

Abstract
Explosive advances in next‐generation sequencer (NGS) and computational analyses have enabled exploration of somatic protein‐altered mutations in most cancer types, with coding mutation data intensively accumulated. However, there is limited information on somatic mutations in non‐coding regions, including introns, regulatory elements and non‐coding RNA. Structural variants and pathogen in cancer genomes remain widely unexplored. Whole genome sequencing (WGS) approaches can be used to comprehensively explore all types of genomic alterations in cancer and help us to better understand the whole landscape of driver mutations and mutational signatures in cancer genomes and elucidate the functional or clinical implications of these unexplored genomic regions and mutational signatures. This review describes recently developed technical approaches for cancer WGS and the future direction of cancer WGS, and discusses its utility and limitations as an analysis platform and for mutation interpretation for cancer genomics and cancer precision medicine. Taking into account the diversity of cancer genomes and phenotypes, interpretation of abundant mutation information from WGS, especially non‐coding and structure variants, requires the analysis of large‐scale WGS data integrated with RNA‐Seq, epigenomics, immuno‐genomic and clinic‐pathological information.
Keywords: cancer genome, mutational signature, non‐coding mutation, structural variant, whole genome sequencing
1. INTRODUCTION
Cancer is essentially a disease of the genome which evolves and progresses with accumulations of somatic mutations, including copy‐number alterations (CNA) and structural variants (SV), and epigenomic alterations with and without some hereditability (germline variants).1, 2 A number of familial cancer segregation studies and loss‐of‐heterozygosity (LOH) analyses on cancer tissues have identified germline and somatic mutations of several classical tumor suppressor genes, such as RB1, TP53 and APC,2, 3 and copy‐number analysis has found some oncogenes and underlying oncogenic activators, such as HER2/ERBB2 and MYC.1, 4 Some of these oncogenic mutations have been successfully targeted for molecular therapy, and specific and recurrent mutations of these oncogenes are now used to predict sensitivity to therapy, prognosis and residual disease.1, 2
Explosive advances in next‐generation sequencer (NGS) and computational analyses handling massive data have enabled us to comprehensively analyze cancer genome profiles at research and clinical levels, such as targeted sequencing for hundreds of genes, whole exome sequencing (WES), RNA sequencing (RNA‐Seq) and whole genome sequencing (WGS).5, 6 So far, to explore cancer genomic alterations and their diversity, more than 50 000 cancer genomes have been sequenced and accumulated worldwide, including The Cancer Genome Atlas (TCGA)2, 7 and The International Cancer Genome Consortium (ICGC),8 and hundreds of millions of cancer patients will have their genome sequenced by 2030. In these projects so far, WES is the main platform for cancer genome sequencing and vast amounts of mutational data in protein‐coding regions have been accumulated for all types of common and rare human tumors. These systematic studies of these cancer genome data have reveled scores of new cancer genes and pathways,2 and saturation analysis suggests that most driver genes with frequent mutation in cancer have been almost elucidated.9, 10 Researchers are now focusing on a “long tail” of rare mutated driver genes2, 11 and rare variants of the driver genes, in addition to validating these functional or clinical implications by integrating with clinical data and using functional assays. Pan‐cancer analysis on WES data demonstrated that carcinogen‐exposed cancer, such as melanoma and lung cancer, have far higher numbers of somatic mutations in coding regions among common cancers, while pediatric tumors and leukemia have much fewer mutations and only several protein‐altered mutations are present in their whole coding regions.12, 13 TCGA and ICGC provide comprehensive mutational data of coding regions in more than 20 000 cancers and the COSMIC database14 has been extensively curating mutations from targeted sequencing and WES, summarizing coding mutations for more than 1 000 000 cancer samples. However, there is limited information on somatic mutations in non‐coding regions spanning 98% of the human genome, which includes untranslated regions (UTR), introns, promoters, regulatory elements, non‐coding functional RNA, repetitive regions and mitochondrial genomes. Somatic structural variants (SV) including large deletions/insertions, inversion, duplication, translocation and pathogen (virus) integration in cancer genomes also remain widely unexplored (Figure 1A). WGS approaches can cover all of these unexplored mutations (Table 1) and help us to better understand the “whole” landscape of cancer genomes and elucidate the functions of these unexplored human genomic regions (Figure 1A). This approach combined with mathematical analysis and other omics analysis can clarify the underlying carcinogenesis and achieve molecular sub‐classification of cancer, which facilitates discovery of genomic biomarkers and personalized cancer medicine. This review describes recent technical approaches for cancer WGS and the future direction of cancer WGS, and discusses its utility and limitations as an analysis platform and for mutation interpretation for cancer genomics and cancer precision medicine.
Figure 1.
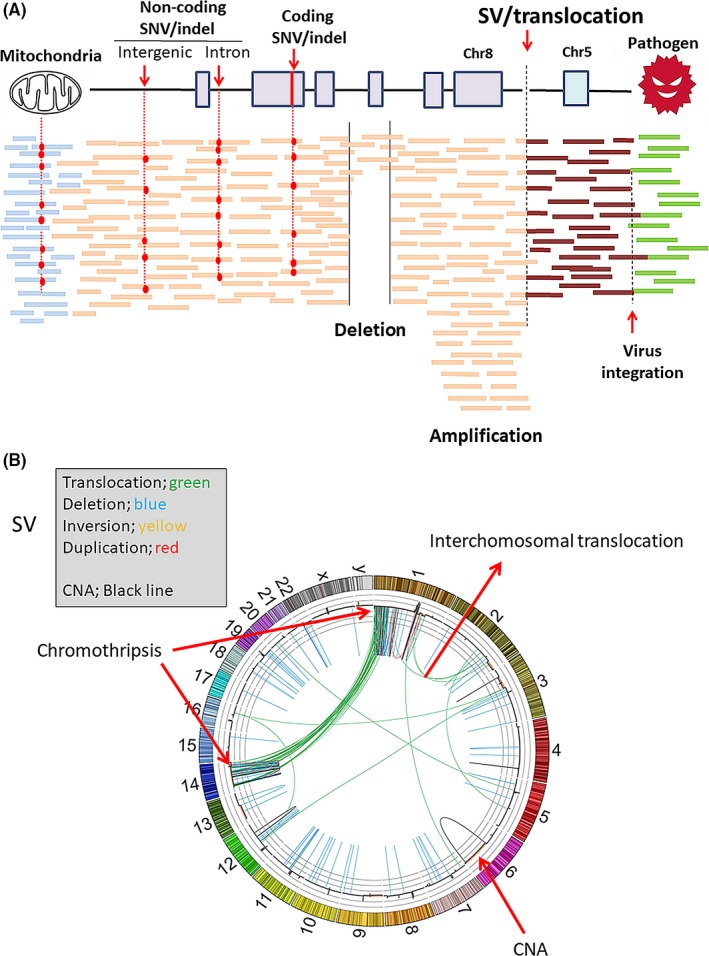
A, Whole genome sequencing (WGS) by next‐generation sequencer (NGS) can detect non‐coding mutations, structural variants (SV), including copy number alterations (CNA), mitochondria mutations and pathogen detection, as well as protein‐coding mutations; B, A representative Circos plot of cancer genome structure from WGS analysis, which indicates SV and CNA in all human chromosomes (1‐22+XY). Chromothripsis was observed in chromosomes 1 and 14. SNV, single nucleotide variants
Table 1.
Detectable mutations by each analysis platform for cancer genome
| DNA chip | Target‐Seq | Exome | RNA‐Seq | WGS | |
|---|---|---|---|---|---|
| Coding SNV | ▵ | ○ | ▵ | ○ | |
| Coding indels | ▵ | ○ | ▵ | ○ | |
| Splicing alteration | ▵ | ○ | ○ | ||
| Promoter mutation | ○ | ||||
| Regulatory regions | ○ | ||||
| Copy‐number alteration | ○ | ▵ | ○ | ||
| Structural variant | ▵ (fusion) | ○ | |||
| Pathogen | ○ | ○ | |||
| Mitochondria | ▵ | ▵ | ○ | ||
| Mutational signature | ▵ | ○ | |||
| Neo‐antigen/HLA | ○ | ○ | ○ | ||
| Sequence (Gb) | — | 0.5‐1 | 10 | 5‐10 | 90‐150 |
| Assay cost ($) | 100 | 200‐500 | 500 | 500 | 1000 |
SNV, single nucleotide variants; WGS, whole genome sequencing.
2. NEXT‐GENERATION SEQUENCER TECHNOLOGY AND WHOLE GENOME SEQUENCING ANALYSIS
Detectable mutations by each of the genomic analysis platforms (DNA chip, target sequencing for 100 genes, WES, RNA‐Seq and WGS) and their performances are summarized in Table 1. WES analysis captures protein‐coding exons spanning approximately 50 Mb (1%‐2%) of the human genome by in‐solution hybridization,15 microarray capturing or PCR amplification, and usually sequences approximately 100 × sequence depth for each sample, which is more accurate than 30 × WGS, because the accuracy of mutation calling by NGS is primarily dependent on the sequencing depth. However, some capturing bias expected is, for example due to difficulty detecting complicated or repetitive genomic regions as well as non‐targeted regions. On the other hand, WGS is technically straightforward. DNA is randomly fragmented by physical shearing, and 30‐50× sequence depth (90‐150 Gb) of each human whole genome is usually sequenced for both cancer and normal genomes,5, 6 which can cover 99% of the entire human genome. Common NGS technology reads are 100‐150 bp for both ends of a 500‐600‐bp DNA fragment,16 but WGS by NGS is still dependent on PCR, with PCR bias, indicating that GC‐rich or AT‐rich regions are difficult to sequence. PCR‐free protocol was recently developed, which shows less GC bias and is more comprehensive than the PCR protocol, although some μg DNA is required as an input for library preparation. The largest limitation of the present 2nd NGS technology (Illumina SBS technology)16 is its short‐read length (100‐250 bp). Around 50% of the 3‐Gb human genome is occupied by repetitive regions and pseudogenes in its 50%, and when short sequence reads are aligned to the redundant reference genome, alignment errors can occur around these repetitive or complicated regions, leading to mutation calling errors.17 The 3rd generation NGS technologies such as PacBio SMART single molecule sequencing18 and nanopore sequencing19 can yield 10 kb and longer reads without PCR bias and are promising for analysis for human WGS; however, they currently suffer from a high error rate (5% and more) in each read and are still prohibitively expensive for WGS analysis, considering that the cost of human WGS was just below $1000 in 2017 (Table 1).
3. COMPUTATIONAL ANALYSIS FOR CANCER WHOLE GENOME SEQUENCING
One of the most challenging issues for cancer WGS is computational analysis. Cancer WGS is required to produce more than 90‐150 Gb ×2 (cancer and normal DNA) of sequence data, corresponding to approximately one terabyte for raw data. Large computer resources are required to handle WGS datasets and to perform alignment and variant calling promptly for thousands of cancer WGS. Academic genome centers are usually increasing their computer resources for WGS, but it would be not sufficient in these academia resources for analysis on tens of thousands of WGS dataset. Cloud computing systems can solve these problems and facilitate the sharing of genomic data globally, although there are technical problems with data transfer and ethical and legal issues in some areas.20
A representative set of computational pipelines and the analysis workflow for cancer WGS are shown in Figure 2. As an initial step, raw sequence data (90‐150 Gb ×2) from NGS (FASTQ files) are aligned to the 3‐Gb human reference sequence (hg19 or new hg38) by BWAmem and other programs, producing BAM files, and PCR duplicates are removed from the BAM file (usually a few percent). Somatic mutations are called by several algorithms specific to somatic mutation types, such as single nucleotide variants (SNV), short indels, CNA and SV, which statistically compares variant allele fractions (VAF) in the cancer genome with those in the normal genome.21, 22, 23 Accuracy is primarily dependent on the sequence depth in each genomic region. The other important factor for accurate analysis is considering alignment or mapping error. Taking account of the complexity and redundancy of the human genome, especially non‐coding regions, alignment error can occur with high frequency when short reads are aligned to repetitive and redundant regions.17, 23 The most serious problem of WGS is that its result is dependent on these mutation call algorithm and each pipeline calls different somatic mutations, especially in low‐depth and complicated regions and somatic short indels.23 The ICGC working group23 performed an extensive benchmark exercise of more than 10 analysis pipelines all of the world and evaluated the consistency of the mutation calling methods. Somatic indel calling showed a high level of inconsistency, while SNV and SV calls showed comparative consistency among the pipelines, concluding that somatic mutation calling remains an unsolved problem. The working group proposed guidelines for computational analysis for cancer WGS.23 For germline variants which are related with cancer risk and hereditary cancer diagnosis, another calling pipeline is required because only normal genome sequencing data is analyzed and VAF is around 50% basically. HaplotyperCaller of GATK (https://software.broadinstitute.org/gatk/) is commonly used for germline variant calling, including SNV and indels from WGS.
Figure 2.
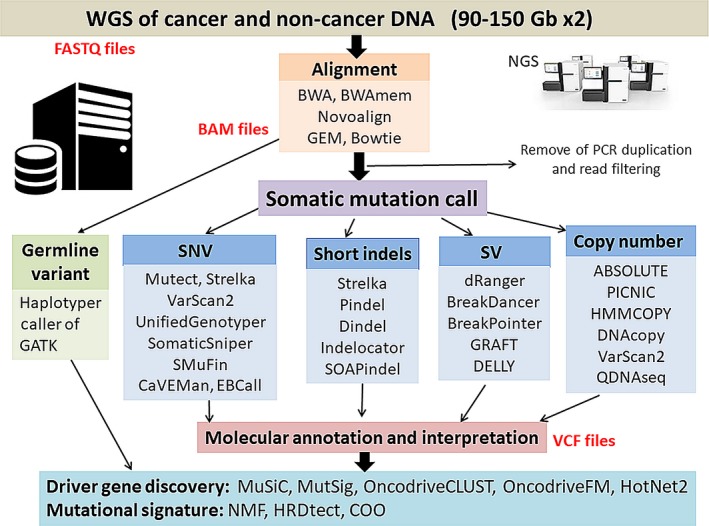
A representative set of computational tools for cancer whole genome sequencing (WGS) analysis. As an initial step, raw sequence data (90‐150‐Gb ×2: FASTQ files) from next‐generation sequencer (NGS) of cancer genome and normal genome are aligned to the 3‐Gb human reference sequence (3 Gb), producing BAM files. PCR duplication is removed from the BAM file (usually a few percent). Somatic mutations are called by several types of algorithms specific to mutation types (SNV, short indels, CNA, SV and others), comparing variant allele numbers in cancer genomes with those in normal genomes by statistical analysis and creating a list of somatic mutations (VCF files). Germline variant call including SNV and indels is commonly performed from sequencing data of normal genomes using other software, HaplotypeCaller of GATK. SNV, single nucleotide variants; SV, structural variants
4. MUTATIONS IN CODING REGIONS AND SPLICING SITES
Whole genome sequencing can detect somatic SNV and short indels (1‐10 bp) in coding regions and splicing sites near the exon‐intron junctions as well as WES. WGS can detect somatic mutations in intronic regions, whose impact is difficult to evaluate and interpret.2, 24 However, combined analysis with RNA‐Seq data can evaluate the impact of deep intronic and synonymous mutations and investigate the transcriptional or functional consequences of the genomic alterations (Figure 3).25, 26 In addition to mutations in exon‐intron junction sites (the GU‐AG consensus sites), mutations in deep intronic regions can generate new splice‐donor or acceptor sites, giving rise to new splicing forms. Synonymous mutations in coding regions and intronic regions can alter exonic motifs that regulate splicing and the functions of cancer‐related genes.27 Systematic combined analysis of WGS and RNA‐Seq is required to interpret these non‐coding mutations.26
Figure 3.
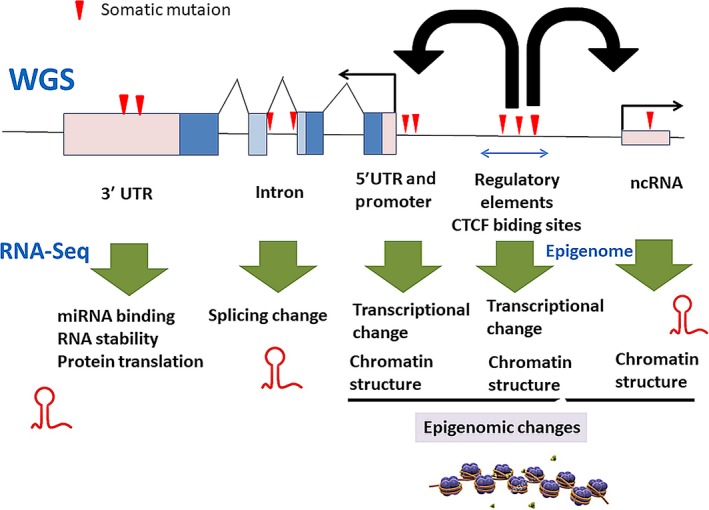
Non‐coding mutations and gene expression in whole genome sequencing (WGS) and RNA‐Seq. Intronic mutations can affect splicing forms. Mutations in 5′UTR and promoter regions can alter transcriptional activity, and regulatory elements such as enhancers, silencers or insulators in intergenic regions can affect chromatin structure and transcriptional activity. Mutations in 3′UTR can alter RNA stability and protein translation through changes in miRNA binding and other mechanisms. Mutations in non‐coding RNA, especially miRNA and lincRNA, may change the interaction of coding RNA/proteins and regulatory elements, and alter chromatin structure
5. MUTATIONS IN NON‐CODING REGIONS
Pre‐mRNA of protein‐coding genes usually contain extensive noncoding sequences, in the form of introns, 5′‐untranslated regions (5′‐UTR) and 3′‐untranslated regions (3′‐UTR) (Figure 3). They are involved with regulation of RNA transcription, splicing and protein translational processes.28 Mutations in 3′‐UTR tend to occur in cancer driver genes29 and are likely to control RNA stability and protein translation through miRNA binding (Figure 3). The human genome contains genes encoding approximately 20 000 non‐coding RNA (ncRNA), including tRNA, ribosomal RNA, microRNA and long non‐coding RNA (lincRNA).30 These functional ncRNA are expected to contribute to chromatin structures, transcription regulation, RNA splicing and the translational machinery.30, 31 Some studies32, 33 report that somatic mutations are accumulated in lincRNA NEAT1 and MALAT1, which are located nearby and are involved with cancer invasions. Furthermore, intergenic regions contain various regulatory element sequences that are crucial to regulate gene expression and associated chromatin structure. Genome‐wide association studies (GWAS) for cancers have identified several hundreds of cancer‐predisposing loci and germline variants, many of which are located in intergenic regions, expecting that they are involved with regulatory elements controlling gene expression around these loci.34 Extrapolations from the ENCODE project,35 Roadmap Epigenomics Consortium36 and FAMTOM30 suggest that 20%‐40% of the human genome could be regulatory elements. Efforts have shifted toward finding interactions between genomic variants and regulatory proteins by ChIP‐Seq, or open chromatin elements (structures), which can indicate where there are active regulatory sequences in a cell type‐specific or cancer‐specific manner.35, 36
Whole genome sequencing analysis in melanoma samples revealed hotspot mutations in the TERT promoter, located −124 bp and −146 bp upstream from the translation start ATG site and conferred enhanced TERT promoter activity.37 These promoter mutations were frequently detected in glioblastoma, bladder cancer, thyroid cancer, liver cancer and melanoma, although the strength of association between these TERT promoter mutations and TERT expression is variable among cancer types.38 In a subset of T‐cell ALL, somatic mutations in non‐coding regions introduced binding motifs for the MYB transcriptional factor and created a super‐enhancer upstream of the TAL1 oncogene.39 Recent systematic or statistical analysis for non‐coding somatic mutations using WGS datasets from TCGA and ICGC indicated that several non‐coding regions are frequently mutated, such as the promoters or regulatory elements of PLEKHS1, WDR74, TFPI2 and BCL6.32, 40, 41 There are a lot of CTCF/cohesion‐binding sites (CBS) across the human genome, which function as insulators to regulate gene expression of nearby genes. Multiple cancer types accumulate CBS mutations42 and these mutations may be involved in the generation of double‐strand breaks and SV as well.43 To identify non‐coding mutations and to interpret their impacts and consequences, more systematic approaches are required by integrating many datasets targeting non‐coding regulatory regions, such as ENCODE,34 FANTOM,30 ChIP‐Seq datasets (epigenomic data) and gene expression datasets (RNA‐Seq).
6. COPY NUMBER ALTERATIONS
Copy number alterations (CNA), affecting large DNA segments (10 kb and more), are one of the most common landmarks of cancer genomes, and lead to activation of oncogenes and inactivation of tumor suppressor genes located in focal CNA. The CNA‐associated oncogenes and tumor suppressors include the focal amplifications of 8q24.21 (MYC), 11q13.3 (CCND1), 7p11.2 (EGFR), 17q12 (ERRB2 = HER2) and 7q31.2 (MET), whereas focal deletion involved 13q14.2 (RB1), 9p21.3 (CDKN2A) and 10q23.31 (PTEN).44, 45 Array CGH and DNA or SNP chip analyses efficiently detect gain or loss of CNA in cancer genomes and we should discuss CNA in a comprehensive cancer genome analysis. The challenge is to identify the oncogene and tumor suppressor gene targets of the driver CNA, which often encompass many genes and elucidate the functional roles of CNA.44, 45 CNA affect not only protein‐coding genes: copy number gains of noncoding regions harboring super‐enhancers near some oncogenes such as KLF5 and MYC are associated with overexpression of these cancer‐related genes.46 Computational tools for WES can detect CNA, but its resolution is not high and sometimes it is difficult to detect specific CNA because exon capturing does not cover many recurrent CNA regions and there is some bias. On the other hand, WGS can analyze CNA in a non‐biased way by counting reads mapped to specific genomic regions in cancer/normal DNA. Even low‐depth WGS (×0.1) can efficiently detect CNA in cancer genomes.47 The theological base of non‐invasive prenatal genetic testing (NIPT) is that CNA of a fetus are detected in plasma of pregnant women,48 and this method of low‐depth WGS is applied to cancer patients as a liquid biopsy or ctDNA (circulating tumor DNA) analysis. Indeed, NIPT by WGS can detect CNA of cancer in low‐depth WGS of plasma from cancer‐bearing pregnant women.49
7. STRUCTURE VARIANTS
Distinct rearrangements or SV in leukemia and sarcoma lead to the activation of proto‐oncogene products or creation of cancer‐specific fusion genes, some of which are clinical diagnostic tools, such as STY‐SSX1 fusion in synovial sarcoma and EWS‐FLI1 fusion in Ewing sarcoma.50 Philadelphia chromosome in CML,51 translocation between chromosome 9q34 and 22q11 gives rise to the BCR‐ABL fusion gene, to which the ABL kinase inhibitor imatinib for first time successful targeted in CML. A small inversion on 2p creates the EML4‐ALK fusion gene, which is found in 1%‐2% of lung adenocarcinomas, and kinase inhibitors are targeting such a kinase fusion gene as ALK in lung cancer.52 SV involving ROS1 at 6q22 (mainly translocation) and RET1 at chromosome 10q11.2 (mainly inversion) were also identified in a small percentage of lung cancers with unique clinical and pathological features. They produce fusion kinases as driver genes and are molecular targets for lung cancer53, 54; 40%‐70% of prostate cancers were found to have SV involving ERG at 21q22 and multiple ETS family genes, producing TMPRSS2‐ERG and ETS family gene fusions.55 Recent analysis for medulloblastoma found recurrent SV activated GFIB proto‐oncogene by enhancer hijacking.56 In liver and kidney cancers, the promoter regions of TERT are frequently affected by SV, inducing TERT overexpression.32 SV affects CD274 (PD‐L1) genes in specific types of lymphoma,57 inducing the stability of PD‐L1 and associated with immune escape of cancer cell.
8. MUTATIONAL ANALYSIS IN REPEAT OR REPETITIVE REGIONS
Repetitive sequences comprise approximately 50% of the human genome. These sequences are highly variable and have been used for genome linkage mapping and diagnosis of cancer with DNA mismatch‐repair deficiency as a microsatellite instability (MSI) test. It is still difficult to analyze mutations and variants in such repetitive regions using WGS and NGS approaches because of alignment issues for short‐read sequences. A recent study analyzed MS mutations in approximately 1000 WGS data across 23 cancer types and identified genes in DNA repair and oncogenic pathways recurrently subject to MSI and uncovered non‐coding loci that frequently display MSI.58 Transposable genetic elements, which are an abundant component in the human genome, can replicate and insert copies of themselves at other locations.23 These transposons played a major role in a driving force for genomic evolution and diversity.59 Several studies have analyzed transposon‐mediated somatic mutations and SV by using cancer WGS data and have identified 4‐5 somatic retrotransposon insertions per tumor.59, 60 These somatic retrotransposon insertions tend to occur in genes that are commonly mutated in cancer and can change their expressions.
9. PATHOGEN DETECTION AND INTEGRATION
Viral and bacterial infection and following chronic inflammation are the strongest etiological factors of cancer development. Hepatitis B virus (HBV) or hepatitis C virus (HCV) infection are linked to liver cancer. Human papillomavirus (HPV) infection initiates and promotes carcinogenesis of the cervix. Helicobactor pylori and Epstein‐Barr virus (EBV) infection are involved with gastric cancer development. Hence, it is important in cancer genomes to detect DNA or RNA sequences derived from known and unknown pathogens (virus and bacteria) leading to chronic inflammation and WGS can detect genomic integrations of pathogens to the host human genome. Technically, unaligned reads to the human genome sequences are extracted and accumulated from WGS or RNA‐Seq data and they can be matched to known pathogen genome sequences with or without pre‐assembling. Especially for tumors in digestive organs, bacteria detection and metagenome analysis of gut flora are important for understand the genome‐environmental interaction in tumor development and therapy resistance, such as Fusobacterium in colorectal cancer61 and Gammaproteobacteria in pancreatic cancer.62 WGS of liver cancers detected several integration sites of the HBV DNA genome (3 kb), which preferentially integrated to the genomic regions of the TERT and MLL4 loci.32, 63 The HPV DNA genome and its integrations are detected by WGS for cervical cancer64 and head and neck cancer. Several rare cancers have a strong viral component, such as EBV in Burkitt lymphoma65 and nasopharyngeal carcinoma, and RNA retrovirus HTLV‐1 in adult T‐cell leukemia/lymphoma.66 Adeno‐associated virus (AAV) is also reported to be integrated in liver cancer genome,32 although its pathogenesis is unclear. These viral integrations/interactions are likely to lead to local genomic instability followed by copy number changes, overexpression around the integration sites, and human‐human or human‐virus gene fusion events, in addition to oncoproteins derived from viral genome.
10. MUTATIONS IN MITOCHONDRIA GENOME
Whole exome sequencing is not designed to capture the 16‐kb mitochondria genome, which includes 13 protein‐coding genes that are equipped with all the elements necessary for their own protein synthesis.67 The proteins encoded by mitochondrial DNA (mtDNA) genes work with other nuclear genes to form the respiratory chain complexes that are the main energy production system in cells. The involvement of mitochondria in carcinogenesis has long been suspected and altered energy metabolism is a common feature of cancer.68 Some studies have examined mtDNA copy numbers in individual cancer types68 or from a collection of WES data69 and demonstrated that there is selective pressure against deleterious coding mutations in mtDNA, supporting that functional mitochondria are required in tumor cells. These studies also observe a strong mutational strand bias, compatible with endogenous replication‐coupled errors as the major source of mutations. Transmission of mtDNA to the nuclear genome occurs in neoplastically transformed cells and mitochondrial‐nuclear genome fusions occur at a similar rate per base pair of DNA as interchromosomal nuclear SV.70
11. MUTATIONAL SIGNATURE
Somatic mutations in cancer are the consequence of multiple mutational processes, including the intrinsic infidelity of the DNA replication machinery, exogenous or endogenous mutagen exposures, enzymatic modification of DNA, and defective DNA repair. Different mutational processes generate unique combinations of mutation types, termed “mutational signatures.”12 Each of the mutational signature patterns is associated with each cancer etiology in a tissue‐specific manner. For example, C>A/G>T transversions such as R293S are the most frequent substitutions in the TP53 gene in liver cancer developed through aflatoxin exposure.71 Unlike WES, WGS detects thousands of somatic SNV in common cancers, and recent comprehensive mutational searches and non‐negative matrix factorization mathematical analysis have extracted more than 30 mutational signatures for cancer genomes, which are shown in the COSMIC database (http://cancer.sanger.ac.uk/cosmic/signatures). Researchers have attempted to implicate each of these mutational signatures in biological and epidemiological aspects.12 Among these established mutational signatures, some are established to be associated with specific mutational processes. Signature 1 represents a clock‐like mutational process (aging) and is observed in all types of cancer.72 Signature 24 represents the signature associated with aflatoxin,32, 71 Signature 22 with aristolochic acid (which is contained in Chinese herbal products),73 Signature 4 with smoking exposure,32, 74 Signature 3 with the defect of DNA double‐strand break repair associated with BRCA1/2 mutation, and Signature 6 with DNA‐mismatch repair defects (Figure 4).75 By observing genome‐wide somatic mutational signatures from cancer WGS data, we may presume the etiological factors for individual cancer development among the multiple internal (aging and intrinsic DNA repair) and external (environmental exposure) etiological steps in carcinogenesis.
Figure 4.
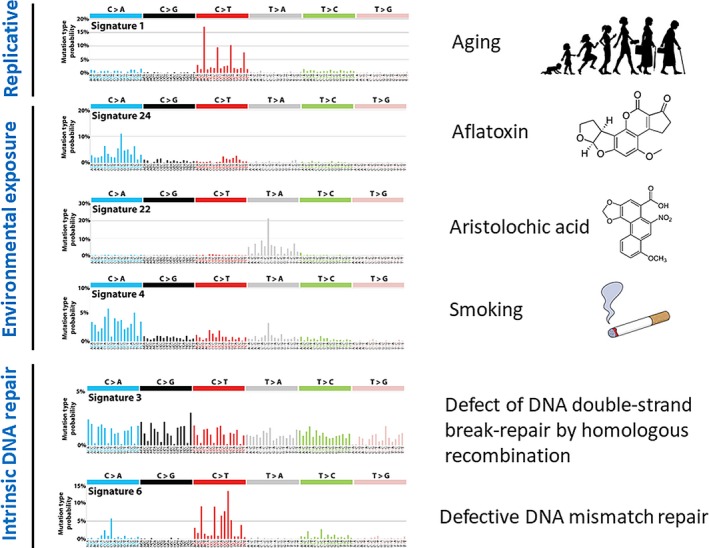
Mutational signature and etiological factors in COSMIC database. The profile of each signature is displayed using the 6 substitution subtypes: C>A, C>G, C>T, T>A, T>C and T>G. Furthermore, each of the substitutions is examined by incorporating information on the bases immediately 5′ and 3′ to each mutated base, generating 96 possible mutation types. NMF analysis of cancer WGS in the COSMIC database (http://cancer.sanger.ac.uk/cosmic/signatures) demonstrates 30 mutational signatures at present, and 6 representative signatures are shown with their associated etiological factors for cancer development (aging, environmental exposures and defect of intrinsic DNA repair)
12. GENOMIC INSTABILITY AND CELL‐OF‐ORIGIN PREDICTION BY MUTATIONAL SIGNATURE
Whole genome sequencing analysis of cancer revealed a distinct signature pattern, termed chromothripsis,76 in which, 1 or a few chromosomes in 1 cell produce dozens to hundreds of clustered SV (Figure 1b). The mechanism for such complicated SV is that at 1 or more carcinogenic stage, distinct chromosomes or genomic regions become fragmented into many segments which are then pieced together inaccurately by DNA repair mechanisms.76 Chromothriptic signatures were detected in cancers arising from patients with inherited p53 mutations,77 suggesting that this event is associated with the functions of p53 and genomic stability in various DNA damage response signaling pathways. WGS analysis for breast, ovarian and pancreatic cancers indicated that signatures were related with inactivation of DNA maintenance genes (BRCA1, BRCA2 and PALB2), and also related with high response to DNA‐damaging agents and PARP inhibitors.75, 78 Recent studies have demonstrated that somatic substitution, insertion/deletion and SV patterns, or “mutational signatures,” are associated with BRCA1/BRCA2 dysfunction. By combining this signature information from WGS, homologous recombination deficiency associated with BRCA1/2 deficiency was evaluated79. Polak et al80 predicted cell‐of‐origin (COO) from WGS data for cancer by comparing the genomic distribution and signature of somatic mutations to 424 epigenetic features that were measured by the Epigenome Roadmap consortium,36 which were derived from 106 different cell types. The genomic distribution of chromatin features corresponding to the tumor's cell type of origin is strongly associated with local mutation density, and they chose the tissue showing the most significant enrichment as the most likely tissue of origin for individual cancer whole genome.
13. IMMUNO‐GENOMIC ANALYSIS FROM CANCER GENOME SEQUENCING
Immunotherapy using immune checkpoint inhibitors and emerging new therapies have already shown great promise in some types of cancers. Genomic biomarkers have been extensively investigated by genome sequencing analysis on pre‐treated or recurrent cancer specimens. The overexpression or genetic alteration of PD‐L1 (CD274) is likely to be associated with response to anti‐PD‐1/PD‐L1 agents in some types of cancer, such as lymphoma.57, 81 Mutation load at whole genome level, which is associated with greater neo‐antigen presentation, is a good genomic marker in melanoma, lung cancer and MSI‐positive colorectal cancer. Genome‐wide CNA pattern, aneuploidy, was reported to be correlated with reduced infiltrating immune cells with tumor, which was evaluated by RNA‐Seq, and with reduced response to immunotherapy.82 Several mutations involved with IFN‐gamma pathway and HLA presentations such as HLA, B2M, JAK1/2 83, 84 are likely to be related to the resistance of immune check‐point inhibitors, but tumor immunology and mechanisms of immune check‐point inhibitors are quite complicated and diverse. To understand immuno‐genomics of cancer and to explore genomic markers to predict the response of immune therapy, comprehensive immune “signature” analysis, including the quantity and quality of immune cells and neoantigen signatures, is required from WGS and RNA‐Seq data on a number of pre‐treatment and post‐treated tumor specimens and immune cells.
14. CONCLUSION AND FUTURE DIRECTION OF CANCER WHOLE GENOME SEQUENCING
As sequencing costs continue to decrease and computer resources expand, WGS analysis for cancer genome research and clinical utilities will become more common and more sophisticated. Cancer WGS provides abundant information to understand the biology underlying the cancer genome and the function of unexplored non‐coding regions and SV in the human genome. There is much potential for transcriptional or functional consequences of SV and non‐coding mutations and they should be further explored by integrative analysis of RNA‐Seq and multi‐omics analysis with DNA methylation data,78 protein expression data, and chromatin structure85 or epigenome data to interpret mutational consequences and to understand the biology and immunology of cancer. Taking into account the diversity of cancer genomes and phenotypes, interpretation of the mutational data from cancer WGS will also require the analysis of much more WGS data and integration with multi‐omics data, functional data, immuno‐genomic data and clinic‐pathological data in a larger sample set.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
ACKNOWLEDGMENTS
We would like to thank technical staff at RIKEN‐IMS and Professor Miyano and his fellows at the Human Genome Center, Institute of Medical Science, The University of Tokyo for their great effort in cancer genome sequencing for the ICGC project. The super‐computing resource “SHIROKANE” was provided by the Human Genome Center, The University of Tokyo (http://sc.hgc.jp/shirokane.html).
Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109:513–522. https://doi.org/10.1111/cas.13505
REFERENCES
- 1. Stratton M, Campbell PJ, Futreal A. The cancer genome. Nature. 2009;458:719‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17‐37. [DOI] [PubMed] [Google Scholar]
- 3. Kinzler KW, Vogetstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159‐170. [DOI] [PubMed] [Google Scholar]
- 4. King CR, Kraus M, Aaronson SA. Amplification of a novel v‐erbB related gene in human mammary carcinoma. Science. 1985;229:974‐976. [DOI] [PubMed] [Google Scholar]
- 5. Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second‐generation sequencing. Nat Rev Genet. 2010;11:685‐696. [DOI] [PubMed] [Google Scholar]
- 6. Nakagawa H, Wardell CP, Furuta M, Taniguchi H, Fujimoto A. Cancer whole genome sequencing: present and future. Oncogene. 2015;34:5943‐5950. [DOI] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. International Cancer Genome Consortium , Hudson TJ, Anderson W, et al. International network of cancer genome projects. Nature. 2010;464:993‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lowrence M, Stojanov P, Mermel C, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leiserson MD, Vandin F, Wu H, et al. Pan‐cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47:106‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laurence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature. 2013;499:214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805‐D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gnirke A, Melnikov A, Maguire J, et al. Solution hybrid selection with ultra‐long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Treangen TJ, Salzberg SL. Repetitive DNA and next‐generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13:36‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eid J, Fehr A, Gray J, et al. Real‐time DNA sequencing form single polymerase molecule. Science. 2009;323:133‐138. [DOI] [PubMed] [Google Scholar]
- 19. Giordano F, Aigrain L, Quail MA, et al. De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci Rep. 2017;7:3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dove ES, Joly Y, Tassé AM, et al. Genomic cloud computing: legal and ethical points to consider. Eur J Hum Genet. 2015;23:1271‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Q, Jia P, Li F, et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boutros PC, Ewing AD, Ellrott K, et al. Global optimization of somatic variant identification in cancer genomes with a global community challenge. Nat Genet. 2014;46:318‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alioto TS, Buchhalter I, Derdak S, et al. A comprehensive assessment of somatic mutation detection in cancer using whole‐genome sequencing. Nat Commun. 2015;6:10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2015;34:1‐14. [DOI] [PubMed] [Google Scholar]
- 25. Xiong HY, Alipanahi B, Lee LJ, et al. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shiraishi Y, Fujimoto A, Furuta M, et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE. 2014;9:e114263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324‐1335. [DOI] [PubMed] [Google Scholar]
- 28. Zhao W, Pollack JL, Blagev DP, Zaitlen N, McManus MT, Erle DJ. Massively parallel functional annotation of 3′ untranslated regions. Nat Biotechnol. 2014;32:387‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oikonomou P, Goodarzi H, Tavazoie S. Systematic identification of regulatory elements in conserved 3′ UTRs of human transcripts. Cell Rep. 2014;7:281‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. FANTOM Consortium . A promoter‐level mammalian expression atlas. Nature. 2014;507:462‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1:391‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fujimoto A, Furuta M, Totoki Y, et al. Whole genome mutational landscape and characterization of non‐coding and structural mutations in liver cancer. Nat Genet. 2016;48:500‐509. [DOI] [PubMed] [Google Scholar]
- 33. Rheinbay E, Parasuraman P, Grimsby J. Recurrent and functional regulatory mutations in breast cancer. Nature. 2017;547:55‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Freedman ML, Monteiro AN, Gayther SA, et al. Principles for the post‐GWAS functional characterization of cancer risk loci. Nat Genet. 2011;43:513‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roadmap Epigenomics Consortium . Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vinagre J, Almeida A, Pópulo H, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. [DOI] [PubMed] [Google Scholar]
- 39. Mansour MR, Abraham BJ, Anders L, et al. An oncogenic super‐enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014;346:1373‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46:1258‐1263. [DOI] [PubMed] [Google Scholar]
- 41. Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome‐wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46:1160‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Katainen R, Kashyap DK, Pitkänen E, et al. CTCF/cohesion‐binding sites are frequently mutated in cancer. Nat Genet. 2015;47:818‐821. [DOI] [PubMed] [Google Scholar]
- 43. Canela A, Maman Y, Jung S, et al. Genome organization drives chromosome fragility. Cell. 2017;170:507‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature. 2010;463:899‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zack TI, Scumacher SE, Csarter SL, et al. Pan‐cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X, Choi PS, Francis JM, et al. Identification of focally amplified lineage‐specific super‐enhancers in human epithelial cancers. Nat Genet. 2016;48:176‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heitzer E, Ulz P, Belic J, et al. Tumor‐associated copy number changes in the circulation of patients with prostate cancer identified through whole‐genome sequencing. Genome Med. 2013;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wong FC, Lo YM. prenatal diagnosis innovation: genome sequencing of maternal plasma. Annu Rev Med. 2016;67:419‐432. [DOI] [PubMed] [Google Scholar]
- 49. Amant F, Verheecke M, Wlodarska I, et al. Presymptomatic identification of cancers in pregnant women during noninvasive prenatal testing. JAMA Oncol. 2015;1:814‐819. [DOI] [PubMed] [Google Scholar]
- 50. Oda Y, Tsuneyoshi M. Recent advances in the molecular pathology of soft tissue sarcoma: implications for diagnosis, patient prognosis, and molecular target therapy in the future. Cancer Sci. 2009;100:200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Groffen J, Stephenson JR, Heisterkamp N, et al. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell. 1984;36:93‐94. [DOI] [PubMed] [Google Scholar]
- 52. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448:561‐566. [DOI] [PubMed] [Google Scholar]
- 53. Kohno T, Ichikawa H, Totoki Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:375‐377. [DOI] [PubMed] [Google Scholar]
- 54. Takeuchi K, Soda M, Togashi Y, et al. KIF5B‐RET fusions in lung adenocarcinoma. Nat Med. 2012;18:378‐381.22327623 [Google Scholar]
- 55. Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644‐648. [DOI] [PubMed] [Google Scholar]
- 56. Northcott PA, Lee C, Zichner T, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature. 2014;511:428‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant PD‐L1 expression through 3′‐UTR disruption in multiple cancers. Nature. 2016;534:402‐406. [DOI] [PubMed] [Google Scholar]
- 58. Cortes‐Ciriano I, Lee S, Park WY, Kim TM, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee E, Iskow R, Yang L, et al. Cancer Genome Atlas Research Network . Landscape of somatic retrotransposition in human cancers. Science. 2012;337:967‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shukla R, Upton KR, Muñoz‐Lopez M, et al. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013;153:101‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kostic AD, Gevers D, Pedamallu CS, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Geller LT, Barzily‐Rokni M, Danino T, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sung WK, Zheng H, Li S, et al. Genome‐wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765‐769. [DOI] [PubMed] [Google Scholar]
- 64. Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cao S, Strong MJ, Wang X, et al. High‐throughput RNA sequencing‐based virome analysis of 50 lymphoma cell lines from the cancer cell line encyclopedia project. J Virol. 2015;89:713‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cook LB, Melamed A, Niederer H, et al. The role of HTLV‐1 clonality, proviral structure and genomic integration site in adult T cell leukemia/lymphoma. Blood. 2014;123:3925‐3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61:667‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Reznik E, Miller ML, Şenbabaoğlu Y, et al. Mitochondrial DNA copy number variation across human cancers. Elife. 2016;5:pii: e10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stewart JB, Alaei‐Mahabadi B, Sabarinathan R, et al. Simultaneous DNA and RNA mapping of somatic mitochondrial mutations across diverse human cancers. PLoS Genet. 2015;11:e1005333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ju YS, Tubio JM, Mifsud W, et al. Frequent somatic transfer of mitochondrial DNA into the nuclear genome of human cancer cells. Genome Res. 2015;25:814‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:29‐431. [DOI] [PubMed] [Google Scholar]
- 72. Alexandrov LB, Jones PH, Wedge DC, et al. Clock‐like mutational processes in human somatic cells. Nat Genet. 2015;47:1402‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Poon SL, Pang T, McPherson JR, et al. Genome‐wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med. 2013;5:197ra101. [DOI] [PubMed] [Google Scholar]
- 74. Alexandrov LB, Ju Y, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature. 2016;534:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Korbel JO, Campbell PJ. Criteria for inference of chromothripsis in cancer genomes. Cell. 2013;152:1226‐1236. [DOI] [PubMed] [Google Scholar]
- 77. Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148:59‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Waddell N, Pajic M, Patch A, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Davies H, Glodzik D, Morganella S, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23:517‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Polak P, Karlić R, Koren A, et al. Cell‐of‐origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ansell SM, Lesokhin AM, Borrello I, et al. PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer‐associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zaretsky JM, Garcia‐Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med. 2016;375:819‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dekker J, Marti‐Renom MA, Mirny LA. Exploring the three‐dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]