

Abstract
Metformin is a biguanide widely prescribed as an antidiabetic drug for type 2 diabetes mellitus patients. The purpose of the present study was to observe the effects of the new metformin derivative, HL156A, on human oral cancer cell and to investigate its possible mechanisms. It was observed that HL156A significantly decreased FaDu and YD‐10B cell viability and colony formation in a dose‐dependent way. HL156A also markedly reduced wound closure and migration of FaDu and YD‐10B cells. We observed that HL156A decreased mitochondrial membrane potential and induced reactive oxygen species (ROS) levels and apoptotic cells with caspase‐3 and ‐9 activation. HL156A inhibited the expression and activation of insulin‐like growth factor (IGF)‐1 and its downstream proteins, AKT, mammalian target of rapamycin (mTOR), and ERK1/2. In addition, HL156A activated AMP‐activated protein kinase/nuclear factor kappa B (AMPK‐NF‐κB) signaling of FaDu and YD‐10B cells. A xenograft mouse model further showed that HL156A suppressed AT84 mouse oral tumor growth, accompanied by down‐regulated p‐IGF‐1, p‐mTOR, proliferating cell nuclear antigen (PCNA) and promoted p‐AMPK and TUNEL expression. These results suggest the potential value of the new metformin derivative HL156A as a candidate for a therapeutic modality for the treatment of oral cancer.
Keywords: HL156A, metformin derivative, migration, mitochondrial membrane potential, oral cancer
1. INTRODUCTION
Oral squamous cell carcinoma (OSCC), one of the top 10 most commonly occurring cancers worldwide, has a high mortality rate. In spite of recent improvements and advances in medicine such as surgery, radiation, and chemotherapy, the 5‐year survival rate for oral cancer patients has remained at 50% over the past 5 decades.1, 2 Several clinical applications of surgery, radiation, and chemotherapy are standard treatments for early OSCC, which result in effective tumor control. However, many severe side‐effects and/or toxicity appear in patients receiving those treatments.3 Therefore, an alternative therapeutic strategy has been demanded.
Recently, metformin has captured the attention of scientists because of its potential for cancer therapy, although it is primarily widely used as an effective type 2 diabetes medication.4 Preclinical and clinical studies have shown that metformin significantly prevents cancer incidence, slows tumor progression and improves survival rates with certain types of malignancies including breast cancer, prostate cancer, and salivary adenocarcinoma.5, 6, 7, 8, 9, 10 Among the possible underlying mechanisms of metformin, AMP‐activated protein kinase (AMPK) activation is considered one of the most important because AMPK can interact with several other signaling pathways and transcription factors, such as AKT, nuclear factor kappa B (NF‐κB), and mammalian target of rapamycin (mTOR).11, 12, 13 However, the precise mechanism for the multiple anticancer effects of metformin remains to be elucidated. Additionally, despite possessing many outstanding properties, limited cell penetration capacity as a result of its hydrophilic nature makes metformin unable to meet the requirements for use as an anticancer drug.11
HL156A is a derivative of metformin synthesized from pyrrolidine that is capable of inducing AMPK activation. It was reported that HL156A has protective effects against peritoneal and liver fibrosis.14, 15 It was also reported that HL156A inhibited LPS‐induced inflammation of macrophages. In addition, HL156A inhibits not only smad3‐dependent signaling activated by high glucose conditions but also epithelial‐mesenchymal transition (EMT).15 A recent study showed that HL156A in combination with temozolomide inhibited the invasive properties of glioblastoma and increased the survival rate in a xenograft in vivo model.16
In the present study, the effects of HL156A on the progression of oral cancer cells were examined. Our results suggest the potential abilities of the new metformin derivative HL156A as a candidate for a therapeutic modality for the treatment of oral cancer.
2. MATERIALS AND METHODS
2.1. Cell culture and reagents
For an OSCC cell line, FaDu, YD‐10B, and AT64 cells were purchased from Korea Cell Line Bank (KCLB, Seoul, Korea). Human hypopharyngeal squamous cell carcinoma (FaDu) cells were maintained in a complete DMEM (Welgene Inc., Gyeongsanbuk‐do, Korea), containing 10% FBS, 100 units/mL penicillin and 100 μg/mL streptomycin. Human oral squamous cell carcinoma (YD‐10B) and mouse oral squamous cell carcinoma (AT84) cells were cultured in RPMI‐1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were maintained at 37°C in a humidified 5% CO2/95% air atmosphere. HL156A is a derivative of phenyl biguanide, which is designed and synthesized by Hanall Biopharma Inc. (Seoul, Korea). The detailed procedure of HL156A synthesis and structure was described in a previous study.15, 16
2.2. Cell proliferation assay
Cell proliferation was assessed using an MTT assay. The cells were seeded in a 96‐well plate at a density of 2 × 103 cells/well. The next day, cells were treated with different concentrations of HL156A (10‐50 μmol/L) for 24 hours. After incubation, the cells were washed twice with PBS, and 500 μg/mL MTT (Sigma‐Aldrich, St Louis, MO, USA) was added to the wells. The MTT solution was removed after 4 hours of incubation at 37°C. A mixture of 0.01 mol/L glycine and DMSO (Sigma‐Aldrich) was added to each well. Absorbance was measured at 540 nm using a DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA).
2.3. Soft agar colony formation assay
Briefly, cells (1 × 104 cells) were exposed to different concentrations of HL156A in 1 mL of 0.3% basal medium Eagle's agar containing 10% FBS and were plated in 60‐mm plates containing 0.6% low‐melting temperature agarose. The cells were allowed to form colonies for 14 days at 37°C in a humidified atmosphere containing 5% CO2. The colonies were fixed with 4% paraformaldehyde for 30 min at room temperature and were then stained with 0.04% crystal violet for 30 min at room temperature. The cell colonies were scored using an IX2‐SLP inverted microscope (Olympus, Tokyo, Japan).
2.4. Annexin V‐FITC/propidium iodide (PI) double staining and cell cycle analysis
Cell cycle distribution was analyzed using flow cytometry. Briefly, 1 × 106 cells were harvested, washed in PBS and then fixed in 70% alcohol for 30 min at 4°C. After 3 washes in cold PBS, the cells were resuspended in 1 mL PBS solution containing 50 μL of 1 mg/mL PI and 1 unit DNase‐free RNase for 30 min at 37°C. The samples were then analyzed to determine the DNA content using a FACScan analyzer (Beckman Coulter Inc., Fullerton, CA, USA). For cell death analysis, Annexin V‐FITC/PI staining was carried out by incubating the cells for 15 min at room temperature (RT) in a binding buffer (10 mmol/L HEPES, 140 mmol/L NaCl, 2.5 mmol/L CaCl2, pH 7.4) containing saturated concentrations of Annexin V‐FITC and PI. After incubation, the cells were pelleted and analyzed in a FACScan analyzer (Beckman Coulter Inc.).
2.5. Western blot analysis
Total protein from the cells was obtained using RIPA buffer (50 mmol/L Tris‐Cl (pH 7.5), 150 mmol/L NaCl, 0.5% sodium deoxycholate, 1% NP‐40, 0.1% SDS and 1 mmol/L EDTA) containing a protease inhibitor cocktail (1 μg/mL aprotinin and leupeptin). Cell lysates (30 μg) were subjected to SDS‐PAGE and then transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membranes were blocked with 5% skim milk for 1 hours and incubated with the primary antibodies. Antibodies against pCDK1 (sc101654), Cyclin B1 (sc245), caspase‐3 (sc7148), caspase‐9 (sc8355), caspase‐7 (sc28295), PARP‐1 (sc1561), SOD‐1 (sc11407), NOS1 (sc5302), Nrf‐2 (sc722), p70S6K (sc230), p‐p70S6K (sc8416), p‐mTOR (sc 101738), NF‐κB (sc109), p‐NF‐κB (sc101752), MMP2 (sc13594), MMP9 (sc6840) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibody against AMPK (2532S), p‐AMPK (2535S), phosphorylated insulin‐like growth factor‐1 receptor (pIGF‐1R) (3024S), AKT (9272S), pAKT (4060S), ERK1/2 (9102S), p‐ERK1/2(9101S), and p‐GSK‐3α/β (8566S) and the HRP‐conjugated secondary antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). After being washed twice, the membranes were incubated with the corresponding secondary antibodies for 1 hours (dilution ratio 1:5000). Protein signals were detected with a Luminescent image analyzer (LAS‐1000; Fujifilm, Tokyo, Japan).
2.6. Caspase activity assay
Caspase‐3 and ‐9 activities were assessed using a caspase‐3 or ‐9 colorimetric assay kit (Abcam, Cambridge, MA, USA) according to the manufacturer's protocol. Briefly, the cells were collected and resuspended in lysis buffer containing 50 mmol/L HEPES, pH 7.4, 0.1% CHAPS, 1 mmol/L DTT, 0.1 mmol/L EDTA and 0.1% Triton X‐100. Following incubation for 30 min on ice, cell lysate was centrifuged at 11 000 g for 10 min at 4°C, and the protein concentration in the supernatants was measured using the Bradford dye method. The supernatants were incubated with reaction buffer containing 2 mmol/L Ac‐DEVD‐AFC for caspase‐3 and LEHD‐AFC for caspase‐9 (Abcam) in a caspase assay buffer at 37°C with 10 mmol/L DTT for 30 min. Caspase activity was determined by measuring the absorbance at 405 nm.
2.7. Mitochondrial membrane potential
Mitochondrial membrane potential was analyzed by flow cytometry using a JC‐1 mitochondrial membrane potential detection kit (Biotium Inc., Hayward, CA, USA). JC‐1 exhibits potential‐dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (530 nm, FL‐1 channel) to red (590 nm, FL‐2 channel). After different treatments, oral cancer cells were incubated in JC‐1 reagent working solution (Biotium Inc.) for 15 min at 37°C, washed once with PBS and then resuspended in staining buffer and analyzed with a flow cytometer or fluorescence microscope (Olympus).
2.8. Reactive oxygen species formation detection
Determination of reactive oxygen species (ROS) levels was based on the oxidation of dihydroethidium (DHE). Cells were seeded to reach 70%‐80% confluency and incubated with HL156A for 3, 6, and 12 hours. Cells were then treated with DHE (10 mmol/L) for 30 min at 37°C in the dark. The cells were then washed twice and harvested in PBS. Fluorescence of DHE was detected with a fluorescence microscope (IX‐71; Olympus) at the excitation/emission wavelength 510/595 nm.
2.9. Wound‐healing motility assay
Cells were allowed to grow in a culture dish overnight and a scratch ~3 mm wide was created in the monolayer using a pipette tip. After being washed twice with PBS, the cells were treated with or without HL156A, and images were captured after 24 hours. Cells were imaged in 5 random microscopic fields per well using an Olympus IX2‐SLP inverted microscope (Olympus) at ×100 magnification.
2.10. Migration assay
Cell migration was determined using a modified 2‐chamber migration assay with a pore size of 8 mm. For the migration assay, cells suspended in 200 μL serum‐free medium were seeded on the upper compartment of a 12‐well Transwell culture chamber, and 600 μL complete medium was added to the lower compartment. After incubation at 37°C, migratory cells in the medium in the lower chamber were quantified by measuring the absorbance at optical density (OD) 595 nm.
2.11. In vivo mice xenograft experiments
Mouse oral cancer AT84 cells were treated with or without 20 μmol/L HL156A for 24 hours. Cells (3 x 106 cells per mouse) were injected s.c. into the left flank of 3‐week‐old male C3H mice (Samtaco Bio, Sungnam, Korea) in each group (n = 5 or 7). Bodyweight was measured every 2 days during the experiment. Three weeks later, tumor volume was measured with a caliper and calculated using the formula V = (ab 2)/2, where a was the longest diameter and b was the shortest diameter of the tumor. All mice were killed on day 21, and the tumors were removed, weighed, and subjected to further analysis. Formalin‐fixed paraffin‐embedded tissues from AT84 xenografted tumors were used for immunohistochemical staining of p‐IGF‐1, p‐mTOR, p‐AMPK, and PCNA expression.
2.12. Statistical analysis
All experiments were carried out at least in triplicate. Results are expressed as the mean ± standard deviation (SD). Student's t test and one‐way analysis of variance (ANOVA) were used to determine the significant difference between the control and experimental groups. P‐values less than .05 were considered statistically significant.
3. RESULTS
3.1. HL156A suppresses oral cancer cell growth
To investigate the effects of HL156A on oral cancer cell proliferation, we carried out MTT assays with different concentrations (10 to 50 μmol/L) of HL156A in FaDu and YD‐10B cells. HL156A significantly decreased the growth of FaDu cells in a concentration‐dependent way (Figure 1A). In FaDu cells, 40 μmol/L HL156A resulted in cell growth inhibition rates of 45% at 24 hours. Similarly, cell growth inhibition in YD‐10B cells was also observed with HL156A treatment (Figure 1B).
Figure 1.
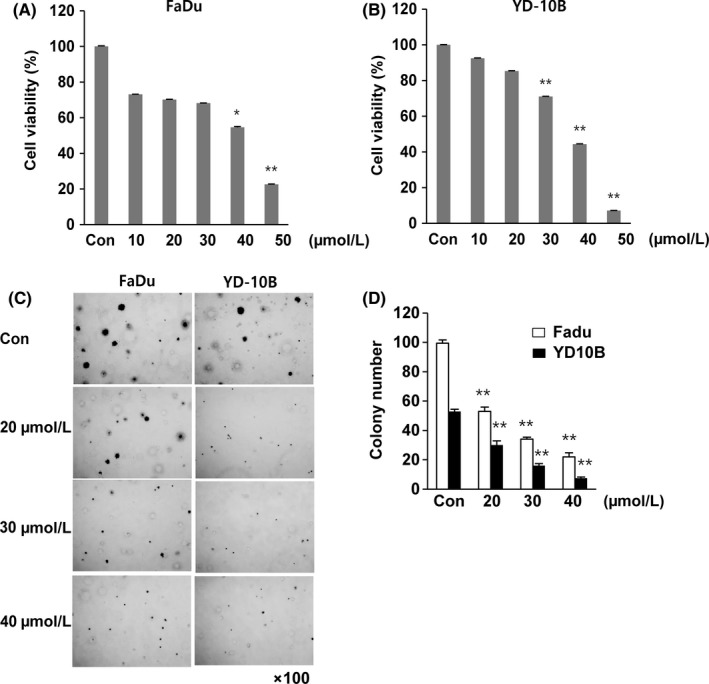
Effect of treatment with HL156A on cell proliferation of oral cancer cell lines. A,B, Cell viability was assessed after 24 hours of HL156A treatment at concentrations ranging from 10 to 50 μmol/L in the human oral cancer cell lines FaDu and YD‐10B using an MTT assay. Data are expressed as means ± SD of the results from three separate experiments (*P < .05 and **P < .01). C,D, Evaluation of colony formation of HL156A‐treated cells. Colony formation was assessed 14 days after HL156A treatment at various concentrations, and cells were stained with crystal violet at the end of the experiment. Images were taken with an inverted microscope at ×100 magnification. Colony quantification was determined by microplate area scan at optical density 550 nm
To further confirm the effect of HL156A on cell proliferation, a soft agar colony formation assay was carried out. The number of colonies observed was appreciably reduced compared with the control untreated FaDu and YD‐10B cells (Figure 1C). Moreover, the size of the colonies was also reduced. At 40 μmol/L, HL156A markedly decreased the clonogenicity to approximately 25% and 13% compared to the control in both cell lines, respectively (Figure 1D). Thus, the results showed that HL156A inhibits the colony‐forming ability of oral cancer cells.
3.2. HL156A induces G2/M cell cycle arrest and cell apoptosis
It was hypothesized that HL156A could induce alterations in cell cycle regulation. Using flow cytometry, we analyzed the effect of HL156A on cell cycle progression. As shown in Figure 2A, HL156A treatment caused a reduction in the G1 phase population and an increase in the G2/M population in both FaDu and YD‐10B cells. In YD‐10B cells, the percentage of cells in G1 phase decreased from 53% to 5.2% after 60 μmol/L HL156A treatment for 24 hours. The proportion of cells in G0 phase increased from 0.39% to 24.2% over this same interval. In FaDu and YD‐10B cells, western blotting analysis showed that the levels of phospho‐CDK1 and cyclin B were decreased by HL156A in a time‐dependent way (Figure 2B). These results suggest that the effects of HL156A on G2/M cell cycle progression were associated with the inhibition of cell growth.
Figure 2.
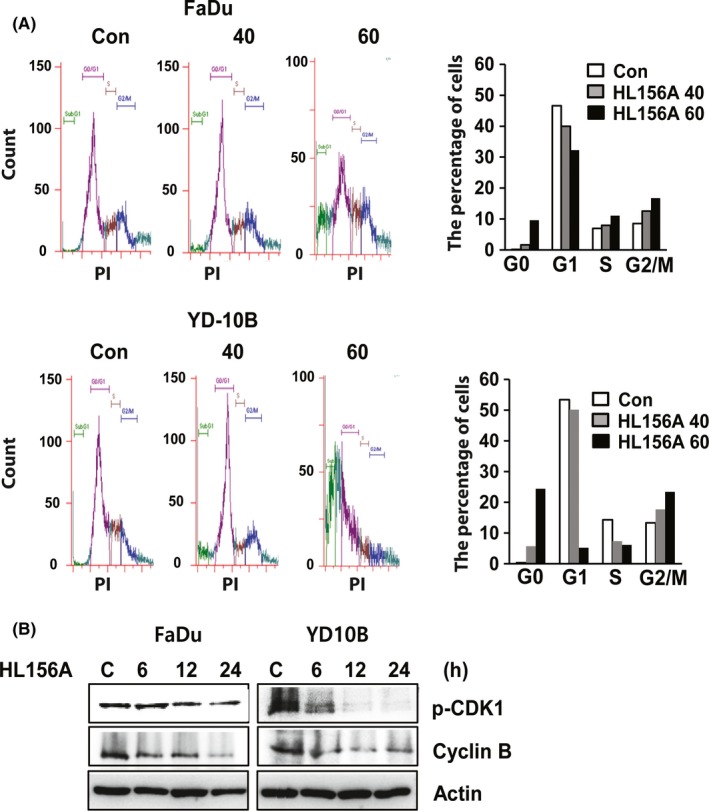
HL156A induces cell cycle arrest at the G2/M phase. A, Cells were incubated with HL156A (40 or 60 μmol/L) for 24 hours and then subjected to flow cytometry to measure cell cycle distribution. Percentage of cells in each cell cycle phase was graphed. B, Immunoblotting of cell cycle‐related proteins in FaDu and YD‐10B cells treated with 40 μmol/L HL156A for the indicated times. Lysates of the above cells were subjected to western blotting with phospho‐CDK1 and cyclin B antibodies. β‐actin served as an internal control
To determine whether HL156A induces cell death in FaDu and YD‐10B cells, we quantified apoptosis with flow cytometry using an Annexin v‐FITC/PI double staining assay. As shown in Figure 3A, after 24 hours of HL156A treatment (40 or 60 μmol/L), there was a significant decrease in the number of living cells; 93.5% of the control cells were alive, whereas only 60.6% or 6.6% of the HL156A‐treated FaDu cells were alive, respectively, whereas treatment with 40 μmol/L or 60 μmol/L HL156A stimulated a noteworthy increase in the proportion of apoptotic cells (32.9% with 40 μmol/L and 85.3% with 60 μmol/L) compared to the untreated control. Similarly, a marked number of dying cells were observed in the HL156A‐treated YD‐10B cells. Consistent with this observation, pro‐caspase‐3, ‐7, ‐9, and the inactive form poly (ADP‐ribose) polymerase (PARP) were decreased in cells treated with HL156A compared with the control in a time‐dependent way (Figure 3B). In addition, caspase‐3 and caspase‐9 activity was progressively induced by HL156A treatment in a time‐dependent way (Figure 3C,D).
Figure 3.

HL156A induces apoptosis in FaDu and YD‐10B cells. A, Cells were incubated with HL156A (40 or 60 μmol/L) for 24 hours. Annexin V‐positive apoptotic cells were assessed by flow cytometric analysis after staining with Annexin V/propidium iodide (PI). B, Effect of HL156A on the activity of caspase‐3, ‐7, and ‐9 and poly (ADP‐ribose) polymerase (PARP). The cells were treated with HL156A, and cell lysates were subjected to western blotting using antibodies against pro‐caspase‐3, ‐7, and ‐9 and PARP. Numbers represent relative density normalized to actin. C,D, FaDu and YD‐10B cells were treated with HL156A (40 μmol/L) for the indicated times and assayed for caspase‐3 and caspase‐9 activity as described in Materials and Methods
3.3. HL156A reduces mitochondrial membrane potential and promotes ROS formation
Loss of mitochondrial membrane potential can lead to the release of apoptotic‐inducing factors and caspase activators, such as cytochrome c.17 To determine the changes in mitochondrial membrane potential after exposure to HL156A, cells were assessed using a Mitochondrial Membrane Potential Assay (Biotium Inc.) with JC‐1. JC‐1 is a fluorescent cationic dye that can selectively enter into mitochondria and reversibly change color from red to green as the membrane potential decreases. Interestingly, green fluorescence was increased in HL156A‐treated cells, suggesting that HL156A led to a reduction in membrane potential (Figure 4A).
Figure 4.
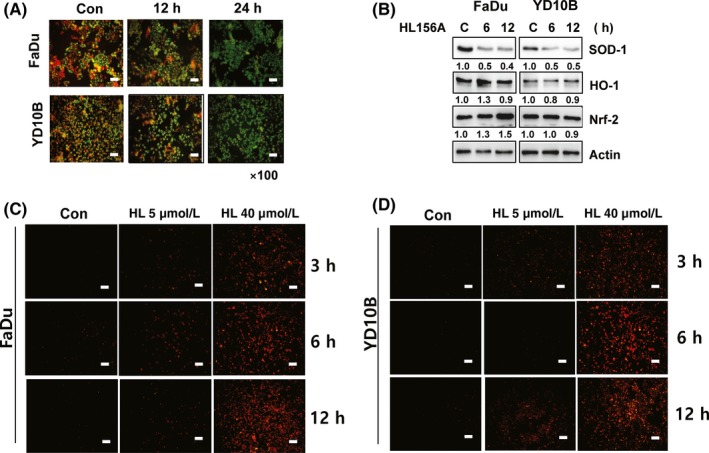
Mitochondrial membrane potential and reactive oxygen species (ROS) production in cells treated with HL156A. A, FaDu and YD‐10B cells were treated with 40 μmol/L HL156A for 12 or 24 hours. Changes in mitochondrial membrane potential were monitored by loading cells with the fluorescent probe JC‐1 followed by analysis with fluorescence microscopy. Scale bar, 50 μm. B, Effect of HL156A on SOD‐1 and NRF/HO‐1 expression. FaDu and YD‐10B cells were treated with HL156A for the indicated times. Total cell lysates were immunoblotted with antibodies against SOD‐1, NRF‐2, and HO‐1. Numbers represent relative density normalized to actin. C,D, ROS were measured in FaDu and YD‐10B cells treated with HL156A (5 or 40 μmol/L) for 3, 6, and 12 hours. Cells were loaded with dihydroethidium (5 mmol/L) and viewed using fluorescence microscopy. Scale bar, 50 μm
To further explore the link between ROS formation and the antiproliferative effects of HL156A, expression of antioxidant proteins was examined. Treatment with HL156A inhibited the level of SOD‐1 in both cells in a time‐dependent way (Figure 4B). However, the expression of Nrf‐2 and its downstream target HO‐1 was not changed during treatment with HL156A. It has been reported that mitochondrial malfunction leads to accumulation of ROS.18 Intracellular ROS production was assessed with fluorescence microscopy using the oxidation‐sensitive dye DHE. Our results showed that, in FaDu and YD‐10B cells, the level of ROS was time‐ and/or dose‐dependently increased upon treatment with HL156A, suggesting that ROS play a significant role in HL156A‐mediated antiproliferation and/or apoptosis (Figure 4C,D).
3.4. Effects of HL156A on insulin‐ and AMPK signaling pathways
To examine the signaling mechanisms which might regulate apoptosis generation by HL156A, we focused attention on the insulin‐like growth factor (IGF)/AKT/mTOR and AMPK pathways. Because, previous studies have revealed that metformin exerts anti‐tumor effect through the inhibition of PI3K/AKT/mTOR signaling and the activation of AMPK signaling.12
To investigate the molecular mechanisms underlying the effects of HL156A on oral cancer cells, we examined the activation of insulin pathways. Phospho‐IGF‐1 levels decreased significantly after 12‐h incubation in the presence of HL156A in FaDu and YD‐10B cells (Figure 5A). We found that HL156A treatment inhibited AKT and ERK phosphorylation/activation and repressed phosphorylation of mTOR and p70S6K in a time‐dependent way. Our results indicate that HL156A could suppress activation and/or expression of IGF‐I, leading to repression of phosphorylation of AKT and mTOR, which subsequently repressed phosphorylation of p70S6K. HL156A also inhibits AKT/ERK1/2 signal transduction pathways that are important for cell proliferation as shown by a decrease in the expression of p‐ERK1/2 (Thr202/Tyr204) after 6 hours of treatment (Figure 5A). Furthermore, western blot results showed that p‐AMPK was increased in HL156A‐treated FaDu and YD‐10B cells whereas total AMPK remained unaltered (Figure 5B). AMPK is involved in the expression of inflammatory cytokines through NF‐κB.19 Therefore, we examined the expression of NF‐κB subunit p65 (NF‐κB p65) in both types of cells treated with HL156A for 6 to 24 hours. HL156A decreased the expression of phospho NF‐κB‐p65 in FaDu and YD‐10B cells in a time‐dependent way.
Figure 5.
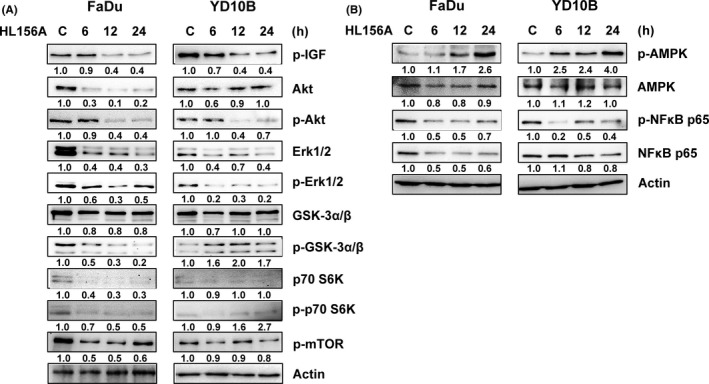
HL156A regulates the insulin‐like growth factor (IGF)/AKT/mammalian target of rapamycin (mTOR) pathway and AMP‐activated protein kinase/nuclear factor kappa B (AMPK/NF‐κB) signaling in FaDu and YD‐10B cells. A, Cells were treated with HL156A (40 μmol/L) for the indicated times, and the total protein was subjected to western blotting analysis using antibodies against IGF‐1R, AKT, mTOR, p70S6K, ERK1/2, and GSK‐3. B, Expression and activation of AMPK and NF‐κB were detected with western blotting. Numbers represent relative density normalized to actin
3.5. HL156A decreases migration capacity
Migration is one of the most important features of cancer. Therefore, to test whether HL156A modulates cell motility, we carried out a wound‐healing experiment. Restoration of FaDu and YD‐10B cells treated with 40 μmol/L HL156A slowly closed the scratch wound compared with the control cells at 24 hours or 36 hours after initiation of the scratch (Figure 6A).
Figure 6.
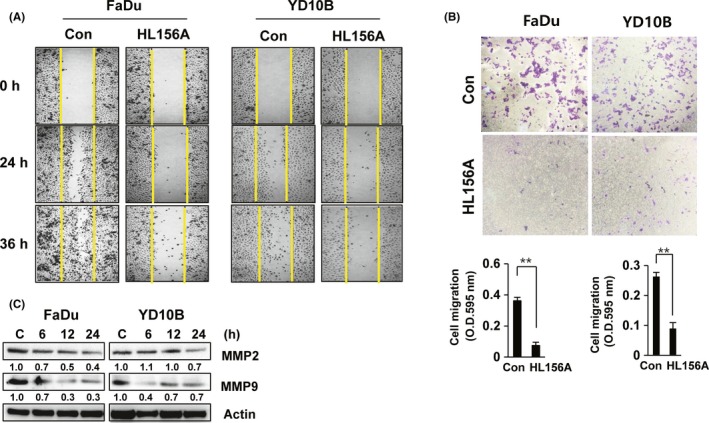
HL156A inhibits cell migration and MMP2 and MMP9 expression. A, Wound‐healing assay shows the closure rates of cells treated with HL156A. Images of wound‐closure rates at the indicated time‐points. B, Effect of HL156A on cell migration. Cells were treated with 40 μmol/L HL156A for 22 hours. Migration assays were carried out as described in the Materials and Methods section. Graphs show the quantitative evaluation of the migration rates. The results represent the averages of three independent experiments. C, Expression of MMP2 and MMP9 in HL156A‐treated cells. FaDu and YD‐10B cells were treated with HL156A (40 μmol/L) for the indicated times and subjected to western blotting
To confirm whether HL156A inhibits cell migration, we carried out in vitro Matrigel Transwell chamber assays. When the FaDu and YD‐10B cells were treated with HL156A, cell migration was significantly inhibited. HL156A, at a concentration of 40 μmol/L, decreased the migration of FaDu and YD‐10B cells to 85% and 70% that of the control cells after 22 hours of treatment (Figure 6B).
Several studies have indicated that MMP signaling pathways play an important role in tumor invasion and migration.20, 21 To elucidate the mechanism by which HL156A inhibits migration in FaDu and YD‐10B cells, we monitored the expression of MMP2 and MMP9. Levels of MMP2 and MMP9 were reduced by HL156A treatment in a time‐dependent way, with 50% inhibition in 40 μmol/L HL156A‐treated FaDu and YD‐10B cells (Figure 6C).
3.6. HL156A suppresses tumor growth in a xenograft mouse model
In support of the identified in vitro antiproliferative effects of HL156A, a tumor xenograft model was used to examine alterations in the pathological characteristics in vivo. C3H mice were randomized and s.c. engrafted with mouse oral cancer AT84 cells treated in vitro with or without HL156A (20 μmol/L). As shown in Figure 7, HL156A significantly inhibited tumor growth up to 82% relative to the control group at the end of the experiment (21 days). No systemic toxicity, including bodyweight changes, or other apparent adverse effects were observed in the animals throughout the study period (Figure 7A). Histological differences between the HL156A‐treated and control groups were examined using H&E staining. Tumor tissues treated with HL156A had larger areas of extensive cell changes showing nuclear pyknosis and cytoplasmic eosinophilia. In immunohistochemistry assay, tumors from the HL156A‐treated group showed decreased levels of phosphorylated IGF‐1 and mTOR compared to control. Consistent with our western blot findings, HL156A increased the expression of p‐AMPK compared with control (Figure 7C). The antitumoral effects of HL156A in the in vivo study were also investigated by examining the expression of PCNA, a cell proliferation marker, and by TUNEL staining to assess the number of apoptotic cells using immunohistochemistry. The number of PCNA‐positive cells was significantly decreased in HL156A‐treated tumor sections compared with tumors from control mice. The number of TUNEL‐positive cells was dramatically increased in HL156A‐treated tumor tissues, suggesting that the effect of HL156A on inhibition of tumor growth and the induction of apoptotic cell death is consistent with the antitumor activity of HL156A in vivo. In addition, AT84 cells (7 × 106 cells per mouse) were injected s.c. into the left flank region of 3‐week‐old male C3H mice. HL156A (30 mg/kg) and metformin (100 mg/kg) were given orally every day throughout the duration of the experiment (3 weeks). We observed that tumor growth in HL156A‐ or metformin‐treated mice was not dramatically inhibited during the experimental period but relatively smaller tumor volumes were observed compared to control (Figure S1).
Figure 7.

HL156A inhibits tumor growth in a mouse AT84 xenograft model. A,B, Inhibition of tumor growth. Mouse oral cancer AT84 cells (1 × 106) treated with 20 μmol/L HL156A were injected s.c. into the left flanks of male C3H mice (n = 7 per group). Tumor growth was assessed by monitoring the mass volume 2 times a week. Bodyweight was monitored and plotted against time for the HL156A (closed squares) and control groups (closed circles). Tumor volume was measured at 20 days and calculated using the formula V = (ab 2)/2 where a was the largest diameter and b was the shortest diameter of the tumor. **P < .01 compared to untreated control. C–E, Immunohistochemistry of tumor sections. Immunohistochemistry for phosphorylated insulin‐like growth factor (p‐IGF), phosphorylated mammalian target of rapamycin (p‐mTOR), AMP‐activated protein kinase (AMPK), proliferating cell nuclear antigen (PCNA) or TUNEL was carried out on paraffin‐embedded tumor sections. Graphs show the quantitative evaluation of the number of PCNA‐ or TUNEL‐positive cells
4. DISCUSSION
Previous studies suggest that metformin inhibits the proliferation and growth of various types of cancer.3, 22, 23, 24 In addition, metformin is also associated with decreased incidence of breast, liver, pancreatic, and colorectal cancers in diabetic patients.6, 7, 11, 12 However, the pathways that contribute to the anticancer effects of metformin have still not been elucidated. Currently, researchers want to know whether metformin or its derivatives may have additional uses in other diseases, including cancer.
Although metformin contributes to the anticancer effects, delivery of metformin to the tissue is limited. Specifically, metformin has limitations as an anticancer drug because its hydrophilic nature prevents it from entering cells.25 HL156A overcomes this shortcoming and shows generally high bioavailability. For example, the prominent survival benefit of in vivo combination treatment with HL156A may be attributable to the excellent penetration ability of HL156A.16 In addition, a number of studies have shown the specific effect of metformin on various human cancers. High concentrations of metformin (up to 10‐15 mmol/L) have been shown to inhibit proliferation of various human cancer cell lines such as lung cancer cells (up to 15 mmol/L), endometrial cancer cells (up to 10 mmol/L), hepatocellular carcinoma cells (up to 10 mmol/L), and head and neck squamous carcinoma cells (5‐20 mmol/L).26, 27, 28 HL156A exerted more potent inhibitory effects on the proliferation of glioblastoma cells than metformin. HL156A showed approximately 100‐fold more potent effects compared to metformin. In a glioblastoma xenograft model, HL156A showed a comparable degree of inhibitory effect on in vivo tumor growth at the 30 mg/kg dose to that of metformin at 100 mg/kg.16, 29 We also found that HL156A inhibits cell growth at lower concentrations (≤40 μmol/L) than metformin in oral squamous carcinoma cells.
In our study, we found that the new metformin derivative HL156A significantly reduced growth of human oral squamous cell carcinoma FaDu and YD‐10B cells. HL156A led to a reduction in colony formation in a dose‐dependent way. Moreover, the inhibitory properties of HL156A observed were markedly augmented when HL156A was used to treat oral cancer cells, even with a dose as low as 40 μmol/L. The cell cycle was inhibited at the G2/M phase in the HL156A‐treated cells that were analyzed. Consistent with these results, HL156A treatment led to a remarkable decrease in cyclin B and cyclin‐dependent kinase (CDK) 1 activity in FaDu and YD‐10B cells. In addition, expression of CDK4, CDK6, CDK2, cyclin A, and cyclin E protein was inhibited in HL156A‐treated cells (data not shown). These results suggest that the effects of HL156A on cell cycle progression were associated with inhibition of cell growth and cell death induction.
Interestingly, we first found that HL156A‐mediated apoptotic cell death was possibly associated with promotion of intracellular oxidative stress. Consistent with previous reports regarding the relationship between the anticancer potency of metformin and the generation of ROS,30 our results showed that treatment with HL156A led to a reduction in mitochondrial membrane potential as well as an increase in ROS production in a time‐ and dose‐dependent way. Furthermore, mitochondrial antioxidant proteins, such as SOD‐1, were also down‐regulated, but not the Nrf/Ho‐1 pathway. The imbalance between excessive ROS production and SOD‐1 depletion may cause oxidative stress for oral cancer cells. This, combined with the reduction in membrane potential, resulted in the release of pro‐apoptotic factors, such as cytochrome c, and the stimulation of caspases, which ultimately ends in apoptosis.17 Our study showed that HL156A gradually increased cell apoptosis, as evidenced by the increased activity of caspase‐3, ‐7, ‐9, and poly (ADP‐ribose) polymerase (PARP).
Another important point is that HL156A exerted more potent inhibitory effects on the migration of oral cancer cells than metformin. Wound healing assays and migration assays showed invasion and/or migration properties of HL156A. We also detected down‐regulation of MMP‐2 and MMP‐9 protein levels in a time‐dependent way following HL156A treatment. A previous study showed that anticancer drugs markedly suppressed migration and invasion in malignant cancer cells and HUVEC, and the effect was partially MMP‐dependent.20, 21, 31 Our results showed that HL156A is involved in the inhibition of FaDu and YD‐10B cell migration through their ability to breakdown the extracellular matrix, especially MMP‐2 and MMP‐9.
Recent studies showed that IGF‐1 activates two main downstream signaling pathways, the PI3K/AKT/mTOR and the Ras‐Raf‐MEK/ERK pathways.32 Activation of the PI3K/AKT/mTOR pathway is known to promote cell proliferation and cell cycle progression and to reduce apoptosis, which ultimately leads to a competitive growth advantage, metastatic competence, angiogenesis, and therapy resistance.33, 34 AKT can directly phosphorylate and activate mTOR. mTOR plays a major role in carcinogenesis, and activation of this pathway is associated with cancer progression.34 In the present study, HL156A was involved in mediating the antiproliferation effect through inhibiting the IGF/AKT/mTOR/p70S6K pathway in FaDu and YD‐10B cells. In addition, HL156A blocked phosphorylation of the AKT substrate glycogen synthase kinase 3b and ERK1/2. A previous study showed that suppression of AKT/ERK signaling resulted in apoptosis, indicating that the activation of AKT/ERK promoted survival under cellular stress.35 Collectively, these results strongly suggest that IGF/AKT/mTOR and AKT/ERK pathways play an important role in maintaining the viability of oral cancer cells. Our results show that the effects of HL156A were partially induced by inhibiting these pathways, IGF/AKT/mTOR and/or AKT/ERK.
Herein, we also found that HL156A importantly induced activation of AMPK by phosphorylation together with suppression of NF‐κB p65 in FaDu and YD‐10B cells. A number of studies have shown indirect suppression of AMPK on NF‐κB signaling by its downstream mediators, namely SIRT1, the Forkhead box O (FoxO) family, and peroxisome proliferator‐activated receptor γ co‐activator 1α (PGC‐1α).36 The HL156A‐mediated decrease in NF‐κB p65 phosphorylation may be accompanied by AMPK activation. These results may suggest a role for HL156A in inhibiting the activation of NF‐κB as well as in inhibition of the expression of proteins/cytokines that regulate inflammation. The importance of these signaling pathways in the pathogenesis of cancer development is suggested from a number of studies.
A recent study showed that HL156A in combination with the chemotherapeutic agent temozolomide (TMZ) inhibited the invasive properties of glioblastoma and increased the survival rate in a xenograft mouse model.13 In our study, consistent with this, HL156A significantly reduced the growth of mouse oral squamous cell carcinoma AT84 xenograft tumors. In the immunohistochemistry assay, the number of proliferative cells was dramatically decreased, whereas the number of apoptotic cells was considerably increased. Importantly, p‐IGF‐1 and p‐mTOR expression were weak in control group tissues compared with HL156A‐treated tumor tissues. In contrast, p‐AMPK expression was stronger in HL156A‐treated tumor tissues. Overall, our results showed that induction of apoptosis by HL156A may be associated with inhibition of cell proliferation through decreasing the mitochondrial membrane potential or IGF‐1/AKT/mTOR signaling or inducing AMPK signaling in FaDu and YD‐10B cells.
Collectively, the results of the present study suggest that HL156A may have the potential to arrest the progression of oral cancer through suppression of the IGF/AKT/mTOR pathway or MMP‐2 and MMP‐9 pathways. In addition, AMPK activity, at least in part, is also required for the above‐mentioned tumor suppression. In conclusion, the results clarify the beneficial effect of HL156A in reducing oral cancer development.
ACKNOWLEDGMENT
This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by Ministry of Science, ICT and Future Planning (no. 2015005588).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Lam TG, Jeong YS, Kim S‐A, Ahn S‐G. New metformin derivative HL156A prevents oral cancer progression by inhibiting the insulin‐like growth factor/AKT/mammalian target of rapamycin pathways. Cancer Sci. 2018;109:699–709. https://doi.org/10.1111/cas.13482
Funding information
National Research Foundation of Korea (NRF) (Grant/Award Number: ‘2015005588’).
REFERENCES
- 1. Chin D, Boyle GM, Porceddu S, Theile DR, Parsons PG, Coman WB. Head and neck cancer: past, present and future. Expert Rev Anticancer Ther. 2006;6:1111‐1118. [DOI] [PubMed] [Google Scholar]
- 2. Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5:311‐316. [DOI] [PubMed] [Google Scholar]
- 3. Hopper C, Kübler A, Lewis H, Tan IB, Putnam G. mTHPC‐mediated photodynamic therapy for early oral squamous cell carcinoma. Int J Cancer. 2004;111:138‐146. [DOI] [PubMed] [Google Scholar]
- 4. Del Barco S, Vazquez‐Martin A, Cufí S, et al. Metformin: multi‐faceted protection against cancer. Oncotarget. 2011;2:896‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akinyeke T, Matsumura S, Wang X, et al. Metformin targets c‐MYC oncogene to prevent prostate cancer. Carcinogenesis. 2013;4:2823‐2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amin S, Mhango G, Lin J, et al. Metformin improves survival in patients with pancreatic ductal adenocarcinoma and pre‐existing diabetes: a propensity score analysis. Am J Gastroenterol. 2016;111:1350‐1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thompson AM. Molecular pathways: preclinical models and clinical trials with metformin in breast cancer. Clin Cancer Res. 2014;20:2508‐2515. [DOI] [PubMed] [Google Scholar]
- 8. Hadad SM, Hardie DG, Appleyard V, Thompson AM. Effects of metformin on breast cancer cell proliferation, the AMPK pathway and the cell cycle. Clin Transl Oncol. 2014;16:746‐752. [DOI] [PubMed] [Google Scholar]
- 9. Colquhoun AJ, Venier NA, Vandersluis AD, et al. Metformin enhances the antiproliferative and apoptotic effect of bicalutamide in prostate cancer. Prostate Cancer Prostatic Dis. 2012;15:346‐352. [DOI] [PubMed] [Google Scholar]
- 10. Guo Y, Yu T, Yang J, et al. Metformin inhibits salivary adenocarcinoma growth through cell cycle arrest and apoptosis. Am J Cancer Res. 2015;5:3600‐3611. [PMC free article] [PubMed] [Google Scholar]
- 11. Daugan M, Dufaÿ Wojcicki A, d'Hayer B, Boudy V. Metformin: An anti‐diabetic drug to fight cancer. Pharmacol Res. 2016;113:675‐685. [DOI] [PubMed] [Google Scholar]
- 12. Sośnicki S, Kapral M, Węglarz L. Molecular targets of metformin antitumor action. Pharmacol Rep. 2016;68:918‐925. [DOI] [PubMed] [Google Scholar]
- 13. Hadad SM, Fleming S, Thompson AM. Targeting AMPK: a new therapeutic opportunity in breast cancer. Crit Rev Oncol Hematol. 2008;67:1‐7. [DOI] [PubMed] [Google Scholar]
- 14. Lee HS, Shin HS, Choi J, et al. AMP‐activated protein kinase activator, HL156A reduces thioacetamide‐induced liver fibrosis in mice and inhibits the activation of cultured hepatic stellate cells and macrophages. Int J Oncol. 2016;49:1407‐1414. [DOI] [PubMed] [Google Scholar]
- 15. Ju KD, Kim HJ, Tsogbadrakh B, et al. HL156A, a novel AMP‐activated protein kinase activator, is protective against peritoneal fibrosis in an in vivo and in vitro model of peritoneal fibrosis. Am J Physiol Renal Physiol. 2016;310:F342‐F350. [DOI] [PubMed] [Google Scholar]
- 16. Choi J, Lee JH, Koh I, et al. Inhibiting stemness and invasive properties of glioblastoma tumorsphere by combined treatment with temozolomide and a newly designed biguanide (HL156A). Oncotarget. 2016;7:65643‐65659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gottlieb E, Armour SM, Harris MH, Thompson CB. Mitochondrial membrane potential regulates matrix configuration and cytochrome c release during apoptosis. Cell Death Differ. 2003;10:709‐717. [DOI] [PubMed] [Google Scholar]
- 18. Wang CH, Wu SB, Wu YT, Wei YH. Oxidative stress response elicited by mitochondrial dysfunction. Implication in the pathophysiology of aging. Exp Biol Med. 2013;238:450‐460. [DOI] [PubMed] [Google Scholar]
- 19. Moiseeva O, Deschênes‐Simard X, St‐Germain E, et al. Metformin inhibits the senescence‐associated secretory phenotype by interfering with IKK/NF‐κB activation. Aging Cell. 2013;12:489‐498. [DOI] [PubMed] [Google Scholar]
- 20. Nabeshima K, Inoue T, Shimao Y, Sameshima T. Matrix metalloproteinases in tumor invasion: role for cell migration. Pathol Int. 2002;52:255‐264. [DOI] [PubMed] [Google Scholar]
- 21. Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16‐27. [DOI] [PubMed] [Google Scholar]
- 22. Provinciali N, Lazzeroni M, Cazzaniga M, Gorlero F, Dunn BK, DeCensi A. Metformin: risk‐benefit profile with a focus on cancer. Expert Opin Drug Saf. 2015;14:1573‐1585. [DOI] [PubMed] [Google Scholar]
- 23. Wu L, Zhu J, Prokop LJ, Murad MH. Pharmacologic therapy of diabetes and overall cancer risk and mortality: a meta‐analysis of 265 studies. Sci Rep. 2015;5:10147‐10157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bao B, Azmi AS, Ali S, Zaiem F, Sarkar FH. Metformin may function as anti‐cancer agent via targeting cancer stem cells: the potential biological significance of tumor‐associated miRNAs in breast and pancreatic cancers. Ann Transl Med. 2014;2:59‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Menendez JA, Quirantes‐Pine R, Rodriguez‐Gallego E, et al. Oncobiguanides: Paracelsus’ law and nonconventional routes for administering diabetobiguanides for cancer treatment. Oncotarget. 2014;5:2344‐2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ashinuma H, Takiguchi Y, Kitazono S. Antiproliferative action of metformin in human lung cancer cell lines. Oncol Rep. 2012;28:8‐14. [DOI] [PubMed] [Google Scholar]
- 27. Cantrell LA, Zhou CX, Mendivil A, Malloy KM, Gehrig PA, Bae‐Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation‐implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. 2012;11:1374‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sesen J, Dahan P, Scotland SJ, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS ONE. 2015;10:e0123721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haugrud AB, Zhuang Y, Coppock JD, Miskimins WK. Dichloroacetate enhance apoptotic cell death via oxidative damage and attenuates lactate production in metformin‐treated breast cancer cells. Breast Cancer Res Treat. 2014;147:539‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johansson N, Ahonen M, Kähäri VM. Matrix metalloproteinases in tumor invasion. Cell Mol Life Sci. 2000;57:5‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cui QL, Almazan G. IGF‐I‐induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src‐like tyrosine kinases. J Neurochem. 2007;100:1480‐1493. [DOI] [PubMed] [Google Scholar]
- 33. Guerrero‐Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35:515‐524. [DOI] [PubMed] [Google Scholar]
- 34. Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143:3050‐3060. [DOI] [PubMed] [Google Scholar]
- 35. Jiao Q, Zou L, Liu P, et al. Xanthoceraside induces apoptosis in melanoma cells through the activation of caspases and the suppression of the IGF‐1R/Raf/MEK/ERK signaling pathway. J Med Food. 2014;17:1070‐1078. [DOI] [PubMed] [Google Scholar]
- 36. Green CJ, Pedersen M, Pedersen BK, Scheele C. Elevated NF‐κB activation is conserved in human myocytes cultured from obese type 2 diabetic patients and attenuated by AMP‐activated protein kinase. Diabetes. 2011;60:2810‐2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials