

Abstract
A long-standing challenge in drug development is the identification of the mechanisms of action of small molecules with therapeutic potential. A number of methods have been developed to address this challenge, each with inherent strengths and limitations. We here provide a brief review of these methods with a focus on chemical-genetic methods that are based on systematically profiling the effects of genetic perturbations on drug sensitivity. In particular, application of these methods to mammalian systems has been facilitated by the recent advent of CRISPR-based approaches, which enable one to readily repress, induce, or delete a given gene and determine the resulting effects on drug sensitivity.
Graphical Abstract
Main text
The importance of identifying molecular targets
Small molecules provide critical tools both for basic biological discovery and therapeutic benefit. Traditionally, such small molecules were frequently derived from natural or man-made products with interesting physiological effects, but with advances in combinatorial chemical synthesis and screening methodologies, active compounds are now largely identified from cell-based phenotypic screens or in vitro screens for inhibition of a defined target. Regardless of origin, central to the utility of such a compound is an understanding of the molecular target(s) through which the compound exerts its physiological effect as well as a comprehensive understanding of off-target effects. For drug candidates, this knowledge is critical for subsequent development toward increased efficacy and for selection of patient populations that might respond most effectively to treatment. Understanding how drugs act is becoming particularly important with the interest in precision medicine efforts1, in which therapies are precisely targeted at the genetic and environmental background of a patient and which therefore require drugs with high specificity for well-defined targets. For chemical probes, a lack of definitive functional characterization can severely confound results, limiting their utility2.
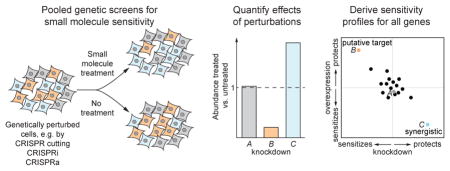
Given the importance of target identification (also termed target deconvolution), it is perhaps not surprising that a number of experimental and computational methods have been developed to address it, each with its advantages and blind spots. Many of these methods have been reviewed in detail previously3, 4, 5, 6, 7, 8, 9. Experimental strategies largely fall into three major categories: chemical-genetic methods, affinity-based biochemical methods, and comparative profiling methods. We here provide a brief review of these methods while focusing in more depth on hypothesis-free chemical-genetic screening methods, which rely on systematically profiling the effect of genetic perturbations on sensitivity to small molecules (Figure 1). After pioneering work in yeast10, 11, these chemical-genetic methods have now come to fruition in human cells with the advent of CRISPR-based genome-wide screening approaches5, 12.
Figure 1.

Schematic of chemical-genetic screen. A library of cells each with a specific genetic perturbation, such as deletion, knockdown, or overexpression of a gene, is exposed to a compound of interest, either in pooled or arrayed format. Quantification of the abundance of library members before and after treatment reveals the effect of each genetic perturbation on sensitivity. In this example, deletion of gene B confers hypersensitivity; thus, gene B likely operates in the pathway targeted by the compound. By contrast, deletion of gene Z confers resistance; thus, gene Z might be required for uptake of the compound.
Development of chemical-genetic strategies for target identification in yeast
The central tenet of chemical-genetic strategies is that sensitivity to a small molecule or drug is influenced by the expression level(s) of its molecular target(s)10, 11. This link between chemical sensitivity and gene dosage was clearly established in yeast: cells carrying a loss-of-function mutation in a specific pathway were typically found to be sensitive to drugs targeting that pathway13, and inversely, increasing the dosage of a drug’s molecular target by expression from a multicopy plasmid conferred resistance14, 15. These observations implied that for drugs with unknown mechanisms of action, target hypotheses should emerge from identification of genes whose expression levels modulate sensitivity. The next challenge was to rapidly and comprehensively identify such genes.
Methods to address this challenge first came about in yeast, specifically Saccharomyces cerevisiae, fueled by the completion of its genome sequence over 20 years ago16 and the ensuing rapid advent of functional genomics methods. As part of efforts to understand the functions of poorly characterized genes, libraries of diploid strains carrying heterozygous deletions of genes were created17, 18. Such deletions result in reduced gene dosage; thus, strains with deletions in a gene or pathway targeted by a drug should exhibit hypersensitivity to the drug. Indeed, this relationship held for several known drug:target pairs such as tunicamycin:ALG7 and benomyl:TUB1.19
At the same time, the development of methods to generate barcoded strain libraries20 coupled with the advent of microarray technology21, 22, which enabled quantification of the abundance of each individual strain in these libraries, allowed for parallel pooled screens in many conditions. Thus, drug sensitivity as a function of gene dosage could be determined for many genes and drugs simultaneously by exposing a pooled library of barcoded heterozygous deletion strains to a drug and quantifying the change in abundance of each strain after the treatment (Figure 1). In a proof-of-principle implementation of this methodology, termed haploinsufficiency profiling (HIP), tunicamycin sensitivity was profiled for a panel of strains carrying heterozygous deletions of selected essential genes, which revealed depletion of ALG7 heterozygotes, among others19. Genome-wide HIP has since been applied to reveal the molecular targets and mechanisms of action as well as modifiers of sensitivity of a wide diversity of drugs including the cancer therapeutics 5-fluorouracil and methotrexate and the antifungal agent fenpropimorph23, 24.
In separate implementations of this concept, HIP screens were performed in arrayed format, in which strains carrying individual heterozygous deletions are robotically arrayed onto agar plates supplemented with compounds of interest and colony size is monitored to quantify growth rate.25 Alternative approaches to modulating gene function or expression were also developed, such as isolating temperature-sensitive alleles with compromised function or tagging the 3′-UTRs of genes with antibiotic markers, which destabilizes the corresponding mRNAs and thereby reduces expression levels (decreased abundance by mRNA perturbation, DAmP)26. Similar to HIP, profiling the chemical sensitivities of temperature-sensitive or DAmP strains allows for identification of genes targeted by small molecules27, 28, 29.
Methods to reduce but not entirely eliminate expression are required for essential genes, as by definition homozygous deletion of such genes is lethal. For non-essential genes, however, both alleles can be deleted. Profiling drug sensitivities of libraries of such homozygous deletion mutants (homozygous profiling, HOP), in analogy to the HIP approach, can provide additional information on drug mechanisms of action. In particular, although HOP rarely reveals the direct target of a drug because most cytotoxic drugs target essential genes, HOP can identify modifiers of sensitivity and mechanisms of resistance such as efflux pumps30, 31. More importantly, the measured sensitivity phenotypes can be used to generate profiles of genetic dependencies for each drug, which are similar for drugs with similar mechanisms of action. Thus, after generating reference profiles for a set of drugs with known mechanisms of action, a clustering or pattern matching approach can be employed to classify poorly characterized drugs by similarity of their genetic dependencies30, 32. This pattern matching approach was recently extended to phenotypes derived from both HIP and HOP for a large panel of >3,000 chemicals, generating an array of fitness signatures that allowed for large-scale assignment of molecular mechanisms of action33.
Loss-of-function profiling approaches such as HIP and HOP have enjoyed substantial success in chemical genetics efforts. A potential drawback to such approaches, however, is that sensitivity or resistance to a drug upon reduced expression of a gene can stem from pleiotropic or indirect effects rather than direct interaction of the gene product with the drug. A complementary approach therefore is to profile the effect of targeted overexpression of genes on drug sensitivity (multicopy suppression profiling, MSP), as increased levels of the molecular target of a drug often lead to resistance14 (with exceptions for subunits of multi-protein complexes or for cases in which the drug-target complex mediates toxicity such as for some topoisomerase-I inhibitors). Such MSP screens were used, for example, to demonstrate that phenylaminopyrimidine targets protein kinase C (Pkc1)34 and to identify genes in the TOR pathway by profiling rapamycin sensitivity35.
Combining deletion and overexpression profiling approaches further increases the sensitivity in identifying molecular targets. In particular, genes directly on the pathway targeted by a small molecule are expected to have anti-correlated phenotypes in deletion and overexpression profiling, whereas individual loss-of-function or gain-of-function screens may be prone to false negatives or false positives (see above). Indeed, in a proof-of-principle study, integration of HIP, HOP, and MSP data readily identified the molecular targets of several small molecules with unknown mechanisms of action with improved sensitivity over any of the individual approaches36.
Yeast was a natural test bed for these chemical-genetic approaches, and further efforts are ongoing, including the mapping of structure-function relationships and drug-drug interactions as well as the profiling of human alleles without yeast counterparts via heterologous expression. Despite the advantages of yeast as a model system, it has intrinsic limitations in identifying the molecular targets of drug candidates or probes for use in human cells. In particular, yeast has a cell wall that influences drug activity, many essential genes or their isoforms or drug binding pockets are not conserved between yeast and humans, and yeast cannot recapitulate the complex organization of a multicellular organism. Thus, with the intellectual groundwork of chemical-genetic approaches laid by these pioneering efforts, attention turned increasingly to implementation in human cells.
Functional genomics tools for chemical-genetic screens in human cells
The past decade has witnessed the rapid development of functional genomics tools for human cells such as RNA interference (RNAi), barcoded open reading frame (ORF) libraries, and most recently CRISPR-based methods37, 38, 39, 40, 41. Briefly, RNAi relies on short RNAs complementary to target mRNAs that are introduced into cells to mediate degradation of these mRNAs and thereby reduce target gene expression37, 38. ORF libraries overexpress human ORFs from standardized expression vectors with a strong constitutive promoter39. Finally, in CRISPR-based approaches, an effector protein such as Cas9 is programmed with a short-guide RNA (sgRNA) that directs the effector to a DNA locus of interest via sequence complementarity. Cas9 then introduces a double-strand break, which triggers DNA repair mechanisms that in protein coding regions frequently result in frameshift mutations40, 41. Other CRISPR-based approaches rely on a catalytically inactivated mutant of Cas9 (dCas9)42 that is essentially a highly programmable DNA-binding protein and can be used to deliver transcription factors to target loci to mediate knockdown or overexpression without DNA cutting41. With the availability of these tools to systematically perturb gene expression, the concepts developed from chemical-genetic efforts in yeast can now be applied directly in human cells.
RNAi and CRISPR-based methods are readily amenable to pooled screening experiments akin to those developed for yeast. In particular, when the expression constructs for short-hairpin RNAs (shRNAs) in RNAi or sgRNAs in CRISPR-based methods are stably inserted into the genome, for example by lentiviral transduction, the DNA copies can directly serve as molecular barcodes because their sequences encode the identities of the targeted genes. In a pooled screen, a library of shRNA or sgRNA expression constructs, with elements targeting genes of interest such as all protein-coding genes in the genome, is introduced into cells such that each cell stably integrates one construct (Figure 2A). The abundance of all shRNAs or sgRNAs in the cell population is then quantified, most commonly by next-generation sequencing, both at the outset of the experiment and after growth under a condition of interest such as drug-induced selective pressure. This quantification reveals the effect of expression of a given shRNA or sgRNA, and by proxy the effect of perturbation of the targeted gene, on growth in this condition43. In such pooled screens, a large number of genetic perturbations can be queried rapidly in a single experiment, allowing for coverage of the entire genome, and all cells are grown in identical conditions, avoiding batch effects. Thereby, pooled screens enable both the identification of genes required for growth in the absence of other selective pressures and chemical-genetic profiling.
Figure 2.

Schematic of combined CRISPRi/a screens.
A: Schematic of pooled CRISPRi and CRISPRa screens for drug sensitivity. Other pooled screens can be conducted analogously using the corresponding cell lines and libraries.
B: Comparing knockdown and overexpression data such as those from combined CRISPRi and CRISPRa screens reveals genes with anti-correlated sensitivity phenotypes. These genes are likely to be directly involved in the pathway targeted by the drug.
Indeed, shortly after the development of large-scale shRNA libraries, shRNA screens were used to map out modifiers of sensitivity to small molecules44, 45, 46. For example, a screen for sensitivity to Nutlin-3, an MDM2 inhibitor, revealed that cytotoxicity depends on TP53BP144 and genome-scale shRNA screens were used to map out the genetic dependencies of sensitivity to established drugs including etoposide45 and imatinib45. Subsequent studies highlighted how shRNA screens can be used to identify the molecular targets of drugs de novo: after a small-molecule screen had identified a promising anti-leukemia agent (STF-118804) with unknown mechanism of action, a genome-scale shRNA screen revealed that knockdown of nicotinamide phosphoribosyl transferase (NAMPT) strongly sensitized cells to STF-11880447. Subsequent validation confirmed that STF-118804 is a specific NAMPT inhibitor. Similarly, a reporter-based shRNA screen revealed that ISRIB, a compound found to enhance cognitive memory in mice48, inhibits the integrated stress response by triggering dimerization and activation of eIF2B49, a finding that was independently made by screening for ISRIB-resistant mutants50. Despite these successful examples and additional advances in library construction and pooled screening, shRNA methodologies suffer from technical limitations including off-target effects and limited knockdown efficiency51, which confounds results and limits sensitivity and thereby has hampered their widespread application in characterizing the mechanisms of action of drug candidates.
CRISPR-based screening techniques have proven to be more specific and efficacious in most instances41, 52, 53 and are similarly applicable to chemical-genetic screening. For example, early results from CRISPR cutting-mediated loss-of-function screens for resistance against 6-thioguanine54, etoposide54, and vemurafenib55 recapitulated the known mechanisms of action, validating the ability of such screens to identify targets and genetic dependencies of known drugs. CRISPR cutting generally results in complete loss-of-function of target genes, whereas RNAi reduces target levels but does not ablate them. This mechanistic difference was noted in a recent study, in which combined CRISPR cutting screens and shRNA screens were used to determine that the antiviral compound GSK983 inhibits dihydroorotate dehydrogenase56. Although some genes appeared as hits in both screens, the majority only scored in one or the other, highlighting strengths and limitations of both approaches: CRISPR cutting screens for drug sensitivity do not perform well for essential genes, as loss of these genes is lethal, but the high efficacy of CRISPR cutting allows for accurate determination of sensitivity phenotypes of non-essential genes, even if the phenotypes are weak in magnitude. By contrast, shRNA screening can be used to probe phenotypes of essential genes but may miss weaker phenotypes.
An alternative to RNAi and CRISPR cutting that combines some of the strengths of both is provided by CRISPR-mediated transcription interference (CRISPRi), in which a fusion protein of catalytically inactivated Cas9 (dCas9)42, 57 and the KRAB transcriptional repressor is targeted to promoters of genes58. CRISPRi consistently mediates strong transcriptional repression (85–>99%) of target genes and allows for determination of both sensitizing and protective phenotypes of essential genes59, 60, 61, 62, making it an ideal technique for chemical-genetics efforts.
Similarly, the programmable DNA binding activity of dCas9 allowed for the development of CRISPR-based methods for overexpression of target genes (CRISPRa), in which transcriptional activators such as VP64 (a tetra-repeat of the viral transcription activation domain VP16) are delivered via dCas9. In a recent comparison of CRISPRa methods63, three methods in particular were found to consistently mediate strong overexpression of target genes (10–10,000x): the SunTag method59, 64, the SAM method65, and the VPR method66. All of these methods rely on dCas9-mediated recruitment of multiple transcription activator domains. Briefly, in the SunTag method, dCas9 fused to ten copies of a GCN4 peptide epitope is co-expressed with the cognate single-chain variable fragment (scFv) fused to VP64, effectively recruiting ten copies of VP6459, 64. The key advance of the SAM method is an sgRNA constant region engineered to contain MS2 aptamers, allowing for recruitment of transcription factors via the MS2 coat protein (MCP). In the current implementation of SAM, a dCas9-VP64 fusion protein is co-expressed with MCP fused to both the NF-κB trans-activating subunit p65 and the activation domain of human heat shock factor 1 (MCP-p65-HSF1)65. In the VPR method, dCas9 is fused to a tripartite transcription activator consisting of VP64, p65, and the replication and transcription activator (Rta) of Eppstein-Barr virus66. Finally, a VP64-dCas9-VP64 fusion protein also appears to mediate strong CRISPRa activity63, 67. As is typical for CRISPR-based methods, sgRNAs can be rapidly generated against all genes in the genome, enabling genome-wide screens, and each sgRNA sequence can serve as a molecular barcode in pooled screening experiments (see above). Although ORF libraries provide an alternative method to target gene overexpression, such libraries contain elements of broadly varying size, posing issues for pooled lentiviral screening methods.
In general, CRISPR cutting, CRISPRi, and CRISPRa are specific for the target locus of interest and mediate loss-of-function or gain-of-function with high efficiency, with few exceptions: whereas CRISPR cutting may cause non-specific toxicity at amplified loci due to DNA damage responses, CRISPRi and CRISPRa are less specific at bidirectional promoters as they may affect both genes. Indeed, a recent analysis found that CRISPR cutting and CRISPRi yield complementary insights in screens61. For each of the CRISPR screening methods, algorithms have been developed to predict highly active sgRNAs with minimal off-target effects, enabling the generation of compact sgRNA libraries for pooled screening applications60, 68, 69.
Combined CRISPRi and CRISPRa screens for target identification
With CRISPR-based knockdown and overexpression methods available, it had become feasible to perform both loss-of-function and gain-of-function screens in a condition of interest, mirroring those performed in yeast36 and with immediate implications for chemical-genetic screening. Indeed, a comparison of genome-wide sensitivity profiles obtained from proof-of-principle screens with a cholera-diptheria toxin fusion clearly revealed anti-correlated phenotypes for genes targeted by the toxin or on the pathway for toxin entry59. Similarly, measurements of sensitivity to CB-5083, an inhibitor of p97 (VCP), as a function of expression levels of p97 modulated by CRISPRi and CRISPRa revealed a strong correlation70, validating p97 as the target of CB-5083.
These results motivated us to further develop a generalizable chemical-genetic screening strategy for the identification of molecular targets and mechanisms of action of small molecules 62. The strategy uses genome-wide pooled CRISPRi and CRISPRa screens for sensitivity to a compound of interest, with selective pressure induced by several pulses of treatment around LC50, to quantify for each gene how knockdown and overexpression affect sensitivity to the compound. (Figure 2A) Then, genes whose levels directly modulate sensitivity, such as the molecular target, are identified as those with anti-correlated phenotypes in knockdown and overexpression (Figure 2B). Application to rigosertib, a drug that had entered phase III clinical trials for high-risk myelodysplastic syndrome but whose mechanism of action had remained controversial with multiple proposed molecular targets71, 72, 73, highlighted the potential of this approach. The combined CRISPRi/a sensitivity profiles identified microtubule destabilization as rigosertib’s mechanism of action, as opposed to previously proposed mechanisms of action62. Subsequent follow-up experiments, including chemical-genetic comparison to known microtubule destabilizing agents and the isolation of a rigosertib-resistant tubulin mutant, confirmed that rigosertib kills cells by destabilizing microtubules.
A few observations from the rigosertib case are worth highlighting. The microtubule signature emerged in the CRISPRi/a profiles despite functional redundancy among tubulin genes and despite strong pleiotropic effects: knockdown of many essential genes conferred apparent protection against rigosertib, likely because these knockdowns prevent cells from reaching mitosis, when cells are highly dependent on microtubules and thus most sensitive to rigosertib. Overexpression of these genes, however, did not impact rigosertib sensitivity, and thus the protective phenotypes observed using CRISPRi could be attributed to pleiotropy. Such indirect effects likely contributed to the controversy over rigosertib’s mechanism of action, during which evidence for several other targets had been obtained from targeted cellular assays, and they similarly often dominate sensitivity profiles especially if the small molecule targets a central biological process such as cell division or the microtubule network. This work illustrates the potential of the combined CRISPRi/a approach to separate direct and indirect effects on drug sensitivity, which provides a critical advantage over any individual screening approach.
Other approaches to target identification
By and large, additional methods to identify the targets of bioactive compounds are either based on identifying or inferring binding partners, phenotypic comparison to compounds with known mechanisms of action, or screening for resistance-conferring mutations. We here provide a brief summary of these methods; the reader is referenced to other reviews for detailed discussions.
Affinity-based methods
Affinity-based methods to directly identify binding partners of small molecules have been mainstays in target identification for decades. Initially developed to purify enzymes74, these methods rely on derivatization of the small molecule and immobilization on a solid-phase matrix. Incubation of this matrix with cell lysate and separation of non-binders allows for affinity purification and subsequent determination of the identities of binding partners. In one of the earliest examples of this approach, the cellular binding partner for the immunosuppressant FK506 was identified to be the cis-trans peptidyl-prolyl isomerase FKBP75. In a related tour-de-force, cyclophilin was identified as the binding partner of cyclosporin A by fractionation of lysates after incubation with radioactively labeled cyclosporin A76. Widespread implementation of such approaches was long limited by challenges in determining the sequences of the isolated proteins, but the development of high-throughput mass spectrometry techniques coupled with the availability of genomic sequence information now allows for straightforward identification of binding partners by proteomic methods. The resulting chemical proteomics workflow has been applied widely7 and continues to evolve. For example, furnishing small molecules with both a photoactivatable diazirine crosslinker and an alkyne group allows for photo-crosslinking to non-covalent binding partners followed by affinity purification and mass spectrometry analysis. A panel of such molecules was recently employed in an elegant fragment-based screening approach that enabled mapping of thousands of small molecule-protein interactions77. Developments in multiplexed MS have furthermore given rise to the related proteome-wide thermal profiling approach, in which the melting curves of all proteins are determined in the presence and absence of a small molecule and proteins that are stabilized by the small molecule are inferred to be binders78, 79.
Comparative profiling
Comparative profiling methods rely on matching phenotypes obtained with a compound in question to those obtained with a set of reference compounds with known mechanisms of action, with the expectation that compounds with similar mechanisms of action will elicit similar phenotypes. In a large-scale effort in the 1980s, the National Cancer Institute assembled a reference set of 60 cancer cell lines (NCI-60) and profiled sensitivity of each cell line to a large panel of compounds80, 81. Indeed, compounds with similar mechanisms of action showed similar patterns of sensitivity, and comparison to these patterns revealed, for example, halichondrin B as an inhibitor of microtubule polymerization. Steady profiling of additional compounds has produced large-scale data sets that are rich resources for target identification.
Phenotypes for comparative profiling can also be derived from chemical-genetic profiling (see above), which has been used to identify or validate drug targets both in yeast32 and in human cells62, 82, or from various other methods such as transcriptional profiling or high-content microscopy. Methods to compare transcriptional profiles were again pioneered in yeast, both by comparing expression profiles of cells treated with panels of drugs83 or by comparing expression profiles from drug-treated cells with those from deletion strains, with the assumption that inhibiting a protein with a drug would be equivalent to deletion of the gene84. Improvements in transcriptome profiling methodologies led to large-scale efforts to comprehensively profile the transcriptional responses of human or other mammalian cell lines to large panels of small molecules8. These efforts have resulted in public databases such as the Connectivity Map85 that can be queried with transcriptional data derived using a compound of interest. The ability to read out both genetic perturbations and transcriptomes from pools of cells using recently developed single-cell sequencing methodologies such as perturb-seq now additionally allows for simultaneous comparative profiling and chemical-genetic screening for hundreds to thousands of candidate genes86, 87, 88, 89. A recent study further demonstrated that even basal gene expression, i.e. expression in the absence of treatment, can be used to predict mechanisms of action. In particular, a gene whose basal expression correlates strongly with sensitivity to a small molecule across cell lines frequently encodes the molecular target, a transporter, or an enzyme that activates the small molecule90. Other comparative profiling approaches have used phenotypes measured by high-content microscopy such as various measurements of cell morphology and cellular contents9, 91. Many other implementations of comparative profiling have been developed that are increasingly relying on computational inference and machine learning trained on multi-dimensional datasets.
Resistant mutants
All of the discussed methods readily generate hypotheses for the molecular target of a compound of interest, but require subsequent verification that the compound indeed exerts its physiological effect through this target. One of the most compelling demonstrations of physiological relevance is the identification of a mutation in the target that confers resistance to the compound. This concept has been used to directly identify molecular targets of compounds by screening for resistant mutants and then identifying the resistance-conferring mutation. In particular, this approach has been widely employed in bacteria, which are haploid by nature and carry small genomes that can be mutated to near-saturation, as well as in yeast. In one of the most influential examples, the TOR kinases TOR1 and TOR2 were discovered in yeast by characterizing rapamycin-resistant yeast mutants92. Saturation screens, however, have been more difficult to implement in higher eukaryotes due to the difficulty in generating dominant resistant mutations as well as the subsequent identification in genomes containing billions of base pairs. Approaches to overcome these challenges exist: the use of near-haploid cell lines can mitigate the former challenge; the latter challenge was recently addressed by identifying resistance-conferring mutations by exome sequencing or RNA-seq rather than genome sequencing, which drastically reduces the required sequencing space93, 94, 95.
Strengths and limitations of target identification strategies
Each of the approaches described here comes with a set of inherent strengths and caveats, consideration of which is critical to successful implementation. At the same time, no single approach will provide the perfect solution, and instead a set of complementary approaches are needed to ultimately identify the molecular target of a small molecule in question.
Improvements in mass spectrometry technology such as increases in throughput and sensitivity, new statistical toolboxes, and workflows to reduce background have made affinity-based methods increasingly robust. Such approaches can, for example, distinguish different isoforms of binding partners, which other methods for target identification are largely insensitive to. To successfully identify binding partners, however, chemical proteomics workflows require derivatization of the compound, which is frequently a bottleneck or may introduce artifacts by changing the binding profile. In addition, detecting binding to membrane proteins or other biochemically poorly behaved proteins and complexes can be challenging, as they can be difficult to maintain in a native state. These concerns are somewhat alleviated in the thermal profiling approach, but this approach may fail if small molecule binding does not stabilize its binding partner or if there is a route to unfolding even when the small molecule is bound. Vice versa, low-affinity but high-abundance binding partners in the cell can complicate deconvolution of the functional target and off-target binders or even prevent detection of the true target. More generally, affinity-based methods test binding and not necessarily activity; the highest-affinity binding partner need not be the target through which a small molecule exerts its physiological effect.
Comparative profiling methods generally do not require compound derivatization or specialized cell models, as endogenous phenotypes can be used as a readout. Such methods are particularly powerful once a large array of compounds has been profiled to generate reference data sets for accurate pattern matching and are amenable to deep learning approaches to identify more complex relationships between small molecules or biological pathways. Such data sets, however, may not be available in the model of interest. More broadly, inhibition of diverse targets in the cell may result in similar phenotypes, for example due to pleiotropic effects; in such cases it can be challenging to identify the proximal mechanism of action. This approach may also fail for compounds with novel mechanisms of action as by concept this approach relies on comparing phenotypes to those of compounds with known mechanism of action.
Chemical-genetic approaches do not require reference data sets or small molecule derivatization and probe function rather than binding and therefore are less prone to identifying non-functional binding partners. In human cells, CRISPR approaches are relatively easy to implement and mediate robust knockout, knockdown, or overexpression. The cell lines generated thus far as well as the libraries are freely available. Nonetheless, loss-of-function and gain-of-function approaches individually are prone to false negatives and false positives. Loss-of-function approaches, for example, may fail in cases of redundancy or pleiotropy, whereas overexpression of a drug target may not provide resistance if the target functions as part of a multiprotein complex. Combining loss-of-function and gain-of-function screens can mitigate these caveats36, 62. In particular, the combined data provide a filter to separate direct and indirect effects and can reveal targets in the presence of redundancy or pleiotropy (Figure 2B). Even if the direct molecular target does not score strongly in such screens, as was the case in the rigosertib screen, the availability of sensitivity phenotypes for all genes in the genome typically identifies the process targeted by the compound. Two key requirements need to be fulfilled for the screening approach: a model system amenable to genetic manipulation needs to be available to implement the CRISPR effectors, and the compound of interest must generate a selectable phenotype. In the simplest manifestation, this phenotype may be growth, but more complex phenotypes such as expression levels of a reporter gene, cell migration, or differentiation can also be probed. As long as these requirements are fulfilled, the combined CRISPRi/a approach provides a high-throughput and sensitive method to probe small molecule mechanisms of action.
Regardless of approach, independent validation of the putative mechanism of action is usually required, most compellingly by identification of a resistant mutant. Direct identification of such mutants is technically challenging especially in diploid organisms in which they need to be dominant to be identified, but target hypotheses generated from other target identification approaches can greatly reduce the search space. For non-essential putative molecular targets, the resistant mutant may be a homozygous deletion mutant; in other cases rational structure-guided approaches or random mutagenesis approaches such as those enabled by newly developed CRISPR-mediated base editors96, 97 may prove successful.
Perspective
The rapid development of high-throughput functional genomics tools has enabled large-scale chemical-genetic screening efforts to identify the molecular targets and mechanisms of action of therapeutic candidates and chemical probes. Newer CRISPR-based approaches in particular can be applied in a wide variety of cellular models, including cancer cell lines or primary cells as well as animal models. The underlying principles furthermore can be readily combined with emerging methods for targeted mutagenesis such as CRISPR cutting98 or base editing96, 97 to enable target identification with amino acid resolution. With the ability to probe drug mechanism of action directly and in vivo, chemical-genetic approaches provide natural complements to other approaches in target identification such as affinity-based methods.
Application of the approaches described here is not limited to eukaryotic models, which have classically been the main targets of drug development efforts. Implementations in bacteria are simultaneously revealing drug mechanisms of action and new biology, for example by comparative profiling of drug sensitivity in the Escherichia coli Keio knockout collection99, by chemical-genetic screening of Bacillus subtilis strains carrying CRISPRi-mediated knockdowns of essential genes100, or by microscopic profiling of drug-treated cells101. With increasing appreciation for the central role of bacteria in human health and disease, such efforts may prove critical toward developing treatments for microbiome-associated deficiencies or infections with multidrug-resistance bacteria. Regardless of ultimate target, there now is a large suite of tools at our disposal to usher in a new era in the development of precisely targeted therapeutics.
Acknowledgments
We thank E. Costa and J. Hussmann for critical reading of the manuscript and C. Gross and members of the Weissman lab for helpful discussions. MJ was funded by National Institutes of Health (NIH) post-doctoral fellowship F32 GM116331. JSW is funded by NIH grants P50 GM102706, U01 CA168370, and R01 DA036858. JSW is a Howard Hughes Medical Institute Investigator. JSW has filed a patent application related to CRISPRi and CRISPRa screening (PCT/US15/40449) and is a founder of KSQ Therapeutics, a CRISPR functional genomics company.
Glossary
- Chemical-genetic screen
Systematic profiling of small molecule sensitivity for a library of strains or cell lines carrying genetic perturbations.
- Haploinsufficiency profiling (HIP)
Chemical-genetic screen in a library of yeast heterozygous deletion strains.
- Homozygous deletion profiling (HOP)
Chemical-genetic screen in a library of yeast homozygous deletion strains.
- Multicopy suppression profiling (MSP)
Chemical-genetic screen in a library of yeast strains with multicopy expression plasmids.
- RNA interference (RNAi)
Inhibition of gene expression by a complementary short RNA.
- CRISPR
Clustered regularly interspaced short palindromic repeats, a bacterial and archaeal adaptive immune system that mediates degradation of DNA or RNA molecules based on sequence complementarity using effector proteins and short RNAs.
- Cas9/dCas9
A CRISPR effector protein that binds to target DNA via sequence complementarity of an associated short RNA and cleaves the target DNA. dCas9 is a nuclease-dead variant that binds but does not cleave.
- Short-guide RNA (sgRNA)
a short RNA, generated by fusion of a targeting sequence and a constant region, that binds Cas9 and directs it to target DNA.
- CRISPR interference (CRISPRi)
transcription repression mediated by dCas9 or fusion proteins of dCas9 or other CRISPR effectors.
- CRISPR activation (CRISPRa)
transcription activation mediated by fusion proteins of dCas9 or other CRISPR effectors.
- Pooled screen
A screen in which cells with different genetic perturbations are grown in a single pool and phenotypes are determined by quantifying cells with each perturbation at the end of the screen, for example by deep sequencing.
References
- 1.National Research Council. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease. 2011. [PubMed] [Google Scholar]
- 2.Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frederiksen M, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Müller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Ross R, Roth B, Schapira M, Schreiber SL, Shoichet B, Sundström M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–541. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong F, Cheung AK, Huang SA. Chemical genetics-based target identification in drug discovery. Annu Rev Pharmacol Toxicol. 2012;52:57–78. doi: 10.1146/annurev-pharmtox-010611-134639. [DOI] [PubMed] [Google Scholar]
- 4.Schenone M, Dančík V, Wagner BK, Clemons PA. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nijman SMB. Functional genomics to uncover drug mechanism of action. Nat Chem Biol. 2015;11:942–948. doi: 10.1038/nchembio.1963. [DOI] [PubMed] [Google Scholar]
- 6.Chan JNY, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol Sci. 2010;31:82–88. doi: 10.1016/j.tips.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 8.Palchaudhuri R, Hergenrother PJ. Transcript profiling and RNA interference as tools to identify small molecule mechanisms and therapeutic potential. ACS Chem Biol. 2011;6:21–33. doi: 10.1021/cb100310h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carpenter AE. Image-based chemical screening. Nat Chem Biol. 2007;3:461–465. doi: 10.1038/nchembio.2007.15. [DOI] [PubMed] [Google Scholar]
- 10.Smith AM, Ammar R, Nislow C, Giaever G. A survey of yeast genomic assays for drug and target discovery. Pharmacol Ther. 2010;127:156–164. doi: 10.1016/j.pharmthera.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho CH, Piotrowski J, Dixon SJ, Baryshnikova A, Costanzo M, Boone C. Combining functional genomics and chemical biology to identify targets of bioactive compounds. Curr Opin Chem Biol. 2011;15:66–78. doi: 10.1016/j.cbpa.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 12.Kampmann M. Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem Commun (Camb ) 2017;53:7162–7167. doi: 10.1039/c7cc02349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–1068. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 14.Rine J, Hansen W, Hardeman E, Davis RW. Targeted selection of recombinant clones through gene dosage effects. Proc Natl Acad Sci U S A. 1983;80:6750–6754. doi: 10.1073/pnas.80.22.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Launhardt H, Hinnen A, Munder T. Drug-induced phenotypes provide a tool for the functional analysis of yeast genes. Yeast. 1998;14:935–942. doi: 10.1002/(SICI)1097-0061(199807)14:10<935::AID-YEA289>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 16.Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, Louis EJ, Mewes HW, Murakami Y, Philippsen P, Tettelin H, Oliver SG. Life with 6000 genes. Science. 1996;274:567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 17.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Véronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Yu K, Zimmermann K, Philippsen P, Johnston M, Davis RW. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 18.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian K, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang C, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 19.Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, Davis RW. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21:278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- 20.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet. 1996;14:450–456. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 21.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 22.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 23.Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, Garrett-Engele P, Rush CM, Bard M, Schimmack G, Phillips JW, Roberts CJ, Shoemaker DD. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116:121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 24.Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, Chu AM, Jordan MI, Arkin AP, Davis RW. Chemogenomic profiling: Identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci U S A. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baetz K, McHardy L, Gable K, Tarling T, Rebérioux D, Bryan J, Andersen RJ, Dunn T, Hieter P, Roberge M. Yeast genome-wide drug-induced haploinsufficiency screen to determine drug mode of action. Proc Natl Acad Sci U S A. 2004;101:4525–4530. doi: 10.1073/pnas.0307122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, Weissman JS, Krogan NJ. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 27.van Pel DM, Stirling PC, Minaker SW, Sipahimalani P, Hieter P. Saccharomyces cerevisiae genetics predicts candidate therapeutic genetic interactions at the mammalian replication fork. G3 (Bethesda) 2013;3:273–282. doi: 10.1534/g3.112.004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Z, Costanzo M, Heisler LE, Paw J, Kaper F, Andrews BJ, Boone C, Giaever G, Nislow C. Yeast barcoders: A chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat Methods. 2008;5:719–725. doi: 10.1038/nmeth.1231. [DOI] [PubMed] [Google Scholar]
- 30.Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- 31.Lee W, St Onge RP, Proctor M, Flaherty P, Jordan MI, Arkin AP, Davis RW, Nislow C, Giaever G. Genome-wide requirements for resistance to functionally distinct DNA-damaging agents. PLoS Genet. 2005;1:e24. doi: 10.1371/journal.pgen.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho C, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126:611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 33.Lee AY, St Onge RP, Proctor MJ, Wallace IM, Nile AH, Spagnuolo PA, Jitkova Y, Gronda M, Wu Y, Kim MK, Cheung-Ong K, Torres NP, Spear ED, Han MKL, Schlecht U, Suresh S, Duby G, Heisler LE, Surendra A, Fung E, Urbanus ML, Gebbia M, Lissina E, Miranda M, Chiang JH, Aparicio AM, Zeghouf M, Davis RW, Cherfils J, Boutry M, Kaiser CA, Cummins CL, Trimble WS, Brown GW, Schimmer AD, Bankaitis VA, Nislow C, Bader GD, Giaever G. Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science. 2014;344:208–211. doi: 10.1126/science.1250217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luesch H, Wu TYH, Ren P, Gray NS, Schultz PG, Supek F. A genome-wide overexpression screen in yeast for small-molecule target identification. Chem Biol. 2005;12:55–63. doi: 10.1016/j.chembiol.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 35.Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, Schreiber SL. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat Chem Biol. 2006;2:103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- 36.Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, Proctor M, Shokat KM, Zhang C, Davis RW, Giaever G, St Onge RP, StOnge RP, Nislow C. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4:498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- 37.Hannon GJ. RNA interference. Nature. 2002;418:418244a. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 38.Mohr SE, Smith JA, Shamu CE, Neumüller RA, Perrimon N. RNAi screening comes of age: Improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L, Vidal M, Hahn WC, Hill DE, Root DE. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright AV, Nuñez JK, Doudna JA. Biology and applications of CRISPR systems: Harnessing nature’s toolbox for genome engineering. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016;85:227–264. doi: 10.1146/annurev-biochem-060815-014607. [DOI] [PubMed] [Google Scholar]
- 42.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kampmann M, Bassik MC, Weissman JS. Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci U S A. 2013;110:2317. doi: 10.1073/pnas.1307002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brummelkamp TR, Fabius AWM, Mullenders J, Madiredjo M, Velds A, Kerkhoven RM, Bernards R, Beijersbergen RL. An shRNA barcode screen provides insight into cancer cell vulnerability to MDM2 inhibitors. Nat Chem Biol. 2006;2:202–206. doi: 10.1038/nchembio774. [DOI] [PubMed] [Google Scholar]
- 45.Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, Mermel C, Barbie DA, Awad T, Zhou X, Nguyen T, Piqani B, Li C, Golub TR, Meyerson M, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:9053–9058. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matheny CJ, Wei MC, Bassik MC, Donnelly AJ, Kampmann M, Iwasaki M, Piloto O, Solow-Cordero DE, Bouley DM, Rau R, Brown P, McManus MT, Weissman JS, Cleary ML. Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem Biol. 2013;20:1352–1363. doi: 10.1016/j.chembiol.2013.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, Walter P. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, Verschueren E, Johnson JR, Krogan NJ, Fraser CS, Weissman JS, Renslo AR, Walter P. Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. Elife. 2015;4:e07314. doi: 10.7554/eLife.07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, Ron D. Stress responses. mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science. 2015;348:1027–1030. doi: 10.1126/science.aaa6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaelin WG. Molecular biology. use and abuse of RNAi to study mammalian gene function. Science. 2012;337:421–422. doi: 10.1126/science.1225787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kampmann M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem Biol. 2017 doi: 10.1021/acschembio.7b00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deans RM, Morgens DW, Ökesli A, Pillay S, Horlbeck MA, Kampmann M, Gilbert LA, Li A, Mateo R, Smith M, Glenn JS, Carette JE, Khosla C, Bassik MC. Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol. 2016;12:361–366. doi: 10.1038/nchembio.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife. 2016;5:e19760. doi: 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenbluh J, Xu H, Harrington W, Gill S, Wang X, Vazquez F, Root DE, Tsherniak A, Hahn WC. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat Commun. 2017;8:15403. doi: 10.1038/ncomms15403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jost M, Chen Y, Gilbert LA, Horlbeck MA, Krenning L, Menchon G, Rai A, Cho MY, Stern JJ, Prota AE, Kampmann M, Akhmanova A, Steinmetz MO, Tanenbaum ME, Weissman JS. Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Mol Cell. 2017;68:223e6. doi: 10.1016/j.molcel.2017.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chavez A, Tuttle M, Pruitt BW, Ewen-Campen B, Chari R, Ter-Ovanesyan D, Haque SJ, Cecchi RJ, Kowal EJK, Buchthal J, Housden BE, Perrimon N, Collins JJ, Church G. Comparison of Cas9 activators in multiple species. Nat Methods. 2016;13:563–567. doi: 10.1038/nmeth.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, Iyer PRE, Lin S, Kiani S, Guzman CD, Wiegand DJ, Ter-Ovanesyan D, Braff JL, Davidsohn N, Housden BE, Perrimon N, Weiss R, Aach J, Collins JJ, Church GM. Highly efficient Cas9-mediated transcriptional programming. Nat Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakraborty S, Ji H, Kabadi AM, Gersbach CA, Christoforou N, Leong KW. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Reports. 2014;3:940–947. doi: 10.1016/j.stemcr.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, Gozdecka M, Ohnishi S, Cooper J, Patel M, McKerrell T, Chen B, Domingues AF, Gallipoli P, Teichmann S, Ponstingl H, McDermott U, Saez-Rodriguez J, Huntly BJP, Iorio F, Pina C, Vassiliou GS, Yusa K. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon M, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou H, Wustrow D, Rolfe M. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reddy MVR, Venkatapuram P, Mallireddigari MR, Pallela VR, Cosenza SC, Robell KA, Akula B, Hoffman BS, Reddy EP. Discovery of a clinical stage multi-kinase inhibitor sodium (E)-2-{2-methoxy-5-[(2′,4′,6′-trimethoxystyrylsulfonyl)methyl]phenylamino}acetate (ON 01910. na): Synthesis, structure-activity relationship, and biological activity. J Med Chem. 2011;54:6254–6276. doi: 10.1021/jm200570p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MVR, Ueno L, Hart JR, Vogt PK, Mulholland D, Guha C, Aggarwal AK, Reddy EP. A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell. 2016;165:643–655. doi: 10.1016/j.cell.2016.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, Stephen AG, Holderfield M, Morrison DK. Inhibition of ras/raf/MEK/ERK pathway signaling by a stress-induced phospho-regulatory circuit. Mol Cell. 2016;64:875–887. doi: 10.1016/j.molcel.2016.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cuatrecasas P, Wilchek M, Anfinsen CB. Selective enzyme purification by affinity chromatography. Proc Natl Acad Sci U S A. 1968;61:636–643. doi: 10.1073/pnas.61.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harding MW, Galat A, Uehling DE, Schreiber SL. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature. 1989;341:758–760. doi: 10.1038/341758a0. [DOI] [PubMed] [Google Scholar]
- 76.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science. 1984;226:544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 77.Parker CG, Galmozzi A, Wang Y, Correia BE, Sasaki K, Joslyn CM, Kim AS, Cavallaro CL, Lawrence RM, Johnson SR, Narvaiza I, Saez E, Cravatt BF. Ligand and target discovery by fragment-based screening in human cells. Cell. 2017;168:541e29. doi: 10.1016/j.cell.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, Nordlund P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341:84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 79.Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S, Kuster B, Nordlund P, Bantscheff M, Drewes G. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346:1255784. doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 80.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;81:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 81.Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 82.Jiang H, Pritchard JR, Williams RT, Lauffenburger DA, Hemann MT. A mammalian functional-genetic approach to characterizing cancer therapeutics. Nat Chem Biol. 2011;7:92–100. doi: 10.1038/nchembio.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marton MJ, DeRisi JL, Bennett HA, Iyer VR, Meyer MR, Roberts CJ, Stoughton R, Burchard J, Slade D, Dai H, Bassett DE, Hartwell LH, Brown PO, Friend SH. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat Med. 1998;4:1293–1301. doi: 10.1038/3282. [DOI] [PubMed] [Google Scholar]
- 84.Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- 85.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 86.Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, Pak RA, Gray AN, Gross CA, Dixit A, Parnas O, Regev A, Weissman JS. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1882e21. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, Adamson B, Norman TM, Lander ES, Weissman JS, Friedman N, Regev A. Perturb-seq: Dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1866e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, Amit I. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell. 2016;167:1896e15. doi: 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 89.Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, Bock C. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017;14:297–301. doi: 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, Soule CK, Alexander B, Li A, Montgomery P, Kotz JD, Hon CS, Munoz B, Liefeld T, Dančík V, Haber DA, Clish CB, Bittker JA, Palmer M, Wagner BK, Clemons PA, Shamji AF, Schreiber SL. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–116. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perlman ZE, Slack MD, Feng Y, Mitchison TJ, Wu LF, Altschuler SJ. Multidimensional drug profiling by automated microscopy. Science. 2004;306:1194–1198. doi: 10.1126/science.1100709. [DOI] [PubMed] [Google Scholar]
- 92.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 93.Wacker SA, Houghtaling BR, Elemento O, Kapoor TM. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol. 2012;8:235–237. doi: 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kasap C, Elemento O, Kapoor TM. DrugTargetSeqR: A genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat Chem Biol. 2014;10:626–628. doi: 10.1038/nchembio.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smurnyy Y, Cai M, Wu H, McWhinnie E, Tallarico JA, Yang Y, Feng Y. DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells. Nat Chem Biol. 2014;10:623–625. doi: 10.1038/nchembio.1550. [DOI] [PubMed] [Google Scholar]
- 96.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hess GT, Tycko J, Yao D, Bassik MC. Methods and applications of CRISPR-mediated base editing in eukaryotic genomes. Mol Cell. 2017;68:26–43. doi: 10.1016/j.molcel.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. Phenotypic landscape of a bacterial cell. Cell. 2011;144:143–156. doi: 10.1016/j.cell.2010.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo B, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell. 2016;165:1493–1506. doi: 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nonejuie P, Burkart M, Pogliano K, Pogliano J. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc Natl Acad Sci U S A. 2013;110:16169–16174. doi: 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]