

Abstract
The branched-chain aminotransferase is a pyridoxal 5′-phosphate (PLP)-dependent enzyme responsible for the final step in the biosynthesis of all three branched-chain amino acids, L-leucine, L-isoleucine, and L-valine, in bacteria. We have investigated the mechanism of inactivation of the branched-chain aminotransferase from Mycobacterium tuberculosis (MtIlvE) by D- and L-cycloserine. D-Cycloserine is currently used only in the treatment of multidrug–drug-resistant tuberculosis. Our results show a time-and concentration-dependent inactivation of MtIlvE by both isomers, with L-cycloserine being a 40-fold better inhibitor of the enzyme. Minimum inhibitory concentration (MIC) studies revealed that L-cycloserine is a 10-fold better inhibitor of Mycobacterium tuberculosis growth than D-cycloserine. In addition, we have crystallized the MtIlvE-D-cycloserine inhibited enzyme, determining the structure to 1.7 Å. The structure of the covalent D-cycloserine-PMP adduct bound to MtIlvE reveals that the D-cycloserine ring is planar and aromatic, as previously observed for other enzyme systems. Mass spectrometry reveals that both the D-cycloserine- and L-cycloserine-PMP complexes have the same mass, and are likely to be the same aromatized, isoxazole product. However, the kinetics of formation of the MtIlvE D-cycloserine-PMP and MtIlvE L-cycloserine-PMP adducts are quite different. While the kinetics of the formation of the MtIlvE D-cycloserine-PMP complex can be fit to a single exponential, the formation of the MtIlvE L-cycloserine-PMP complex occurs in two steps. We propose a chemical mechanism for the inactivation of D- and L-cycloserine which suggests a stereochemically determined structural role for the differing kinetics of inactivation. These results demonstrate that the mechanism of action of D-cycloserine’s activity against M. tuberculosis may be more complicated than previously thought and that D-cycloserine may compromise the in vivo activity of multiple PLP-dependent enzymes, including MtIlvE.
Graphical abstract
The most recent report released by the World Health Organization (WHO) disclosed that tuberculosis (TB) is now the leading cause of human mortality by an infectious disease.1 The causative agent of TB, Mycobacterium tuberculosis, resulted in approximately 1.5 million deaths in 2015 and is a major source of morbidity on HIV-positive patients.1 The length of treatment (6–24 months) and the difficulties accessing effective drugs in developing countries, combined with the emergence of antibiotic resistant strains, are major challenges facing the fight against TB.2 Therefore, a more thorough knowledge of the resistance mechanisms employed by M. tuberculosis against common antitubercular drugs, as well as the validation of new targets for the development of new drugs, is an urgent and unmet global health need.
D-Cycloserine (DCS) is an orally available, broad-spectrum antibacterial that was isolated in 1955 from a Streptomyces species.3 It was shown in 1960 to inhibit both the Staphylococcus aureus alanine racemase (AR) and D-ala-D-ala ligase (DAL), enzymes involved in the synthesis of the peptidoglycan component of bacterial cell walls, and was the first antibiotic whose mechanism of action was shown to be due to enzyme inhibition.4 DCS has been shown to be an irreversible inhibitor of bacterial D-amino acid transaminase,5 and the structure of the covalent pyridoxal 5′-phosphate (PLP)-DCS complex has been determined.6 The structure revealed that the DCS ring was intact and aromatic. This finding was also confirmed for the inactivation of γ-aminobutyric (GABA) aminotransferase by L-cycloserine (LCS).7 Structures of the DCS- and LCS-inhibited forms of dialkylglycine decarboxylase8 and of the LCS-inhibited forms of methionine γ-lyase9 have also been reported.
DCS is presently used almost exclusively as a second-line drug to treat multidrug-resistant TB (MDR-TB),10,11 due to its toxicity related to its activity as a mammalian neuronal NMDA receptor agonist and an increase in the levels of inhibitory neurotransmitters.12,13 The targets of DCS in M. tuberculosis were proposed to be similar to those in S. aureus; the PLP-dependent AR and DAL.11 Early studies using genetic selection revealed that resistance to DCS was the result of mutations in the alanine-glycine-D-serine permease and the D-ala-D-ala ligase.14 Overexpression of the gene encoding alanine racemase was shown to confer resistance to DCS in Mycobacterium smegmatis.15 DCS was subsequently shown to be a slow, tight-binding inhibitor of the M. tuberculosis D-ala-D-ala ligase.11 Two reports of metabolomic profiling of DCS-treated mycobacterial cell extracts both concluded that D-ala-D-ala ligase was the primary, and lethal, target of DCS.16,17 A recent study demonstrated that clinical strains with low-level resistance to DCS mapped to mutations in the ald gene, encoding alanine dehydrogenase.18
Branched-chain amino acids are biosynthesized in M. tuberculosis in a manner similar to other eubacteria and by enzymes that have all been annotated in the genome.19 We have previously reported the three-dimensional structure of the ilvE-encoded (Rv2210c) branched-chain aminotransferase (BCAT) from M. tuberculosis (MtIlvE).20 The PLP-dependent enzyme was crystallized in complex with pyridoxamine 5′-phosphate (PMP) and is a homodimer. MtIlvE catalyzes the L-glutamate-dependent amination of α-ketoisocaproate, α-keto-methyl-oxopentanoic acid, and α-ketoisovalerate to generate the three branched-chain amino acids (BCAAs) L-leucine, L-isoleucine, and L-valine.21 The enzyme is essential for growth under multiple growth conditions.22,23 Carbonyl reagents such as O-alkylhydroxylamines were inhibitors of the M. tuberculosis enzyme, the best exhibiting a Ki value versus L-leucine of 21 μM and an MIC value against M. tuberculosis of 78 μM.24 The human BCAAT isozymes (cytosolic [hBCATc] and mitochondrial [hBCATm]; 58% sequence identity) are differentially inhibited by gabapentin.25 MtIlvE shares approximately 34% sequence identity with hBCATm but is not inhibited by gabapentin.20
In the present study, we examined the ability of DCS and LCS to inhibit MtIlvE. Our results show that both isomers inhibit the enzyme in a time- and concentration-dependent manner. However, LCS binds more tightly, and inhibits more rapidly, than does DCS and is a better inhibitor of mycobacterial growth. We crystallized the DCS-inhibited enzyme, and solved the structure to 1.7 Å. The structure of the enzyme-bound PMP-DCS adduct reveals that the DCS ring is intact and aromatic, as previously observed. Using absorbance spectra and mass spectrometry, we identified the MtIlvE LCS-PMP final complex as identical to that of the MtIlvE DCS-PMP complex. Based on our findings, we propose a mechanism of inhibition of MtIlvE by each of the cycloserine isomers based on stereochemical, kinetic, and structural evidence.
RESULTS AND DISCUSSION
Inactivation of MtIlvE by DCS and LCS
Mechanism-based inhibitors are a class of substrate analogs that lead to irreversible inhibition. Enzyme inactivation is caused by an initial chemical step identical to that normally occurring in catalysis followed by subsequent steps resulting in the formation of a stable dead-end complex, creating an irreversible protein–inhibitor complex.26,27 The highly conserved catalytic mechanism of PLP-dependent enzymes makes them particularly amenable to mechanism-based inhibitor design and enzyme inactivation.6,26–30 Due to their involvement in so many essential biological processes, a number of PLP-dependent enzymes have been targeted as potential drug targets.3,11–13
The preincubation of MtIlvE with different concentrations of either DCS or LCS led to a decrease of enzymatic activity over time (Figure 1). The semilogarithmic plots of the remaining activity as a function of time of inactivation in the presence of different concentrations of DCS and LCS indicates that the inactivation follows pseudo-first-order kinetics. Kitz and Wilson replots (insets, Figure 1) are linear for both DCS and LCS, suggesting that at least one molar equivalent of inhibitor binds to one molecule of enzyme for inactivation.31 In the case of the DCS-inhibited enzyme, the Kitz and Wilson replot suggests the formation of a low affinity inhibitor–enzyme complex which intercepts at the origin.29,32 This prevents an accurate estimation of KI and kinact values when using a linear regression method.29,32 No activity is recovered from the inactivated enzyme after several hours of dialysis, arguing that the inactivation is irreversible (data not shown). In comparison to DCS, LCS is a more efficient inhibitor of MtIlvE. The KI and kinact values observed for LCS are 88 μM and 4.5 × 10−4 s−1, respectively. The stereospecificity displayed by MtIlvE, which is an L-amino acid aminotransferase, explains the more rapid inactivation by LCS with the second-order rate constant of inactivation kinact/KI_LCS = 7.32 M−1 s−1 being greater than kinact/KI_DCS = 0.12 M−1 s−1. LCS is also a significantly better inhibitor than DCS of the PLP-dependent serine palmitoyl-transferase from Sphingomonas paucimobili, an enzyme that catalyzes the condensation of L-serine with palmitoyl-CoA in the biosynthetic pathway of sphingolipids.29 LCS has been exploited in several model systems,8,33 including seizure models, due to its ability to increase intracerebral GABA levels.34
Figure 1.
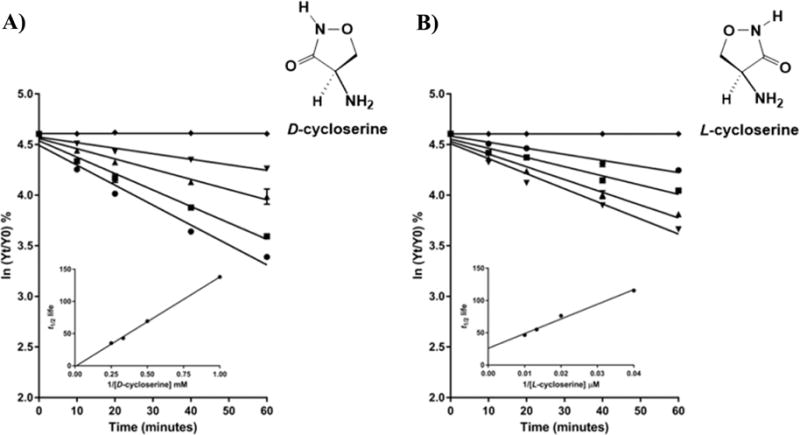
Mechanism-based inhibition of MtIlvE by (A) DCS and (B) LCS. (A) ln % of remaining activity of MtIlvE inhibited over time and increasing concentrations of DCS as follows: (⧫) control, (▼) 1 mM, (▲) 2 mM, (■) 3 mM, and (●) 4 mM. (B) ln % of remaining activity of MtIlvE inhibited over time and increasing concentrations of LCS as follows: (⧫) control, (●) 25 μM, (■) 50 μM, (▲) 75 μM, and (▼) 100 μM. The insets represent the Kitz-Wilson replots and describe the t1/2 life. The second-order rate constant kinact/KI is calculated as 1/slope.
We tested other PLP-dependent enzyme inactivators including propargylglycine, an inhibitor of cystathionine γ-synthase;28 gabaculine, an inhibitor of GABA aminotransferase and ornithine decarboxylase;27 and difluoromethylornithine (DFMO), the inhibitor of ornithine decarboxylase.35 The preincubation of MtIlvE with 10 mM of each of these inhibitors for over 1 h did not lead to any decrease in the activity of MtIlvE (data not shown). It has been previously shown that gabapentin, a potent inhibitor of the hBCATc, has no effect on the activity of MtIlvE.24 Given our results, it is interesting that neither human BCAT isozyme is inhibited by DCS.36 This clearly demonstrates that despite the sequence similarities shared among MtIlvE and the human BCATs, especially hBCATm, the M. tuberculosis enzyme has the potential for selective inhibition.
Determination of MIC Values for LCS and DCS
In order to compare the inhibitory effects of LCS and DCS on in vivo growth of M. tuberculosis, growth inhibition experiments were carried out using an auxotrophic (ΔpanCD; requires pantothenate for growth) strain, mc26230, derived from M. tuberculosis H37Rv. The MIC value of each compound was established as the lowest concentration of LCS or DCS that fully inhibits the growth of the bacteria after 15 days. DCS displayed a MIC value of 2.3 μg/mL using this strain (Table S1). Previous studies on wild-type M. tuberculosis found MIC values for DCS equal to 15 μg/mL,18 while MDR-TB clinical isolates exhibited MIC values ranging from 32 to 64 μg/mL.37 In general, laboratory strains are more susceptible to antibiotics than clinically isolated strains, such as MDR-TB strains. Similar experiments revealed that LCS exhibits a ∼10-fold better inhibitory growth effect than DCS, with a MIC value of 0.3 μg/mL (Table S1). Previous studies reported an increases in the MIC values in the presence of added L- and D-alanine,17 and we observed similar increases with the pantothenate auxotroph. The addition of either L-isoleucine, L-leucine, or L-valine, or all three, had no effect on the measured MIC values for either DCS or LCS (Table S1).
Electrospray Ionization–Mass Spectrometric Analysis of MtIlvE DCS and LCS Complexes
To assess the identities of the final MtIlvE complexes formed with LCS and DCS, we performed liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS). The purified enzyme in the PLP form served as a control. The PLP released from the active enzyme yielded a peak at m/z 248, corresponding to the PLP cofactor, and a peak at m/z 150, corresponding to the loss of the phosphate group from the cofactor [M + H − 98]+ (Figure S1).
The overnight incubation of MtIlvE with 5 mM DCS or LCS leads to the complete inactivation of the protein. The covalent cycloserine-PMP complexes were then examined by LC-ESI-MS and confirmed by MS/MS. A peak at m/z 332 was observed for complexes generated by incubation with both DCS and LCS (Figure 2), corresponding to the predicted mass of the PMP-isoxazole covalent complexes (m/z 332.06, Scheme 1). The fragmentation data (MS/MS) for the DCS and LCS inhibited complexes were identical to each other (Figure S2) and similar to those reported previously for the LCS-inhibited γ-aminotransferase.7 Both DCS and LCS–cofactor complexes are fragmented in a way that makes it impossible to distinguish between the tautomeric PMP-isoxazole, internal aldimine, or ketimine forms of the adduct. However, previous reports of the concerted 1,3-prototropic shift mechanism of MtIlvE21 and the structural studies presented below suggest the formation of a stable, aromatic PMP-isoxazole adduct.
Figure 2.
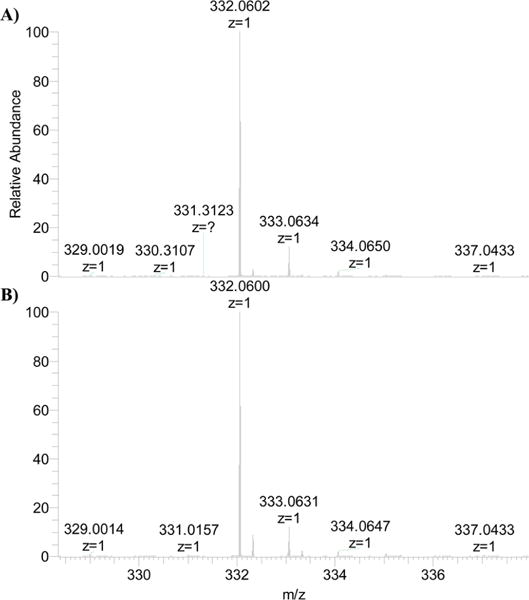
ESI-MS analysis of intact molecular weights of MtIlvE-PLP inhibited overnight by (A) DCS and (B) LCS. The peak at m/z 332.06 represents, in both cases, the presence of a noncovalent protein inhibitor complex where the cycloserine ring is intact.
Scheme 1.
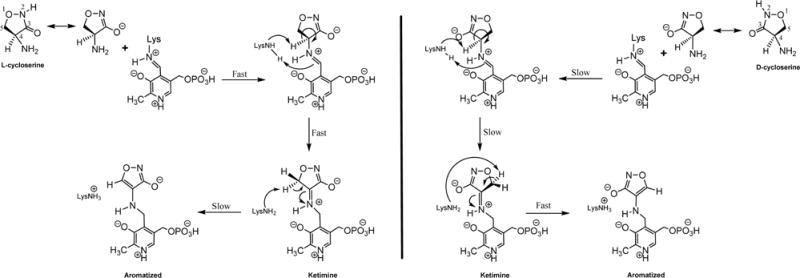
Proposed Aromatization Mechanism of Inactivation of MtIlvE by LCS (left) and DCS (right)
UV–vis Analysis of MtIlvE Inactivated by DCS and LCS
Enzymes containing the PLP cofactor can be kinetically analyzed using UV–vis spectrophotometry, since the absorbance is dependent on the chemical nature of the cofactor (PLP, PMP or quininoid). PLP, bound as the internal aldimine to Lys204 in MtIlvE, displays a characteristic UV–vis spectra with a visible absorbance maximum at 415 nm.21 The time course of the UV–vis spectral changes accompanying the transamination reaction catalyzed by MtIlvE starts with a PLP Schiff-base or internal aldimine (λmax ≈ 415 nm) that is converted to the next stable enzyme form, PMP (λmax ≈ 330 nm). A quininoid intermediate often observed at 490 nm is absent in the reaction catalyzed by MtIlvE due to its concerted 1,3-prototropic shift mechanism.21 In the presence of 1 mM DCS, a similar pattern is observed (Figure 3A). In 60 min, the conversion of the 415 nm peak representing the internal aldimine is reduced and a 330 nm peak indicative of a PMP derivative appears. There is a very clear isosbestic point observed at ca. 350 nm, and a plot of the A420 vs time can be fit to a first-order decrease of [E-PLP]. This result suggests that the MtIlvE-bound cofactor forms a complex with DCS to generate a covalent DCS-PMP complex (λmax = 330 nm) where the 2-oxazolidine ring is intact and has aromatized, as reported for other enzyme systems.15,38
Figure 3.
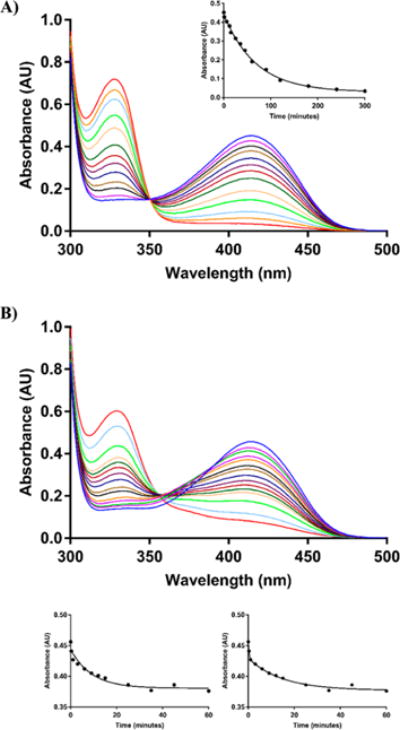
UV–vis analysis of MtIlvE inhibition by the cycloserine isomers over time. The upper blue lines on (A) and (B) represent MtIlvE-PLP in the absence of inhibitor. All the other lines represent the spectra of MtIlvE in the presence of (A) 1 mM DCS at 1, 6, 12, 15, 25, 35, 45, 60, 90, 120, 180, and 300 min, respectively. The inset in (A) represents the change in absorbance at λmax ≈ 415 nm over time. The data was fit to a single exponential equation; and (B) 0.5 mM LCS at 1, 6, 25, 45, 90, 120, 180, 240, 300, 360, 420, 600, 1050, and 1665 min, respectively. The two lower panels represent the change in absorbance at λmax ≈ 415 nm over time. The data on the left lower panel was fit to a single exponential equation. The right lower panel was fit to a double exponential equation. In both (A) and (B), the peak at λmax ≈ 415 nm (internal aldimine) is reduced over time, while the PMP (λmax ≈ 330 nm) peak is increased. The appearance of the PMP in the DCS inhibited enzyme happens approximately 5-fold faster than with LCS.
Significant differences in the time course of inhibition of MtIlvE by LCS are observed spectrophotometrically in the UV–vis region (Figure 3B). The addition of 0.5 mM LCS to MtIlvE led to a rapid (∼10 min) but modest decrease of the internal aldimine peak (415 nm) with a corresponding modest increase at approximately 370 nm and without a corresponding increase at 330 nm. An isosbestic point at approximately 380 nm was reported for the time course of LCS inhibition of serine palmitoyltransferase from S. paucimobili.29 These spectral changes were proposed to be due to the formation of an oxime intermediate, and a similar spectrum was observed for GABA aminotransferase inhibited by hydroxylamine.33 However, this transient intermediate is slowly replaced at longer times (>20 min) by spectral features equivalent to those observed during reaction of the enzyme with DCS. At these longer times of reaction, the spectrophotometric changes are interpretable as the conversion of the PLP form of the enzyme into a PMP-LCS adduct identical to that observed for DCS and exhibiting a clear isosbestic point indicative of this conversion. If we again plot the A420 as a function of time, it is clear that a first-order decrease in the absorbance at times less than 8–10 min is not observed, while a two-exponential fit conforms to all the data. The kinetic and stereochemical interpretation of these differences will be discussed below.
Overall Structure of MtIlvE-PLP-DCS
MtIlvE incubated with DCS until complete inactivation of the enzyme was achieved was crystallized, and the structure of MtIlvE-PMP-DCS was solved to 1.7 Å. Data collection and refinement statistics are summarized in Table 1. The complex crystallized with two monomers in the asymmetric unit, covalently linked by an interchain disulfide bond. Tremblay et al. previously solved the MtIlvE-PMP structure and showed that MtIlvE forms a dimer in solution.20 Extremely low r.m.s. displacement values for the Cα atom positions (0.18 Å) is observed when the MtIlvE-PMP structure is superimposed onto the MtIlvE-PMP-DCS structure, indicating both structures are extremely similar.
Table 1.
Data Collection and Refinement Statistics
| data collection | MtIlvE-PLP-cycloserine |
|---|---|
| PDB ID | |
| Space Group | P212121 |
| Unit Cell Dimensions | |
| a, b, c (Å) | 91.9; 80.5; 81.3 |
| α, β, γ (deg) | 90.0; 90.0; 90.0 |
| Resolution Range (Å) | 50.0–1.70 |
| Wavelength (Å) | 0.97931 |
| Redundancy (Highest shell) | 3.8 (3.6) |
| Rmerge (%) (Highest shell) | 6.8 (54.2) |
| CC(1/2) (Highest shell) | 99.8 (79.7) |
| I/σI (Highest shell) | 12.5 (2.4) |
| Completeness (%) (Highest shell) | 91.1 (98.1) |
| Total Reflections (Unique) | 442066 (117641) |
| Refinement | |
| Rwork/Rfree | 17.6/20.9 |
| Number of non-hydrogen atoms | 5655 |
| Protein | 4994 |
| Water | 617 |
| Ligand | 44 |
| Average B-factors (Å2) | 24.0 |
| Protein | 23.1 |
| Water | 30.7 |
| Ligand | 19.4 |
| Wilson B factor | 19.5 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (deg) | 1.206 |
| Ramachandran plot | |
| Favored (%) | 98.0 |
| Outliers (%) | 0.3 |
The MtIlvE structure displays the characteristic type IV fold of PLP-dependent enzymes,20 with each monomer consisting of a small and a large domain that define the two active sites at the domain interfaces. The first domain includes both N- and C-terminal residues (residues 34–173 and 362–368) forming three α-helices and eight β-strands while the second domain is composed of residues 182 to 362, displaying nine β-strands and three α-helices (Figure 4). A rigid α-helix between residues E340 and R354 connects the two domains. A flexible loop (residues 173–182) should also link both domains, but no electron density was observed and the loop was not built into the MtIlvE-PMP-DCS model.
Figure 4.
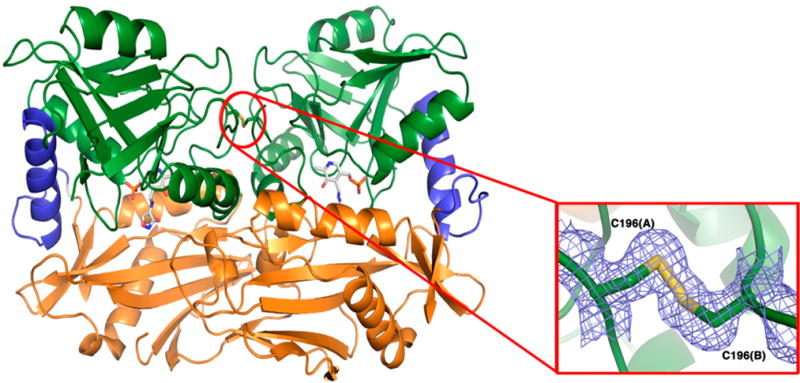
Overall structure of MtIlvE homodimer in complex with PMP-DCS. The two domains of MtIlvE are colored in orange (residues 34–173 and 363–368) and green (residues 182–362), respectively. An α-helix (residues 340–354, colored in blue), as well as a flexible loop (residues 173–182, poorly resolved in this structure), connect the two domains. The PLP-cycloserine molecule is rendered as sticks colored by CPK, with carbon atoms in white. A red circle highlights the presence of a disulfide bond covalently linking the two monomers. Close-up view (inset) of the interchain disulfide bond observed in the MtIlvE-PMP-DCS structure. The cysteines C196 are involved in the formation of a disulfide bond located at the homodimer interface. The electron density of a 2Fo-Fc map (blue) is shown contoured at 1σ. The two residues are represented as sticks colored by CPK, with carbon and sulfur atoms colored in green and yellow, respectively.
As previously observed, the N-terminal portion of the protein is highly disordered and electron density is only observable from H35 in monomer A and Y34 in monomer B in the MtIlvE-PMP-DCS structure. Additionally, a loop consisting of residues G174 to I181, which connect the two domains of MtIlvE, is poorly resolved in this structure. This linker loop is located near the active site and is highly flexible due to the presence of two glycine residues (G179-G180). This linker loop has been shown to play a significant role in the entrance and exit of the substrates/products.39,40 In contrast to aspartate aminotransferase, that exhibits a conformational change involving the small domain closing onto the active site upon binding of the substrate,41–44 only the linker loop between the small and large domains moves to close the active site in BCAT enzymes.39,40,45
Cys196 Disulfide Bond
In the MtIlvE-PMP structure, Tremblay et al. observed cysteine residues (Cys196) that were located at the homodimer interface 3.6 Å away from its identical symmetry-mate residue.20 There was no evidence in that structure that the two cysteine residues form a disulfide bond, but based on their separation, it was clear that they could under oxidative conditions. In contrast, the MtIlvE-PMP-DCS structure exhibits clear 2Fo-Fc electron density supporting the presence of a disulfide linkage between the Cys196 residues from each monomer. The distance between the Sγ atoms in the intersubunit disulfide bond is 2.05 Å, an appropriate bond length distance. The formation of an intersubunit disulfide bond had previously been hypothesized to increase the stability of the MtIlvE dimer under the oxidative conditions observed within the macrophages.20 Cysteine residues have been shown to play a significant role in the regulation of catalytic activity of hBCATm.45–47 A C315XXC318 motif is conserved among all mammalian BCAT enzymes and the formation of a C315–C318 disulfide bond leads to a loss in activity of hBCATm. The cysteine residues involved in the disulfide bridge in the MtIlvE-PMP-DCS structure are structurally located in a different region, and C196 is not a conserved residue among other BCAT enzymes (Figure S3).
Enzyme Interaction with PMP-Cycloserine Adduct
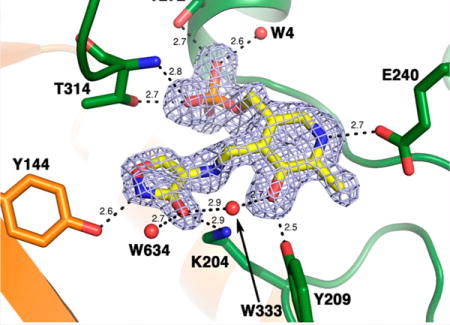
Clear 2Fo-Fc and Fo-Fc electron density, corresponding to a PMP-D-cycloserine adduct, was observed in the active site of MtIlvE-PMP-DCS. Only an intact ring form of cycloserine could account for the Fo-Fc electron density observed. This result is in agreement with the mass spectrometry data. The adduct appears tightly bound within the MtIlvE active site as supported by the low B-factors observed (less than 25 Å2). The pyridoxamine moiety of the PMP-cycloserine derivative lies in the same conformation as PMP in the MtIlvE-PMP model (Figure 5A). The cycloserine ring replaces a water molecule (W423) in the previously solved apo structure. Similar interactions to the ones previously observed in the MtIlvE-PMP active site are observed in this new structure (Figure 5B,C). The N1 atom of the pyridine moiety interacts with E240 via hydrogen bonding and the ring hydroxyl group (O3) is forming a hydrogen bond with the phenolic moiety of Y209, as well as a water molecule (W333). The oxygen atoms of the phosphate moiety form multiple polar interactions with several residue side chains (R101, T272, and T314), backbone nitrogen groups (I271 and T314), and water molecules W4 and W41 (Figure 5B,C). The exocyclic N atom of cycloserine lies close (2.8 Å) to K204 (Figure 5C).
Figure 5.
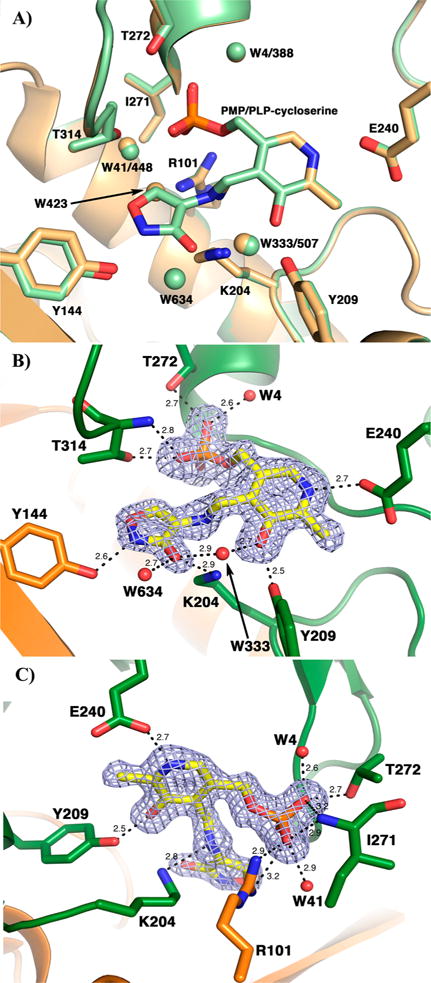
Active site of the MtIlvE-PLP-DCS structure. (A) Superimposition of the MtIlvE-PMP (orange) and MtIlvE-PLP-DCS (green) structures. Residues interacting directly with either PMP or PLP-DCS adduct are representing as sticks colored by CPK whereas the water molecules are rendered as spheres. When two water molecule positions overlap, the first number corresponds to the MtIlvE-PLP-DCS structure and the second to the MtIlvE-PMP structure. W423 was replacing the cycloserine ring in the MtIlvE-PMP structure. (B,C) The electron density of a Fo-Fc omit map (blue) is shown contoured at 3σ, the PLP-cycloserine molecule was omitted during map calculation. PLP-cycloserine is represented as sticks colored by CPK, with carbon atoms in yellow. The residues participating in hydrogen bonding (dashes) with PLP-cycloserine are rendered as sticks, while the water molecules (W) interacting with the ligand are represented as spheres. The distances are given in angstroms.
The cycloserine ring appears tilted compared to the plane of the pyridine ring. The geometry indicates an sp3 hybridization of the C4A atom, confirming the identity of the PMP form of the cofactor. Additionally, the five-membered ring is planar and is consistent with the presence of an aromatic, isoxazole ring. The cycloserine portion of the adduct makes polar interactions with residues within the active site. N2 forms a hydrogen bond (2.6 Å) with the phenolic group of Y144. The C3–O atom (presumably as the alkoxide) of the cycloserine isoxazole ring interacts with the catalytic residue K204 (2.9 Å), as well as two water molecules (W333 and W634). This structure is similar to the previously reported structure of D-amino acid aminotransferase crystallized after reaction with DCS.48 Superposition of the two structures reveals a low overall r.m.s. displacement (1.8 Å), and a detailed comparison of the active site reveals a large number of conserved interaction between the PMP-DCS complex and side chain residues (Figure S4).
Mechanistic Analysis of the Inhibition of MtIlvE by DCS and LCS
The very different kinetic behavior of the two stereoisomers of cycloserine was somewhat unexpected considering the identity of the final products of the pyridoxal adducts with the two compounds. LCS was a significantly more efficient inhibitor of MtIlvE than DCS and also more effective in inhibiting the growth of M. tuberculosis. The high-resolution structure of MtIlvE-PMP-DCS allows us to propose a detailed chemical mechanism for the inhibition by the two isomers and speculate about the relative kinetics of the steps leading up to the final aromatized products. While both LCS and DCS bind as analogs of the natural amino donor, L-glutamate, LCS is likely to bind and assume a catalytically appropriate orientation more rapidly. As shown in Scheme 1, the N2–C3 N-hydroxyamide function of the 2-oxazolidine ring is most likely bound as the ionized C3 hydroxyl anion, mimicking the carboxyl group of an L-amino acid. The stereochemistry of LCS at the equivalent of the Cα center, coupled with the position of the K204 side chain that functions to remove the Cα proton below the plane of the PLP ring, and the requirement that the Cα-H bond be oriented perpendicular to the plane of the conjugated external aldimine for maximal stereoelectronic overlap allows us to draw the precatalytic PLP-LCS complex as shown (Figure S5). In this orientation, the C3 hydroxyl anion can interact electrostatically with R101 and is on the “C5-phosphate” side of the PLP ring and the C4-αH is within 2.7 Å of K204 in a model of the precatalytic complex. Removal of the αH is expected to be fast from this complex, generating the ketimine intermediate, in which the orientation of the oxazolidine ring is now fixed due to the restricted rotation about the cofactor–inhibitor imine linkage. These two steps, external aldimine formation followed by deprotonation to generate the ketimine, may be the rapid steps that are reported on in the early (<10 min) spectral traces of inactivation by LCS (Figure 3B). This argues that the final step, C5–H abstraction to generate the aromatized and stable 2-isoxazole adduct, is slow from the LCS-generated ketimine.
In contrast, the time course of the UV–vis changes accompanying DCS inhibition (Figure 3A) of MtIlvE suggest that the opposite stereochemistry is associated with changes in the relative rates of these same steps discussed above. DCS may bind initially in the “LCS orientation” with the C3 hydroxyl anion on the “C5-phosphate” side of the pyridoxal ring, but must ultimately rotate to allow for C4-αH removal by K204, placing the C3 hydroxyl anion on the “C3-hydroxyl” side of the pyridoxal ring. This is likely to make the formation of the precatalytic external aldimine conformation slower than with LCS. Proton abstraction, accompanied by a 1,3 prototropic shift, generates the ketimine, which differs from the ketimine generated from LCS in the orientation of the oxazole ring and the positioning of the C5 protons. Proton abstraction from C5 of the oxazole ring leads to the final aromatized PMP–isoxazole covalent complex. Since the stereochemical differences are lost as a result of Cα-H abstraction, as are any rotational restrictions in the PMP-LCS and PMP-DCS adducts, it is unclear whether the crystallographically observed orientation of the ring for the PMP-DCS adduct (Figure 5B) is also not adopted by the PMP-LCS adduct after ring aromatization.
CONCLUSION
Branched-chain aminotransferases are responsible for essential processes in mammalian catabolism and eubacterial biosynthesis. Significant efforts have been made toward the development of lead compounds capable of inhibiting the activity of the hBCATm, an enzyme that has been implicated in human obesity.49 In addition to its role in the biosynthesis of L-isoleucine, L-leucine, and L-valine, MtIlvE is implicated in the biosynthesis of L-methionine and catabolism of aromatic amino acids,21,24 suggesting that MtIlvE is a suitable therapeutic target.
While the essential, and lethal, targets of DCS in M. tuberculosis have been demonstrated,11,18 our data suggest that the branched chain amino acid aminotransferase, MtIlvE, is a bona fide target of both DCS and LCS. DCS inhibits several PLP-dependent enzymes in multiple organisms,6,8,12,29,50 including the M. tuberculosis alanine racemase and D-ala-D-ala ligase involved in peptidoglycan biosynthesis. The rate of development of resistance to DCS is very low compared to other antitubercular drugs,14,15,18 which has been attributed to the overexpression of DAL and AR.10,14,15
The unusually robust growth-inhibiting activity of LCS compared to DCS suggests that this compound may have a different, and broader, range of inhibitable targets, including MtIlvE. Of the known DCS targets, the effects of LCS are likely due to the inhibition of AR since D-ala-D-ala ligase is not inhibited by LCS.17 Our MIC data show that both D- and L-alanine substantially increase the MIC value for LCS supporting the conclusion that alanine racemase as the primary target under these conditions. While our biochemical and structural data clearly support MtIlvE as being inhibitable by both DCS and LCS, under the conditions used, a similar “protective” effect of added branched-chain amino acids is not observed. Instead, a pleiotropic effect on multiple, essential PLP-dependent enzymes, including MtIlvE may be the reason for the robust activity of LCS against M. tuberculosis. While the current drug design principle of “one drug-one target” has been effective, perhaps “death by a thousand cuts” might be equally effective as an antibacterial therapeutic paradigm.
MATERIALS AND METHODS
Reagents
All chemicals were of analytical or reagent grade and did not undergo further purification. DCS and LCS were purchased from Sigma-Aldrich.
Expression and Purification of MtIlvE
MtIlvE (encoded by the recombinant pET28a(+)::ilvE plasmid) was expressed and purified using the protocol previously reported.20 The protein was dialyzed against 50 mM Tris HCl, pH 8.0 (Buffer A). Glycerol (50% v/v) was added to the protein sample for long-term storage at −20 °C. The protein concentration was determined by absorbance spectroscopy at a wavelength of 280 nm using the theoretical extinction coefficient (ε280= 56 380 M−1 cm−1) calculated with the ProtParam function on the ExPASy server.51
Activity Assay and Inactivation by D- and L-Cycloserine
MtIlvE activity was measured continuously using a coupled spectrophotometric assay including bovine liver glutamate dehydrogenase (GDH; Sigma), excess reduced nicotinamide adenine dinucleotide (NADH) and ammonia as previously described.21 The activity was monitored by the decrease in absorbance at 340 nm (ε340 = 6220 M−1 cm−1) on a UV-2450 Shimadzu spectrophotometer.
Inactivation assays were performed by incubating 40 nM of MtIlvE-PLP form with DCS (1 mM, 2 mM, 3 mM, and 4 mM) or LCS (25 μM, 50 μM, 75 μM, and 100 μM). The remaining activity was measured at different time points (0, 10, 20, 40, and 60 min) after addition of the assay mixture. The latter contained 15 mM L-glutamate, 2 mM α-ketoisocaproate, 40 nM PLP, 1 mM ammonium chloride, 160 μM NADH, and 8 units of GDH in a total reaction volume of 1 mL. The reaction was initiated by the addition of aliquots from the inactivation mixture. All measurements were performed in duplicate at 25 °C using buffer A (50 mM Tris HCl, pH 8.0).
A percentage of the total activity in the absence of inhibitor or time zero was calculated. Kitz and Wilson plots of ln % remaining activity versus time were linear.52 All data points are from an average of duplicate or triplicate measurements, and all data were analyzed by nonlinear regression using the SigmaPlot software (Systat Software, Inc.) and fitting to eq 153
| (1) |
where E(0) represents the activity prior to addition of the inhibitor, E(t) is the activity after addition of either DCS or LCS at time = t, kapp is the inactivation rate at any inhibitor concentration = [I], KI is an estimate of inhibitor binding affinity, and kinact is the first-order inactivation rate constant. By plotting 1/kapp versus 1/[I], a straight line with an intercept of 1/kinact and slope of KI/kinact was obtained.
UV–vis Spectroscopy of MtIlvE Inhibition by DCS and LCS
MtIlvE was incubated with α-ketoisocaproate for 60 min to ensure that all active sites of the enzyme were converted into the internal aldimine (Schiff-base) form. The protein was dialyzed against buffer A to remove excess PLP and leucine formed during the incubation. The concentration of PLP bound to the enzyme was determined using absorbance spectroscopy at a wavelength of 415 nm (ε415 = 4900 M−1 cm−1) as previously calculated.21 UV–vis time course experiments were carried out using 100 μM MtIlvE and the spectrophotometer was blanked with buffer A. One milliliter quartz cuvettes were used, and spectra from 550 to 240 nm were recorded upon the addition of 1 mM DCS or 0.5 mM LCS at different times.
Mass Spectrometry
MtIlvE-PLP form (100 μM) was incubated with 5 mM LCS or DCS overnight at RT in buffer A. An Amicon device (10K molecular weight cutoff) was used to remove the cycloserine excess by centrifugal filtration at 4 °C for 15 min at 4000 rpm. A negative control was carried out without addition of either drug. LC-ESI-MS was performed in order to confirm the molecular weight of the final complexes MtIlvE-DCS and -LCS. A Thermofinnigan, LTQ Orbitrap Velos mass spectrometer was used at 15 000 resolution (at m/z 400) in positive ionization mode and scanned from 200 to 500 m/z. The mobile phase A was composed of 5% v/v acetonitrile/0.1% v/v formic acid, and the mobile phase B consisted of 100% v/v acetonitrile/0.1% v/v formic acid. We used an HPLC Cogent Diamond Hydride 150 mm × 2.1 mm column. Step gradient was as follows: 0 to 5 min at 2% phase B; 5 to 6 min 20% phase B; 6 to 8 min 40% phase B; 10 to 12 min 60% phase B; 12 min 80% phase B; 12 to 19 min 90% phase B; and held to 90% up to 30 min. Approximately 50 mL of sample without further processing was injected. After 4.5 min of desalting to waste, the tubing outlet from the column was connected to the mass spectrometer. Samples started being collected at 5 min. Elution time was 27 min.
Determination of MIC Value for DCS and LCS
In vivo growth inhibition studies were performed using a BSL2-safe derivative strain of M. tuberculosis H37Rv called mc26230 (H37Rv ΔpanCD), which is auxotroph strain for pantothenate (pan−).54 The auxotroph strain was maintained in Middlebrook 7H9 broth (Difco) supplemented with 10% v/v ADC (5% v/v bovine serum albumin [BSA], 2% v/v dextrose, 5% v/v catalase) 0.025% v/v tyloxapol, and 50 μg/mL of pantothenate. For the rescue experiments, 1 mM of the following amino acids were individually added to the cultures: L-alanine, D-alanine, L-glutamate, L-isoleucine (I), L-leucine (L), L-valine (V), and ILV. Both DCS and LCS were dissolved in water to a stock concentration of 0.5 mg mL−1 and sterilized by filtration. From logarithmically growing cultures, aliquots were transferred to supplemented 7H9 media (as described above) to an OD600 = 0.01. Following standard 1.5-fold dilutions, dose–response assays were performed in triplicate using a 96-well plate (Corning Incorporated Costar) in 100 μL volume with final concentrations ranging from 135 to 0.012 μg/mL. Cultures without LCS or DCS served as positive controls and cultures with 100 μg/mL kanamycin served as negative controls. After 15 days of incubation at 37 °C, 30 μL of resazurin dye (filter sterilized, 0.2 mg mL−1 concentration) was added to each well and incubated for 6 h under shaking and the plates were imaged. The MIC (minimum inhibitory concentration) was defined as the lowest concentration that completely inhibits the growth of mc26230 at day 15, as indicated by resazurin dye staining.
Crystallization
MtIlvE (20 mM HEPES, pH 7.5) containing an N-terminal His6-tag was concentrated to 6 mg mL−1 using an Amicon Ultracentrifugal filter. The sample was incubated with 0.2 mM PLP and 10 mM DCS for 3 h at 25 °C. The inactivated protein was screened in a 1:1 ratio against the commercially available screen MCSG suite (Microlytic). The screening was performed utilizing the sitting-drop vapor diffusion technique in a 96-well plate format (Intelli-Plates 96, Art Robbins). Reservoir solutions and drops were dispensed using a Crystal Gryphon robot (Art Robbins). The crystal plates were incubated at 20 °C. The screening yielded single crystals grown against a well solution containing 0.1 M Bis-Tris, pH 6.5, and 25% w/v polyethylene glycol 3350. The crystals were obtained after approximately six to eight weeks of incubation. The crystal was soaked with 25 mM D-cycloserine for 10 min prior to flash cooling in liquid nitrogen.
Data Collection and Structure Determination
Diffraction data were collected at the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at the Advance Photon Source Argonne National Laboratory (APS-ANL, IL) at 0.97931 Å wavelength radiation. Diffraction data were indexed, integrated, and scaled to 1.7 Å using the XDS program package.55 Molecular replacement was carried out with the software Phaser-MR using PDB 3HT5 as the search model.20,56 Two molecules per asymmetric unit were observed. Using the solution obtained by molecular replacement, the refine tool available in the PHENIX suite was used to perform rigid body refinement simulated annealing, positional and B-factor refinements.50 The model was manually corrected using COOT.57 Clear 2Fo-Fc and Fo-Fc electron density maps corresponding to a PLP-cycloserine adduct were observed. The geometric restraints of the PLP-cycloserine adduct, represented by the three-letter code DCS, were determined using eLBOW (electronic Ligand Builder and Optimization Workbench) in PHENIX.58 MOLPROBITY was used to assess the quality of the structure.59 Two residues were observed in the disallowed region of the Ramachandran plot (V317 in each monomer).60 The structure was deposited in the Protein Data Bank (PDB accession code 5U3F).
Supplementary Material
Acknowledgments
We would like to thank S. Cameron from Albert Einstein College of Medicine for insightful discussions. We thank E. Nieves from the proteomics facility at the Albert Einstein College of Medicine for his help with the LC-ESI-MS experiments using an LTQ Orbitrap Velos Mass Spectrometer System (Supported by a grant from NIH: S10-RR-029398). We thank M. Toney for careful reading of the manuscript. We thank S. Almo and his laboratory for the use of their X-ray crystallography equipment. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
Funding
This work was supported by a grant (NIH AI060899) to J.S.B. and Science Without Boarders fellowship - CAPES, Coordenaçã Aperfeiçoamento de Pessoal de Ńivel Superior, Brazil - to T.M.A.F. (325–13–9). Structure file has been deposited with the accession code: PDB ID 5U3F.
ABREVIATIONS
- AR
alanine racemase
- BCAAs
branched-chain amino acids
- BCATs
branched-chain aminotransferases
- DAL
D-alanine-D-alanine ligase
- DCS
D-cycloserine
- DFMO
difluoromethylor-nithine
- GABA
γ-aminobutyric acid
- GDH
glutamate dehydrogenase
- LCS
L-cycloserine
- MDR
multidrug-resistant
- MtIlvE
branched-chain aminotransferase IlvE
- NADH
nicotinamide adenine dinucleotide
- PLP
pyridoxal 5′-phosphate
- PMP
pyridoxamine 5′-phosphate
- TB
tuberculosis
- XDR
extensively drug-resistant
- WHO
World Health Organization
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.7b00142.
Comparison of the minimal inhibitory concentration between DCS and LCS; ESI-MS analysis; Sequence alignment charts; Superimposition of structural diagrams (PDF)
ORCID
John S. Blanchard: 0000-0002-9195-4402
Notes
The authors declare no competing financial interest.
References
- 1.World Health Organization releases. 2015 global report on tuberculosis. Breathe. 2015;11:244–244. [Google Scholar]
- 2.Franco TMA, Rostirolla DC, Ducati RG, Lorenzini DM, Basso LA, Santos DS. Biochemical characterization of recombinant guaA-encoded guanosine monophosphate synthetase (EC 6.3.5.2) from Mycobacterium tuberculosis H37Rv strain. Arch Biochem Biophys. 2012;517:1–11. doi: 10.1016/j.abb.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Neuhaus FC. Antibiotics I, Mechanism of Action. Springer, Verlag; New York: 1967. pp. 40–83. [Google Scholar]
- 4.Strominger J, Ito E, Threnn RH. Competitive Inhibition of Enzymatic Reactions by Oxamycin. J Am Chem Soc. 1960;82:998–999. [Google Scholar]
- 5.Soper TS, Manning JM. Different modes of action of inhibitors of bacterial D-amino acid transaminase. A target enzyme for the design of new antibacterial agents. J Biol Chem. 1981;256:4263–4268. [PubMed] [Google Scholar]
- 6.Peisach D, Chipman DM, Van Ophem PW, Manning JM, Ringe D. D-Cycloserine Inactivation of D-Amino Acid Aminotransferase Leads to a Stable Noncovalent Protein Complex with an Aromatic Cycloserine-PLP Derivative. J Am Chem Soc. 1998;120:2268–2274. [Google Scholar]
- 7.Olson GT, Fu MM, Lau S, Rinehart KL, Silverman RB. An Aromatization Mechanism of Inactivation of γ-Aminobutyric acid Transaminase for the Antibiotic L-cycloserine. J Am Chem Soc. 1998;120:2256–2267. [Google Scholar]
- 8.Malashkevich VN, Strop P, Keller JW, Jansonius JN, Toney MD. Crystal structures of dialkylglycine decarboxylase inhibitor complexes. J Mol Biol. 1999;294:193–200. doi: 10.1006/jmbi.1999.3254. [DOI] [PubMed] [Google Scholar]
- 9.Kuznetsov NA, Faleev NG, Kuznetsova AA, Morozova EA, Revtovich SV, Anufrieva NV, Nikulin AD, Fedorova OS, Demidkina TV. Pre-steady-state kinetic and structural analysis of interaction of methionine gamma-lyase from Citrobacter freundii with inhibitors. J Biol Chem. 2015;290:671–681. doi: 10.1074/jbc.M114.586511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong W, Chen L, Xie J. Molecular basis underlying Mycobacterium tuberculosis D-cycloserine resistance. Is there a role for ubiquinone and menaquinone metabolic pathways? Expert Opin Ther Targets. 2014;18:691–701. doi: 10.1517/14728222.2014.902937. [DOI] [PubMed] [Google Scholar]
- 11.Prosser GA, de Carvalho LP. Reinterpreting the mechanism of inhibition of Mycobacterium tuberculosis D-alanine:D-alanine ligase by D-cycloserine. Biochemistry. 2013;52:7145–7149. doi: 10.1021/bi400839f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dann OT, Carter CE. Cycloserine Inhibition of Gamma-Aminobutyric-Alpha-Ketoglutaric Transaminase. Biochem Pharmacol. 1964;13:677–684. doi: 10.1016/0006-2952(64)90002-4. [DOI] [PubMed] [Google Scholar]
- 13.Yew WW, Wong CF, Wong PC, Lee J, Chau CH. Adverse neurological reactions in patients with multidrug-resistant pulmonary tuberculosis after coadministration of cycloserine and ofloxacin. Clin Infect Dis. 1993;17:288–289. doi: 10.1093/clinids/17.2.288. [DOI] [PubMed] [Google Scholar]
- 14.David HL. Resistance to D-cycloserine in the tubercle bacilli: mutation rate and transport of alanine in parental cells and drug-resistant mutants. Appl Microbiol. 1971;21:888–892. doi: 10.1128/am.21.5.888-892.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caceres NE, Harris NB, Wellehan JF, Feng Z, Kapur V, Barletta RG. Overexpression of the D-alanine racemase gene confers resistance to D-cycloserine in Mycobacterium smegmatis. J Bacteriol. 1997;179:5046–5055. doi: 10.1128/jb.179.16.5046-5055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halouska S, Fenton RJ, Zinniel DK, Marshall DD, Barletta RG, Powers R. Metabolomics analysis identifies D-Alanine-D-Alanine ligase as the primary lethal target of D-Cycloserine in mycobacteria. J Proteome Res. 2014;13:1065–1076. doi: 10.1021/pr4010579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prosser GA, de Carvalho LP. Metabolomics Reveal D-Alanine:D-Alanine Ligase As the Target of D-Cycloserine in Mycobacterium tuberculosis. ACS Med Chem Lett. 2013;4:1233–1237. doi: 10.1021/ml400349n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desjardins CA, Cohen KA, Munsamy V, Abeel T, Maharaj K, Walker BJ, Shea TP, Almeida DV, Manson AL, Salazar A, Padayatchi N, O’Donnell MR, Mlisana KP, Wortman J, Birren BW, Grosset J, Earl AM, Pym AS. Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat Genet. 2016;48:544. doi: 10.1038/ng.3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 20.Tremblay LW, Blanchard JS. The 1.9 A structure of the branched-chain amino-acid transaminase (IlvE) from Mycobacterium tuberculosis. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2009;65:1071–1077. doi: 10.1107/S1744309109036690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amorim Franco TM, Hegde S, Blanchard JS. The chemical mechanism of the branched-chain aminotransferase IlvE from Mycobacterium tuberculosis. Biochemistry. 2016;55:6295–6303. doi: 10.1021/acs.biochem.6b00928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 24.Venos ES, Knodel MH, Radford CL, Berger BJ. Branched-chain amino acid aminotransferase and methionine formation in Mycobacterium tuberculosis. BMC Microbiol. 2004;4:39. doi: 10.1186/1471-2180-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutson S. Structure and function of branched chain aminotransferases. Prog Nucleic Acid Res Mol Biol. 2001;70:175–206. doi: 10.1016/s0079-6603(01)70017-7. [DOI] [PubMed] [Google Scholar]
- 26.Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, di Salvo ML, Borri-Voltattorni C, Bossa F, Mozzarelli A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr Med Chem. 2007;14:1291–1324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 27.Shah SA, Shen BW, Brunger AT. Human ornithine aminotransferase complexed with L-canaline and gabaculine: structural basis for substrate recognition. Structure. 1997;5:1067–1075. doi: 10.1016/s0969-2126(97)00258-x. [DOI] [PubMed] [Google Scholar]
- 28.Johnston M, Jankowski D, Marcotte P, Tanaka H, Esaki N, Soda K, Walsh C. Suicide inactivation of bacterial cystathionine gamma-synthase and methionine gamma-lyase during processing of L-propargylglycine. Biochemistry. 1979;18:4690–4701. doi: 10.1021/bi00588a033. [DOI] [PubMed] [Google Scholar]
- 29.Lowther J, Yard BA, Johnson KA, Carter LG, Bhat VT, Raman MCC, Clarke DJ, Ramakers B, McMahon SA, Naismith JH, Campopiano DJ. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol BioSyst. 2010;6:1682–1693. doi: 10.1039/c003743e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rando RR. Irreversible inhibition of aspartate aminotransferase by 2-amino-3-butenoic acid. Biochemistry. 1974;13:3859–3863. doi: 10.1021/bi00716a006. [DOI] [PubMed] [Google Scholar]
- 31.Hegde SS, Blanchard JS. Kinetic and mechanistic characterization of recombinant Lactobacillus viridescens FemX (UDP-N-acetylmuramoyl pentapeptide-lysine N-6-alanyltransferase) J Biol Chem. 2003;278:22861–22867. doi: 10.1074/jbc.M301565200. [DOI] [PubMed] [Google Scholar]
- 32.Parsons ZD, Gates KS. Redox Regulation of Protein Tyrosine Phosphatases: Methods for Kinetic Analysis of Covalent Enzyme Inactivation. Methods Enzymol. 2013;528:129–154. doi: 10.1016/B978-0-12-405881-1.00008-2. [DOI] [PubMed] [Google Scholar]
- 33.Olson GT, Fu MM, Lau S, Rinehart KL, Silverman RB. An aromatization mechanism of inactivation of gamma-aminobutyric acid aminotransferase for the antibiotic L-cycloserine. J Am Chem Soc. 1998;120:2256–2267. [Google Scholar]
- 34.Vogel KR, Pearl PL, Theodore WH, McCarter RC, Jakobs C, Gibson KM. Thirty years beyond discovery-Clinical trials in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. J Inherited Metab Dis. 2013;36:401–410. doi: 10.1007/s10545-012-9499-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: the native structure and the structure in complex with alpha-difluoromethylornithine. Biochemistry. 1999;38:15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- 36.Hall TR, Wallin R, Reinhart GD, Hutson SM. Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J Biol Chem. 1993;268:3092–3098. [PubMed] [Google Scholar]
- 37.Singh R. Determination of Minimun Inhibitory Concentration of Cycloserine in Multidrug Resistant Mycobacterium tuberculosis Isolates. Jordan J Biol Sci. 2014;7:139–145. [Google Scholar]
- 38.Bharath SR, Bisht S, Harijan RK, Savithri HS, Murthy MR. Structural and mutational studies on substrate specificity and catalysis of Salmonella typhimurium D-cysteine desulfhydrase. PLoS One. 2012;7:e36267. doi: 10.1371/journal.pone.0036267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okada K, Hirotsu K, Hayashi H, Kagamiyama H. Structures of Escherichia coli branched-chain amino acid aminotransferase and its complexes with 4-methylvalerate and 2-methylleucine: induced fit and substrate recognition of the enzyme. Biochemistry. 2001;40:7453–7463. doi: 10.1021/bi010384l. [DOI] [PubMed] [Google Scholar]
- 40.Yennawar NH, Conway ME, Yennawar HP, Farber GK, Hutson SM. Crystal structures of human mitochondrial branched chain aminotransferase reaction intermediates: ketimine and pyridoxamine phosphate forms. Biochemistry. 2002;41:11592–11601. doi: 10.1021/bi020221c. [DOI] [PubMed] [Google Scholar]
- 41.McPhalen CA, Vincent MG, Jansonius JN. X-ray structure refinement and comparison of three forms of mitochondrial aspartate aminotransferase. J Mol Biol. 1992;225:495–517. doi: 10.1016/0022-2836(92)90935-d. [DOI] [PubMed] [Google Scholar]
- 42.Jager J, Moser M, Sauder U, Jansonius JN. Crystal structures of Escherichia coli aspartate aminotransferase in two conformations. Comparison of an unliganded open and two liganded closed forms. J Mol Biol. 1994;239:285–305. doi: 10.1006/jmbi.1994.1368. [DOI] [PubMed] [Google Scholar]
- 43.Rhee S, Silva MM, Hyde CC, Rogers PH, Metzler CM, Metzler DE, Arnone A. Refinement and comparisons of the crystal structures of pig cytosolic aspartate aminotransferase and its complex with 2-methylaspartate. J Biol Chem. 1997;272:17293–17302. [PubMed] [Google Scholar]
- 44.Okamoto A, Higuchi T, Hirotsu K, Kuramitsu S, Kagamiyama H. X-ray crystallographic study of pyridoxal 5′-phosphate-type aspartate aminotransferases from Escherichia coli in open and closed form. J Biochem. 1994;116:95–107. doi: 10.1093/oxfordjournals.jbchem.a124509. [DOI] [PubMed] [Google Scholar]
- 45.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Human mitochondrial branched chain aminotransferase: structural basis for substrate specificity and role of redox active cysteines. Biochim Biophys Acta, Proteins Proteomics. 2003;1647:61–65. doi: 10.1016/s1570-9639(03)00051-7. [DOI] [PubMed] [Google Scholar]
- 46.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Identification of a peroxide-sensitive redox switch at the CXXC motif in the human mitochondrial branched chain aminotransferase. Biochemistry. 2002;41:9070–9078. doi: 10.1021/bi020200i. [DOI] [PubMed] [Google Scholar]
- 47.Yennawar NH, Islam MM, Conway M, Wallin R, Hutson SM. Human mitochondrial branched chain aminotransferase isozyme: structural role of the CXXC center in catalysis. J Biol Chem. 2006;281:39660–39671. doi: 10.1074/jbc.M607552200. [DOI] [PubMed] [Google Scholar]
- 48.Peisach D, Chipman DM, Van Ophem PW, Manning JM, Ringe D. D-cycloserine inactivation of D-amino acid aminotransferase leads to a stable noncovalent protein complex with an aromatic cycloserine-PLP derivative. J Am Chem Soc. 1998;120:2268–2274. [Google Scholar]
- 49.Borthwick JA, Ancellin N, Bertrand SM, Bingham RP, Carter PS, Chung CW, Churcher I, Dodic N, Fournier C, Francis PL, Hobbs A, Jamieson C, Pickett SD, Smith SE, Somers DO, Spitzfaden C, Suckling CJ, Young RJ. Structurally Diverse Mitochondrial Branched Chain Aminotransferase (BCATm) Leads with Varying Binding Modes Identified by Fragment Screening. J Med Chem. 2016;59:2452–2467. doi: 10.1021/acs.jmedchem.5b01607. [DOI] [PubMed] [Google Scholar]
- 50.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kitz R, Wilson IB. Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 53.Maurer TS, Fung HL. Comparison of methods for analyzing kinetic data from mechanism-based enzyme inactivation: Application to nitric oxide synthase. AAPS PharmSci. 2000;2:68. doi: 10.1208/ps020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain P, Hsu TD, Arai M, Biermann K, Thaler DS, Nguyen A, Gonzalez PA, Tufariello JM, Kriakov J, Chen B, Larsen MH, Jacobs WR. Specialized Transduction Designed for Precise High-Throughput Unmarked Deletions in Mycobacterium tuberculosis. mBio. 2014;5:e01245–14. doi: 10.1128/mBio.01245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lambert RJW, Pearson J. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J Appl Microbiol. 2000;88:784–790. doi: 10.1046/j.1365-2672.2000.01017.x. [DOI] [PubMed] [Google Scholar]
- 56.Kabsch W. XDS. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mccoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moriarty NW, Grosse-Kunstleve RW, Adams PD. electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr, Sect D: Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis IW, Murray LW, Richardson JS, Richardson DC. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.