

Abstract
Background
The efficacy of antibiotics against M. tuberculosis has been shown to be influenced by experimental media conditions. Investigations of M. tuberculosis growth in physiological conditions have described an environment that is different from common in vitro media. Thus, elucidating the interplay between available nutrient sources and antibiotic efficacy has clear medical relevance. While genome-scale reconstructions of M. tuberculosis have enabled the ability to interrogate media differences for the past 10 years, recent reconstructions have diverged from each other without standardization. A unified reconstruction of M. tuberculosis H37Rv would elucidate the impact of different nutrient conditions on antibiotic efficacy and provide new insights for therapeutic intervention.
Results
We present a new genome-scale model of M. tuberculosis H37Rv, named iEK1011, that unifies and updates previous M. tuberculosis H37Rv genome-scale reconstructions. We functionally assess iEK1011 against previous models and show that the model increases correct gene essentiality predictions on two different experimental datasets by 6% (53% to 60%) and 18% (60% to 71%), respectively. We compared simulations between in vitro and approximated in vivo media conditions to examine the predictive capabilities of iEK1011. The simulated differences recapitulated literature defined characteristics in the rewiring of TCA metabolism including succinate secretion, gluconeogenesis, and activation of both the glyoxylate shunt and the methylcitrate cycle. To assist efforts to elucidate mechanisms of antibiotic resistance development, we curated 16 metabolic genes related to antimicrobial resistance and approximated evolutionary drivers of resistance. Comparing simulations of these antibiotic resistance features between in vivo and in vitro media highlighted condition-dependent differences that may influence the efficacy of antibiotics.
Conclusions
iEK1011 provides a computational knowledge base for exploring the impact of different environmental conditions on the metabolic state of M. tuberculosis H37Rv. As more experimental data and knowledge of M. tuberculosis H37Rv become available, a unified and standardized M. tuberculosis model will prove to be a valuable resource to the research community studying the systems biology of M. tuberculosis.
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0557-y) contains supplementary material, which is available to authorized users.
Keywords: M. tuberculosis, Genome-scale reconstruction, Environmental condition, Antibiotic resistance
Background
The success of M. tuberculosis as a pathogen has been partially attributed to its unique metabolic capabilities. The metabolic network of M. tuberculosis has evolved to withstand and navigate the harsh environment imposed by the alveolar macrophage. Most bacteria cannot thrive in this hypoxic, acidic and nutrient-limited condition, yet it is in this harsh environment that M. tuberculosis encounters and evolves resistance to antibiotics. Elucidating the robust properties of metabolism that enable M. tuberculosis pathogenicity and drug resistance evolution has become a key area of research.
Recent studies have demonstrated that the choice of experimental media conditions plays an important role in understanding physiologically-relevant phenotypes of M. tuberculosis [1]. Commonly used experimental media conditions such as Middlebrook 7H9 are known to be much different from the physiological environment. For example, despite extensive research describing fatty acids as a key carbon source within the macrophage environment, most studies forgo the inclusion of fatty acids in the media, opting instead for glucose or glycerol [2]. Perhaps it is no surprise then that differences between the in vitro and in vivo environments have been shown to affect antibiotic screening results [3–11]. In particular, it has been shown that hypoxic or nutrient limited conditions alter the metabolism of M. tuberculosis to a nonreplicating, drug-resistant state [5–7]. Specific mechanism-changing effects between in vitro and in vivo conditions have been shown to occur for many antibiotics [9, 10].
While it is understood that differences in experimental media conditions lead to phenotypic variations, dissecting a mechanistic understanding of these different phenotypes remains challenging. Genome-scale models (GEMs) of metabolisms have emerged as powerful tools to computationally probe the effect of media composition on a cell’s phenotype [12]. For the past 10 years, GEMs have provided a mechanistic basis for exploring the metabolic capabilities of M. tuberculosis on the systems-level. GEMs of M. tuberculosis have helped interrogate a variety of biological phenomena, from understanding the transcriptional regulatory network [13] to elucidating metabolic interactions between M. tuberculosis and the alveolar macrophage [14].
While new M. tuberculosis H37Rv GEMs have enabled novel insights, they have been constructed from different base models resulting in divergent representations of the metabolic network. For example, gene-protein-reaction rules (GPRs) (i.e., the Boolean relationship between a gene, or set of genes, and the corresponding reaction(s)) differ within reactions shared amongst models (e.g., The GPR of Rv0904c differs between iOSDD and iSM810). In addition to variation in network topology, divergent GEMs have a variety of identifiers used for metabolite and reaction names, making them difficult to compare and build from (e.g., “R#” reaction identifier nomenclature used in most models built primarily off of GSMN-TB). While such differences may seem insignificant, the presence of multiple divergent M. tuberculosis H37Rv reconstructions hinders progress and may result in future wasted efforts [15].
Here, we present iEK1011, a new GEM of M. tuberculosis H37Rv that unifies, standardizes, and updates previous divergent GEMs of this model organism. We assess the performance of iEK1011 to that of previous GEMs through gene essentiality prediction on two different datasets. iEK1011 is further characterized by performing simulations that examine the model’s predictions in physiological conditions and interrogate differences between in vitro and in vivo media conditions. Finally, in order to provide a comprehensive platform for elucidating antibiotic resistance (AMR), we integrate knowledge derived from experimental literature into iEK1011.
Results
Workflow for updating, unifying, and standardizing previous reconstructions of M. tuberculosis
In order to ensure a comprehensive unification, we first gathered and compared available reconstructions of M. tuberculosis H37Rv. Since the first two M. tuberculosis H37Rv reconstructions released in 2007 [16, 17], a total of 9 reconstructions have been built (Table 1). Most models were largely based off of either iNJ661 [16] or GSMN-TB [17]. Specifically, out of the most recent M. tuberculosis reconstructions, sMtb [18], iSM810 [13] and gal2015 [19] were primarily built from GSMN-TB while iOSDD [20] was built from iNJ661.
Table 1.
Summary of existing genome-scale models of M. tuberculosis. iAB-AMØ-1410-Mt-661 has over 2000 genes because it combines an updated version of iNJ661 with a macrophage model
| Model | Year | Genes | Reactions | Metabolites | Reference |
|---|---|---|---|---|---|
| iNJ661 | 2007 | 661 | 1025 | 826 | [16] |
| GSMN-TB | 2007 | 726 | 856 | 645 | [17] |
| MMF-RmwBo | 2009 | 776 | 1108 | ??? | [56] |
| HQMTB | 2009 | 686 | 607 | 734 | [57] |
| iNJ661v | 2010 | 663 | 1049 | 838 | [58] |
| iAB-AMØ-1410-Mt-661 | 2010 | 2071 | 4489 | 3400 | [14] |
| MergedTBmodel | 2012 | 917 | 1400 | 1017 | [59] |
| GSMN-TB1.1 | 2013 | 759 | 876 | 667 | [60] |
| iOSDD890 | 2014 | 890 | 1152 | 961 | [20] |
| sMtb | 2014 | 915 | 1192 | 929 | [18] |
| gal2015 | 2015 | 760 | 965 | 754 | [19] |
| iSM810 | 2015 | 810 | 938 | 723 | [13] |
| iNJ661mu | 2016 | 672 | 1057 | 846 | [61] |
| iEK1011 | 2017 | 1011 | 1228 | 998 | This study |
The model provided by Garay et al. was given the name of gal2015 because it is unnamed in the original publication
Using a variety of both quantitative and qualitative criteria (e.g., standardized identifiers, gene essentiality predictions, mass balanced reactions; see Methods), iOSDD and sMtb were chosen as the base reconstructions for the unification process (Fig. 1a). The recently developed M. tuberculosis H37Rv BioCyc Database [21] provided an additional reconstruction resource to supplement the standardized draft model. The reconstruction process was performed following a clear workflow (Fig. 1a): the base models were mapped to standardized BiGG identifiers [22], joined into a draft model of shared reactions and unified by assessing model disagreements. The resulting unified draft model was then expanded through manual curation of new biochemical knowledge. Thus, the reconstruction process was iterative and involved constant re-evaluation of model content (see Methods).
Fig. 1.
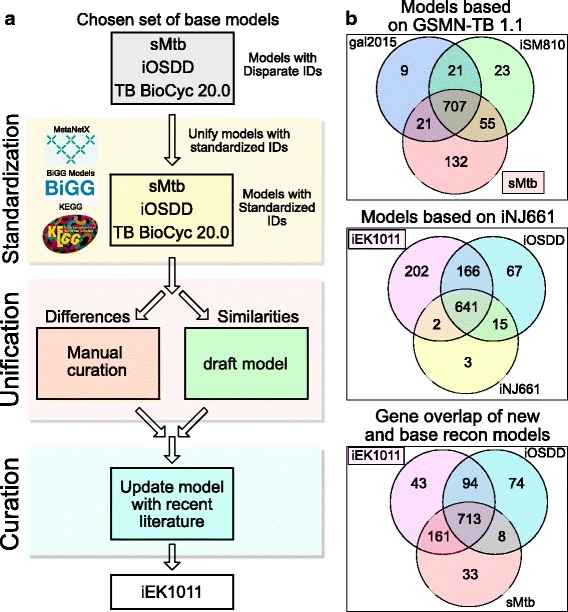
a Workflow of reconstruction process. A draft GEM model was built from the TB BioCyc 20.0 database and mapped to BIGGs IDs along with sMtb and iOSDD (see Additional file 2). The models were then unified by first joining the similarities between them, followed by manual curation of model differences literature and database validation. b Overlap of genes across different model sets. The model that covers most of the models within the particular set is enclosed by a box
The resulting unified and updated reconstruction of M. tuberculosis H37Rv, named iEK1011, contains 1011 genes, 1228 reactions, and 998 metabolites. iEK1011 encapsulates the majority of genes in the previous models based on either iNJ661 or GSMN-TB (Fig. 1b). iEK1011 accounts for 96% of sMtb genes (874 of 915 genes) and 91% of iOSDD genes (807 of 890). A total of 151 unique genes from iOSDD, iNJ661, gal2015, iSM810, and sMtb were not accounted for in iEK1011 (see Additional file 1) either due to insufficient evidence necessary to resolve major inconsistencies across models or lack of confidence in gene annotation.
In addition to unifying previous reconstructions, iEK1011 incorporated 39 new genes absent from previous models. In particular, sulfur metabolism was updated by adding the cysO-dependent biosynthesis of L-cysteine, which connects molybdenum metabolism with sulfur metabolism through the use of moeZ in both pathways [23]. New pathways and reactions include heme uptake [24], tuberculosinol biosynthesis [25], ergothioneine biosynthesis [26, 27], and mycobilin biosynthesis [28]. The resulting unified reconstruction of M. tuberculosis, iEK1011, provides a biochemically-derived knowledge-base that can be functionally assessed computationally.
Functional assessment of iEK1011
iEK1011 was converted to a mathematical model to examine the functional capabilities of the improved reconstruction and to quantitatively compare it with previous reconstructions. The primary tool for evaluating genome-scale reconstructions of M. tuberculosis H37Rv has been in silico gene essentiality testing. Therefore, we used gene essentiality as a metric for evaluating and comparing the performance of iEK1011. Gene essentiality predictions across previous M. tuberculosis H37Rv reconstructions were determined using the same data and quantitative score used in evaluating the predictive ability of iSM810 [13]. In addition to the commonly used gene essentiality dataset by Griffin et al. [29], a recent gene essentiality dataset by DeJesus et al. [30] was also utilized in our model comparisons. The primary differentiating feature between the datasets was the media condition used to generate them (see Additional file 1). Using these gene-essentiality datasets, we evaluated and compared the ability of five models (iNJ661, iOSDD, sMtb, iSM810, and iEK1011) to predict gene essentiality.
When using the Griffin dataset, we found that iEK1011 increases the prediction of true positives (i.e., the model correctly predicts growth for the gene knockout when the gene is annotated as non-essential) by 23% (579) (Fig. 2a) over sMtb (470), which had the largest number of true positives amongst the previous models. iEK1011 gene essentiality predictions decrease the number of false negatives (i.e., the model incorrectly predicts no growth for the gene knockout when the gene is annotated as non-essential) by 11.4% (31) (Fig. 2a) over iSM810 (35), which had the least number of false negatives amongst the previous models.
Fig. 2.
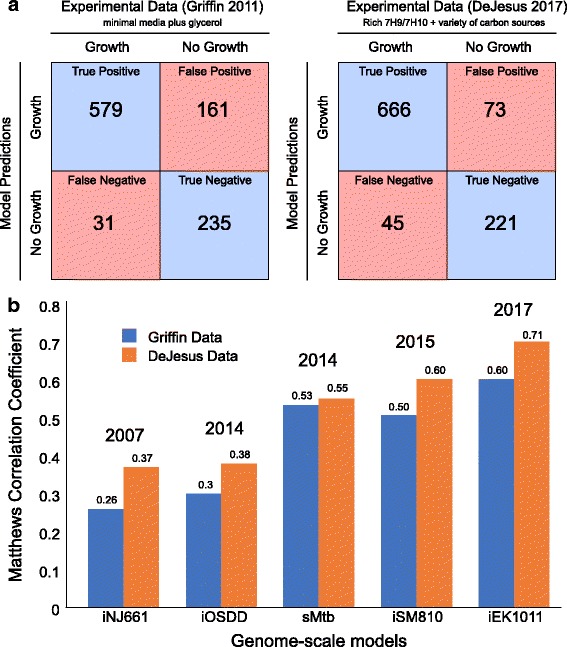
Gene Essentiality Prediction Comparisons. a Model-predicted gene essentiality results compared to both the Griffin et al. and deJesus et al. essentiality experimental datasets. b Gene essentiality performance using the Matthews Correlation Coefficient. iSM810 and sMtb, which were both built off of GSMN-TB 1.1, significantly outperform iNJ661 and iOSDD. iEK1011 outperforms all models on both gene essentiality datasets
With respect to the more recent DeJesus essentiality dataset, iEK1011 increases the number of true positives by 24% (666) (Fig. 2a) over sMtb (538), and iEK1011 increases the number of true negatives by 11% (221) (Fig. 2a) over sMtb (199). iEK1011 decreases the number of false positives by 14% (73) over sMtb (83), and increases the number of false negatives by one (45) over iSM810 (44). The increase in one more false positive over iSM810 is due to having 9 genes tested in the false negative category that are not contained in iSM810. Moreover, relating specific groups, such as false negatives or true positives, against other models with a different number of genes may not correctly represent the changes due to significant differences in class sizes.
In order to account for the variations in class sizes amongst models, we calculated the Matthews Correlation Coefficient (MCC) for each model’s prediction on both gene essentiality datasets (Fig. 2b). iEK1011 scores the highest on both datasets with an MCC of 0.60 and 0.71 on the Griffin and DeJesus dataset, respectively (Fig. 2b) (see Additional file 1). These iEK1011 MCC values are a 6% and 18% increase over the previous best model MCC’s of sMtb and iSM810 on the Griffin and DeJesus dataset, respectively.
Although the DeJesus essentiality dataset is more recent than the Griffin dataset by 6 years, the media condition used in determining essentiality on the DeJesus dataset was not as well defined because it utilized oleic-albumin-dextrose-catalase (OADC) in middlebrook 7H10/7H9 media supplemented with a variety of carbon sources [30]. The contents of OADC are not well defined primarily because of albumin, which may supplement amino acids to M. tuberculosis. The extent of OADC’s impact remains unknown, which ultimately hinders the ability to rigorously define the inputs for GEMs, which are crucial components of COBRA methods [31]. Conversely, the media used in Griffin was well defined as minimal media supplemented with glycerol [29]. Therefore, the increase in MCC by 6% over sMtb on the Griffin essentiality data should be evaluated with more confidence than the significantly higher percent increase in MCC over all models on the DeJesus dataset. Thus, the gene essentiality results presented above demonstrate improved predictive capability of iEK1011 over previous M. tuberculosis GEMs.
iEK1011 qualitatively recapitulates flux states indicative of physiologically relevant media conditions
While the gene essentiality predictions are a useful metric to evaluate model quality, we prioritized the model’s ability to recapitulate M. tuberculosis behavior described in the literature. Specifically, an emphasis was placed on central carbon metabolism given its distinctive usage in M. tuberculosis and recent emergence as an unexpected research frontier [32]. In addition, we focused on M. tuberculosis studies involving conditions relevant to pathogenicity [1]. Therefore, we compared simulations between two conditions relevant to the purpose of this study: Lowenstein-Jensen media, representing in vitro drug testing conditions; and an in vivo nutrient condition approximated from the literature that attempts to replicate the pathogenic state. We used Flux Variability Analysis (FVA) [33] and randomized sampling [34] to characterize and compare the fluxes between the two media conditions.
Taking advantage of recent studies investigating nitrogen metabolism within the context of M. tuberculosis pathogenicity [35–38], we set the in vivo nitrogen sources to be composed of nitrate, aspartate, asparagine, glutamate, urea, and glutamine (Fig. 3, see Additional file 1). Under hypoxic in vivo conditions, iEK1011 predicts use of nitrate in a respiratory role as opposed to a nitrogen source where it is taken in and reduced to nitrite by narG, and then exported out of the cell, a finding consistent with previous experiments [37]. The chosen in vivo carbon sources include fatty acids (both even and odd chain), cholesterol, CO2, and Alanine. Fatty acids were chosen as the primary source of carbon in vivo due to the vast amount of literature evidence supporting the claim that M. tuberculosis uses host-derived fatty acids [2, 39, 40]. iEK1011 catabolizes fatty acids through beta-oxidation, which generates acetate (even chain fatty acid catabolism), propionyl-CoA (odd chain fatty acid catabolism) and acetyl-CoA (Fig. 3). Although CO2 was incorporated due to evidence showing it being fixated by M. tuberculosis in an approximated in vivo environment [38], iEK1011 was not predicted to fixate CO2 due to a net gluconeogenic flux through phosphoenolpyruvate carboxykinase - a simulation result also found in Beste et al. [38]. Alanine was included as a nutrient due to evidence describing it to be in abundant quantities within the alveolar macrophage and being imported from the macrophage [38].
Fig. 3.
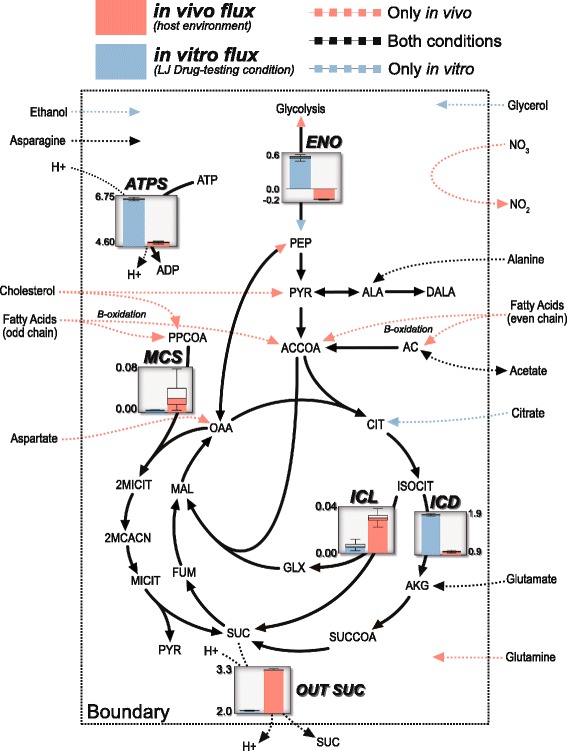
Metabolic map of flux differences through central carbon metabolism in iEK1011 between approximate in vitro and in vivo conditions. The media conditions are represented by nutrients outside of the dotted boundary line. Box plots graphically depict flux differences in the sampled solution spaces between in vivo and in vitro media conditions
The differences in flux state simulations predicted by iEK1011 between the two conditions recapitulate key behavior described in the literature. Specifically, in the approximated in vivo condition involving hypoxia and growth on fatty acids, model-predicted flux decreases through TCA with an accompanying increase in succinate secretion (Fig. 3). iEK1011 predicts the secretion of succinate to allow optimal growth in these conditions because it removes an intracellular proton, allowing for membrane potential related reactions such as oxidative phosphorylation to proceed. This mechanism has been previously described to be specific and essential in M. tuberculosis hypoxia adaptation [41]. Thus, iEK1011 can recapitulate known physiological phenomena using stoichiometry alone.
In addition to succinate secretion, iEK1011 simulates the activation of both the glyoxylate shunt and the methylcitrate cycle in response to both hypoxia and growth on fatty acids [41, 42]. Although the median flux values are low (Fig. 3) (based on markov chain monte carlo sampling of the solution space [34]), FVA simulations show maximum flux values through methylcitrate cycle and glyoxylate shunt to have a threefold and twofold increase in in vivo media conditions relative to in vitro conditions, respectively (see Additional file 1). Furthermore, the metabolic model does not account for the toxic effect of glyoxylate and propionate which has been shown to necessitate flux through glyoxylate shunt and methylcitrate cycle. While iEK1011 simulations do not account for characteristics like toxicity, the examples outlined above show that iEK1011 is capable of qualitatively recapitulating key phenomena uncovered in recent years.
iEK1011 as a computational knowledge base for interrogating features of antibiotic resistance
We have shown that iEK1011 is a valuable source of computational inquiry through gene essentiality predictions and its ability to recapitulate phenomena described in the literature. In addition to providing a computational platform, GEMs are fundamentally a knowledge-base that are capable of contextualizing a variety of concepts that extend beyond the genome-scale metabolic network [31]. Taking advantage of this ability to incorporate abstractions, we translate knowledge derived from experimental investigations of antibiotic resistance (AMR) evolution into a format that can be integrated into GEMs.
Using the extensive literature on the mechanism of AMR evolution in M. tuberculosis, we curated a relational table between antibiotics, genes, and metabolic reactions for eight different antibiotics (Table 2). The genes associated with a particular antibiotic are those known to be central to AMR evolution (i.e., mutations in the genes that code for the reactions often confer resistance to specific drugs). Displaying AMR genes on a metabolic map of iEK1011 portrays relationships that would be difficult to comprehend without a GEM (Fig. 4). Notably, we found that the close topological relationships between para-aminosalicylic acid, ethambutol, D-cycloserine, and pyrazinamide may hint at pleiotropic effects (i.e., mutations that affect multiple phenotypes) of resistance conferring mutations on the efficacy of different antibiotics.
Table 2.
Table of antibiotics and the associated genes whose mutations confer antibiotic resistance
| Drug | Gene | iEK1011 reaction | Reference |
|---|---|---|---|
| Ethambutol | embABC | EMB | [62] |
| ubiA | DCPT | [43] | |
| aftA | AFTA | [43] | |
| D-cycloserine | alr | ALAR | [63] |
| ddl | ALAALAR | [64] | |
| ald | ALAD_L, GXRA | [44] | |
| Isoniazid | katG | CAT | [65] |
| inhA | FAS | [66] | |
| fabG1 | MYCSacp56/58/50 | [67] | |
| Benzothiazinones | dprE1 | DCPE | [68] |
| PAS | thyA | TMDS | [51] |
| ribD | FOLR2, ASPRAUR, DHPPDA2 | [51] | |
| folC | DHFS, THFGLUS | [51] | |
| Pyrazinamide | pncA | NNAM | [69] |
| Ethionamide | mshC | CIGAMS | [52] |
| Rifampicin | drrABC | PDIMAT, PPDIMAT | [70] |
Fig. 4.
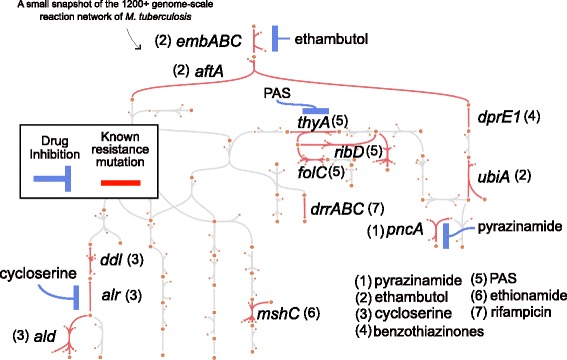
Escher map of arabinogalactan-peptidoglycan complex biosynthesis with known resistance-conferring genes mapped. The gene-antibiotic relation is indicated by the number placed proximal to the gene. The mechanistic effect by the antibiotic is indicated by the blue line. No blue line is shown for mutations in which the gene-antibiotic relation remains unclear (i.g., mshC, drrBC), Escher-usable maps were built for multiple subsystems in iEK1011 (see Additional file 4)
In order to incorporate specific antibiotic pressures into iEK1011, we evaluated each antibiotic and associated a biochemical objective function that approximates the evolutionary drivers of selection (Table 3). In the case of ethambutol, it has been shown that flux-increasing mutations in ubiA confer resistance by increasing the production of decaprenylphosphoryl-b-D-arabinose (DPA), which outcompetes ethambutol for embB binding spots [43]. Therefore, in a GEM, the evolutionary pressure imposed by ethambutol can be approximated as a metabolic objective where the production of DPA is maximized (Table 3). A total of four antibiotics were associated with approximated objective functions representing evolutionary forces (see Methods for further reasoning of the choice of objective function).
Table 3.
List of objective functions related to the evolutionary drivers of antibiotic resistance
| Drug | Objective | Reaction in iEK1011 | Reference |
|---|---|---|---|
| Ethambutol | MAX DPA production | decda__tb_c → □ | [43] |
| PAS | MAX Tetrahydrofolate production | thf_c → □ | [51] |
| D-cycloserine | MAX L-Alanine Production | ala__L_c → □ | [44] |
| Ethionamide | MIN mycothiol production | msh_c → □ | [52] |
The abbreviations are as follows: PAS para-aminosalicylic acid, MAX maximize, MIN minimize
Taking advantage of the translation of antibiotic features to formats amenable by iEK1011, we simulated the evolutionary pressures induced by antibiotics and calculated the maximum and minimum fluxes for the AMR-associated reactions in both in vivo and in vitro conditions through FVA (see Additional file 1).
There were few differences in relative flux for a specific drug objective between these conditions. However, those that were uncovered highlighted potential impacts of environmental/media composition differences. In particular, we see major differences in the fluxes that correspond to optimizing the approximated ethambutol-induced evolutionary pressure (see Fig. 5). Furthermore, this ethambutol flux is correlated with fluxes induced by the approximated d-cycloserine objective. Closer inspection of the uptake differences driving these differential flux states points to L-alanine as a key environmental influence. In particular, the differential fluxes within the cases of ethambutol resistance-conferring genes ubiA (DCPT) and embB (EMB), as well as d-cycloserine resistance-conferring genes of alr (ALAR), ald (ALAD_L), and ddlA (ALAALAr), exemplify the differential effect of environmental L-alanine availability. Notably, L-alanine has been shown to be an important substrate in the macrophage environment (Beste 2013). While L-alanine and other amino acids may be available in LJ drug-testing media due to utilization of egg base or bovine serum, our analysis only accounted for metabolites that were explicitly stated in defined quantities within the media conditions. With respect to the efficacy of antibiotics, these results suggest that d-cycloserine and ethambutol may be less effective in vivo due to increased availability of L-alanine, which is a key precursor reaction catalyzed by AMR genes targeted by d-cycloserine and ethambutol, whereas in vitro conditions may increase susceptibility to ethambutol. In both cases, the significant decrease in model-predicted maximum ALAD_L (ald) flux is in line with studies describing the deleterious mutations in ald that confer resistance to D-cycloserine [44]. Altogether, iEK1011 provides a knowledge base for relating antibiotic resistance features through genome-scale metabolic network analysis.
Fig. 5.
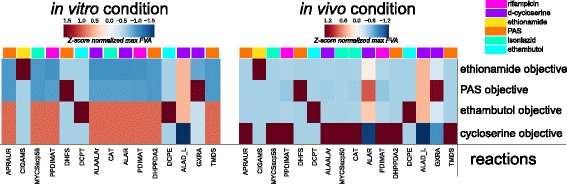
Heatmaps of maximum FVA values for for a matrix representing FVA values for the curated AMR reactions across simulations of different drug-specific objective functions (see Table 2 for curated list of AMR genes and their associated iEK1011 reactions, see Table 3 for drug-specific objectives)
Discussion
The divergence of M. tuberculosis H37Rv reconstructions has created an unnecessary obstacle in contextualizing the increasing growth of biochemical data for this troublesome pathogen. In order to address experimental insights to pathogenic conditions and alleviate roadblocks for future reconstruction efforts of M. tuberculosis H37Rv, we built a unified and updated GEM of M. tuberculosis, iEK1011. We tested the predictive potential of iEK1011 by comparing gene essentiality predictions with previous models and showed that iEK1011 outperforms previous models. We further assessed the predictive capabilities of iEK1011 by comparing simulated flux states between in vitro drug testing and approximate in vivo media conditions. Comparisons recapitulate specific phenomena indicative of biochemical flux states seen in physiological conditions. We incorporated antibiotic resistance knowledge in iEK1011, which enabled a network-based perspective of multi-antibiotic resistance evolution.
iEK1011 unified previous M. tuberculosis H37Rv reconstructions and encompassed the majority of genes within the two divergent groups of reconstructions. Additionally, iEK1011 incorporates new pathways including the incorporation of ergothioneine biosynthesis. This addition will aid a quantitative elucidation of the relationships between sulfur metabolism, bioenergetic homeostasis, and redox balance [27]. As a unified, standardized, and updated model, iEK1011 provides a base for future models of M. tuberculosis H37Rv.
Functional assessment of previous M. tuberculosis H37Rv reconstructions through gene essentiality predictions showed that iEK1011 achieves a higher MCC than previous models on two different datasets. While the two datasets were crucial in both assessing and driving iEK1011 reconstruction, experimental gene essentiality datasets derived from physiologically-relevant conditions are warranted for understanding the human-restricted lifestyle of M. tuberculosis.
Using iEK1011, we qualitatively determined differences in biochemical states between in vitro and approximated in vivo conditions. We showed that iEK1011 successfully recapitulates specific phenomena described in physiologically-relevant studies of M. tuberculosis. Future reconstruction efforts may target iEK1011’s lack of predicted CO2 fixation [38] and account for compartmentalized co-metabolism of multiple substrates [45, 46]. iEK1011 may provide a base for future host-pathogen integrated reconstructions that leverage valuable experimental data.
An integrated knowledge-base of genome-scale metabolism and antibiotic resistance components may enable new perspectives for understanding and combating M. tuberculosis H37Rv. We translated experimental knowledge of AMR genes and specific adaptation mechanisms to formats amenable to iEK1011. Comparing simulations of these AMR features between in vitro and in vivo conditions emphasized the potential impact of hypoxia and L-alanine availability on the pressures induced by antibiotics. Future constraint-based analysis of M. tuberculosis AMR may leverage new experimental approaches, such as those that have analyzed changes in essentiality under antibiotic exposure [47].
Taken together, iEK1011 is a new, comprehensive and predictive constraint-based model of M. tuberculosis H37Rv. In this study, we computationally demonstrate that in vivo nutrient sources absent from in vitro media significantly alter the flux state of central carbon metabolism. As experimental insights to M. tuberculosis pathogenicity and antibiotic resistance continue to grow, this GEM will provide a foundation to connect disparate data types and knowledge.
Methods
Reconstruction of iEK1011
Choosing a base reconstruction
A variety of both quantitative and qualitative criteria was considered in determining which model would provide the base for the new model. The determining criterion included the amount of curated data, extent of previous model unification, gene essentiality predictions, standardized identifiers, cross-references to databases, and quality of physical representations, such as the use of an extracellular compartment and mass balanced reactions. Based on this criterion, the reconstruction of iEK1011 was based on the unification of iOSDD, sMtb, and portions of the M. tuberculosis H37Rv BioCyc Database [21] (Fig. 1a).
With regards to the selection criterion, sMtb was chosen as the primary base model. Notably, sMtb performed the best amongst the previous models in gene essentiality predictions (Fig. 2b). In addition, sMtb included metabolite formulas, an extracellular compartment, and cross-references to databases. Both iSM810 and gal2015, which were both built off of GSMN-TB 1.1, lacked standardized identifiers (i.e., reactions identifiers were arbitrarily named R1, R2, etc.), metabolite formulas, and an extracellular compartment (i.e. inputs into the model could be utilized without being transported across the membrane). The lack of chemical formulas disables the assessment of mass conservation, which is a defining feature of constraint-based modeling. Furthermore, an extracellular compartment is key in distinguishing between what goes into the media and what is being transported across the membrane. While iOSDD performed well in categories related to component descriptions, it was based on iNJ661 and thus had a lower gene essentiality score, as previously shown [13]. Despite the low gene essentiality score, we utilized iOSDD as a representative for the models based on iNJ661. In this study, we show through gene essentiality predictions that the integration of iOSDD with sMtb results in a 6% increase in gene essentiality predictions.
The reconstruction process was straightforward (Fig. 1a). The base models were first algorithmically mapped to standardized BiGG identifiers (see Additional file 2) [22]. Identifiers that could not be mapped by the algorithm were manually assigned identifiers that follow the BiGGs format. Importantly, BiGGs was chosen as the standardization platform due to being a centralized repository for high-quality models. Once a standardized basis for identifiers was established, a draft reconstruction was built from the set of reactions shared across the standardized models. The differences between reactions across the models were manually assessed through literature references and added to the draft reconstruction. Once the models were unified into the draft reconstruction, manual curation of new biochemical knowledge was incorporated in the reconstruction. The reconstruction process was iterative and involved constant re-evaluation of model components. iEK1011 is provided at the BiGG website [22] initialized with m7H10 media as well as in JSON format with five different media initializations media as well as in JSON format with five different media initializations (see Additional file 3).
Updating the reconstruction
The model was updated with newly characterized metabolic processes, standardized identifications, and mass balanced reactions. In addition, detailed and designable metabolic maps of M. tuberculosis metabolism were manually built and provided in the supplement (see Additional file 4) in order to help in silico simulation and reconstruction efforts as well as provide access to systems biology research for non-computational biologists. Specifics on using the escher maps are described in the section titled “Escher Flux Maps”.
Before any updating took place, sMtb identifiers for metabolites and reactions were mapped to standardized identifiers in the BiGG Models database [22]. In addition to sMtb, the BioCyc M. tuberculosis H37Rv version 20.0 database was approximately converted to a cobra model - standardizing it first to BiGGs IDs, then MetaNetx, and then BioCyc identifiers if no BiGGs or Metanetx reference mapping was available. When an sMtb component had no equivalent BiGGs identification, a new identifier was created that followed the BiGG’s nomenclature. The updated reconstruction utilizes data from Tuberculist, 2016 TB BioCyc database, and manually curated literature sources. New pathways and major GPR updates include Tuberculosinol biosynthesis, oxidized GTP and dGTP detoxification, Heme uptake and degradation, GlgE pathway update, glucosylglycerate biosynthesis I, included essential genes Rv3805c and Rv2673 in MAP complex biosynthesis, and others (see Additional file 1). In addition to incorporating updates from the new BioCyc database, we re-curated pathways that had inconsistencies across divergent models.
Description of GAM and NGAM parameters
Our model includes both growth-associated (GAM) and non-growth (NGAM) associated ATP maintenance parameters. NGAM quantifies the energy required by Mtb to maintain itself in a given environment while GAM quantifies growth energy requirements not accounted for in the metabolic model. For iEK1011, the GAM was chosen to be 60 mmol gDw− 1, which was the same as the GAM used in previous M. tuberculosis H37Rv reconstructions of iNJ661, iAB-AMØ-1410-Mt-661, and iOSDD. For comparison, the GAM used for sMtb—a model built from the GSMN-TB line of reconstructions—was 57 mmol gDw− 1. For the NGAM, iEK1011 uses a value of 3.15 mmol gDw− 1 h− 1, which was taken from the E. coli model [48]. For comparison, the NGAM used in sMtb was set to 0.1 mmol gDw− 1 h− 1, while the NGAM used in iSM810 was 1 mmol gDw− 1 h− 1. We are not aware of any datasets available for M. tuberculosis that enables a rigorous evaluation of the NGAM parameter, such as those used for E. coli [49] (i.e., quantitative substrate uptake rates for different substrates).
In order to assess the sensitivity of our chosen NGAM, we recomputed the gene essentiality using an NGAM value of 1.0 and 0.01. With respect to our previous NGAM of 3.15, the NGAMs of 1.0 and 0.1 result in very similar values (see Additional file 5: Table S1). We hope that future experimental efforts will enable a better parameterization of GAM and NGAM in genome-scale reconstructions of M. tuberculosis.
Future directions
Inconsistencies between fatty acid metabolic pathways amongst models
There is a major difference in fatty acid metabolism between the previous models. ACP metabolites, holo enzymes, FAS metabolites, and other metabolites within the fatty acid metabolic pathways make relating and joining these pathways very challenging. iEK1011 primarily uses the form of the fatty acid pathways described in iOSDD, which significantly departs from the GSMN-TB based models and sMtb. We computationally profiled switches between forms taken in iOSDD and those in sMtb and found minor differences in simulations. The key feature maintained across divergent models was that the odd and even chain fatty acids described in the media went into the methylcitrate cycle and TCA, respectively.
In order to help future efforts in coming up with a consensus fatty acid metabolic pathway for a M. tuberculosis reconstruction, we have additionally included models named “sMtb_mapped_model.json” and “biocyc_sMtb_joined.json” (see Additional file 4) that have the fatty acid pathways of sMtb and BioCyc mapped to BiGGs identifiers.
Lumped reactions that violate mass conservation
We have tabulated out the lumped reactions in iEK1011 that violate mass conservation (Additional file 5: Table S2). These reactions were taken from previous models and are lumped in order to join parallel pathways. Although these reactions are kept in iEK1011, we hope that future reconstructions of M. tuberculosis H37Rv will work towards having all reactions mass balanced. Despite these disadvantages, we believe that iEK1011 is a step in the correct direction and allows for convergent and additive knowledge.
Blocked reactions in iEK1011
Out of the 1228 reactions in iEK1011, 157 are blocked (i.e., reactions that can’t carry flux even with all exchange reactions open). We have provided a list of these 157 blocked reactions (see Additional file 1). The subsystem with the highest frequency of blocked reactions is “Cofactor and Prosthetic Group Biosynthesis” (34 reactions), followed by “Exchange” (21 reactions) and “Transport” (10 reactions). Thus, future efforts should target the blocked reactions in the subsystem of “Cofactor and Prosthetic Group Biosynthesis” in order to optimize the reconstruction process.
Gene essentiality predictions
Gene essentiality predictions were determined using the same data and quantitative score used in evaluating the predictive ability of iSM810 [13]. The gene essentiality dataset was acquired from Griffin et al. [29]. If the Griffin essentiality confidence score was less than 0.1, the gene was determined to be essential. A growth cutoff of 20% of optimal growth was chosen to determine whether the in silico knockout was essential or not (i.e. if it was less than 20% of optimal growth, the gene was determined to be essential).
In addition to the Griffin gene essentiality dataset, we also evaluated the performance of the models in using a recent gene essentiality dataset acquired from DeJesus et al. [30]. A cutoff of 20% was used for the DeJesus dataset for the gene annotations of GD (growth defect), ES (essential), and ESD (essential domain). If growth was above 20% of optimal growth, the gene was said to be NE (non-essential) and GA (growth advantage). The Matthews Correlation Coefficient was used to score the quality of each model’s prediction, given by the following equation:
where TP (True Positive) represents the event where the model correctly simulates growth when a gene is nonessential. TN (True Negative) represents the event where the model correctly simulates no-growth when a gene is essential. FP (False Positive) represents the event where the model simulates no growth with the gene knockout when the gene is in fact non-essential. FN (False Negative) represents the event where the model simulates growth when the gene is in fact essential. While the Griffin et al. essentiality dataset is older, we utilized it due to having a more defined media conditions and was previous used in previous M. tuberculosis H37Rv reconstruction studies. The default objective function (“biomass”) was used across all models. Differences in MCC values between this study and that in Ma et al. [13] are due to differences in growth cutoff thresholds and media conditions. Despite inconsistencies, the values remained similar and did not change the resulting 6% increase in gene essentiality by iEK1011. Specifics regarding which genes are falsely predicted in all models, which pathways iEK1011 fails, which pathways iEK1011 performs correct predictions are provided (see iEK1011_supplementary.xlsx sheets titled “All models incorrect”, “iEK Wrong and sMtb Correct”, and “iEK Correct and sMtb Wrong”).
We further verified that the increase in accuracy for the iEK model is due to model curation as opposed to the addition of new non-essential genes by comparing only the gene essentiality predictions of the genes shared across all models. In particular, only 472 genes are shared across all the models. Using this set of genes for all model gene essentiality predictions, we find that iEK1011 still increases accuracy over other models (see Additional file 5: Table S4). Specifically, iEK1011 achieves 57% MCC on the Griffin dataset while sMtb achieves 50% MCC. On the de Jesus dataset, iEK1011 achieves 66% MCC while iSM810 achieves 55%. In the first case, iEK1011 has + 8 TP, + 9 TN, − 9 FP, and − 8 FN over sMtb. In the second case, iEK1011 has + 2 TP, + 25 TN, − 25 FP, and − 2 FN over iSM810. These results show that the increase in accuracy is due to model curation. This analysis has been appended to the provided ipython notebook “reproducible_modeling.ipynb”, and the results are attached in the supplementary excel file “iEK1011_supplementary.xlsx” under the sheets labeled “Shared essentiality predictions the Griffin et al. dataset” and “Shared essentiality predictions the deJesus dataset”, under sheets 9 and 10, respectively.
Flux variability analysis and sampling of in vitro and in vivo conditions
All constraint-based simulations of iEK1011 were done using the python constraint-based modeling package, COBRApy [50]. While the linear program is guaranteed to find the global optimum, the flux state solution to this optimization problem may not be unique, leading to the alternate optimal flux states. To account for this, we ran Flux Variability Analysis (FVA) in both the Lowenstein-Jensen media and approximated in vivo conditions using the “biomass” objective function (see Additional file 1). FVA gives the maximum and minimum amount of flux a reaction can take on. In addition to FVA, we sampled the solution space of iEK1011 on both media conditions using markov-chain monte-carlo sampling (MCMC) package available in cobrapy.
Furthermore, for both FVA and MCMC sampling, we allowed for solutions within 95% of the optimal value. The growth rate for both simulations were approximately the same to allow for a meaningful quantitative flux value comparisons.
Approximation of literature-derived evolutionary forces of antibiotic-resistance evolution
A more in depth reasoning for the choice of objective function is described below for each antibiotic.
Ethambutol
It has been shown that flux-increasing mutations in ubiA confer resistance to ethambutol by increasing the production of decaprenylphosphoryl-b-D-arabinose (DPA), which outcompetes ethambutol for embB bindings spots [43]. Therefore, we approximate the evolutionary force of adaptation as maximizing the production of DPA.
D-cycloserine
Analogous to the mechanism of ethambutol, it has been shown that loss-of-function mutations in ald confer resistance to the d-cycloserine by increasing the pool of Alanine (i.e. ald no longer converts Alanine to pyruvate), thereby competitively inhibiting d-cycloserine [44].
Para-aminosalicylic acid (PAS)
Mutations in folC, ribD, and thyA have been shown to confer resistance to PAS [51]. It was suggested that thyA mutations are selected in order to decrease the utilization of folates. In addition, it was suggested that the up-regulation of ribD occurs as an alternative when dfrA is inhibited.
Ethionamide
It has been shown that mycothiol biosynthesis is essential for ethionamide susceptibility [52]. We approximate ethionamide resistance is minimizing the production of mycothiol. It is worth noting that the objective defined for ethionamide is a much looser approximation than the other objectives defined before.
Comparison of FVA across different drug objective simulations
For both in vivo and in vitro media conditions, we simulated each of the drug objectives and compared the maximum and minimum fluxes of the reactions catalyzed by the curated antibiotic resistance genes (see Table 3 main text). The maximum and minimum fluxes for each reaction were determined by FVA (described above) allowing for solutions within 95% of optimum. Furthermore, iEK1011 was constrained to produce at least 20% of biomass growth (i.e., the lower bound of the “biomass” reaction was set to frac*max_biomass_growth, where max_biomass_growth is the optimum value of iEK1011 when maximizing biomass on either in vivo or in vitro conditions), and frac is the percentage of biomass to maintain while optimizing the other objective functions. All simulations can be performed using the provided IPython notebook (see Additional file 6).
Escher flux maps
In order to help future in silico simulations and reconstruction efforts of M. tuberculosis, we have provided detailed, manually constructed escher maps of central carbon metabolism, nitrogen metabolism, sulfur metabolism, and arabinogalactan-peptidoglycan complex biosynthesis [53] (see Additional file 4). Escher maps are designable metabolic maps that allow for the integration of different data types with a genome-scale metabolic model. Furthermore, the provided Escher maps will hopefully enable non-computational biologists to explore the complex network of M. tuberculosis in an intuitive manner; simply download the json map file(s), go to http://escher.github.io/, and load in the json map. If the model is loaded in as well, you will be able to modify reactions on the map or build your own. In addition, you can view the flux states generated by iEK1011 on the Escher map.
Additional files
Sheet 1: Reactions in iEK1011 and details of reactions. Sheet 2: Metabolites in iEK1011 and details of metabolites. Sheet 3: Genes present in previous models but not accounted for in iEK1011. Sheet 4: Gene Essentiality Predictions with the Griffin et al. dataset. Sheet 5: Gene Essentiality Predictions with the deJesus dataset. Sheet 6: Description of media conditions used in study. Sheet 7: Maximum FVA values for AMR genes across different objectives on in vivo media. Sheet 8: Maximum FVA values for AMR genes across different objectives on in vitro media. Sheet 9: “Shared” Essentiality Predictions with the Griffin et al. dataset. Sheet 10: “Shared” Essentiality Predictions with the deJesus et al. dataset. Sheet 11: List of blocked reactions in iEK1011. Sheet 12: “All models incorrect” - List of genes that were incorrectly predicted across all models and their corresponding iEK1011 subsystems. Sheet 13: “iEK Wrong and sMtb Correct” - List of incorrect iEK1011 predictions that were correctly predicted by sMtb. Sheet 13: “iEK Correct and sMtb Wrong” - List of correct iEK1011 predictions that were incorrectly predicted by sMtb. (XLSX 258 kb)
A conversion of the sMtb model to BiGGs identifiers (JSON 476 kb)
iEK1011 models: Genome-scale models of iEK1011 in json format initialized with different media conditions. iEK1011_griffinEssen_media.json - Used for essentiality testing on Griffin et al. dataset. iEK1011_deJesusEssen_media.json - Used for essentiality testing on deJesus et al. dataset. iEK1011_m7H10_media.json - iEK1011 initialized with Middlebrook 7H10 media. iEK1011_drugTesting_media.json - Used for simulating on Lowenstein-Jensen media. iEK1011_inVivo_media.json - Used for simulating on approximated physiological media. (ZIP 310 kb)
Escher Maps: Contains four escher maps of M. tuberculosis metabolic subsystems. Central_carbon.json. Arabinogalactan_peptidoglycan_complex.json. Nitrogen.json. Sulfur_and_folate.json (ZIP 236 kb)
Table S1. Table describing changes in gene essentiality predictions according to changes in GAM and NGAM values that were utilized across different genome-scale reconstructions of M. tuberculosis .Table S2. List of reactions in iEK1011 that violate the law of mass conservation. Table S3. Examples of false negatives computed by iEK1011 on the DeJesus et al. gene essentiality dataset that are not within the iSM810 model, and reasoning for its inclusion [54, 55]. Table S4. Gene essentiality predictions using the shared set of 472 genes. (DOCX 17 kb)
An ipython notebook that runs the simulations described in this study. (IPYNB 637 kb)
Funding
This research was supported by the NIH NIAID grant (1-UO1-AI124316–01).
Abbreviations
- AMR
Antimicrobial resistance
- DPA
Decaprenylphosphoryl-b-D-arabinose
- FVA
Flux variability analysis
- MCC
Matthews correlation coefficient
- PAS
Para-aminosalicylic acid
Authors’ contributions
EK reconstructed the network, performed the analysis, and drafted the manuscript. BOP and JM conceived the study and revised the manuscript. YS, JTY, CN, SP, SG, and BG revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0557-y) contains supplementary material, which is available to authorized users.
Contributor Information
Erol S. Kavvas, Email: ekavvas@eng.ucsd.edu
Yara Seif, Email: yseif@eng.ucsd.edu.
James T. Yurkovich, Email: jyurkovich@ucsd.edu
Charles Norsigian, Email: cnorsigi@eng.ucsd.edu.
Saugat Poudel, Email: sapoudel@ucsd.edu.
William W. Greenwald, Email: billgreenwald@ucsd.edu
Sankha Ghatak, Email: sankhaghatak@gmail.com.
Bernhard O. Palsson, Phone: +18585345668, Email: palsson@ucsd.edu
Jonathan M. Monk, Phone: +18585345668, Email: jmonk@ucsd.edu
References
- 1.Cumming BM, Steyn AJC. Metabolic plasticity of central carbon metabolism protects mycobacteria. Proc Natl Acad Sci U S A. 2015;112:13135–13136. doi: 10.1073/pnas.1518171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee W, VanderVen BC, Fahey RJ, Russell DG. Intracellular mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J Biol Chem. 2013;288:6788–6800. doi: 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, et al. Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med. 2014;92:139–149. doi: 10.1007/s00109-013-1100-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell DG, Barry CE, Flynn JL. Tuberculosis: what we don’t know can, and does, hurt us. Science. 2010;328:852–856. doi: 10.1126/science.1184784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci U S A. 2013;110:E2510–E2517. doi: 10.1073/pnas.1309171110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062–2069. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wayne LG, Sohaskey CD. Nonreplicating persistence of mycobacterium tuberculosis 1. Ann Rev Microbiol. 2001;55:139–163. doi: 10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- 8.Mitchison DA, Coates ARM. Predictive in vitro models of the sterilizing activity of anti-tuberculosis drugs. Curr Pharm Des. 2004;10:3285–3295. doi: 10.2174/1381612043383269. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Mitchison D. The curious characteristics of pyrazinamide: a review. Int J Tuberc Lung Dis. 2003;7:6–21. [PubMed] [Google Scholar]
- 10.Prosser GA, de Carvalho LPS. Metabolomics reveal d-alanine:d-alanine ligase as the target of d-Cycloserine in mycobacterium tuberculosis. ACS Med Chem Lett. 2013;4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, et al. A chemical genetic screen in mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat Commun. 2010;1:57. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Brien EJ, Monk JM, Palsson BO. Using genome-scale models to predict biological capabilities. Cell. 2015;161:971–987. doi: 10.1016/j.cell.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma S, Minch KJ, Rustad TR, Hobbs S, Zhou S-L, Sherman DR, et al. Integrated modeling of gene regulatory and metabolic networks in mycobacterium tuberculosis. PLoS Comput Biol. 2015;11:e1004543. doi: 10.1371/journal.pcbi.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bordbar A, Lewis NE, Schellenberger J, Palsson BØ, Jamshidi N. Insight into human alveolar macrophage and M tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. doi: 10.1038/msb.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monk J, Nogales J, Palsson BO. Optimizing genome-scale network reconstructions. Nat Biotechnol. 2014;32:447–452. doi: 10.1038/nbt.2870. [DOI] [PubMed] [Google Scholar]
- 16.Jamshidi N, Palsson BØ. Investigating the metabolic capabilities of mycobacterium tuberculosis H37Rv using the in silico strain iNJ661 and proposing alternative drug targets. BMC Syst Biol. 2007;1:26. doi: 10.1186/1752-0509-1-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beste DJV, Hooper T, Stewart G, Bonde B, Avignone-Rossa C, Bushell ME, et al. GSMN-TB: a web-based genome-scale network model of mycobacterium tuberculosis metabolism. Genome Biol. 2007;8:R89. doi: 10.1186/gb-2007-8-5-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rienksma RA. Suarez-Diez M, spina L, Schaap PJ, martins dos Santos VAP. Systems-level modeling of mycobacterial metabolism for the identification of new (multi-)drug targets. Semin. Immunol. 2014;26:610–622. doi: 10.1016/j.smim.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Garay CD, Dreyfuss JM, Galagan JE. Metabolic modeling predicts metabolite changes in mycobacterium tuberculosis. BMC Syst Biol. 2015;9:57. doi: 10.1186/s12918-015-0206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vashisht R, Bhat AG, Kushwaha S, Bhardwaj A, OSDD Consortium. Brahmachari SK. Systems level mapping of metabolic complexity in mycobacterium tuberculosis to identify high-value drug targets. J Transl Med. 2014;12:263. doi: 10.1186/s12967-014-0263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caspi R, Billington R, Ferrer L, Foerster H, Fulcher CA, Keseler IM, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–D480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King ZA, Lu J, Dräger A, Miller P, Federowicz S, Lerman JA, et al. BiGG models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44:D515–D522. doi: 10.1093/nar/gkv1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatzios SK, Bertozzi CR. The regulation of sulfur metabolism in mycobacterium tuberculosis. PLoS Pathog. 2011;7:e1002036. doi: 10.1371/journal.ppat.1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci U S A. 2011;108:5051–5056. doi: 10.1073/pnas.1009516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Layre E, Lee HJ, Young DC, Martinot AJ, Buter J, Minnaard AJ, et al. Molecular profiling of mycobacterium tuberculosis identifies tuberculosinyl nucleoside products of the virulence-associated enzyme Rv3378c. Proc Natl Acad Sci U S A. 2014;111:2978–2983. doi: 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richard-Greenblatt M, Bach H, Adamson J, Peña-Diaz S, Li W, Steyn AJC, et al. Regulation of Ergothioneine biosynthesis and its effect on mycobacterium tuberculosis growth and infectivity. J Biol Chem. 2015;290:23064–23076. doi: 10.1074/jbc.M115.648642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saini V, Cumming BM, Guidry L, Lamprecht DA, Adamson JH, Reddy VP, et al. Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of mycobacterium tuberculosis. Cell Rep. 2016;14:572–585. doi: 10.1016/j.celrep.2015.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nambu S, Matsui T, Goulding CW, Takahashi S, Ikeda-Saito M. A new way to degrade heme: the mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J Biol Chem. 2013;288:10101–10109. doi: 10.1074/jbc.M112.448399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio. 2017;8. Available from: 10.1128/mBio.02133-16 [DOI] [PMC free article] [PubMed]
- 31.Palsson BØ. Systems Biology: Constraint-based Reconstruction and Analysis. Cambridge: Cambridge University Press; 2015.
- 32.Rhee KY, de Carvalho LPS, Bryk R, Ehrt S, Marrero J, Park SW, et al. Central carbon metabolism in mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 2011;19:307–314. doi: 10.1016/j.tim.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Megchelenbrink W, Huynen M, Marchiori E. optGpSampler: an improved tool for uniformly sampling the solution-space of genome-scale metabolic networks. PLoS One. 2014;9:e86587. doi: 10.1371/journal.pone.0086587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gouzy A, Larrouy-Maumus G, Wu T-D, Peixoto A, Levillain F, Lugo-Villarino G, et al. Mycobacterium tuberculosis nitrogen assimilation and host colonization require aspartate. Nat Chem Biol. 2013;9:674–676. doi: 10.1038/nchembio.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gouzy A, Larrouy-Maumus G, Bottai D, Levillain F, Dumas A, Wallach JB, et al. Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PLoS Pathog. 2014;10:e1003928. doi: 10.1371/journal.ppat.1003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gouzy A, Poquet Y, Neyrolles O. Nitrogen metabolism in mycobacterium tuberculosis physiology and virulence. Nat Rev Microbiol. 2014;12:729–737. doi: 10.1038/nrmicro3349. [DOI] [PubMed] [Google Scholar]
- 38.Beste DJV, Nöh K, Niedenführ S, Mendum TA, Hawkins ND, Ward JL, et al. 13C-flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular mycobacterium tuberculosis. Chem Biol. 2013;20:1012–1021. doi: 10.1016/j.chembiol.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7:e1002093. doi: 10.1371/journal.ppat.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muñoz-Elías EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006;8:10–22. doi: 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- 41.Eoh H, Rhee KY. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2013;110:6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eoh H, Rhee KY. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of mycobacterium tuberculosis on fatty acids. Proc Natl Acad Sci U S A. 2014;111:4976–4981. doi: 10.1073/pnas.1400390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Safi H, Lingaraju S, Amin A, Kim S, Jones M, Holmes M, et al. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-[beta]-D-arabinose biosynthetic and utilization pathway genes. Nat Genet. 2013;45:1190–1197. doi: 10.1038/ng.2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desjardins CA, Cohen KA, Munsamy V, Abeel T, Maharaj K, Walker BJ, et al. Genomic and functional analyses of mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat Genet. 2016;48:544–551. doi: 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Carvalho LPS, Fischer SM, Marrero J, Nathan C, Ehrt S, Rhee KY. Metabolomics of mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem Biol. 2010;17:1122–1131. doi: 10.1016/j.chembiol.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Zimmermann M, Kogadeeva M, Gengenbacher M, McEwen G, Mollenkopf H-J, Zamboni N, et al. Integration of Metabolomics and Transcriptomics Reveals a Complex Diet of Mycobacterium tuberculosis during Early Macrophage Infection. mSystems. 2017;2. Available from: 10.1128/mSystems.00057-17. [DOI] [PMC free article] [PubMed]
- 47.Xu W, DeJesus MA, Rücker N, Engelhart CA, Wright MG, Healy C, et al. Chemical Genetic Interaction Profiling Reveals Determinants of Intrinsic Antibiotic Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017;61. Available from: 10.1128/AAC.01334-17. [DOI] [PMC free article] [PubMed]
- 48.Orth JD, Conrad TM, Na J, Lerman JA, Nam H, Feist AM, et al. A comprehensive genome-scale reconstruction of Escherichia coli metabolism--2011. Mol Syst Biol. 2011;7:535. doi: 10.1038/msb.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat Biotechnol. 2017;35:904–908. doi: 10.1038/nbt.3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ebrahim A, Lerman JA, Palsson BO, Hyduke DR. COBRApy: COnstraints-based reconstruction and analysis for python. BMC Syst Biol. 2013;7:74. doi: 10.1186/1752-0509-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, et al. para-Aminosalicylic acid is a prodrug targeting dihydrofolate reductase in mycobacterium tuberculosis. J Biol Chem. 2013;288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vilchèze C, Av-Gay Y, Attarian R, Liu Z, Hazbón MH, Colangeli R, et al. Mycothiol biosynthesis is essential for ethionamide susceptibility in mycobacterium tuberculosis. Mol Microbiol. 2008;69:1316–1329. doi: 10.1111/j.1365-2958.2008.06365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King ZA, Dräger A, Ebrahim A, Sonnenschein N, Lewis NE, Palsson BO. Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput Biol. 2015;11:e1004321. doi: 10.1371/journal.pcbi.1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morita YS, Sena CBC, Waller RF, Kurokawa K, Sernee MF, Nakatani F, et al. PimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteria. J Biol Chem. 2006;281:25143–25155. doi: 10.1074/jbc.M604214200. [DOI] [PubMed] [Google Scholar]
- 55.Cai L, Zhao X, Jiang T, Qiu J, Owusu L, Ma Y, et al. Prokaryotic expression, identification and bioinformatics analysis of the mycobacterium tuberculosis Rv3807c gene encoding the putative enzyme committed to Decaprenylphosphoryl-d-arabinose synthesis. Indian J Microbiol. 2014;54:46–51. doi: 10.1007/s12088-013-0418-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Colijn C, Brandes A, Zucker J, Lun DS, Weiner B, Farhat MR, et al. Interpreting expression data with metabolic flux models: predicting mycobacterium tuberculosis mycolic acid production. PLoS Comput Biol. 2009;5:e1000489. doi: 10.1371/journal.pcbi.1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kalapanulak S. High quality genome-scale metabolic network reconstruction of mycobacterium tuberculosis and comparison with human metabolic network: application for drug …. era.lib.ed.ac.uk. 2009. [Google Scholar]
- 58.Fang X, Wallqvist A, Reifman J. Development and analysis of an in vivo-compatible metabolic network of mycobacterium tuberculosis. BMC Syst Biol. 2010;4:160. doi: 10.1186/1752-0509-4-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chindelevitch L, Stanley S, Hung D, Regev A, Berger B. MetaMerge: scaling up genome-scale metabolic reconstructions with application to mycobacterium tuberculosis. Genome Biol. 2012;13:r6. doi: 10.1186/gb-2012-13-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lofthouse EK, Wheeler PR, Beste DJV, Khatri BL, Wu H, Mendum TA, et al. Systems-based approaches to probing metabolic variation within the mycobacterium tuberculosis complex. PLoS One. 2013;8:e75913. doi: 10.1371/journal.pone.0075913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Puniya BL, Kulshreshtha D, Mittal I, Mobeen A, Ramachandran S. Corrigendum: integration of metabolic modeling with gene co-expression reveals transcriptionally programmed reactions explaining robustness in mycobacterium tuberculosis. Sci Rep. 2016;6:24916. doi: 10.1038/srep24916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sreevatsan S, Stockbauer KE, Pan X, Kreiswirth BN, Moghazeh SL, Jacobs WR, Jr, et al. Ethambutol resistance in mycobacterium tuberculosis: critical role of embB mutations. Antimicrob Agents Chemother. 1997;41:1677–1681. doi: 10.1128/aac.41.8.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caceres NE, Harris NB, Wellehan JF, Feng Z, Kapur V, Barletta RG. Overexpression of the D-alanine racemase gene confers resistance to D-cycloserine in mycobacterium smegmatis. J Bacteriol Am Soc Microbiol. 1997;179:5046–5055. doi: 10.1128/jb.179.16.5046-5055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neuhaus FC, Lynch JL. The enzymatic synthesis of d-ALANYL-d-alanine. 3. On the inhibition of d-ALANYL-d-alanine SYNTHETASE by the antibiotic d-CYCLOSERINE. Biochemistry. 1964;3:471–480. doi: 10.1021/bi00892a001. [DOI] [PubMed] [Google Scholar]
- 65.Musser JM, Kapur V, Williams DL, Kreiswirth BN, Van Soolingen D, Van Embden JDA. Characterization of the catalase-peroxidase gene (katG) and inhA locus in isoniazid-resistant and-susceptible strains of mycobacterium tuberculosis by automated DNA sequencing: restricted array of mutations associated with drug resistance. J Infect Dis. 1996;173:196–202. doi: 10.1093/infdis/173.1.196. [DOI] [PubMed] [Google Scholar]
- 66.Rozwarski DA, Grant GA, Barton DH, Jacobs WR, Jr, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 67.Torres JN, Paul LV, Rodwell TC, Victor TC, Amallraja AM, Elghraoui A, et al. Novel katG mutations causing isoniazid resistance in clinical M. Tuberculosis isolates. Emerg Microbes Infect. 2015;4:e42. doi: 10.1038/emi.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, et al. Benzothiazinones kill mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 70.Li G, Zhang J, Guo Q, Wei J, Jiang Y, Zhao X, et al. Study of efflux pump gene expression in rifampicin-monoresistant mycobacterium tuberculosis clinical isolates. J Antibiot. 2015;68:431–435. doi: 10.1038/ja.2015.9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sheet 1: Reactions in iEK1011 and details of reactions. Sheet 2: Metabolites in iEK1011 and details of metabolites. Sheet 3: Genes present in previous models but not accounted for in iEK1011. Sheet 4: Gene Essentiality Predictions with the Griffin et al. dataset. Sheet 5: Gene Essentiality Predictions with the deJesus dataset. Sheet 6: Description of media conditions used in study. Sheet 7: Maximum FVA values for AMR genes across different objectives on in vivo media. Sheet 8: Maximum FVA values for AMR genes across different objectives on in vitro media. Sheet 9: “Shared” Essentiality Predictions with the Griffin et al. dataset. Sheet 10: “Shared” Essentiality Predictions with the deJesus et al. dataset. Sheet 11: List of blocked reactions in iEK1011. Sheet 12: “All models incorrect” - List of genes that were incorrectly predicted across all models and their corresponding iEK1011 subsystems. Sheet 13: “iEK Wrong and sMtb Correct” - List of incorrect iEK1011 predictions that were correctly predicted by sMtb. Sheet 13: “iEK Correct and sMtb Wrong” - List of correct iEK1011 predictions that were incorrectly predicted by sMtb. (XLSX 258 kb)
A conversion of the sMtb model to BiGGs identifiers (JSON 476 kb)
iEK1011 models: Genome-scale models of iEK1011 in json format initialized with different media conditions. iEK1011_griffinEssen_media.json - Used for essentiality testing on Griffin et al. dataset. iEK1011_deJesusEssen_media.json - Used for essentiality testing on deJesus et al. dataset. iEK1011_m7H10_media.json - iEK1011 initialized with Middlebrook 7H10 media. iEK1011_drugTesting_media.json - Used for simulating on Lowenstein-Jensen media. iEK1011_inVivo_media.json - Used for simulating on approximated physiological media. (ZIP 310 kb)
Escher Maps: Contains four escher maps of M. tuberculosis metabolic subsystems. Central_carbon.json. Arabinogalactan_peptidoglycan_complex.json. Nitrogen.json. Sulfur_and_folate.json (ZIP 236 kb)
Table S1. Table describing changes in gene essentiality predictions according to changes in GAM and NGAM values that were utilized across different genome-scale reconstructions of M. tuberculosis .Table S2. List of reactions in iEK1011 that violate the law of mass conservation. Table S3. Examples of false negatives computed by iEK1011 on the DeJesus et al. gene essentiality dataset that are not within the iSM810 model, and reasoning for its inclusion [54, 55]. Table S4. Gene essentiality predictions using the shared set of 472 genes. (DOCX 17 kb)
An ipython notebook that runs the simulations described in this study. (IPYNB 637 kb)