

Abstract
Solute carrier family members control essential physiological functions and are tightly linked to human diseases. Solute carrier family 35 member F2 (SLC35F2) is aberrantly activated in several malignancies. However, the biological function and molecular mechanism of SLC35F2 in papillary thyroid carcinoma (PTC) are yet to be fully explored. Here, we showed that SLC35F2 was prominently upregulated in PTC tissues at both protein and mRNA expression level compared with matched adjacent normal tissues. Besides, the high expression of SLC35F2 was significantly associated with lymph node metastasis in patients with PTC. CRISPR/Cas9‐mediated knockout of SLC35F2 attenuated the tumorigenic properties of PTC, including cell proliferation, migration and invasion and induced G1 phase arrest. In contrast, ectopic expression of SLC35F2 brought about aggressive malignant phenotypes of PTC cells. Moreover, SLC35F2 expedited the proliferation and migration of PTC cells by targeting transforming growth factor‐β type I receptor (TGFBR1) and phosphorylation of apoptosis signal‐regulating kinase 1 (p‐ASK‐1), thereby activating the mitogen‐activated protein kinase signaling pathway. The malignant behaviors induced by overexpression of SLC35F2 could be abrogated by silencing of TGFBR1 using a specific inhibitor. We conducted the first study on SLC35F2 in thyroid cancer with the aim of elucidating the functional significance and molecular mechanism of SLC35F2. Our findings suggest that SLC35F2 exerts its oncogenic effect on PTC progression through the mitogen‐activated protein kinase pathway, with dependence on activation of TGFBR‐1 and apoptosis signal‐regulating kinase 1.
Keywords: cell cycle, mitogen‐activated protein kinase pathway, papillary thyroid carcinoma, solute carrier family 35 member F2, tumorigenesis
1. INTRODUCTION
The incidence of thyroid cancers has increased steadily in recent years. It accounts for 96% of endocrine system cancers and is one of the most rapidly increasing cancers in China.1, 2, 3 Papillary thyroid carcinomas (PTC) are the predominant type of thyroid cancer and are slow growing and well‐differentiated. Although a majority of PTC exhibit indolent behavior, a subset of PTC behave aggressively, with early‐dissemination to local lymph nodes and oppression to organs, suggesting that some important determinants of clinical behavior exist.4 Therefore, identification of molecular markers that could predict the metastatic potential of these lesions early on and new prevention strategies are urgently needed.
Solute carrier family 35 member F2 (SLC35F2) belongs to the solute carrier family, which is a superfamily of membrane‐bound carriers involved in transportation of all kinds of substrates, such as metabolites, cofactors, vitamins, nutrients, ions and drugs.5 The SLC gene superfamily controls essential physiological functions and its disturbance may result in diseases like malignancies. For example, Xu et al found that amplification of SLC12A5 plays a strong tumorigenic role and is associated with poor prognosis of patients with colorectal cancer.6 SLC39A6 promotes aggressiveness of esophageal squamous carcinoma cells by increasing intracellular levels of zinc and activates phosphatidylinositol 3‐kinase (PI3K) pathway.7 SLC35, a group of nucleotide sugar transporters, currently comprises at least 31 homologous molecular species from SLC35A to SLC35G. SLC35A2 functions as a UDP‐galactose transporter, whose substrate is required for galactosylation. In addition, SLC35D2 has been reported to translocate UDP‐glucose into the Golgi as a substrate for heparan sulfate synthesis.8 However, only one‐third of the whole solute carrier family members have been studied in detail. Little is known about most of the members in the SLC35 family, and SLC35F2 is one of them. The expression of SLC35F2 was initially found in ataxia telangiectasia.9 Subsequently, Nishimura et al discovered that it had the highest expression level in human adult salivary glands (detected by RT‐PCR).10 Bu et al report that SLC35F2 was highly expressed in non‐small cell lung cancer (NSCLC) tissues, was associated with the pathological stage and had prognostic value in NSCLC.11 Nyquist et al found that SLC35F2 expression was linked to intratumor androgen levels and was regulated by the androgen receptor axis signaling in prostate cancer.12 Recently, a study verified SLC35F2 as a cargo of anti‐cancer drug YM155 and determined that lack of SLC35F2 was capable of conferring drug resistance.13 Therefore, SLC35F2 could be used as a clinical biomarker for YM155 susceptibility.
Currently, very little is known regarding SLC35F2 expression in many types of cancer and its role in carcinogenesis and related pathways of carcinogenesis. In the present study, we, for the first time, uncovered the function of SLC35F2 in PTC. This protein is highly expressed in PTC tissues compared with the adjacent normal tissues and is positively correlated with lymph node metastasis. Ectopic expression of SLC35F2 evokes malignant phenotypes of PTC cells, such as proliferation, migration and invasion in vitro and in vivo, while SLC35F2 abrogation shows the opposite effects. Both gain‐of‐function and loss‐of‐function studies verify the pivotal roles of SLC35F2 in cell cycle control by regulating multiple G1/S transition‐related proteins. Moreover, our finding provides new insight into the mechanism of the SLC35F2/transforming growth factor‐β type I receptor (TGFBR1)/apoptosis signal‐regulating kinase 1 (ASK‐1) axis in contributing to the activation of MAPK pathway and suggests that SLC35F2 and its related signaling pathways could be novel targets for the treatment of PTC.
2. MATERIALS AND METHODS
2.1. Tissue samples
All tissues were obtained from Huashan Hospital of Fudan University from patients diagnosed with PTC who underwent resection between 2015 and 2016. The patients enrolled in this study had not received radioiodine therapy or chemotherapy before surgery. A total of 42 pairs of fresh PTC tissue samples and their corresponding non‐tumorous thyroid tissues were collected during surgery and frozen into liquid nitrogen immediately for further research. The study was approved by the ethics committee of Huashan Hospital, Fudan University and all patients provided written informed consent.
2.2. Cell culture
BCPAP and KTC‐1 cells were kindly provided by Stem Cell Bank, Chinese Academy of Sciences. Cultured cells were maintained in a humidified, 5% CO2 atmosphere at 37°C. BCPAP and KTC‐1 cells were maintained in RPMI 1640 (Gibco, USA) supplemented with 10% FBS (Gibco, USA) and 1% non‐essential amino acids (NEAA, Gibco, USA). Human immortalized thyroid cells, Nthy‐ori 3‐1, were preserved in our laboratory and grown in DMEM supplemented with 10% FBS (Gibco, USA).
2.3. RNA extraction and real‐time PCR
Total RNA from thyroid cancer cells was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and first‐strand cDNA was synthesized using PrimeScript RT Reagent Kit (TaKaRa Biotechnology, Shiga, Japan) at 37°C for 15 minutes, 85°C for 5 seconds and then at 4°C. Quantitative real‐time PCR was subsequently performed using an ABI 7900 instrument (Applied Biosystems, Foster City, CA, USA). The relative expression levels were determined using the 2−ΔΔCt method. Each experiment was performed in triplicate.
2.4. Immunohistochemistry
Immunohistochemistry analysis and HE staining were performed according to standard protocols. After treatment with 3% hydrogen peroxide for 30 minutes in a 37°C incubator, antigen retrieval was performed by incubating the slides in a microwave oven for 15 minutes in 0.01 mol/L citrate buffer (pH 6.0). The immunohistochemistry score was calculated as previously described.14 In brief, the expression of SLC35F2 was scored according to the percentage of positive‐stained tumor cells and the intensity of staining. The final immunoreactivity score was obtained by 2 independent researchers by multiplying the percentage and the score for each case was from 0 to 9.
2.5. Western blot
Cell lysates were harvested by RIPA lysis buffer on ice. An equal amount of 20 μg total cell lysates were separated on 8%‐12% SDS‐PAGE gel and electrophoretically transferred to polyvinylidene fluoride (PVDF) membranes. Following blocking with 5% BSA at room temperature for 2 hours, membranes were then incubated with primary antibodies overnight at 4°C. Finally, membranes were detected with the SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). The following antibodies were used to explore protein expression: anti‐SLC35F2 antibodies (AV43971, Sigma‐Aldrich), anti‐β‐actin (#4970), anti‐CDK2 (#2546), anti‐CDK4 (#12790), anti‐CDK6 (#13331), anti‐Cyclin D1 (#2978), anti‐Cyclin D3 (#2936), anti‐p27 Kip1 (#3686), anti‐p21 Waf1/Cip1 (#2947), anti‐p18 INK4C (#2896), anti‐ERK1/2 (#4695), anti‐p‐ERK1/2 (Thr202/Tyr204, #4370), anti‐JNK (#9252), anti‐p‐JNK (Thr183/Tyr185, #4668), anti‐p38 MAPK (#8690), anti‐p‐p38 MAPK (Thr180/Tyr182, #4511), anti‐ASK‐1 (#8662), anti‐phosphorylation of apoptosis signal‐regulating kinase 1 (p‐ASK‐1) (Thr845, #3765) (Cell Signaling Technology) and anti‐TGFBR1 (CY2905, Abways, China).
2.6. Establishment of solute carrier family 35 member F2 knockout and overexpression cell lines
For knockout of SLC35F2, the targeted gRNA expression oligos were introduced into the lentiCRISPR v2 vector, with an empty lentiCRISPR v2 vector used as a control. Constructed vectors were confirmed by DNA sequencing. These plasmids were transfected into 293T packaging cells to generate lentivirus, which was then used to infect BCPAP and KTC‐1 cell lines. Meanwhile, a full‐length of SLC35F2 cDNA was sub‐cloned into the pCDH plasmid, which was transfected into 293T cells using Lipofectamine 2000 (Invitrogen) reagents. The lentivirus supernatant was used to infect target cell lines. For selection of stable SLC35F2 knockout and overexpression cell lines, cells were infected with lentivirus for 48 hours and further selected with puromycin (2.0 μg/mL) for 14 days. The efficiency of SLC35F2 deletion was confirmed by western blot. Overexpression of SLC35F2 was determined through quantitative RT‐PCR (qRT‐PCR) and western blot.
2.7. Cell proliferation and colony formation
Proliferation ability of thyroid cancer cells was measured using Cell Counting Kit‐8 reagent (CCK8, Dojindo, Japan). Approximately, 1 × 103 cells were seeded per well into 96‐well cell culture plates; 10 μL CCK‐8 reagents were added to each well and incubated for 2 hours at 37°C. Absorbance was read at 450 nm on a microplate reader (Multiskan GO, Thermo Scientific). For colony formation assay, parental or SLC35F2 knockout cells (500 cells/well) were plated into the 35‐mm dish and cultured for 14 days. The medium was changed at 3‐day intervals. Cells on the plates were fixed with 4% paraformaldehyde and stained with crystal violet.
2.8. Flow cytometry for apoptosis and cell cycle
A PE Annexin V Apoptosis Detection Kit (BD Biosciences, Rockville, MD, USA) was applied to measure cell apoptosis. Cells were gently trypsinized and washed with cold PBS twice, resuspended in 500 μL Annexin V binding buffer; then 5 μL 7‐AAD and 2.5 μL Annexin V were added to each sample, which were incubated in the dark for 15 minutes at room temperature, and analyzed with a FACSCaliber flow cytometer (BD Biosciences). For cell cycle analysis, cells were washed with PBS twice and fixed with 70% ethanol at 4°C overnight. After centrifugation, cells were washed and resuspended in 500 μL BD Pharmingen PI/RNase staining buffer (BD Biosciences). Then cells were incubated for 15 minutes at room temperature and analyzed on the same flow cytometer.
2.9. Cell migration and invasion assay
For transwell migration and invasion assays, cells were suspended in RPMI 1640 without serum in the upper chamber and medium supplemented with 20% FBS was used as a chemoattractant in the bottom chamber. Then cells were incubated at 37°C for 24 hours (migration assay) or 48 hours (invasion assay). Cells remaining on the upper chamber were removed by cotton swab and the migrated or invaded tumor cells were fixed by 4% paraformaldehyde and stained by 0.1% crystal violet solution. At least 5 visual fields under the microscope were photographed each time and the number of cells was calculated using ImageJ software.
2.10. Animal models
All animal procedures were performed under the approval of the Animal Ethics Committee of Fudan University. Sixteen female BALB/c nude mice (Shanghai SLAC Laboratory Animal, China) aged 5 weeks were housed in a 12 hours light/12 hours dark cycle with free access to sterilized murine diet and water. The mice were randomly divided into 4 groups (n = 4 per group) and were subcutaneously injected with BCPAP cells (3 × 106 cells/mouse in 150 μL PBS) in their left flanks. Tumor growth was measured every 4 days. Tumor volume was calculated with a caliper and evaluated using the formula: (length × width2)/2. Five weeks post‐injection, the mice were killed and the tumors were weighed; IHC and HE staining were performed.
2.11. Statistical analysis
Data are presented as mean ± SD and analyzed using Student's t test and the χ2‐test for comparisons among the groups. A paired t test was used for paired PTC and corresponding normal thyroid samples. Association between the two gene expression levels was analyzed by Pearson correlation test. Statistical analysis was performed with GraphPad Prism 7.0 software (La Jolla, CA, USA). P < .05 was considered a significant statistical difference.
3. RESULTS
3.1. Solute carrier family 35 member F2 overexpression in papillary thyroid carcinoma tissues is positively correlated with lymph node metastasis
By analyzing data from Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/index.html) and GSE3678 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3678),15 we found that SLC35F2 expression was significantly overexpressed in human PTC tissues (Figure 1A). To verify the robustness of data mining results, we first explored the expression of SLC35F2 in PTC cell lines, quantified by qRT‐PCR and western blotting (Figure 1B). SLC35F2 was elevated in 2 PTC cell lines (BCPAP and KTC‐1) compared to that in an immortalized thyroid follicular cell line Nthy‐ori 3‐1. We further detected SLC35F2 expression level in 42 pairs of PTC tissues and their adjacent non‐cancerous tissues using qRT‐PCR and western blotting. The results revealed that both SLC35F2 mRNA and protein levels were markedly upregulated in PTC tissues compared to normal tissues (Figure 1C,D). Then, to unveil the correlation between SLC35F2 expression and patients’ clinicopathological characteristics, patients were divided into 2 groups according to the ratio of SLC35F2 mRNA expression in tumor tissues to adjacent normal tissues (Table 1). Among the 42 PTC cases, 29 (69.0%) patients were defined as the high group with this ratio above 2‐fold and 13 (31.0%) patients were defined as the low group with the ratio below 2‐fold. Strikingly, SLC35F2 expression was closely correlated with the presence of lymph node metastasis (P = .0109). Next, we used immunohistochemistry staining in another cohort containing 40 patients to verify the clinical relevance of SLC35F2 in PTC, consistent with prior findings, IHC analysis of paired PTC and adjacent normal tissues also confirmed its overexpression at the protein level (Figure 1E). Moreover, patients with lymph node metastasis had higher SLC35F2 staining scores than those without lymph node metastasis (Figure S1). Taken together, our results demonstrate that SLC35F2 is an oncoprotein, whose expression is closely associated with lymph node metastasis.
Figure 1.
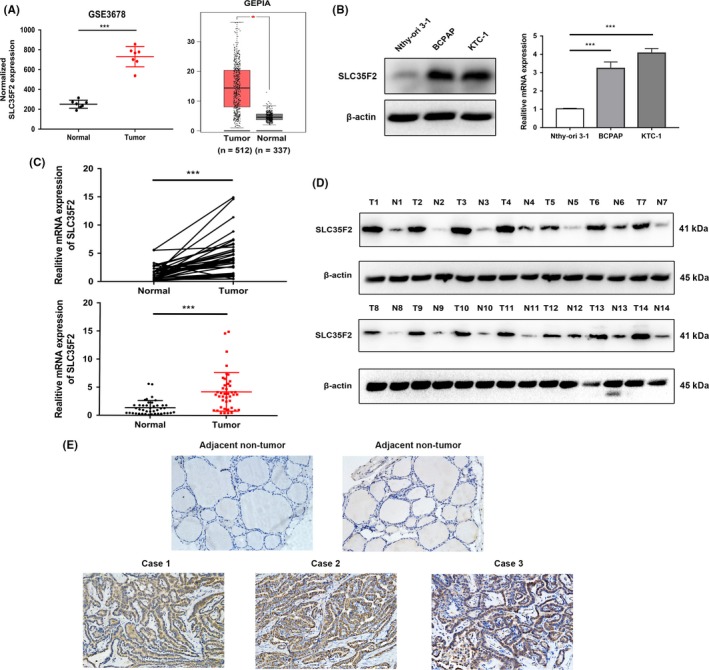
Solute carrier family 35 member F2 (SLC35F2) is frequently upregulated in papillary thyroid carcinoma (PTC) tissues compared to that of adjacent non‐tumor tissues. A, Expression profile of SLC35F2 mRNA in PTC tissues (n = 7) and paired normal thyroid tissues (n = 7; GSE3678) (left panel); expression profile of SLC35F2 mRNA in primary PTC tissues (n = 512) and normal thyroid tissues (n = 337; data from GEPIA) (right panel). B, Western blotting and quantitative RT‐PCR analysis of SLC35F2 expression in human immortalized thyroid follicular cells and PTC cell lines. C, qRT‐PCR analysis of SLC35F2 mRNA expression in 42 PTC samples and paired adjacent non‐tumor tissues. D, SLC35F2 protein level in 14 paired primary PTC tissues and adjacent non‐tumor tissues determined by western blotting. E, Representative immunohistochemistry (IHC) staining images showing the expression of SLC35F2 in PTC tissues and non‐tumor tissues (*P < .05, ***P < .001)
Table 1.
Correlation between solute carrier family 35 member F2 (SLC35F2) expression level and clinicopathological factors in papillary thyroid carcinoma patients
| Characteristics | Number | SLC35F2 expression | P‐value | |
|---|---|---|---|---|
| High group | Low group | |||
| Age | ||||
| <45 | 16 | 10 | 6 | .4715 |
| ≥45 | 26 | 19 | 7 | |
| Gender | ||||
| Male | 12 | 10 | 2 | .2053 |
| Female | 30 | 19 | 11 | |
| T Stage | ||||
| T1 + T2 | 25 | 20 | 5 | .0626 |
| T3 + T4 | 17 | 9 | 8 | |
| Mutlifocality | ||||
| Unifocal | 24 | 18 | 6 | .3353 |
| Multifocal | 18 | 11 | 7 | |
| Lymph node metastasis | ||||
| N0 | 20 | 10 | 10 | .0109a |
| N1a + N1b | 22 | 19 | 3 | |
| TNM stage | ||||
| I | 19 | 11 | 8 | .3264 |
| II | 16 | 13 | 3 | |
| III + IV | 7 | 5 | 2 | |
P < .05.
3.2. Solute carrier family 35 member F2 is required for papillary thyroid carcinoma cell proliferation
To further investigate the biological function of SLC35F2 overexpression in PTC progression, BCPAP and KTC‐1 cell lines that stably knock out or overexpress SLC35F2 were established. We used a lentiCRISPR v2 vector and designed sgRNA against exon 7 of SLC35F2. As CRISPR‐Cas9 knockout system causes indel mutation, the level of SLC35F2 mRNA expression was not significantly changed in SLC35F2‐KO cells. Western blotting confirmed that SLC35F2 protein expression was efficiently downregulated in BCPAP and KTC‐1 cells using the CRISPR/Cas9 System (Figure 2A). Respectively, western blotting and qRT‐PCR verified the overexpression of SLC35F2 in SLC35F2‐OE cells compared to BCPAP and KTC‐1 cells with empty pCDH vector (Figure 2C,E). CCK‐8 assay was carried out to determine the effects of SLC35F2 on cell proliferation and a cell growth curve was constructed. Our results showed that knockout of SLC35F2 remarkably decreased cell viability in PTC cell lines from 24 hours after seeding cells (Figure 2B). In contrast, overexpression of SLC35F2 dramatically accelerated cell growth in BCPAP and KTC‐1 cells (Figure 2D). The above results emphasized a crucial role of SLC35F2 in PTC cell proliferation.
Figure 2.
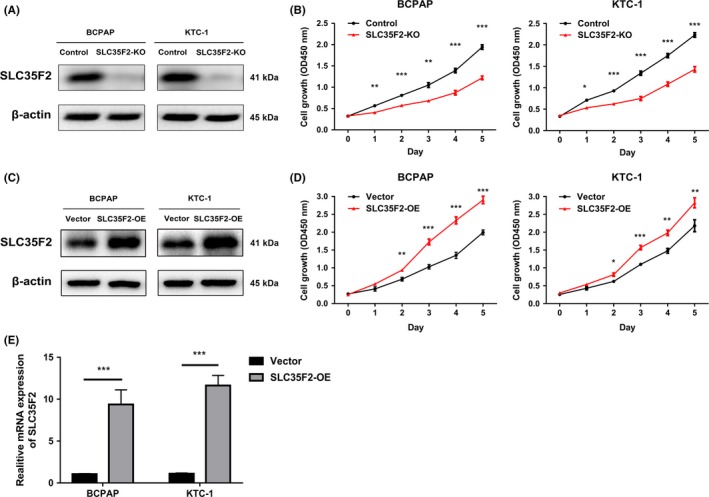
Solute carrier family 35 member F2 (SLC35F2) acts as an oncogene promoting papillary thyroid carcinoma (PTC) cell growth. A, CRISPR/Cas9 mediated knockout efficiency of SLC35F2 in BCPAP and KTC‐1 cells were tested by western blotting. Equal amounts of cell lysates were probed with anti‐SLC35F2 antibody. B, SLC35F2 knockout markedly inhibited PTC cell proliferation quantitated by CCK‐8 assay. C, Overexpression efficiency of SLC35F2 in BCPAP and KTC‐1 cells were confirmed by western blot. D, SLC35F2 overexpression stimulates both BCPAP and KTC‐1 cells proliferation. E, qRT‐PCR analysis of SLC35F2 expression in BCPAP and KTC‐1 cells stably overexpressing SLC35F2 (*P < .05, **P < .01, ***P < .001)
3.3. Solute carrier family 35 member F2 expedites cell cycle coordinating with some critical cyclin‐dependent kinases and cyclins
Given the essential role of SLC35F2 in cell proliferation, we used flow cytometry to analyze the cell cycle distribution and cell apoptosis in SLC35F2 knockout cells to elaborate the effects of SLC35F2 on cell growth. The cell cycle analysis demonstrated that silencing of SLC35F2 increased the percentage of cells in G1 phase, thereby inducing cell cycle arrest at G1 phase, which was in agreement with its inhibitory effects on proliferation (Figure 3A,B). Conversely, ectopic expression of SLC35F2 accelerated G1/S phase transition and increased S‐phase proportion (Figure 3C,D). Considering SLC35F2 could expedite PTC cell growth through promoting G1/S transition, we applied western blotting to investigate whether SLC35F2 exerts its oncogenic activity through modulation of certain cyclins and cyclin‐dependent kinases. As shown in Figure 3E and F, the protein levels of CDK2, CDK4, CDK6, Cyclin D1 and Cyclin D3, key G1/S transition markers, were reduced in the SLC35F2‐KO group compared with the control group. In contrast, knockout of SLC35F2 elevated the expression of cell cycle inhibitors, including p27 Kip1, p21 Waf1/Cip1 and p18 INK4C. The opposite results were observed in the overexpressing SLC35F2 group, which demonstrated that overexpression of SLC35F2 might contribute to quickening cell cycle progression and confer a growth advantage to these cells through interactions with cyclin‐dependent kinases and cyclins. Corroborating the protein levels, our qRT‐PCR results for SLC35F2 knockout in KTC‐1 cells also demonstrated a significant decrease in the expression of genes involved in G1/S cell cycle transition and an increase in genes of cell cycle inhibitors (Figure 3G). Subsequently, the effect of SLC35F2 on cellular apoptosis was explored. However, FACS analysis revealed that there was no significant difference in Annexin V‐positive cells between control and KO groups. The same results were observed between vector and SLC35F2‐OE groups (Figure S2). To summarize, these findings suggest that SLC35F2 promotes PTC cell growth through regulation of cell cycle progression rather than induction of cell apoptosis.
Figure 3.
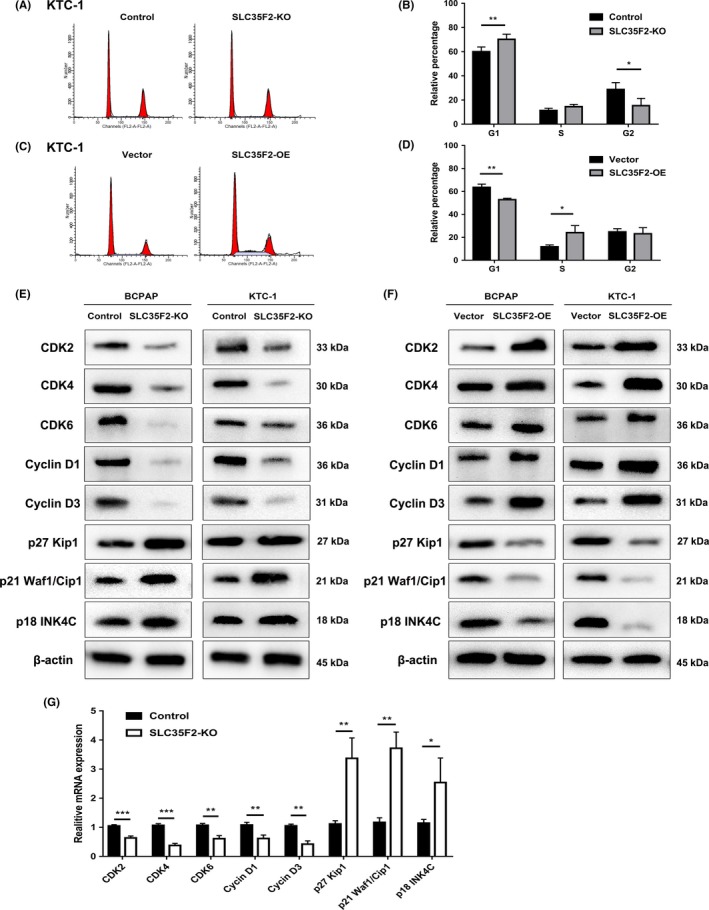
Cyclin‐dependent kinases and cyclins are involved in solute carrier family 35 member F2 (SLC35F2)‐regulated cell cycle progression. A, Effects of SLC35F2 knockout on cell cycle by FACS. B, Cell cycle distribution showed that inhibition of SLC35F2 induced cell cycle arrest at G1 phase. C, Flow cytometry analysis of the cell cycle of SLC35F2 overexpression and empty vector control cells. D, Ectopic expression of SLC35F2 promoted G1/S phase transition and increased S‐phase proportion. E, F, Protein levels of CDK2, CDK4, CDK6, Cyclin D1, Cyclin D3, p27 Kip1, p21 Waf1/Cip 1 and p18 INK4C were detected by western blotting in indicated PTC cells. G, Relative mRNA expression levels of some cell cycle related genes analyzed by qPCR in SLC35F2‐KO group compared to control KTC‐1 cells. Data were plotted relative to expression levels in control cells (*P < .05, **P < .01, ***P < .001)
3.4. Knockout of solute carrier family 35 member F2 inhibited papillary thyroid carcinoma cell migration and invasion in vitro
Tumor metastasis is an important cause of cancer‐related death. As SLC35F2 was closely associated with patients’ lymph node metastasis, we speculated that SLC35F2 could influence PTC cell motility. Transwell experiments with or without Matrigel were used to assess the effect of SLC35F2 on cell invasion and migration, respectively. Both BCPAP and KTC‐1 cells exhibited obvious weakened migration and invasion capabilities, as judged by fewer migrated and invaded cells in the SLC35F2‐KO group (Figure 4A‐D). Our loss‐of‐function experiments collectively suggest that SLC35F2 is required for both PTC cell migration and invasion. We also evaluated if the genetic knockout of SLC35F2 impacts colony‐formation ability. Colony formation assay was performed in a 35‐mm dish. The results revealed that silencing SLC35F2 not only drastically reduced the number of colonies formed by BCPAP and KTC‐1 cells, but decreased the colony size of cancer cells, supporting the idea that SLC35F2 influence PTC cell colony‐formation capacity (Figure 4E,F).
Figure 4.

Silencing of solute carrier family 35 member F2 (SLC35F2) represses the migration, invasion and colony formation abilities. A, Representative microscopic images of the migration assay in SLC35F2‐KO and control cells. B, Quantification of average migrated cells in control vs SLC35F2 knockout cells. Error bars indicate the mean ± SD based on 3 independent experiments. C, Transwell invasion assays performed using BCPAP or KTC‐1 cells showed decreased invasion capacities by the loss of SLC35F2. D, Numbers of average invaded cells in control vs SLC35F2 knockout cells were shown per test. Representative images (E) and quantification (F) of colonies in colony formation assay. Colonies over 100 cells were scored (**P < .01, ***P < .001)
3.5. Solute carrier family 35 member F2 promotes papillary thyroid carcinoma progression through activating transforming growth factor‐β type I receptor/apoptosis signal‐regulating kinase 1/mitogen‐activated protein kinase pathway
So far, the related downstream pathway for SLC35F2 has not been reported. Therefore, we aimed to identify the potential pathways by which SLC35F2 contributes to the malignant phenotype of PTC cells. We performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis on the GSE3678 dataset using DAVID Bioinformatics Resources 6.7.16 A list of genes that have strong co‐expression correlation (Pearson r value >.7 or < −.7) with SLC35F2 were extracted from this dataset (Table S1). GO enrichment analysis showed that these genes were tightly associated with cell proliferation and intracellular signaling cascade, supporting the role of SLC35F2 in PTC development (Figure 5A and Table S1). Meanwhile, KEGG enrichment analysis demonstrated that the MAPK pathway is among the most significantly altered pathways (Figure 5B and Table S1). Therefore, we assessed several critical cellular protein kinases in MAPK pathway. Interestingly, we discovered that suppression of SLC35F2 inhibited the phosphorylation of ERK1/2, p38 MAPK and JNK at protein levels. However, the total ERK1/2, p38 MAPK and JNK were almost unchanged (Figure 5C). The activity of phosphor‐p38 MAPK, phosphor‐JNK and phosphor‐Erk1/2 is directly implicated in the progression of PTC. The phosphorylation status of these proteins represents the activation of MAPK pathway, which is a key molecular signaling pathway involved in thyroid cancer. Similarly, overexpression of SLC35F2 increased protein levels of phosphorylated ERK1/2, p38 MAPK and JNK, and vice versa (Figure 5D). Meanwhile, we observed an increased p‐ERK, p‐p38 MAPK and p‐JNK expression in PTC specimens compared with their normal counterparts (Figure S3). To further clarify the mechanism by which SLC35F2 activates MAPK pathway, we noticed that transforming growth factor‐β type I receptor (TGFBR1) and apoptosis signal‐regulating kinase 1 (ASK‐1), core members of the MAPK pathway, were emerged in the co‐expression gene list (Table S1). According to the TCGA THCA dataset, the expression level of SLC35F2 was positively correlated with TGFBR1 (r = .7225, P < .0001, n = 513) and ASK‐1 (r = .7006, P < .0001, n = 513) (Figure 5e). TGFBR1, a receptor of for TGF‐β ligand, was less studied in PTC. We analyzed the expression of TGFBR1 in our cohort and found that TGFBR1 was upregulated in PTC samples. The clinical relevance between SLC35F2 and TGFBR1 was also investigated and results showed that TGFBR1 expression is strongly linked to SLC35F2 (r = .6840, P < .0001) (Figure 5f). We next tested whether SLC35F2 promotes PTC progression through TGFBR1 and ASK‐1. Western blotting results showed that knockout of SLC35F2 attenuated the protein expression of TGFBR1 and phosphorylated ASK‐1 (Thr845) whereas total ASK‐1 was unchanged, and opposite results were observed in SLC35F2‐OE group (Figure 5G,H). RT‐PCR analysis indicated that SLC35F2 also influenced the expression of TGFBR1 at transcriptional level (Figure S4). To further elucidate the role of TGFBR1 in PTC initiated by SLC35F2, SB431542 (10 μmol/L, Selleckchem), a specific inhibitor of TGFBR1 kinases, was used to inhibit the downstream targets, including ASK‐1, ERK1/2, JNK and p38 MAPK in KTC‐1 cells with SLC35F2 overexpression. We found that SB431542 abolished the ability of SLC35F2‐induced enhanced cell migration and invasion of KTC‐1 cells (Figure 6A,B). Furthermore, SB431542 impaired the effects of SLC35F2 overexpression on cell growth (Figure 6C). We found that elevated p‐ASK‐1, JNK and p38 MAPK phosphorylation can be attenuated by SB431542 in the SLC35F2‐OE group (Figure 6D). In summary, these results present convincing evidence to support our emerging view that SLC35F2 contributes to the progression of PTC through the TGFBR1‐mediated MAPK pathway activation.
Figure 5.
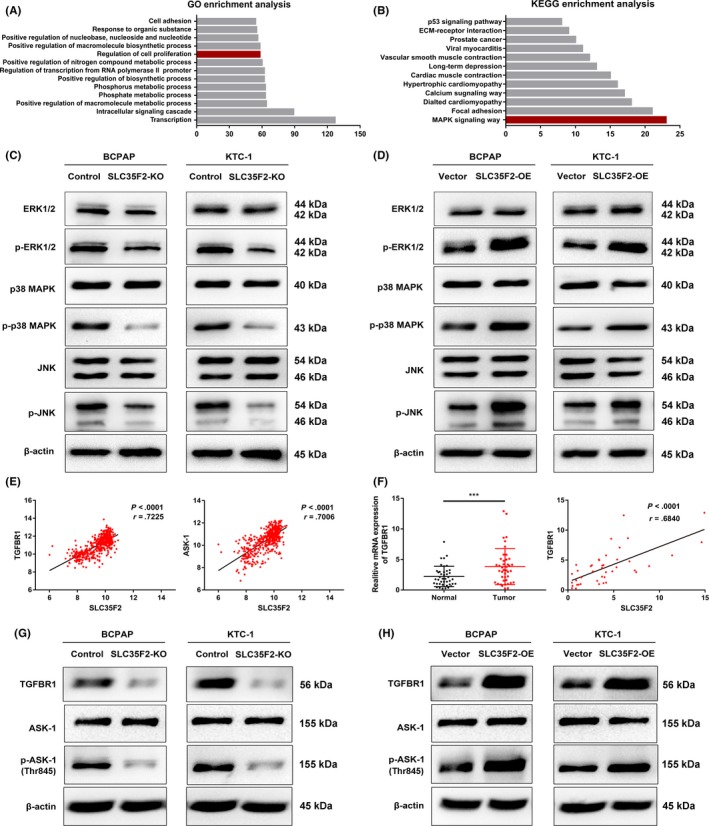
Solute carrier family 35 member F2 (SLC35F2) exerts tumor‐promoting efforts through TGFBR1 and ASK‐1 mediated activation of the MAPK pathway. Genes highly co‐expressed with SLC35F2 are enriched in the MAPK signaling pathway and regulation of cell proliferation using GO (A) and KEGG (B) enrichment analysis. Western blotting was performed to detect the protein expression levels of ERK1/2, p38MAPK, JNK and their phosphorylation forms after SLC35F2 knockout (C) or overexpression (D) compared to their corresponding control cells in BCPAP and KTC‐1 cells. E, Pearson correlation analysis revealed that both TGFBR1 and ASK‐1 were significantly correlated with SLC35F2 in human papillary thyroid carcinoma (PTC) tissues using the TCGA database (n = 513). F, Expression levels of TGFBR1 and clinical relevance between SLC35F2 and TGFBR1 in 42 PTC specimens and their corresponding adjacent normal tissues. (G, H) Inhibition or overexpression of SLC35F2 influences the protein levels of TGFBR1 and phosphorylation of apoptosis signal‐regulating kinase 1 (p‐ASK‐1) (Thr845), while ASK‐1 was not altered
Figure 6.
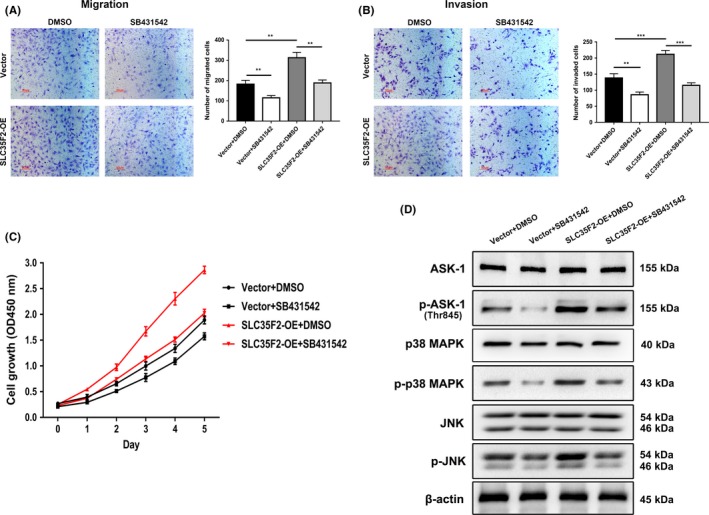
Specific TGFBR1 inhibitor attenuated tumor proliferation and migration induced by solute carrier family 35 member F2 (SLC35F2) overexpression. A, B, Migration and invasion assays were performed in a transwell chamber containing either DMSO or SB431542 in both vector and SLC35F2‐OE cells. SB431542 disrupted the ability of SLC35F2 overexpression to promote cell migration and invasion in KTC‐1 cells. C, SB431542 inhibits cell proliferation in SLC35F2‐OE KTC‐1 cells. D, Western blot was used to detect the level of ASK‐1, phosphorylation of apoptosis signal‐regulating kinase 1 (p‐ASK‐1), p38 MAPK, p‐p38 MAPK, JNK and p‐JNK in the presence or absence of SB431542 in vector and SLC35F2‐OE cells (**P < .01, ***P < .001)
3.6. Knockout of solute carrier family 35 member F2 ameliorates the growth potential of papillary thyroid carcinoma cells in vivo
Finally, to investigate the effects of SLC35F2 on regulation of thyroid cancer progression in vivo, we established xenograft tumor models using BCPAP cells. Equal amountS of BCPAP‐OE and BCPAP‐KO and their corresponding control cells were injected subcutaneously into nude mice. After observing for 36 days, implanted control BCPAP cells exhibited fast tumor formation and significant increases in tumor growth and weight compared to mice implanted with SLC35F2‐KO cells (Figure 7A‐C). In contrast, tumors formed by SLC35F2‐overexpressing cells were larger and heavier than the tumors formed by control cells (Figure 7E‐G). Immunohistochemistry results of SLC35F2‐overexpressing tumors displayed a higher positive rate of Ki‐67 and SLC35F2 (Figure 7H), whereas SLC35F2‐silenced tumors displayed a dramatic reduction in the protein expression of Ki‐67 and SLC35F2 compared to control cells (Figure 7D). These in vivo data are consistent with the above in vitro results, confirming the oncogenic role of SLC35F2 in PTC progression in vivo.
Figure 7.

Solute carrier family 35 member F2 (SLC35F2) contributes to papillary thyroid carcinoma (PTC) tumorigenesis in vivo. A, Representative pictures of tumors formed in nude mice bearing BCPAP cells in SLC35F2‐KO and control group. B, Tumor weights of xenograft tumors between 2 groups are significantly different. C, Tumor volumes were measured on the indicated days and the tumor growth curve is shown. D, Representative images of immunohistochemistry (IHC) and HE staining of SLC35F2 and Ki67 in xenograft tumors of 2 groups. E, Representative images of tumors from mice in SLC35F2‐OE and vector group. Statistical analysis of tumor weights (F) and volumes (G) in SLC35F2‐OE and vector group. H, Sections of xenograft tumor tissues stained with HE as well as IHC staining for SLC35F2 and Ki‐67 (**P < .01, ***P < .001)
4. DISCUSSION
It is generally assumed that approximately 10% of all human genes are transporter‐related, consistent with the biological significance of transporters, channels and pumps.17 A previous study has reported that over 456 SLC transporters in 52 subfamilies constitute the SLC superfamily.18 The SLC transporter family is one of the least studied groups of genes compared to other gene families of similar stature. Technical barriers such as structural biology characterization and non‐available high‐quality antibodies of most of SLC impeded research in this area. Defining their endogenous natural substrates is especially challenging.19 Genome‐wide association studies revealed that approximately 190 different SLC had been found mutated in human disease, including PTC. Solute carrier family 5A (SLC5A), the sodium iodide symporter (NIS), mediates the transport of iodine anion (I−) into thyroid follicular cells to facilitate thyroid hormone biosynthesis.20 Gene polymorphism of SLC5A conferred the risk for differentiated thyroid cancer.21 Hu et al found that the hypermethylation of SLC5A8 has been associated with advanced stage of tumors, multifocality and extrathyroidal invasion in PTC.22 Besides, methylation of SLC5A8 is associated with the BRAF V600E mutation, which is the most frequent genetic event during the development of PTC.23 In the current study, we present both in vitro and in vivo evidence clarifying the role of SLC35F2 in promoting PTC tumorigenesis from research on clinical samples and cell lines. We performed knockout of SLC35F2 by CRISPR/Cas9 in PTC cells and observed that silencing SLC35F2 significantly inhibited proliferation, abolished colony‐formation abilities and attenuated the invasion and metastasis of thyroid cancer cells. Notably, SLC35F2 may serve as an important predictor of lymph node metastasis for PTC patients. However, the underlying mechanism responsible for aberrant expression of SLC35F2 in PTC is still unknown. Epigenetic changes such as histone acetylation or methylation of its promoter might result in elevated expression of SLC35F2 and the mechanism remains to be further investigated.
Accumulating evidence reveals that sustaining proliferative signaling through the deregulated cell cycle is an essential step during carcinogenesis.24, 25 Cyclin‐dependent kinase (CDK)‐cyclin complexes have been described as the critical regulatory enzymes in the cell cycle.26 CDK4 and CDK6 are the main interphase CDK, activated by cyclin D1, D2 and D3, and form CDK4/6‐cyclin D complexes, orchestrating the advance of the cell cycle entry and progression through G1 phase.27 In contrast, CDK inhibitors (CKI), such as the cip/Kip family proteins, p21 Waf/Cip1 and p27 Kip1, bind to and inhibit the activity of the above CDK‐cyclin complexes, which are participated in the negative control of cell cycle progression.28 Numerous CDK inhibitors were expected to be an effective strategy because many tumorigenic events eventually drive proliferation by subverting CDK4 or CDK6 complexes in the G1 phase of the cell cycle.29, 30 Therefore, clarifying the mechanism concerning the dysregulation of cell cycle regulator will be of great importance. Sun et al previously demonstrated a new miR‐144/E2F8/Cyclin D1 regulatory axis controlling PTC development.31 Here, we proved that SLC35F2 could regulate cell cycle progression by accelerating G1/S phase transition without affecting apoptosis of PTC cells, suggesting that SLC35F2 may be a novel modulator of the cell cycle. Furthermore, gain‐of‐function and loss‐of‐function studies have demonstrated that SLC35F2 modulates multiple cyclin‐dependent kinases, including CDK2, CDK4, CDK6, Cyclin D1 and Cyclin D3, and represses p27 Kip1, p21 Waf1/Cip1 and p18 INK4C expression at both mRNA and protein level correspondingly. Strikingly, p21 Waf1/Cip1 was obviously upregulated upon SLC35F2 knockout and vice versa. Elevated p21 Waf1/Cip1 expression inhibited the activity of Cyclin D1 and D3/CDK 4 and 6 for G1/S transition, which possibly explained why the proliferation abilities of PTC were remarkably influenced during this process. We propose that these aberrant expression of cell cycle markers regulated by SLC35F2 may disrupt cell cycle control, in turn promote cell proliferation, and consequently facilitate the development of PTC.
Transforming growth factor‐β pathway is one of the most commonly altered cellular signaling pathways in human cancer, which plays a crucial role as both a tumor suppressor in early stage carcinogenesis and pro‐metastatic factor in advanced stages of tumors.32, 33 TGFβ isoforms exert their cellular effects by binding to the TGF‐β type II receptor (TGFBR2), and this binding facilitates activation of TGFBR1 kinase, which leads to the activation of Smad and non‐Smad pathway, thereby regulating the transcription of specific genes.34, 35 The non‐Smad pathway acts through the intracellular MAPK signaling cascades (ERK1/2, JNK1/2/3 and p38MAPK being the 3 major branches of the stress‐induced activation of the MAPK pathway).36 MAPK cellular signaling pathway, frequently activated in the thyroid carcinogenesis, transmits growth signals from the plasma membrane to the nucleus and plays a central part in promoting cancer cell proliferation and survival.37, 38, 39 Shen et al report that miRNA‐106a directly targeting RARB influence the viability, apoptosis, differentiation of PTC, and alter the iodine uptake function by regulating MAPK pathway.40 Here, we unravel for the first time that SLC35F2 exerts its oncogenic effect on PTC progression mainly through the MAPK pathway, with dependence on the induction and activation of TGFBR‐1 and ASK‐1. Knockout of SLC35F2 decreased TGFBR1 expression, leading to reduction of phosphorylated ASK‐1, ERK1/2, JNK and p38MAPK, further indicating that targeting TGFBR1 can nullify the pro‐tumor signals of MAPK pathway. Conversely, ectopic expression of SLC35F2 induced the opposite expression of these proteins. Moreover, inhibition of TGFBR1 can rescue p‐ASK‐1, p‐JNK and p‐p38 MAPK expression and malignant phenotypes, such as proliferation, migration and invasion in SLC35F2 overexpressing cells. ASK‐1, also known as MAP3K5, participates in stress‐induced apoptosis. Overexpression of ASK‐1 activates JNK and p38MAPK, and induces apoptosis through the mitochondrial cell death pathway.41 However, elevated p‐ASK‐1 (phosphorylation at Thr845) by SLC35F2 did not affect cell apoptosis. All these points raised the possibility that the elevated phosphorylated‐ASK‐1 provoked other downstream pathways, which compensated the effects of ASK‐1 triggered apoptosis. In addition, we first found that ASK‐1 is the downstream target of TGFBR1 for the reason that using the inhibitor targeting of TGFBR1 can abolish SLC35F2‐induced elevated p‐ASK‐1 expression. Taken together, these results support the hypothesis that activation of MAPK signaling is a critical downstream event responsible for the SLC35F2/TGFBR1/ASK‐1 signal axis in cell proliferation and metastasis of PTC.
Solute carrier family members are closely linked to carcinogenesis in different types of cancer. To the best of our knowledge, this is the first study to present a comprehensive set of clinical and experimental evidence establishing SLC35F2 as an oncogenic factor that facilitates PTC tumor progression and metastasis. In addition, the newly identified SLC35F2/TGFBR1/ASK‐1 axis not only provides novel insight into the cellular mechanisms that mediate MAPK pathway activation in human PTC cells, but also represents a novel, potential therapeutic target for the treatment of PTC.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
He J, Jin Y, Zhou M, et al. Solute carrier family 35 member F2 is indispensable for papillary thyroid carcinoma progression through activation of transforming growth factor‐β type I receptor/apoptosis signal‐regulating kinase 1/mitogen‐activated protein kinase signaling axis. Cancer Sci. 2018;109:642–655. https://doi.org/10.1111/cas.13478
Funding information
Science and Technology Commission of Shanghai Municipality (no. 15411952503); National Nature Science Foundation of China (no. 81602326).
Jing He and Yiting Jin contributed equally to this work.
Contributor Information
Xiuping Liu, Email: xpliu1228@fudan.edu.cn.
Qiang Zou, Email: zouqiang_hs@163.com.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Kitahara CM, Sosa JA. The changing incidence of thyroid cancer. Nat Rev Endocrinol. 2016;12:646‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115‐132. [DOI] [PubMed] [Google Scholar]
- 4. Cabanillas ME, McFadden DG, Durante C. Thyroid cancer. Lancet. 2016;388:2783‐2795. [DOI] [PubMed] [Google Scholar]
- 5. He L, Vasiliou K, Nebert DW. Analysis and update of the human solute carrier (SLC) gene superfamily. Hum Genomics. 2009;3:195‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu L, Li X, Cai M, et al. Increased expression of Solute carrier family 12 member 5 via gene amplification contributes to tumour progression and metastasis and associates with poor survival in colorectal cancer. Gut. 2016;65:635‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheng X, Wei L, Huang X, et al. Solute carrier family 39 member 6 gene promotes aggressiveness of esophageal carcinoma cells by increasing intracellular levels of zinc, activating phosphatidylinositol 3‐kinase signaling, and up‐regulating genes that regulate metastasis. Gastroenterology. 2017;152:1985‐1997. [DOI] [PubMed] [Google Scholar]
- 8. Ishida N, Kawakita M. Molecular physiology and pathology of the nucleotide sugar transporter family (SLC35). Pflugers Arch. 2004;447:768‐775. [DOI] [PubMed] [Google Scholar]
- 9. Stankovic T, Byrd PJ, Cooper PR, et al. Construction of a transcription map around the gene for ataxia telangiectasia: identification of at least four novel genes. Genomics. 1997;40:267‐276. [DOI] [PubMed] [Google Scholar]
- 10. Nishimura M, Suzuki S, Satoh T, Naito S. Tissue‐specific mRNA expression profiles of human solute carrier 35 transporters. Drug Metab Pharmacokinet. 2009;24:91‐99. [DOI] [PubMed] [Google Scholar]
- 11. Bu L, Jiang G, Yang F, Liu J, Wang J. Highly expressed SLC35F2 in non‐small cell lung cancer is associated with pathological staging. Mol Med Rep. 2011;4:1289‐1293. [DOI] [PubMed] [Google Scholar]
- 12. Nyquist MD, Corella A, Burns J, et al. Exploiting AR‐regulated drug transport to induce sensitivity to the survivin inhibitor YM155. Mol Cancer Res. 2017;15:521‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winter GE, Radic B, Mayor‐Ruiz C, et al. The solute carrier SLC35F2 enables YM155‐mediated DNA damage toxicity. Nat Chem Biol. 2014;10:768‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He J, Zhou M, Chen X, et al. Inhibition of SALL4 reduces tumorigenicity involving epithelial‐mesenchymal transition via Wnt/beta‐catenin pathway in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2016;35:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44‐57. [DOI] [PubMed] [Google Scholar]
- 17. Hediger MA, Clemencon B, Burrier RE, Bruford EA. The ABCs of membrane transporters in health and disease (SLC series): introduction. Mol Aspects Med. 2013;34:95‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015;14:29‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cesar‐Razquin A, Snijder B, Frappier‐Brinton T, et al. A call for systematic research on solute carriers. Cell. 2015;162:478‐487. [DOI] [PubMed] [Google Scholar]
- 20. Smanik PA, Liu Q, Furminger TL, et al. Cloning of the human sodium lodide symporter. Biochem Biophys Res Commun. 1996;226:339‐345. [DOI] [PubMed] [Google Scholar]
- 21. Al‐Rasheed MM, Alzahrani AS, Macadam A, Overall A, Gard P, Dzimiri N. The potential role of the sodium iodide symporter gene polymorphism in the development of differentiated thyroid cancer. Gene. 2015;572:163‐168. [DOI] [PubMed] [Google Scholar]
- 22. Hu S, Liu D, Tufano RP, et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J Cancer. 2006;119:2322‐2329. [DOI] [PubMed] [Google Scholar]
- 23. Porra V, Ferraro‐Peyret C, Durand C, et al. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations in classical papillary thyroid carcinomas. J Clin Endocrinol Metab. 2005;90:3028‐3035. [DOI] [PubMed] [Google Scholar]
- 24. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 25. Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220‐5227. [DOI] [PubMed] [Google Scholar]
- 26. Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140:3079‐3093. [DOI] [PubMed] [Google Scholar]
- 27. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153‐166. [DOI] [PubMed] [Google Scholar]
- 28. Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6:56‐64. [DOI] [PubMed] [Google Scholar]
- 29. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tamura K, Mukai H, Naito Y, et al. Phase I study of palbociclib, a cyclin‐dependent kinase 4/6 inhibitor, in Japanese patients. Cancer Sci. 2016;107:755‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun J, Shi R, Zhao S, et al. E2F8, a direct target of miR‐144, promotes papillary thyroid cancer progression via regulating cell cycle. J Exp Clin Cancer Res. 2017;36:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jiang X, Shan J, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 regulates angiogenesis in a transforming growth factor beta‐dependent manner in human osteosarcoma. Cancer Sci. 2015;106:1394‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Budi EH, Duan D, Derynck R. Transforming growth factor‐beta receptors and Smads: regulatory complexity and functional versatility. Trends Cell Biol. 2017;27:658‐672. [DOI] [PubMed] [Google Scholar]
- 35. Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415‐424. [DOI] [PubMed] [Google Scholar]
- 36. Zhang YE. Non‐Smad signaling pathways of the TGF‐beta family. Cold Spring Harb Perspect Biol. 2017;9:pii: a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zaballos MA, Santisteban P. Key signaling pathways in thyroid cancer. J Endocrinol. 2017;235:R43‐R61. [DOI] [PubMed] [Google Scholar]
- 38. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279‐3290. [DOI] [PubMed] [Google Scholar]
- 39. Kamiyama M, Naguro I, Ichijo H. In vivo gene manipulation reveals the impact of stress‐responsive MAPK pathways on tumor progression. Cancer Sci. 2015;106:785‐796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen CT, Qiu ZL, Song HJ, Wei WJ, Luo QY. miRNA‐106a directly targeting RARB associates with the expression of Na(+)/I(−) symporter in thyroid cancer by regulating MAPK signaling pathway. J Exp Clin Cancer Res. 2016;35:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hassan M, Feyen O, Grinstein E. Fas‐induced apoptosis of renal cell carcinoma is mediated by apoptosis signal‐regulating kinase 1 via mitochondrial damage‐dependent caspase‐8 activation. Cell Oncol. 2009;31:437‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials