

Abstract
Prostate cancer can progress from androgen dependence to androgen deprivation resistance with some unknown mechanisms. The current study aims to explore the possible role of pituitary tumor transforming gene1 (PTTG1) in castration‐resistant prostate cancer (CRPC). Initially, we found that PTTG1 expression was significantly increased in androgen‐independent prostate cancer cell lines PC3, DU145 and CRPC specimens compared with that in androgen‐dependent prostate cancer cell line LNCaP and initial prostate cancer specimens. PTTG1 overexpression significantly enhanced the cell survival rate, clonality and tumorigenicity in LNCaP cells upon androgen‐deprivation therapy (ADT). While knockdown of PTTG1 expression significantly elevated the sensitivity of DU145 cells to ADT. The effects of PTTG1 overexpression on LNCaP cells may be ascribed to the induced EMT and increased CD44+ CD24‐ cancer stem cell population. Furthermore, we detected that PTTG1 expression was regulated by interleukin‐6 via activated signal transducer and activator of transcription 3 (STAT3) directly binding to the region −500 to +1 of PTTG1 promoter in LNCaP cells. In conclusion, our results elucidate that interleukin‐6/STAT3 activation can increase PTTG1 expression and, consequently, promote the resistance to ADT in CRPC by inducing EMT and increasing the cancer stem cell population, suggesting that PTTG1 may be a novel therapeutic target for CRPC.
Keywords: cancer stem cell, castration‐resistant prostate cancer, epithelial‐to‐mesenchymal transition, interleukin‐6, pituitary tumor transforming gene 1
1. INTRODUCTION
Prostate cancer (PCa) is the second most common cancer worldwide, and the 6th leading cause of cancer‐related death (258 400 deaths annually).1 Although the PCa incidence in China was reported to be ranked 117th among all nations, it has substantially increased since 2010.2 Nowadays, androgen deprivation therapy (ADT) is the mainstay therapy for advanced prostate cancer in addition to surgery.3 Although ADT is initially effective in suppressing tumor growth and survival, many prostate cancer patients will undergo recurrence and progress on anti‐androgen therapy, reaching a lethal stage termed castration‐resistant prostate cancer (CRPC).4 There is an urgent need to explore the mechanisms of castration resistance, which will provide better intervention strategies for CRPC patients.
Pituitary tumor transforming 1 (PTTG1) is an oncogene that was originally cloned from rat pituitary tumor cells.5 PTTG1 overexpression can interfere with sister chromatid separation during mitosis, leading to chromosome aneuploidy.6 Therefore, PTTG1 is involved in the development and progression in many malignancies.7 In our previous studies, we demonstrated that PTTG1 expression was significantly increased in human prostate cancer metastasis tissue,8 and PTTG1 promoted the proliferation of prostate cancer cells by inhibiting SMAD3.9 Moreover, the PTTG1 mRNA expression was higher in androgen‐independent prostate cancer cell lines PC3 or DU145 than that in androgen‐dependent prostate cancer cell line LNCaP.10 These results indicate that PTTG1 may play an important role in the development of castration resistance in CRPC. However, the exact role and potential mechanisms underlying the involvement of PTTG1 in CRPC are poorly clarified.
A series of studies has demonstrated that development and progression of CRPC are closely related to interleukin‐6 (IL‐6). IL‐6 is abundantly expressed in androgen‐independent prostate cancer cell lines PC3 or DU145. In contrast, IL‐6 expression in androgen‐dependent prostate cancer cell line LNCaP is found to be negative.11 However, the gain of IL‐6 expression in LNCaP cells through lentiviral transfection can lead these cells to be resistant to ADT.12 In addition, IL‐6 levels in plasma and serum from patients with CRPC are significantly higher than those from patients with earlier stages of prostate cancer and healthy individuals.13 Furthermore, it has been postulated that IL‐6 can regulate the expression of androgen‐responsive genes in low androgen conditions and thereby contribute to the transition from hormone‐dependent to CRPC.14 As a novel downstream target gene of androgen receptor,8 we consider whether PTTG1 can be regulated by IL‐6 and is consequently involved in the development of castration resistance in CRPC. The current study aims to verify this hypothesis and further explore the potential mechanisms in IL‐6/PTTG1 modulated castration resistance in CRPC.
2. MATERIALS AND METHODS
2.1. Cell culture
Human LNCaP, PC3 and DU145 prostate cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and routinely tested to exclude mycoplasma contamination. All cells were maintained in RPMI 1640 supplemented with 10% FBS and 1% penicillin‐streptomycin (both were purchased from Gibco‐Life Technologies, Grand Island, NY, USA). All cells were cultured at 37°C in 5% CO2 and 95% air.
The plasmid containing the wild‐type full‐length sequence of PTTG1 and IL‐6, and shRNA targeted to human PTTG1 were produced by Genema (Shanghai, China). LNCaP and DU145 cells were transfected with recombinant lentiviruses and screened by puromycin. LNCaP cells expressing transgene PTTG1 or IL‐6 were named as LNCaP/PTTG1 or LNCaP/IL‐6, and LNCaP cells transfected with control recombinant lentiviruses were nominated as LNCaP/Control. DU145 cells treated with negative control shRNA or shRNA targeted to PTTG1 were named as DU145/ncPTTG1 or DU145/shPTTG1, respectively.
2.2. Clinical samples and immunohistochemical staining
Five prostate tissue specimens from CRPC patients and 5 prostate tissue specimens from patients with initial prostate cancer were collected in the Department of Urology, Second Affiliated Hospital, Third Military Medical University, China. After being fixed with formalin, all samples were embedded in paraffin and serially cut for IHC staining. The experiment was approved by the Institutional Review Board of the Third Military Medical University, China and was performed in accordance with the provisions of the Declaration of Helsinki.15 As previously described,16 IHC staining was performed to investigate PTTG1 expression in the 5 paired prostate tissue specimens. Then, immunoreactive score (IRS) was calculated to measure the protein expression levels of PTTG1, as previous described.16 Briefly, the IRS, which ranged from 0 to 12, was composited by the sub‐scores for the percentage of immunoreactive cells (0‐4) and immunoreactive intensity (0‐3). The percent positivity was scored as “0” (<1%), “1” (1%‐10%), “2” (11%‐50%), “3” (51%‐80%) and “4” (>80%). The staining intensity was scored as “0” (negative), “1” (weak), “2” (moderate) and “3” (strong). IRS was considered negative (0‐1), weakly positive (2‐4), moderately positive (6‐8) and strongly positive (9‐12). The sections were incubated with the PTTG1 primary antibody (dilution 1:200; Abcam).
2.3. Tumor xenografts
All animal studies were reviewed and approved by the Animal Care and Use Committee of the Second Affiliated Hospital and Third Military Medical University. Four‐week‐old male athymic nude mice were purchased from the Experimental Animals Center of the Third Military Medical University (Chongqing, China). Nude mice were surgically castrated 1 week prior to subcutaneous injection with 5 × 106 LNCaP/Control and LNCaP/PTTG1 cells mixed with Matrigel (1:1 [vol/vol]) (n = 15 in each group), or with 2 × 106 DU145/ncPTTG1 and DU145/shPTTG1 cells (n = 15 in each group). After injection, all nude mice also received intraperitoneal injections of bicalutamide (50 mg/kg body weight, 3 times/wk). Tumor volume was calculated every 2 weeks according to the following formula: TV = (Length) × (Width)2/2. The subcutaneous tumor‐bearing mice were killed at the 6th week after implantation and then tumor weights were measured.
2.4. Flow cytometric analysis
Following the established protocol,17 LNCaP cells were trypsinized and washed once with FACs buffer (PBS containing 1.5% BSA and 5 mM EDTA). 106 cells were incubated with 10 μL primary antibodies: anti‐CD24‐FITC and anti‐CD44‐PE (both were purchased from Abcam) in 100 μL buffer at 4°C for 15 minutes. Following incubation, cells were washed once with FACs buffer. For flow cytometric sorting, cells were resuspended in FACs buffer at 2 × 107 cells/mL and separated on either an Aria cell sorter (BD Biosciences, San Jose, CA, USA) or a MoFlo High Performance cell sorter (Dako Cytomation, Carpinteria, CA, USA).
2.5. Luciferase assays
For luciferase assays, PTTG1 promoters, −2000/+1, −1500/+1, −1000/+1 and −500/+1 were cloned into Pmir‐GLO‐P luciferase reporter vector (Promega, Madison, WI, USA), and were named as Pmir/−2000, Pmir/−1500, Pmir/−1000 and Pmir/−500. The following primers were used: forward 5′‐GAAGATCTACCTCAAAACCTCTGTAGATAGA‐3′, 5′‐GAAGATCTATATGGAATCAACTCCTCTCTCTC‐3′, 5′‐GAAGATCTAAGTATTGAGGATAGTCCTAAG‐3′, 5′‐CCGCTCGAGGCGGCGCACTCCTGGTTTAA‐3′ and reverse 5′‐GCGGCGCACTCCTGGTTTAA‐3′. The primers used for generating site mutated promoter plasmid were: forward 5′‐ACGTAAAAAACTAGGATCGTGTAAT‐3′ and reverse 5′‐TTTTTACGTGCCACAAAGTTTGCAAG‐3′ (deleting the cis‐regulatory element in plasmid Pmir/−500). The mutated plasmid was named Pmir/M−500.
Transfections were done using Lipofectamine 2000 Transfection reagents (Invitrogen) according to the manufacturer's instructions. Cells were planted into 24‐well plates, and were transfected with 0.5 μg PTTG1 promoter‐Pmir‐GLO‐P at 70%‐80% confluence. After 24 hours, whole‐cell lysate was collected for reporter detection by the Dual Luciferase Reporter System (Promega). Reactions were measured using an Orion Microplate Luminometer (Berthold Detection System, Tanja Vicentic, Germany). Transfections were performed in triplicate and repeated 3 times to assure reproducibility.
2.6. Statistical analysis
Data are presented as the mean ± SD. All experiments were performed with at least 3 independent replications. Statistical analyses were conducted using SPSS 13.0 software (SPSS, Chicago, IL, USA), and significant difference between each group was analyzed using unpaired 2‐tailed Student's t tests or one‐way ANOVA. Differences were considered to be statistically significant at P < .05.
2.7. Supplemental experimental procedures
Data S1 contains a detailed description of the western blot analysis, quantitative RT‐PCR, in vitro growth assays, clonogenic assays, tumor sphere formation assays and ChIP assays.
3. RESULTS
3.1. Pituitary tumor transforming gene 1 expression was increased in castration‐resistant prostate cancer specimens and androgen‐deprivation therapy‐resistant prostate cancer cells
We detected that PTTG1 mRNA and protein expression levels were significantly increased in PC3 and DU145 cells compared with that in LNCaP cells (Figure 1A‐C). Furthermore, using IHC staining, we examined the PTTG1 expression in 5 paired prostate tissue specimens from patients with CRPC or initial prostate cancer (initial PCa), whose details were listed in Table 1. There were no significant differences of age and Gleason score between patients with CRPC and initial PCa. Average time for ADT in patients with CRPC were 56.0 ± 23.6 months. Subsequently, we found that PTTG1 expression in prostate cancer tissues from CRPC patients was higher than that in prostate cancer tissues from initial PCa patients (Figure 1D,E).
Figure 1.

Pituitary tumor transforming gene1 (PTTG1) expression in prostate cancer cells and specimens. A‐C, PTTG1 protein and mRNA expressions were higher in PC3 and DU145 cells than that in LNCaP cells. D,E, Immunohistochemical analysis demonstrated that PTTG1 expression was increased in castration‐resistant prostate cancer (CRPC) patients compared with initial prostate cancer (PCa) patients. (Data are presented as mean ± SD, *P < .05.)
Table 1.
Details of clinical prostate cancer specimens
| Pca type | Age (y) | Gleason score | Time for ADT (mo) |
|---|---|---|---|
| Initial Pca | 70.8 ± 10.0 | 7.0 ± 1.0 | 0 |
| CRPC | 79.8 ± 2.6 | 8.2 ± 0.8 | 56.0 ± 23.6 |
ADT, androgen‐deprivation therapy; CRPC, castration‐resistant prostate cancer; PCa prostate cancer.
3.2. Pituitary tumor transforming gene 1 overexpression in LNCaP cells promoted the resistance to androgen‐deprivation therapy in vitro and in vivo
To investigate the role of PTTG1 in CRPC development, we first utilized recombinant lentiviruses transfection to reach PTTG1 overexpression in LNCaP cells. Successfully, we found that PTTG1 protein and mRNA expressions were overexpressed in LNCaP/PTTG1 cells compared with LNCaP/Control cells (Figure 2A‐C). As shown in Figure 2D, upon the increasing doses of bicalutamide (1‐5 μM) treatments, LNCaP/Control cells showed higher sensitivity to bicalutamide treatment than LNCaP/PTTG1 cells. 1 μM bicalutamide reduced the cell survival rate of LNCaP/Control cells by more than 45%, while it had little effect on the cell survival rate of LNCaP/PTTG1 cells. Even at a higher concentration of bicalutamide (5 μM), the cell survival rate reduction in LNCaP/PTTG1 cells was only approximately 30% compared with almost 65% reduction in LNCaP/Control cells.
Figure 2.

Pituitary tumor transforming gene1 (PTTG1) overexpression in LNCaP cells led to resistant androgen deprivation. A‐C, PTTG1 protein and mRNA expressions were overexpressed in LNCaP/PTTG1 cells compared with LNCaP/Control cells (Data are presented as mean ± SD, *P < .05 and ***P < .001). D, Upon the different doses of bicalutamide (1‐5 μM) treatments in medium containing complete FBS for 48 h, the cell survival rate in LNCaP/PTTG1 cells was significantly higher than that in LNCaP/Control cells (Data are presented as mean ± SD, **P < .01). E,F, Results in clonogenic assays demonstrated that LNCaP/PTTG1 cells formed higher numbers of colonies when treated with 5 μM bicalutamide and charcoal stripped FBS (CS‐FBS) compared with LNCaP/Control cells (Data are presented as mean ± SD, **P < .01). G, In castrated male nude mice treated with bicalutamide, LNCaP/PTTG1 cells exhibited stronger tumorigenicity than LNCaP/Control cells. H, Tumors were harvested at the 6th week, and tumor weights in LNCaP/PTTG1 group were significantly greater than that in LNCaP/Control group. (Data are presented as mean ± SD, *P < .05.) I, After cell injection, tumor volumes were measured every 2 weeks. Tumor volumes in LNCaP/PTTG1 group were significantly bigger than that in LNCaP/Control group at the 4th and 6th week. (Data are presented as mean ± SD, *P < .05.)
To further test the influence of PTTG1 overexpression on the LNCaP cells clonality upon the treatment with 5 μM bicalutamide or charcoal stripped FBS (CS‐FBS), clonogenic assays in vitro were performed. We found that compared with LNCaP/Control cells, LNCaP/PTTG1 cells formed higher numbers of colonies when treated with 5 μM bicalutamide or CS‐FBS (Figure 2E,F). Consistent with the in vitro studies, PTTG1 overexpression resulted in stronger tumorigenicity in the xenograft model. In castrated male nude mice treated with bicalutamide, LNCaP/PTTG1 cells exhibited higher tumor formation rate than LNCaP/Control cells (6/15 in LNCaP/PTTG1 vs 3/15 in LNCaP/Control) (Figure 2G). Tumor weight and volume in LNCaP/PTTG1 group were also significantly greater than that in LNCaP/Control group (Figure 2H,I).
3.3. Knockdown of pituitary tumor transforming gene 1 expression enhanced the sensitivity of DU145 cells to androgen‐deprivation therapy in vitro and in vivo
We further knocked down PTTG1 expression in DU145 cells to verify the functional role of PTTG1 in CRPC development. Three shRNA targeted to human PTTG1 were used. Compared with the negative control shRNA (ncPTTG1), all the 3 shRNA significantly reduced the PTTG1 mRNA expression in DU145 cells, and such reduction induced by the 3rd shRNA (shPTTG1‐3) was the most significant (Figure 3A). Western blot analysis demonstrated that shPTTG1‐3 also significantly decreased the PTTG1 protein expression in DU145 cells compared with ncPTTG1 (Figure 3B,C). Afterwards, we detected that the increasing doses of bicalutamide (1‐5 μM) had no significant effects on DU145/ncPTTG1 cells, but bicalutamide in concentration of 2.5 and 5 μM significantly decreased the cell survival rate of DU145/shPTTG1‐3 cells (Figure 3D). Furthermore, using clonogenic assays, we found that DU145/ncPTTG1 cells formed more colonies than DU145/shPTTG1‐3 cells upon the treatment with 5 μM bicalutamide or CS‐FBS (Figure 3E,F).
Figure 3.

Knockdown of pituitary tumor transforming gene1 (PTTG1) expression restored the sensitivity of DU145 cells to ADT. A, Three shRNA targeted to human PTTG1 were utilized to knock down PTTG1 expression. Compared with the negative control shRNA (ncPTTG1), all the 3 shRNA significantly decreased the PTTG1 mRNA expression in DU145 cells, and the decrease induced by the 3rd shRNA (shPTTG1‐3) was the most significant. (Data are presented as mean ± SD, ***P < .001.) B,C, shPTTG1‐3 also significantly reduced the PTTG1 protein expression in DU145 cells compared with ncPTTG1 (Data are presented as mean ± SD, ***P < .001.) D, DU145/ncPTTG1 cells showed obvious resistance to bicalutamide (1‐5 μM) treatment, while shPTTG1‐3 significantly restored the sensitivity of DU145 cells to bicalutamide (2.5 and 5 μM) treatment. (Data are presented as mean ± SD, **P < .01 and ***P < .001.) E,F, Furthermore, results in clonogenic assays demonstrated that the numbers of DU145/ncPTTG1 cells formed colony were significantly higher than that of DU145/shPTTG1‐3 cells upon the treatment with 5 μM bicalutamide or CS‐FBS. (Data are presented as mean ± SD, **P < .01.) G, DU145/shPTTG1‐3 cells manifested weaker tumorigenicity than DU145/ncPTTG1 cells in castrated male nude mice treated with bicalutamide. H, At the 6th week, the tumor weights in DU145/shPTTG1‐3 group were significantly lighter than that in DU145/ncPTTG1 group. (Data are presented as mean ± SD, *P < .05.) I, Tumor volumes in DU145/shPTTG1‐3 group were also significantly smaller than those in the DU145/ncPTTG1 group at the 4th and 6th week. (Data are presented as mean ± SD, **P < .01.)
Contrary to PTTG1 overexpression, PTTG1 expression knockdown led to weaker tumorigenicity in the castrated male nude mice treated with bicalutamide. Results showed that tumor formation rate of DU145/shPTTG1‐3 cells was significantly lower than that of DU145/ncPTTG1 cells (8/15 in DU145/shPTTG1‐3 vs 12/15 in DU145/ncPTTG1) (Figure 3G). Tumor weight and volume in DU145/ncPTTG1 group were also significantly greater than that in DU145/shPTTG1‐3 group (Figure 3H,I). These results in gain‐of‐function and loss‐of‐function studies of PTTG1 indicated the functional importance of PTTG1 in the development of ADT resistance.
3.4. Pituitary tumor transforming gene 1 overexpression induced prostate cancer epithelial‐mesenchymal transition and regulated the prostate cancer stem cell population
To explore exact role of PTTG1 in affecting ADT resistance, we first detected the effect of PTTG1 overexpression on epithelial‐mesenchymal transition (EMT). Western blot analysis showed that PTTG1 overexpression upregulated the expressions of N‐cadherin, Vimentin, Snail and downregulated the E‐cadherin expression (Figure 4A,B). Because of EMT is accompanied by the gain of stem cell traits in cancer cell populations,18 we next explored whether the stem cell properties of LNCaP cells were changed by PTTG1 overexpression. Firstly, we found that the protein expressions of cancer stem cell biomarkers, Sox2 and Nanog, were significantly increased by PTTG1 overexpression (Figure 4A,B). Then, we used tumor sphere formation assays to test the changes in sphere‐forming ability of LNCaP cells induced by PTTG1 overexpression. Notably, the sphere numbers in LNCaP/PTTG1 group were significantly increased compared with that in LNCaP/Control group (Figure 4C,D). This result suggested that PTTG1 may be required for self‐renewal of prostate cancer stem cells. Moreover, a previous study reported that prostate cancer stem cells are enriched in CD44+/CD24− cell population.17 Subsequently, using FACS analysis, we detected that PTTG1 overexpression caused an increase in the CD44+/CD24‐ cell population in LNCaP cells (Figure 4E,F). Taken together, these data suggested that PTTG1 plays a pivotal role in maintaining prostate cancer stem cell populations.
Figure 4.
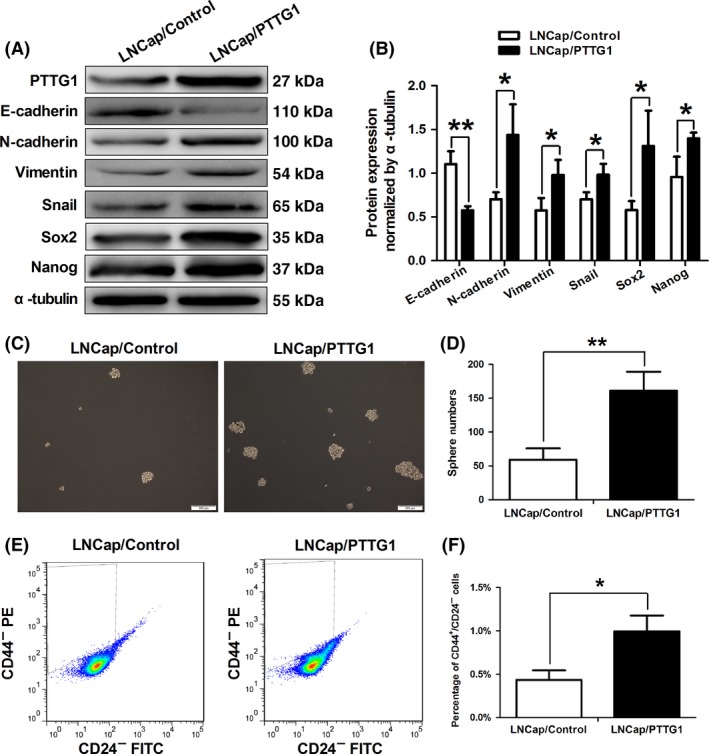
Pituitary tumor transforming gene1 (PTTG1) overexpression induced prostate cancer epithelial‐mesenchymal transition (EMT) and regulated the prostate cancer stem cell population. A,B, Western blotting demonstrated that PTTG1 overexpression resulted in down‐expression of E‐cadherin and up‐expression of N‐cadherin, vimentin, snail, Sox2 and Nanog, which were biomarkers of EMT and stem cells. (Data are presented as mean ± SD, *P < .05 and **P < .01.) C,D, Sphere formation assays were performed in LNCaP/PTTG1 and LNCaP/Control cells. LNCaP/PTTG1 cells formed greater sphere numbers than LNCaP/Control cells (Data are presented as mean ± SD, **P < .01.) E,F, Using fluorescence activated cell sorting (FACS), prostate cancer stem cell population was quantified. CD44+/CD24− cell number in LNCaP/PTTG1 group was significantly increased compared with that in the LNCaP/Control group (Data are presented as mean ± SD, *P < .05.)
3.5. Interleukin‐6 increased pituitary tumor transforming gene 1 expression via activating signal transducer and activator of transcription 3 in LNCaP cells
Furthermore, we attempted to verify whether PTTG1 is regulated by IL‐6 in prostate cancer and to clarify the potential mechanism. We first expressed IL‐6 in LNCaP cells which possess negative IL‐6 expression by lentiviral transfection (Figure 5A‐C). IL‐6 expression significantly increased the protein and mRNA expressions of PTTG1, which can be significantly attenuated by AG490 (a STAT3 blocker, 20 μM) (Figure 5A,B,D). These results suggested that IL‐6 may regulate PTTG1 expression via activating STAT3 in LNCaP cells.
Figure 5.
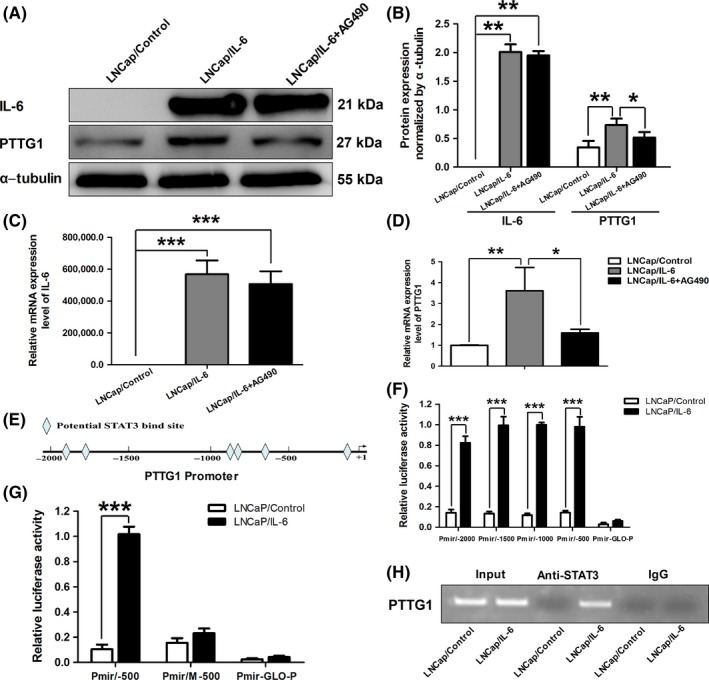
Interleukin‐6 (IL‐6) increased pituitary tumor transforming gene 1 (PTTG1) expression via activating signal transducer and activator of transcription 3 (STAT3) in LNCaP cells. A‐D, IL‐6 was expressed in LNCaP cells with lentiviral transfection. After IL‐6 expression, PTTG1 protein and mRNA expressions were significantly increased in LNCaP/IL‐6 cells. 20 μM AG490 significantly weakened the effect of IL‐6 on PTTG1 expression. (Data are presented as mean ± SD, *P < .05, **P < .01 and ***P < .001.) E, Schematic representation of potential STAT3 binding sites on PTTG1 promoter. F,G, Using luciferase reporter assays, the transcriptional activities of different PTTG1 promoter deletion constructs were quantified in LNCaP/Control and LNCaP/IL‐6 cells. (Data are presented as mean ± SD, ***P < .001.) H, Putative STAT3 binding site sequence of PTTG1 were immunoprecipitated by an anti‐STAT3 antibody in extracts from LNCaP/IL‐6 cells. No signal was detected in the control IgG lane while control PCR reactions show robust amplification of the target sequence in input DNA
In addition, to clarify the potential regulatory mechanism, we first analyzed regulatory regions of PTTG1 gene using a web‐based tool (Consite) for finding cis‐regulatory elements of STAT3 in genomic sequences. We found some putative cis‐regulatory elements of STAT3 binding site in the PTTG1 promoter region (Figure 5E). Then, plasmids Pmir/−2000, Pmir/−1500, Pmir/−1000 and Pmir/−500 were transfected into LNCaP cells in order to measure their transcriptional activities responding to IL‐6 expression. Luciferase assays demonstrated that all PTTG1 promoter fragments including −2000/+1, −1500/+1, −1000/+1 and −500/+1 exhibited strong luciferase activities responding to IL‐6 expression (Figure 5F). These data indicated that the STAT3 response element localized in the region of −500 to +1. Moreover, one putative STAT3 binding site in the promoter region of −500 to +1 was postulated using web‐based tools. To further confirm the responsibility of this putative STAT3 binding site for IL‐6 stimulated transcription, this cis‐regulatory element was deleted by a special primer. As expected, the mutated promoter construct did not respond to IL‐6 expression (Figure 5G). To further confirm that STAT3 mediates androgen‐induced PTTG1 transcription in vivo, we immunoprecipitated cross‐linked chromatin from LNCaP cells with a STAT3 primary antibody. By amplifying region of −500 to +1 in PTTG1 promoter which around STAT3 binding site sequence, the precipitated chromatin was analyzed. As shown in Figure 5H, the putative STAT3 binding site sequence of PTTG1 were immunoprecipitated by the anti‐STAT3 antibody in extracts from LNCaP/IL‐6 cells, indicating that endogenous STAT3 protein can bind to the PTTG1 promoter in the presence of IL‐6 in LNCaP cells. These results suggested that PTTG1 expression can be regulated by IL‐6 via the directly binding of activated STAT3 to PTTG1 promoter.
3.6. Signal transducer and activator of transcription 3 inhibition significantly resensitized the DU145 cells to bicalutamide treatment
Subsequently, we tried to explore whether STAT3 inhibition can resensitize the androgen‐independent cells to ADT. AG490 (20 μM) was used to block STAT3 in DU145 cells. We confirmed that DU145 cells were resistant to 5 μM bicalutamide treatment. However, the combined application with bicalutamide and AG490 significantly reduced the colony numbers of DU145 cells compared with that of DU145 cells treated with single bicalutamide or AG490 (Figure 6A,B). Consistently, we detected that DU145 cells received the combined application with bicalutamide and AG490 possessed lower cell survival rate than that treated with single bicalutamide or AG490 (Figure 6C).
Figure 6.
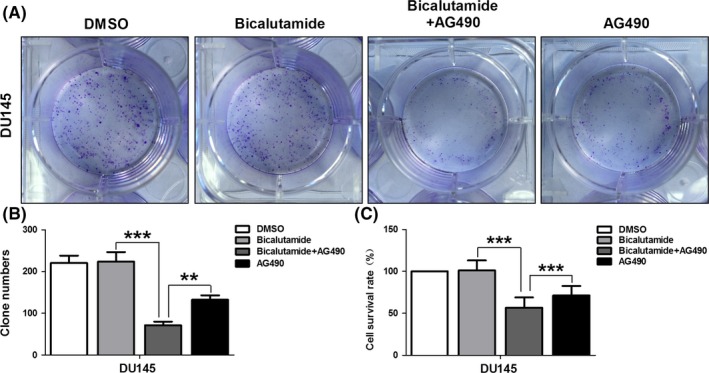
Signal transducer and activator of transcription 3 (STAT3) inhibitor, AG490, reversed bicalutamide resistance in DU145 cells. DU145 cells were treated with 5 μM bicalutamide combined with or without 20 μM AG490, clonogenic assays were performed and cell survival rate was calculated. As expected, we confirmed that DU145 cells were resistant to 5 μM bicalutamide treatment. However, the combined application with bicalutamide and AG490 significantly reduced the colony numbers (A and B) and cell survival rate (C) of DU145 cells compared with that of DU145 cells treated with single bicalutamide or AG490. (Data are presented as mean ± SD, **P < .01 and ***P < .001.)
4. DISCUSSION
In the present study, we found that PTTG1 expression was significantly increased in androgen‐independent prostate cancer cell lines PC3 or DU145 and CRPC specimens. PTTG1 overexpression in LNCaP cells and PTTG1 expression knockdown in DU145 cells significantly promoted and attenuated the resistance to ADT, respectively. Subsequently, we found that PTTG1 overexpression can induce EMT and increase cancer stem cell population in LNCaP cells. Furthermore, we demonstrated that PTTG1 expression can be regulated by IL‐6 via the directly binding of activated STAT3 to PTTG1 promoter in LNCaP cells. Moreover, STAT3 inhibition significantly resensitized the androgen‐independent DU145 cells to bicalutamide treatment. Taken together, these results indicate that PTTG1 can be regulated by IL‐6/STAT3 and participate in the development and progression of CRPC via inducing prostate cancer EMT and increasing the cancer stem cell population.
In breast cancer, PTTG1 expression in specimens from ER‐positive breast cancer patients who recurred after tamoxifen treatment is found to be significantly increased compared with specimens from patients who did not.19 Moreover, in prostate cancer, PTTG1 overexpression is an unfavorable prognostic factor for progression in advanced prostate cancer receiving hormone therapy.20 In the present study, we also found that PTTG1 expression in specimens from CRPC patients was significantly higher than that in specimens from initial PCa patients. Moreover, we reconfirmed the androgen‐independent prostate cancer cell lines PC3 and DU145 possess greater PTTG1 expression than androgen‐dependent prostate cancer cell line LNCaP. Based on these results, we speculate that PTTG1 may play a vital role in the development and progression of endocrine therapy resistance. Moreover, the subsequent gain‐of‐function and loss‐of‐function experiments of PTTG1 further confirmed the functional importance of PTTG1 in the development of ADT resistance.
Many mechanisms have been proposed to explain the CRPC development: hypersensitive pathways, promiscuous receptor, coactivators and corepressors, bypass pathway and prostate cancer stem cells.13 In recent years, a substantial body of evidence suggests that cancer stem cells (CSC) play an important role in the development of CRPC. CSC possess many phenotypic and functional properties associated with normal stem cells and is proposed to be responsible for tumor initiation, progression including castration resistance and metastasis.21 It has been reported that the occurrence and development of CRPC are ascribed to clonal expansion of a small percentage of androgen‐independent cells, which are considered to be the androgen‐independent epithelial stem cells.22 In addition, therapy strategy by combining wnt inhibitor with docetaxel to target CSC can significantly enhance the therapeutic efficacy of CRPC.23 Moreover, Piyush et al24 have successfully identified agents with specific toxicity for epithelial CSC using high‐throughput chemical screening. In the current study, we demonstrated that PTTG1 overexpression significantly enhanced the sphere‐forming ability and increased CSC population in LNCaP cells. Also, the expressions of Sox2 and Nanog which can regulate the gain of CSC phenotypes and properties in tumor cells22 were significantly increased due to PTTG1 overexpression. These data suggest that PTTG1 can affect the development of CRPC via improving CSC stemness. To our knowledge, EMT also plays a pivotal role in the development of CRPC.22 EMT is a developmental process that epithelial‐derived tumor cells reduced intercellular adhesion and acquired a migratory and invasive mesenchymal phenotype.25, 26 Therefore, it has been proved that induction of an EMT in human mammary epithelial cells results in the acquisition of epithelial stem‐cell properties.18 In our present study, we detected that after PTTG1 overexpression, the expression of E‐cadherin (epithelial marker) was downregulated and the expressions of N‐cadherin, Vimentin (mesenchymal markers) and Snail (transcriptional repressors of E‐cadherin) were upregulated, which demonstrated the induction of EMT. In a previous study, it had been shown that PTTG1 can induce EMT and promote expansion of cancer stem cell population in breast cancer.27 Taken together, we suggest that PTTG1 can regulate the CSC stemness via inducing EMT, and facilitate the development of CRPC.
Subsequently, we tried to clarify the potential regulatory mechanism underlying the involvement of PTTG1 in CRPC. We focused on a pleiotropic cytokine IL‐6 due to its important role in the progression of CRPC.14 As we know, the effect of IL‐6 on tumor cell proliferation and metastasis is mediated by STAT3 activation.28, 29 In our study, we found that IL‐6 increased PTTG1 expression via activating STAT3 in LNCaP cells. This result is consistent with that IL‐6 upregulates PTTG1 expression via activating STAT3 in colorectal cancer.30 Meanwhile, we detected that STAT3 binds to special cis‐acting element on the region −500 to +1 of PTTG1 promoter. However, in colorectal cancer, STAT3 binding site is located on the region −2642 to −1717 of PTTG1 promoter.30 These results suggest that STAT3 can regulate PTTG1 expression by binding to different site of PTTG1 promoter in various cancers. Moreover, in accordance with a previous report by others that AG490 reversed the enzalutamide resistance in LNCaP‐s17 Cells which is resistant to ADT,31 we also found that STAT3 inhibition by AG490 significantly resensitized the DU145 cells to bicalutamide treatment. In addition, overexpression of constitutively active STAT3 in LNCaP cells can induce resistance to ADT and knockdown of STAT3 expression can restore the sensitivity of LNCaP‐s17 cells to enzalutamide treatment.31, 32 These data reveal the crucial role of STAT3 in development of ADT resistance. Taken together, we speculate that IL‐6/STAT3 may critically modulate the development of CRPC via regulating PTTG1 expression. Furthermore, more recent studies have suggested that IL‐6/STAT3 activation is involved in EMT induction and stem‐cell properties acquisition in many cancers,33, 34, 35, 36 further proving that PTTG1 can be regulated by IL‐6/STAT3 and play its important role in CRPC via inducing EMT and improving CSC stemness.
Although, our findings illustrate a causative link between PTTG1 and EMT, as well as the gain of stem‐cell properties in prostate cancer cells, we can't conclude that whether this link is ascribe to direct protein interaction or indirect regulation by other factors. In breast cancer, PTTG1 can induce EMT via PI3K/AKT signaling.27 In ovarian epithelial cancer, such effects of PTTG1 are achieved by regulating TGF‐β.37 Therefore, more in‐depth studies are required to uncover the mechanism underlying the regulation of EMT and the gain of stem‐cell properties in prostate cancer cells by PTTG1.
In conclusion, our study has demonstrated that PTTG1 is overexpressed in androgen independent prostate cancer cell lines and CRPC specimens. Furthermore, we found that, as a response gene of IL‐6/STAT3, PTTG1 can promote the resistance to ADT via inducing EMT and improving prostate CSC stemness. These results suggest that PTTG1 plays a vital role in CRPC and that PTTG1 can be regarded as a novel therapeutic target for CRPC.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
Huang S, Liu Q, Liao Q, et al. Interleukin‐6/signal transducer and activator of transcription 3 promotes prostate cancer resistance to androgen deprivation therapy via regulating pituitary tumor transforming gene 1 expression. Cancer Sci. 2018;109:678–687. https://doi.org/10.1111/cas.13493
Funding information
National Natural Science Foundation of China No. 81470989 research project key projects of Second Affiliated Hospital, Third Military Medical University No. 2015YLC06.
Huang and Liu contributed equally to this work.
REFERENCES
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69‐90. [DOI] [PubMed] [Google Scholar]
- 2. Ren SC, Chen R, Sun YH. Prostate cancer research in China. Asian J Androl. 2013;15:350‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karantanos T, Thompson TC. GEMMs shine a light on resistance to androgen deprivation therapy for prostate cancer. Cancer Cell. 2013;24:11‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Patel LR, Barton MC. TRIM‐ing ligand dependence in castration‐resistant prostate cancer. Cancer Cell. 2016;29:776‐778. [DOI] [PubMed] [Google Scholar]
- 5. Pei L, Melmed S. Isolation and characterization of a pituitary tumor‐transforming gene (PTTG). Mol Endocrinol. 1997;11:433‐441. [DOI] [PubMed] [Google Scholar]
- 6. Jallepalli PV, Waizenegger IC, Bunz F, et al. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445‐457. [DOI] [PubMed] [Google Scholar]
- 7. Wondergem B, Zhang Z, Huang D, et al. Expression of the PTTG1 oncogene is associated with aggressive clear cell renal cell carcinoma. Cancer Res. 2012;72:4361‐4371. [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z, Jin B, Jin Y, et al. PTTG1, A novel androgen responsive gene is required for androgen‐induced prostate cancer cell growth and invasion. Exp Cell Res. 2017;350:1‐8. [DOI] [PubMed] [Google Scholar]
- 9. Huang S, Liao Q, Li L, Xin D. PTTG1 inhibits SMAD3 in prostate cancer cells to promote their proliferation. Tumour Biol. 2014;35:6265‐6270. [DOI] [PubMed] [Google Scholar]
- 10. Xin DQ, Zhu XH, Lai YQ, et al. Regulation of expression of pituitary tumor transforming gene 1 (PTTG1) by androgen in prostate cancer. Beijing Da Xue Xue Bao Yi Xue Ban. 2005;37:638‐640. [PubMed] [Google Scholar]
- 11. Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin‐6 regulation of prostate cancer cell growth. J Cell Biochem. 2005;95:497‐505. [DOI] [PubMed] [Google Scholar]
- 12. Feng S, Tang Q, Sun M, Chun JY, Evans CP, Gao AC. Interleukin‐6 increases prostate cancer cells resistance to bicalutamide via TIF2. Mol Cancer Ther. 2009;8:665‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Azevedo A, Cunha V, Teixeira AL, Medeiros R. IL‐6/IL‐6R as a potential key signaling pathway in prostate cancer development. World J Clin Oncol. 2011;2:384‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nguyen DP, Li J, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL‐6). BJU Int. 2014;113:986‐992. [DOI] [PubMed] [Google Scholar]
- 15. World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191‐2194. [DOI] [PubMed] [Google Scholar]
- 16. Ding X, Yang DR, Xia L, et al. Targeting TR4 nuclear receptor suppresses prostate cancer invasion via reduction of infiltrating macrophages with alteration of the TIMP‐1/MMP2/MMP9 signals. Mol Cancer. 2015;14:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer. 2008;98:756‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mani SA, Guo W, Liao MJ, et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghayad SE, Vendrell JA, Bieche I, et al. Identification of TACC1, NOV, and PTTG1 as new candidate genes associated with endocrine therapy resistance in breast cancer. J Mol Endocrinol. 2009;42:87‐103. [DOI] [PubMed] [Google Scholar]
- 20. Cao XL, Gao JP, Wang W, Xu Y, Shi HY, Zhang X. Expression of pituitary tumor transforming gene 1 is an independent factor of poor prognosis in localized or locally advanced prostate cancer cases receiving hormone therapy. Asian Pac J Cancer Prev. 2012;13:3083‐3088. [DOI] [PubMed] [Google Scholar]
- 21. Chen X, Rycaj K, Liu X, Tang DG. New insights into prostate cancer stem cells. Cell Cycle. 2013;12:579‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li P, Yang R, Gao WQ. Contributions of epithelial–mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer. 2014;13:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yun EJ, Zhou J, Lin CJ, et al. Targeting cancer stem cells in castration‐resistant prostate cancer. Clin Cancer Res. 2016;22:670‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high‐throughput screening. Cell. 2009;138:645‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sun Y, Wang BE, Leong KG, et al. Androgen deprivation causes epithelial‐mesenchymal transition in the prostate: implications for androgen‐deprivation therapy. Cancer Res. 2012;72:527‐536. [DOI] [PubMed] [Google Scholar]
- 26. Zhou D, Kannappan V, Chen X, et al. RBP2 induces stem‐like cancer cells by promoting EMT and is a prognostic marker for renal cell carcinoma. Exp Mol Med. 2016;48:e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoon CH, Kim MJ, Lee H, et al. PTTG1 oncogene promotes tumor malignancy via epithelial to mesenchymal transition and expansion of cancer stem cell population. J Biol Chem. 2012;287:19516‐19527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S, Wang N, Brodt P. Metastatic cells can escape the proapoptotic effects of TNF‐alpha through increased autocrine IL‐6/STAT3 signaling. Cancer Res. 2012;72:865‐875. [DOI] [PubMed] [Google Scholar]
- 29. Qu Y, Oyan AM, Liu R, et al. Generation of prostate tumor‐initiating cells is associated with elevation of reactive oxygen species and IL‐6/STAT3 signaling. Cancer Res. 2013;73:7090‐7100. [DOI] [PubMed] [Google Scholar]
- 30. Zhou C, Tong Y, Wawrowsky K, Melmed S. PTTG acts as a STAT3 target gene for colorectal cancer cell growth and motility. Oncogene. 2014;33:851‐861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu C, Zhu Y, Lou W, Cui Y, Evans CP, Gao AC. Inhibition of constitutively active Stat3 reverses enzalutamide resistance in LNCaP derivative prostate cancer cells. Prostate. 2014;74:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee SO, Lou W, Johnson CS, Trump DL, Gao AC. Interleukin‐6 protects LNCaP cells from apoptosis induced by androgen deprivation through the Stat3 pathway. Prostate. 2004;60:178‐186. [DOI] [PubMed] [Google Scholar]
- 33. Li L, Han R, Xiao H, et al. Metformin sensitizes EGFR‐TKI‐resistant human lung cancer cells in vitro and in vivo through inhibition of IL‐6 signaling and EMT reversal. Clin Cancer Res. 2014;20:2714‐2726. [DOI] [PubMed] [Google Scholar]
- 34. Liu H, Ren G, Wang T, et al. Aberrantly expressed Fra‐1 by IL‐6/STAT3 transactivation promotes colorectal cancer aggressiveness through epithelial‐mesenchymal transition. Carcinogenesis. 2015;36:459‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim SY, Kang JW, Song X, et al. Role of the IL‐6‐JAK1‐STAT3‐Oct‐4 pathway in the conversion of non‐stem cancer cells into cancer stem‐like cells. Cell Signal. 2013;25:961‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Albino D, Civenni G, Rossi S, Mitra A, Catapano CV, Carbone GM. The ETS factor ESE3/EHF represses IL‐6 preventing STAT3 activation and expansion of the prostate cancer stem‐like compartment. Oncotarget. 2016;7:76756‐76768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shah PP, Kakar SS. Pituitary tumor transforming gene induces epithelial to mesenchymal transition by regulation of Twist, Snail, Slug, and E‐cadherin. Cancer Lett. 2011;311:66‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials