation activity (Figure 2).17, 19, 20, 30, 31 The active form of HIF‐1α then translocates into the nucleus and interacts with its partner subunit, HIF‐1β. The resultant heterodimer, HIF‐1, induces the transcription initiation of hundreds of genes by binding to their enhancer element, HRE, which is located in or around the promoter region of each HIF‐1‐regulated gene.32, 33
Although PHD and FIH‐1 both function as sensors of oxygen concentrations and down‐regulate the expression and transactivation activity of HIF‐1α with their hydroxylation activities, their Km values differ from each other.34, 35 As the Km value of FIH‐1 is markedly lower than that of PHD, the HIF‐1α protein initially accumulates under mild hypoxic conditions (e.g. treatment with 1% oxygen for 5 hours35, 36) because of decreases in the enzymatic activities of PHD. It then acquires transactivation activity by interacting with p300/CBP under severely hypoxic conditions as a result of the inactivation of FIH‐1 (e.g. treatment with 0.2% oxygen for 5 hours35, 36), eventually showing maximum transcriptional activity.
3. GENETIC, MECHANISTIC, AND FUNCTIONAL ALTERATION‐MEDIATED MECHANISMS
Dioxygenase activities of PHD and FIH‐1 are dependent not only on oxygen, but also on 2OG (also known as α‐ketoglutarate) and ferrous ions (Fe2+); therefore, HIF‐1 becomes active in their absence, even under normoxic conditions. In addition, genetic, mechanistic, and/or functional alterations in genes that potentially influence intracellular levels of oxygen, 2OG, or Fe2+ or that directly influence the rates of the neo‐synthesis or degradation of HIF‐1α have been reported to up‐regulate HIF‐1 activity and subsequently promote the malignant progression of tumors. We herein review the following accumulated information (Figure 3; Tables 1, 2, 3).
Table 1.
Positive and negative regulators of hypoxia‐inducible factor 1 (HIF‐1) functioning at transcriptional initiation, transcript stability, and translational initiation levels
| Functional category/Gene symbol | Positive or negative impact on HIF‐1 activity | Regulatory mechanisms for HIF‐1 activity | Reference |
|---|---|---|---|
| Transcription initiation of the hif‐1α gene | |||
| ISGF3 (STAT1/STAT2/IRF9) | Positive | Binding to the promoter region of the hif‐1α gene to activate transcription initiation | 37 |
| STAT3 | Positive | Binding to the promoter region of the hif‐1α gene to activate transcription initiation | 38 |
| NF‐κB | Positive | Binding to the promoter region of the hif‐1α gene to activate transcription initiation | 39 |
| PI3K/Akt/PKC/HDAC pathway | Positive | Up‐regulating transcription initiation in case of the G13997A mutation in mitochondrial ND6 gene | 40 |
| LY6E | Positive | Activating the PI3K/Akt pathway through a decrease in PTEN expression | 6 |
| HIF‐1 | Positive | Binding to HRE in the promoter regions of the hif‐1α gene for positive autoregulation | 41 |
| NRF‐1 | Negative | Binding to the promoter region of the hif‐1α gene to repress transcription initiation | 42 |
| Stability of the hif‐1α transcript (mRNA) | |||
| P‐bodies (USP52/PAN2) | Positive | Interacting with the 3′‐UTR of HIF‐1α mRNA for its stabilization | 43 |
| Translation initiation of the hif‐1α gene | |||
| PI3K/Akt pathway | Positive | Up‐regulating cap‐dependent and IRES‐dependent translation initiation | 45, 47, 56 |
| YB‐1 | Positive | Binding to the unique secondary structure of the 5′UTR of HIF‐1α mRNA to enhance translation initiation | 48 |
| ATR | Positive | Influencing the cis‐element in the coding region of HIF‐1α mRNA when activated by replicative stress | 49 |
ATR, ataxia telangiectasia and Rad3‐related protein; HDAC, histone deacetylase; HRE, hypoxia‐response element; IRES, internal ribosomal entry site; IRF9, interferon regulatory factor 9; ISGF3, interferon‐stimulated gene factor 3; LY6E, lymphocyte antigen 6 complex locus E; ND6, NADH dehydrogenase subunit 6; NF‐κΒ, nuclear factor kappa B; NRF‐1, NF‐E2‐related factor 1; PAN2, poly(A) nuclease 2; PI3K, phosphoinositide 3‐kinase; PKC, protein kinase C; STAT, signal transducer and activator of transcription; USP52, ubiquitin‐specific protease 52; YB‐1, Y‐box‐binding protein 1.
Table 2.
Positive and negative regulators of hypoxia‐inducible factor 1 (HIF‐1) functioning at protein stability levels
| Functional category/Gene symbol | Positive or negative impact on HIF‐1 activity | Regulatory mechanisms for HIF‐1 activity | Reference |
|---|---|---|---|
| Stability of the HIF‐1α protein by modulating its prolyl hydroxylation status | |||
| PHD1, 2, 3 | Negative | Hydroxylating P402 and P564 of HIF‐1α for ubiquitination | 17, 19, 20 |
| Loss‐of‐function mutant of SDH | Positive | Inactivation of PHD and FIH‐1 through “product inhibition” as a result of the abnormal accumulation of succinate | 24 |
| Loss‐of‐function mutant of FH | Positive | Inactivation of PHD and FIH‐1 through “product inhibition” as a result of the abnormal accumulation of fumarate | 23 |
| Gain‐of‐function mutants of IDH1, 2 | Controversial | L‐2HG inhibits PHD activities leading to an increase in HIF‐1α levels, whereas D‐2HG promotes PHD | 51 |
| L‐2HG inhibits PHD activities leading to an increase in HIF‐1α levels, whereas D‐2HG has no effect | 52 | ||
| IDH3 | Positive | Inactivating PHD through a decrease in 2OG level, when overexpressed aberrantly. | 7 |
| NQO1 | Positive | Binding to HIF‐1α and physically preventing its interaction with PHD | 53 |
| SIRT2 | Negative | Deacetylating HIF‐1α at K709 and enhancing the interaction between HIF‐1α and PHD2 | 56 |
| Stability of the HIF‐1α protein without modulating its prolyl hydroxylation status | |||
| HSP90 | Positive | Protecting HIF‐1α from RACK1‐mediated and PHD‐VHL‐independent proteolysis | 57 |
| GSK‐3 | Negative | Phosphorylating S551, T555, and S589 of HIF‐1α for PHD‐VHL‐independent proteolysis | 58 |
| PLK3 | Negative | Phosphorylating S576 and S657 of HIF‐1α for PHD‐independent proteolysis | 59 |
| RSUME | Positive | SUMOylating HIF‐1α for its stabilization | 62 |
| SENP1 | Positive | deSUMOylating HIF‐1α to inhibit VHL‐mediated ubiquitination in a PHD‐independent way | 63 |
| SET7/9 | Negative | Methylating HIF‐1α at K32 for its ubiquitin‐mediated proteolysis in a PHD‐VHL‐independent way | 64 |
| LSD1 | Positive | Demethylating HIF‐1α at K32 to suppress PHD‐VHL‐independent proteolysis | 65 |
| Stability of the HIF‐1α protein by modulating its ubiquitination status | |||
| VHL | Negative | Ubiquitinating HIF‐1α for its proteasomal degradation | 25, 26, 27 |
| VDU (also known as USP20) | Positive | Deubiquitinating HIF‐1α for its stabilization | 66 |
| USP8 | Positive | Deubiquitinating HIF‐1α for its stabilization | 67 |
| UCHL1 | Positive | Deubiquitinating HIF‐1α for its stabilization | 8, 77 |
D‐2HG, D‐2‐hydroxyglutarate; FH, fumarate hydratase, FIH‐1, factor‐inhibiting HIF‐1; GSK‐3, glycogen synthase kinase 3; HSP, heat‐shock protein; IDH, isocitrate dehydrogenase; LSD1, lysine‐specific demethylase‐1; L‐2HG, L‐2‐hydroxyglutarate; NQO1, NAD(P)H:quinone oxidoreductase 1; PHD, prolyl‐4‐hydroxylase; PLK3, polo‐like kinase 3; RACK1, receptor for activated C kinase; RSUME, RWD‐containing SUMOylation enhancer; SDH, succinate dehydrogenase; SENP, sentrin/SUMO‐specific protease; SIRT, sirtuin; VDU, pVHL‐interacting deubiquitinating enzyme; VHL, von Hippel‐Lindau; 2OG, 2‐oxoglutarate.
Table 3.
Positive and negative regulators of hypoxia‐inducible factor 1 (HIF‐1) functioning at other levels
| Functional category/Gene symbol | Positive or negative impact on HIF‐1 activity | Regulatory mechanisms for HIF‐1 activity | Reference |
|---|---|---|---|
| Nuclear translocation | |||
| Dynein | Positive | Interacting with NLS of HIF‐1α for the nuclear translocation of HIF‐1α | 69 |
| AMPK/HDAC pathway | Positive | Triggering HIF‐1α nuclear localization by deacetylating HSP70 and promoting the HIF‐1α‐HSP90 interaction | 70 |
| MAPK | Negative | Phosphorylating S641 and S643 for the export of HIF‐1α from the nucleus | 71, 72 |
| Heterodimer formation | |||
| CK1δ | Negative | Phosphorylating HIF‐1α at S247 to inhibit heterodimer formation with HIF‐1β | 73 |
| Transactivation activity of the HIF‐1α protein | |||
| FIH‐1 | Negative | Hydroxylating N803 of HIF‐1α to inhibit the interaction between HIF‐1α and p300/CBP | 30, 31 |
| SIRT1 | Negative | Inhibiting the HIF‐1α‐p300/CBP interaction by deacetylating HIF‐1α K647, which is acetylated by PCAF | 74 |
| XBP1 | Positive | Forming a transcriptional complex with HIF‐1α and promoting the recruitment of RNA polymerase II | 76 |
| IDH3 | Positive | Inactivating FIH‐1 through a decrease in 2OG levels when overexpressed aberrantly | 7 |
AMPK, AMP‐activated protein kinase; CBP, CREB‐binding protein; CK1δ, casein kinase 1δ; FIH‐1, factor‐inhibiting HIF‐1; HDAC, histone deacetylase; HSP, heat‐shock protein; IDH, isocitrate dehydrogenase; NLS, nuclear localization signal; PCAF, p300/CBP‐associated factor; SIRT, sirtuin; 2OG, 2‐oxoglutarate.
3.1. Mechanisms regulating HIF‐1α mRNA levels
Transcription initiation is generally regarded as one of the most influential steps affecting the expression levels of genes. It is typically stimulated by one or multiple transcription factors that bind to a cognate enhancer sequence in its target promoter region. Transcription of the HIF‐1α gene is not an exception, although basal levels of HIF‐1α may be assured regardless of oxygen availability. Previous studies reported that each transcription factor, such as the ISGF3 complex, which is composed of STAT1/STAT2/IRF9,37 STAT3,38 or NF‐κB,39 binds to the promoter region of the HIF‐1α gene in order to activate its transcription initiation (Table 1). Moreover, the PI3K/Akt/PKC/HDAC pathway was also shown to be involved in the up‐regulation of HIF‐1α transcription when cells had the G13997A mutation in the mitochondrial ND6 gene40 (Table 1). We also identified LY6E as an upstream activator of HIF‐1α transcription6 (Table 1). Another mechanism may exist whereby tumor‐associated CpG demethylation facilitates the positive autoregulation of HIF‐1α expression at the transcription level. For example, Koslowski et al41 found that the HIF‐1α promoter itself possesses an HRE, which is normally repressed by the methylation of a CpG dinucleotide located in the core element in normal cells, but, in contrast, is frequently and aberrantly activated by demethylation in colon cancer cell lines and primary colon cancer specimens, enabling the binding of HIF‐1α to its own promoter, which results in the autotransactivation of HIF‐1α expression (Table 1). However, negative regulators of the transcription initiation of the HIF‐1α gene have been reported. An example is NRF‐1; after binding to the HIF‐1α promoter region, NRF‐1 represses its transcription initiation42 (Table 1).
In addition to the regulation of transcription initiation efficiency, that for mRNA stability is a key determinant of mRNA expression levels. Regarding HIF‐1α mRNA stability, among processing body (P‐body) components, USP52/PAN2 was found to stabilize HIF‐1α mRNA by interacting with (or at least by co‐localizing with) the 3′‐untranslated region (UTR) of HIF‐1α mRNA, leading to an increase in HIF‐1α protein levels43 (Table 1).
3.2. Mechanisms regulating the translation initiation of the HIF‐1α gene
Translation initiation is another important regulatory step for gene expression, as shown in the regulation of HIF‐1α expression levels. Translation is initiated through a cap‐dependent or IRES‐dependent mechanism.44 Regarding HIF‐1α, the PI3K/Akt pathway was shown to affect and up‐regulate the translational initiation of the HIF‐1α protein45, 46, 47 (Table 1). YB‐1 was also found to directly bind to a unique secondary structure of the 5′UTR of HIF‐1α mRNA and enhance its translation initiation48 (Table 1). Moreover, when activated through replicative stress under hypoxic conditions, ATR was shown to stimulate the translation initiation of the HIF‐1α protein through a cis‐acting element contained in the HIF‐1α open reading frame49 (Table 1).
3.3. Mechanisms regulating HIF‐1α protein stability
The molecular mechanisms regulating the stability of HIF‐1α have the greatest impact on HIF‐1 activity. In around 2000, the factors responsible, such as VHL and PHD, were cloned one after another, as described earlier17, 19, 20, 25, 26 (Figure 1). Several gene products with the potential to influence the effects of these two factors on HIF‐1α were subsequently identified as novel indirect modulators of HIF‐1 activity.
3.3.1. Mechanisms affecting HIF‐1α stability by modulating its prolyl‐hydroxylation status
As PHD need oxygen and 2OG as substrates and Fe2+ and ascorbate as co‐factors for their dioxygenase effects on P402 and P564 of HIF‐1α (Figure 2), gene products that influence the intracellular levels of these molecules have been shown to modulate HIF‐1 activity. 2OG is an intermediary metabolite of the TCA cycle; therefore, functional disorders in three key enzymes in the TCA cycle, SDH, FH, and IDH, were found to affect the activities of PHD and subsequently HIF‐1 as follows23, 24 (Table 2). Loss‐of‐function mutations in SDH and FH, which have been detected in “hereditary paraganglioma” and “hereditary leiomyomatosis and renal cell cancer”, were shown to cause abnormal accumulation of succinate and fumarate, respectively.23, 24 The accumulated metabolites inhibited PHD through product inhibition, whereby the product of an enzymatic reaction directly binds to the enzyme and inhibits its activity for negative feedback regulation, and eventually leads to the stabilization of HIF‐1α23, 24 (Table 2). IDH has three isoforms, IDH1‐3, and their functional disorders or aberrant overexpression influences the function of PHD because of the structural similarity between 2OG and 2HG. Mutations in IDH1/2 have been detected in gliomas or other hematological malignancies.50 Although the mutant forms of IDH1/2 have been shown to produce 2HG, it currently remains unclear whether 2HG activates or inhibits the activity of PHD or is dependent on the enantiomer, D or L (R or S). For example, Koivunen et al51 showed that L‐2HG inhibits PHD activity, leading to an increase in HIF‐1α levels, whereas D‐2HG promotes PHD (Table 1). Meanwhile, Burr et al52 reported similar, but controversial, findings showing that L‐2HG inhibits PHD, whereas D‐2HG has no effect (Table 2). Regarding IDH3, we found that the aberrant activation of wild‐type IDH3 inactivated PHD activity by decreasing intracellular levels of 2‐OG, resulting in the stabilization of HIF‐1α7 (Table 2).
Another mechanism has been reported whereby hydroxylation of the two proline residues is actively regulated in order to control the stability of HIF‐1α without directly inhibiting the enzymatic activities of PHD. NQO1 was recently identified as a responsible gene53 (Figure 3A; Table 2). Although NQO1 was originally cloned as a cytosolic reductase, a recent study showed that it binds to the ODD domain of HIF‐1α, physically prevents HIF‐1α from interacting with PHD, and eventually inhibits the proteasome‐mediated degradation of HIF‐1α.53 In an experimental mouse model, knockdown of NQO1 in human colorectal and breast cancer cell lines resulted in suppression of HIF‐1 signaling and tumor growth. Clinically, overexpression of NQO1 is associated with poor prognosis of patients with various cancers.54, 55
Sirtuin 2, which was originally cloned as an isoform of a family of NAD(+)‐dependent protein deacetylases that regulate cellular metabolism in response to stress, was recently shown to destabilize HIF‐1α using its deacetylase activity56 (Figure 3A; Table 2). SIRT2 has been shown to directly interact with HIF‐1α in order to deacetylate HIF‐1α at K709 and enhance the interaction of HIF‐1α with PHD2. SIRT2 overexpression was consistently confirmed to destabilize HIF‐1α by promoting prolyl‐hydroxylation, whereas the HIF‐1α protein was maintained at high levels when SIRT2 was deficient or when the acetylation‐defective mutation, K709R, was introduced into HIF‐1α.
3.3.2. Mechanisms affecting HIF‐1α stability without modulating its prolyl‐hydroxylation status
Another type of regulatory mechanism of HIF‐1α stability is not mediated by the conventional PHD‐VHL system.
A previous study reported that the molecular chaperone 90 kDa HSP90 and RACK1 competitively function to regulate the stability of HIF‐1α in a PHD‐VHL‐independent way57 (Table 2). As HSP90 works to protect HIF‐1α from the PHD‐VHL‐independent degradation of HIF‐1α, RACK1‐mediated dissociation of HSP90 from the Per‐Arnt‐Sim A (PAS‐A) domain of HIF‐1α and subsequent recruitment of the Elongin‐B/C ubiquitin ligase complex to HIF‐1α may destabilize HIF‐1α.
Accumulating evidence has shown that phosphorylation of specific serine and threonine residues of HIF‐1α, S551, T555, and S589, all of which are located within the ODD domain, plays an important role in the PHD‐VHL‐independent destabilization of HIF‐1α stability. Glycogen synthase kinase 3 was identified as the gene responsible for phosphorylation based on the finding that the pharmacological inhibition or depletion of GSK‐3 induced HIF‐1α, whereas overexpression of GSK‐3β reduced HIF‐1α, and that mutations in the phosphorylation residues, S551A/T555V/S589A, abolished the destabilization of HIF‐1α58 (Figure 3A; Table 2). This conclusion was further supported by the finding that GSK‐3‐mediated destabilization of HIF‐1α was independent of the hydroxylation sites within HIF‐1α (P402/P564/N803) and pVHL.
Other serine residues, S576 and S657, of HIF‐1α were also suggested to be phosphorylated and involved in regulation of HIF‐1α stability.59 The responsible modulator is an evolutionarily conserved serine/threonine kinase, PLK3, which is known to regulate cell cycle progression through the M phase59, 60 (Figure 3A; Table 2). Xu et al reported that PLK3 physically interacted with HIF‐1α under hypoxic conditions and phosphorylated two serine residues, S576 and S657, in order to destabilize HIF‐1α. PLK3 knock‐out MEF were consistently shown to express higher levels of HIF‐1α, whereas the phosphorylation‐resistant mutations, S576A and S657A, stabilized HIF‐1α. In addition, the finding that simultaneous mutations in the prolyl and Plk3‐targeting phosphorylation sites (P402A, P564A, S576A, and S657A) further increased the levels of HIF‐1α strongly suggested that Plk3 decreases HIF‐1α stability in a PHD‐independent way.
The SUMOylation of HIF‐1α is another post‐translational modification that affects HIF‐1α stability; however, it currently remains unclear whether SUMOylation increases or decreases the stability of HIF‐1α. Bae et al61 was the first to report in 2004 that SUMOylation increased HIF‐1α stability. They identified K391 and K477 of HIF‐1α as targets of SUMOylation and showed that overexpression of SUMO‐1 increased HIF‐1α stability (Figure 3A). In 2007, the SUMOylation enzyme for HIF‐1α, RSUME, was identified and found to increase the stability of HIF‐1α62 (Figure 3A; Table 2). However, an opposite function of SUMOylation on the stability of HIF‐1α was reported by Cheng et al63 in an analysis of an isoform of SENP, SENP1 (Figure 3A; Table 2). They showed that SENP1‐mediated deSUMOylation inhibited the interaction between HIF‐1α and VHL, resulting in the stabilization of HIF‐1α, suggesting that SUMOylation leads to HIF‐1α proteolysis. Expression levels of HIF‐1 downstream genes were consistently shown to be markedly reduced in MEF and in mice with the homozygous deletion of SENP1.
Besides the major modifications such as ubiquitination, phosphorylation, or SUMOylation described above, methylation also plays a role in modulating HIF‐1α stability that is irrelevant to PHD. A recent study showed that HIF‐1α is methylated by SET7/9 methyltransferase at K3264 (Figure 3A; Table 2), and LSD1 functions in its demethylation65 (Figure 3A; Table 2). K32 methylation triggers the ubiquitin‐mediated proteolysis of HIF‐1α in a PHD‐VHL‐independent mechanism. Knock‐in mice harboring biallelic methylation‐defective K32A mutations in the HIF‐1α gene showed enhanced retinal angiogenesis and tumor vascularization by HIF‐1α stabilization, further supporting the importance of methylation. Mutations near K32 (S28Y and R30Q) of HIF‐1α, which have been detected in human cancers, have also been reported to be involved in altered HIF‐1α stability affecting K32 methylation.64
3.3.3. Mechanisms affecting HIF‐1α stability by directly modulating its ubiquitination status
A deubiquitinating enzyme potentially acts as a stabilizer of HIF‐1α because ubiquitinated HIF‐1α may be rescued from degradation by deubiquitination.
The first factor identified as a deubiquitinating enzyme for HIF‐1α was VDU2, also known as USP2066 (Table 2). The interaction between HIF‐1α and VDU2 was shown using an immunoprecipitation assay. Molecular and cellular experiments showed that VDU2 interacted with HIF‐1α in order to induce the expression of HIF‐1 downstream genes, such as VEGF, through the deubiquitination and stabilization of HIF‐1α. Previous findings showing that VDU2 had no clear effect on another ubiquitinated short‐lived protein, p53, indicated the target specificity of VDU2. Other deubiquitinating enzymes (e.g. USP8) were also shown to stabilize HIF‐1α67 (Table 2). Furthermore, we also identified another deubiquitinating protein, UCHL1,8 as described below (Table 2).
3.4. Mechanisms regulating nuclear translocation of HIF‐1α
Translocation from the cytosol into the nucleus is another important regulatory step in HIF‐1 activity. HIF‐1α has the ability to shuttle between the nucleus and the cytoplasm.68 Carbonaro et al69 reported that the HIF‐1α protein associates with polymerized microtubules and traffics to the nucleus when its NLS is recognized by the motor protein, dynein (Table 3). In addition to this mechanism, the AMPK‐HDAC pathway deacetylates the cytosolic molecular chaperone, HSP70, promotes the interaction between HIF‐1α and HSP90, and facilitates the rapid nuclear accumulation of HIF‐1α70 (Table 3).
In contrast, the active export of HIF‐1α from the nucleus was found to be important in the regulation of HIF‐1 activity. Phosphorylation of the serine residues, Ser641 and Ser643, which are in close proximity to an atypical nuclear export signal of HIF‐1α (between residues 632 and 639), by MAPK has been reported to block export by chromosome region maintenance 1 protein homologue (CRM1), thereby promoting nuclear accumulation and transcriptional activity68, 71, 72 (Figure 3B; Table 3).
3.5. Mechanisms modulating HIF‐1α and HIF‐1β heterodimer formation
After being stabilized, HIF‐1α forms a heterodimer with the constitutively expressed binding partner, HIF‐1β. Heterodimer formation is also an important regulatory step in the activation of HIF‐1, and CK1δ was found to be involved in this process as a result of this kinase activity on HIF‐1α S24773 (Figure 3B; Table 3). A luciferase assay to evaluate HIF‐1 transcription activity showed that the siRNA‐mediated silencing of CK1δ enhanced, whereas the overexpression of CK1δ inhibited HIF‐1 activity under hypoxic conditions. The phosphorylation‐defective mutation, S247A, enhanced, whereas the corresponding phosphorylation‐mimic mutation, S247Q, inhibited heterodimerization, further suggesting the importance of the CK1δ‐dependent phosphorylation of HIF‐1α.
3.6. Mechanisms regulating the transactivation activity of HIF‐1α
Regulation of the transactivation activity of HIF‐1 is primarily modulated by the hydroxylation of N803 by an oxygen‐dependent dioxygenase other than PHD, namely, FIH‐1 (Figure 2). Asparagine hydroxylation prevents the acetyltransferases, p300/CBP, from interacting with the C‐terminal transactivation domain (C‐TAD) of HIF‐1α and subsequently inhibits the recruitment of RNA polymerase II to the promoter regions of the downstream genes of HIF‐1. Thus, factors modulating this process including not only the asparagine hydroxylation status, but also the efficiency of p300/CBP recruitment have been found to affect the transactivation activity of HIF‐1.
Another isoform of SIRT2, SIRT1, was shown to suppress the transactivation activity of HIF‐1α74 (Figure 3B; Table 3). HIF‐1α is acetylated by PCAF at K647 (Figure 3B; Table 3). SIRT1 deacetylates it and represses HIF‐1α activity by inhibiting the recruitment of p300/CBP. SIRT1 was down‐regulated under hypoxic conditions as a result of reduced NAD(+) levels, which facilitated the acetylation and activation of HIF‐1α.
Yasinska and Sumbayev reported the importance of another post‐translational modification of HIF‐1α, C800 S‐nitrosylation, for increases in the transactivation activity of HIF‐1α75 (Figure 3B). S‐nitrosylation was previously reported to stimulate the recruitment of p300 to the HIF‐1α C‐terminal domain in a GST pull‐down assay. The increase in HIF‐1 activity was not observed when the cysteine residue was substituted with alanine.
Chen et al recently reported that XBP1 is activated in triple‐negative breast cancers (TNBC) and plays a pivotal role in their tumorigenicity and progression.76 From a mechanistic viewpoint, they showed that XBP1 formed a transcriptional complex with HIF‐1α, enhancing the expression of HIF‐1α target transcripts by recruitment of RNA polymerase II (Table 3). Depletion of XBP1 inhibited tumor growth and relapse.
4. SCREENING OF NOVEL UPSTREAM ACTIVATORS OF HIF‐1
We recently established a genetic screening method to investigate novel genes responsible for the activation of HIF‐1, and successfully identified the following three genes as novel activators of HIF‐1: IDH3 (Tables 2 and 3), UCHL1 (Table 2), and LY6E6, 7, 8 (Table 1). We reported that the forced expression of each promoted the malignant progression and/or growth of tumors through a different mechanism for the activation of HIF‐1.
4.1. IDH3‐HIF‐1 axis
Aberrant overexpression of wild‐type IDH3α, α subunit of the IDH3 heterotetramer, significantly decreased 2OG levels and increased the stability and transactivation activity of HIF‐1α in cancer cells by inactivating 2OG‐ as well as the oxygen‐dependent dioxygenases (Tables 2 and 3), PHD and FIH‐1.7 However, the silencing of IDH3α inhibited the Warburg effect and angiogenesis, resulting in a significant delay in tumor growth. Clinically, a database‐based Kaplan‐Meier analysis showed that the expression of IDH3α was associated with the poor postoperative overall survival of lung and breast cancer patients. These findings support the use of IDH3 as a novel diagnostic and treatment target.
4.2. UCHL1‐HIF‐1 axis
Regarding UCHL1, we showed that it stabilizes the HIF‐1α protein by deconjugating the VHL‐mediated ubiquitin from HIF‐1α8 (Table 2). In murine models of pulmonary metastasis, the aberrant overexpression of UCHL1 was shown to promote distant tumor metastases in a HIF‐1‐dependent way. In contrast, the inhibition of the UCHL1‐HIF‐1 axis resulted in significant suppression of metastatic tumor formation. Intratumoral expression levels of UCHL1 were found to correlate with those of HIF‐1α and be strongly associated with the poor overall survival of patients with breast and lung cancers. These findings collectively provide a rationale for using UCHL1 as a prognostic marker and therapeutic target of cancers. In addition, we recently reported that UCHL1 increased the radioresistance of cancer cells by producing the antioxidant, reduced glutathione (GSH), through HIF‐1‐mediated reprogramming of the glucose metabolic pathway.77
4.3. LY6E‐HIF‐1 axis
Regarding LY6E, we showed that it induced HIF‐1α expression principally at the transcription level6 (Table 1). This, in turn, increased the expression levels of proangiogenic factors, such as VEGFA and platelet‐derived growth factor subunit B (PDGFB), by decreasing those of phosphatase and tensin homolog (PTEN) and subsequently activating the PI3K/Akt pathway. The LY6E‐mediated activation of HIF‐1 was found to eventually increase micro blood vessel density and promote the growth of tumor xenografts in in vivo mouse models. Expression levels of LY6E were significantly higher in human breast cancers than in normal breast tissues, and were strongly associated with the poor prognoses of patients with various cancers.
5. CONCLUSION AND PERSPECTIVE
Hypoxia‐inducible factor 1 has been associated with the malignant progression and growth of cancers. Thus, HIF‐1 has attracted considerable attention as a target for cancer therapies and diagnoses, and extensive efforts have been made to elucidate the mechanisms responsible for its activation. Accumulating information suggested that multiple mechanisms, in addition to the well‐established mechanisms, which are mediated by PHD‐dependent hydroxylation, VHL‐dependent ubiquitination, and FIH‐1‐dependent hydroxylation, contribute to the regulation of HIF‐1 activity. Although molecular and cellular research has shown individual mechanisms of action, the mechanistic relationships among some of them remain largely unknown. A clearer understanding of these mechanistic relationships is critical for fully elucidating when, where, and how HIF‐1 becomes active and the application of this information to the development of novel therapeutic strategies for cancers.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
ACKNOWLEDGMENT
Our study was supported by the Joint Usage Program of the Radiation Biology Center, Kyoto University, Japan. S.K. is a Japan Society for the Promotion of Science (JSPS) postdoctoral research fellow, SPD.
Koyasu S, Kobayashi M, Goto Y, Hiraoka M, Harada H. Regulatory mechanisms of hypoxia‐inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018;109:560–571. https://doi.org/10.1111/cas.13483
Sho Koyasu and Minoru Kobayashi contributed equally to this work.
Funding information
H.H. was supported by the program for Precursory Research for Embryonic Science and Technology (PRESTO) from the Japan Science and Technology Agency (JST) (No. JPMJPR14M8), by the Research Project on the Development of New Drugs from the Japan Agency for Medical Research and Development (AMED) (No. 17ak0101084 h0001), by the Bilateral Joint Research Projects from JSPS, by Grants‐in‐Aid for Scientific Research (B) (Nos. 26293276; 17H04261), for Living in Space (No. 16H01640), for Conquering Cancer through Neo‐dimensional Systems Understanding (No. 16H01573), and for Challenging Exploratory Research (Nos. 26670558; 16K15576) from MEXT, Japan, and by the research grant programs of the Princess Takamatsu Cancer Research Fund, the Takeda Science Foundation, the Relay for Life Japan, the Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care, the Suzuken Memorial Foundation, the Tokyo Biochemical Research Foundation, the Japan‐China Medical Association, and the Kobayashi Foundation for Cancer Research. S.K. was supported by a Grant‐in‐Aid for JSPS research fellows (No. 17J07699) from JSPS, Japan. M.K. was supported by a Grant‐in‐Aid for Challenging Exploratory Research (No. 16K15577) from MEXT, Japan. Y.G. was supported by a Grant‐in‐Aid for Young Scientists (B) (No. 17K16433) from JSPS, Japan.
REFERENCES
- 1. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia‐inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci USA. 1991;88:5680‐5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Imagawa S, Goldberg MA, Doweiko J, Bunn HF. Regulatory elements of the erythropoietin gene. Blood. 1991;77:278‐285. [PubMed] [Google Scholar]
- 3. Wang GL, Semenza GL. Purification and characterization of hypoxia‐inducible factor 1. J Biol Chem. 1995;270:1230‐1237. [DOI] [PubMed] [Google Scholar]
- 4. Harada H, Kizaka‐Kondoh S, Li G, et al. Significance of HIF‐1‐active cells in angiogenesis and radioresistance. Oncogene. 2007;26:7508‐7516. [DOI] [PubMed] [Google Scholar]
- 5. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003;3:721‐732. [DOI] [PubMed] [Google Scholar]
- 6. Yeom CJ, Zeng L, Goto Y, et al. LY6E: a conductor of malignant tumor growth through modulation of the PTEN/PI3K/Akt/HIF‐1 axis. Oncotarget. 2016;7:65837‐65848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeng L, Morinibu A, Kobayashi M, et al. Aberrant IDH3alpha expression promotes malignant tumor growth by inducing HIF‐1‐mediated metabolic reprogramming and angiogenesis. Oncogene. 2015;34:4758‐4766. [DOI] [PubMed] [Google Scholar]
- 8. Goto Y, Zeng L, Yeom CJ, et al. UCHL1 provides diagnostic and antimetastatic strategies due to its deubiquitinating effect on HIF‐1alpha. Nat Commun. 2015;6:6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Semenza GL. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med. 2012;18:534‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harada H. How can we overcome tumor hypoxia in radiation therapy? J Radiat Res (Tokyo). 2011;52:545‐556. [DOI] [PubMed] [Google Scholar]
- 11. Harada H. Hypoxia‐inducible factor 1‐mediated characteristic features of cancer cells for tumor radioresistance. J Radiat Res. 2016;57(Suppl 1):i99‐i105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Semenza GL. Oxygen‐dependent regulation of mitochondrial respiration by hypoxia‐inducible factor 1. Biochem J. 2007;405:1‐9. [DOI] [PubMed] [Google Scholar]
- 13. Semenza GL. Mitochondrial autophagy: life and breath of the cell. Autophagy. 2008;4:534‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Semenza GL. Regulation of cancer cell metabolism by hypoxia‐inducible factor 1. Semin Cancer Biol. 2009;19:12‐16. [DOI] [PubMed] [Google Scholar]
- 15. Kizaka‐Kondoh S, Inoue M, Harada H, Hiraoka M. Tumor hypoxia: a target for selective cancer therapy. Cancer Sci. 2003;94:1021‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Semenza GL. Hypoxia‐inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33:207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43‐54. [DOI] [PubMed] [Google Scholar]
- 18. Hirota K, Semenza GL. Regulation of hypoxia‐inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun. 2005;338:610‐616. [DOI] [PubMed] [Google Scholar]
- 19. Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464‐468. [DOI] [PubMed] [Google Scholar]
- 20. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF‐alpha to the von Hippel‐Lindau ubiquitylation complex by O2‐regulated prolyl hydroxylation. Science. 2001;292:468‐472. [DOI] [PubMed] [Google Scholar]
- 21. Yeom CJ, Goto Y, Zhu Y, Hiraoka M, Harada H. Microenvironments and cellular characteristics in the micro tumor cords of malignant solid tumors. Int J Mol Sci. 2012;13:13949‐13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trastour C, Benizri E, Ettore F, et al. HIF‐1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer. 2007;120:1451‐1458. [DOI] [PubMed] [Google Scholar]
- 23. Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143‐153. [DOI] [PubMed] [Google Scholar]
- 24. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF‐alpha prolyl hydroxylase. Cancer Cell. 2005;7:77‐85. [DOI] [PubMed] [Google Scholar]
- 25. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature. 1999;399:271‐275. [DOI] [PubMed] [Google Scholar]
- 26. Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia‐inducible factor requires direct binding to the beta‐domain of the von Hippel‐Lindau protein. Nat Cell Biol. 2000;2:423‐427. [DOI] [PubMed] [Google Scholar]
- 27. Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia‐inducible factor‐1 alpha by the von Hippel‐Lindau tumor suppressor protein. EMBO J. 2000;19:4298‐4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaelin WG Jr. The von Hippel‐Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865‐873. [DOI] [PubMed] [Google Scholar]
- 29. Kaelin WG Jr. The von Hippel‐Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13:680s‐684s. [DOI] [PubMed] [Google Scholar]
- 30. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858‐861. [DOI] [PubMed] [Google Scholar]
- 31. Mahon PC, Hirota K, Semenza GL. FIH‐1: a novel protein that interacts with HIF‐1alpha and VHL to mediate repression of HIF‐1 transcriptional activity. Genes Dev. 2001;15:2675‐2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mimura I, Nangaku M, Kanki Y, et al. Dynamic change of chromatin conformation in response to hypoxia enhances the expression of GLUT3 (SLC2A3) by cooperative interaction of hypoxia‐inducible factor 1 and KDM3A. Mol Cell Biol. 2012;32:3018‐3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High‐resolution genome‐wide mapping of HIF‐binding sites by ChIP‐seq. Blood. 2011;117:e207‐e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4‐hydroxylases that modify the hypoxia‐inducible factor. J Biol Chem. 2003;278:30772‐30780. [DOI] [PubMed] [Google Scholar]
- 35. Koivunen P, Hirsila M, Gunzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4‐hydroxylases. J Biol Chem. 2004;279:9899‐9904. [DOI] [PubMed] [Google Scholar]
- 36. Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286:13041‐13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gerber SA, Pober JS. IFN‐alpha induces transcription of hypoxia‐inducible factor‐1alpha to inhibit proliferation of human endothelial cells. J Immunol. 2008;181:1052‐1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dang EV, Barbi J, Yang HY, et al. Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell. 2011;146:772‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rius J, Guma M, Schachtrup C, et al. NF‐kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF‐1alpha. Nature. 2008;453:807‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koshikawa N, Hayashi J, Nakagawara A, Takenaga K. Reactive oxygen species‐generating mitochondrial DNA mutation up‐regulates hypoxia‐inducible factor‐1alpha gene transcription via phosphatidylinositol 3‐kinase‐Akt/protein kinase C/histone deacetylase pathway. J Biol Chem. 2009;284:33185‐33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koslowski M, Luxemburger U, Tureci O, Sahin U. Tumor‐associated CpG demethylation augments hypoxia‐induced effects by positive autoregulation of HIF‐1alpha. Oncogene. 2011;30:876‐882. [DOI] [PubMed] [Google Scholar]
- 42. Wang D, Zhang J, Lu Y, Luo Q, Zhu L. Nuclear respiratory factor‐1 (NRF‐1) regulated hypoxia‐inducible factor‐1alpha (HIF‐1alpha) under hypoxia in HEK293T. IUBMB Life. 2016;68:748‐755. [DOI] [PubMed] [Google Scholar]
- 43. Bett JS, Ibrahim AF, Garg AK, et al. The P‐body component USP52/PAN2 is a novel regulator of HIF1A mRNA stability. Biochem J. 2013;451:185‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593‐1612. [DOI] [PubMed] [Google Scholar]
- 45. Harada H, Itasaka S, Kizaka‐Kondoh S, et al. The Akt/mTOR pathway assures the synthesis of HIF‐1alpha protein in a glucose‐ and reoxygenation‐dependent manner in irradiated tumors. J Biol Chem. 2009;284:5332‐5342. [DOI] [PubMed] [Google Scholar]
- 46. Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia‐inducible factor 1alpha (HIF‐1alpha) synthesis: novel mechanism for HIF‐1‐mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995‐4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou J, Callapina M, Goodall GJ, Brune B. Functional integrity of nuclear factor kappaB, phosphatidylinositol 3’‐kinase, and mitogen‐activated protein kinase signaling allows tumor necrosis factor alpha‐evoked Bcl‐2 expression to provoke internal ribosome entry site‐dependent translation of hypoxia‐inducible factor 1 alpha. Cancer Res. 2004;64:9041‐9048. [DOI] [PubMed] [Google Scholar]
- 48. El‐Naggar AM, Veinotte CJ, Cheng H, et al. Translational activation of HIF1alpha by YB‐1 promotes sarcoma metastasis. Cancer Cell. 2015;27:682‐697. [DOI] [PubMed] [Google Scholar]
- 49. Fallone F, Britton S, Nieto L, Salles B, Muller C. ATR controls cellular adaptation to hypoxia through positive regulation of hypoxia‐inducible factor 1 (HIF‐1) expression. Oncogene. 2013;32:4387‐4396. [DOI] [PubMed] [Google Scholar]
- 50. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)‐enantiomer of 2‐hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burr SP, Costa AS, Grice GL, et al. Mitochondrial protein lipoylation and the 2‐oxoglutarate dehydrogenase complex controls hif1alpha stability in aerobic conditions. Cell Metab. 2016;24:740‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Oh ET, Kim JW, Kim JM, et al. NQO1 inhibits proteasome‐mediated degradation of HIF‐1alpha. Nat Commun. 2016;7:13593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ji M, Jin A, Sun J, et al. Clinicopathological implications of NQO1 overexpression in the prognosis of pancreatic adenocarcinoma. Oncol Lett. 2017;13:2996‐3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tong YH, Zhang B, Yan YY, et al. Dual‐negative expression of Nrf2 and NQO1 predicts superior outcomes in patients with non‐small cell lung cancer. Oncotarget. 2017;8:45750‐45758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Seo KS, Park JH, Heo JY, et al. SIRT2 regulates tumour hypoxia response by promoting HIF‐1alpha hydroxylation. Oncogene. 2015;34:1354‐1362. [DOI] [PubMed] [Google Scholar]
- 57. Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF‐1alpha and is required for O(2)‐independent and HSP90 inhibitor‐induced degradation of HIF‐1alpha. Mol Cell. 2007;25:207‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flugel D, Gorlach A, Michiels C, Kietzmann T. Glycogen synthase kinase 3 phosphorylates hypoxia‐inducible factor 1alpha and mediates its destabilization in a VHL‐independent manner. Mol Cell Biol. 2007;27:3253‐3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu D, Yao Y, Lu L, Costa M, Dai W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF‐1alpha. J Biol Chem. 2010;285:38944‐38950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ouyang B, Pan H, Lu L, et al. Human Prk is a conserved protein serine/threonine kinase involved in regulating M phase functions. J Biol Chem. 1997;272:28646‐28651. [DOI] [PubMed] [Google Scholar]
- 61. Bae SH, Jeong JW, Park JA, et al. Sumoylation increases HIF‐1alpha stability and its transcriptional activity. Biochem Biophys Res Commun. 2004;324:394‐400. [DOI] [PubMed] [Google Scholar]
- 62. Carbia‐Nagashima A, Gerez J, Perez‐Castro C, et al. RSUME, a small RWD‐containing protein, enhances SUMO conjugation and stabilizes HIF‐1alpha during hypoxia. Cell. 2007;131:309‐323. [DOI] [PubMed] [Google Scholar]
- 63. Cheng J, Kang X, Zhang S, Yeh ET. SUMO‐specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell. 2007;131:584‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim Y, Nam HJ, Lee J, et al. Methylation‐dependent regulation of HIF‐1alpha stability restricts retinal and tumour angiogenesis. Nat Commun. 2016;7:10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Baek SH, Kim KI. Regulation of HIF‐1alpha stability by lysine methylation. BMB Rep. 2016;49:245‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li Z, Wang D, Messing EM, Wu G. VHL protein‐interacting deubiquitinating enzyme 2 deubiquitinates and stabilizes HIF‐1alpha. EMBO Rep. 2005;6:373‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Troilo A, Alexander I, Muehl S, Jaramillo D, Knobeloch KP, Krek W. HIF1alpha deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep. 2014;15:77‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Depping R, Jelkmann W, Kosyna FK. Nuclear‐cytoplasmatic shuttling of proteins in control of cellular oxygen sensing. J Mol Med (Berl). 2015;93:599‐608. [DOI] [PubMed] [Google Scholar]
- 69. Carbonaro M, Escuin D, O'Brate A, Thadani‐Mulero M, Giannakakou P. Microtubules regulate hypoxia‐inducible factor‐1alpha protein trafficking and activity: implications for taxane therapy. J Biol Chem. 2012;287:11859‐11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen S, Yin C, Lao T, et al. AMPK‐HDAC5 pathway facilitates nuclear accumulation of HIF‐1alpha and functional activation of HIF‐1 by deacetylating Hsp70 in the cytosol. Cell Cycle. 2015;14:2520‐2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mylonis I, Chachami G, Samiotaki M, et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia‐inducible factor‐1alpha. J Biol Chem. 2006;281:33095‐33106. [DOI] [PubMed] [Google Scholar]
- 72. Mylonis I, Chachami G, Paraskeva E, Simos G. Atypical CRM1‐dependent nuclear export signal mediates regulation of hypoxia‐inducible factor‐1alpha by MAPK. J Biol Chem. 2008;283:27620‐27627. [DOI] [PubMed] [Google Scholar]
- 73. Kalousi A, Mylonis I, Politou AS, Chachami G, Paraskeva E, Simos G. Casein kinase 1 regulates human hypoxia‐inducible factor HIF‐1. J Cell Sci. 2010;123:2976‐2986. [DOI] [PubMed] [Google Scholar]
- 74. Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia‐inducible factor 1alpha. Mol Cell. 2010;38:864‐878. [DOI] [PubMed] [Google Scholar]
- 75. Yasinska IM, Sumbayev VV. S‐nitrosation of Cys‐800 of HIF‐1alpha protein activates its interaction with p300 and stimulates its transcriptional activity. FEBS Lett. 2003;549:105‐109. [DOI] [PubMed] [Google Scholar]
- 76. Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple‐negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nakashima R, Goto Y, Koyasu S, et al. UCHL1‐HIF‐1 axis‐mediated antioxidant property of cancer cells as a therapeutic target for radiosensitization. Sci Rep. 2017;7:6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang GL, Semenza GL. General involvement of hypoxia‐inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci USA. 1993;90:4304‐4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Salceda S, Caro J. Hypoxia‐inducible factor 1alpha (HIF‐1alpha) protein is rapidly degraded by the ubiquitin‐proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox‐induced changes. J Biol Chem. 1997;272:22642‐22647. [DOI] [PubMed] [Google Scholar]
- 80. Kallio PJ, Wilson WJ, O'Brien S, Makino Y, Poellinger L. Regulation of the hypoxia‐inducible transcription factor 1alpha by the ubiquitin‐proteasome pathway. J Biol Chem. 1999;274:6519‐6525. [DOI] [PubMed] [Google Scholar]