

Abstract
Progressive neuronal death in brainstem nuclei and widespread accumulation of α-synuclein are neuropathological hallmarks of Parkinson’s disease (PD). Reduction of α-synuclein levels is therefore a potential therapy for PD. However, because α-synuclein is essential for neuronal development and function, α-synuclein elimination would dramatically impact brain function. We previously developed conjugated small interfering RNA (siRNA) sequences that selectively target serotonin (5-HT) or norepinephrine (NE) neurons after intranasal administration. Here, we used this strategy to conjugate inhibitory oligonucleotides, siRNA and antisense oligonucleotide (ASO), with the triple monoamine reuptake inhibitor indatraline (IND), to selectively reduce α-synuclein expression in the brainstem monoamine nuclei of mice after intranasal delivery. Following internalization of the conjugated oligonucleotides in monoamine neurons, reduced levels of endogenous α-synuclein mRNA and protein were found in substantia nigra pars compacta (SNc), ventral tegmental area (VTA), dorsal raphe nucleus (DR), and locus coeruleus (LC). α-Synuclein knockdown by ∼20%–40% did not cause monoaminergic neurodegeneration and enhanced forebrain dopamine (DA) and 5-HT release. Conversely, a modest human α-synuclein overexpression in DA neurons markedly reduced striatal DA release. These results indicate that α-synuclein negatively regulates monoamine neurotransmission and set the stage for the testing of non-viral inhibitory oligonucleotides as disease-modifying agents in α-synuclein models of PD.
Keywords: Parkinson’s disease, α-synuclein, siRNA, ASO, intranasal administration, DA neurotransmission, 5-HT neurotransmission, caudate putamen, prefrontal cortex
Graphical Abstract
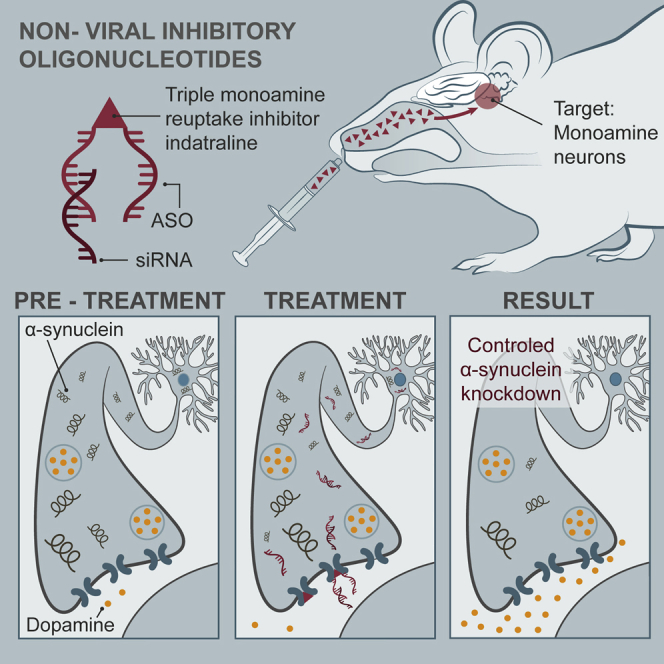
Bortolozzi and colleagues show that the intranasal administration of non-viral conjugated oligonucleotides selectively reduces α-synuclein levels in brainstem monoamine neurons without signs of neurotoxicity. Functionally, this effect translates into an enhanced forebrain monoamine neurotransmission. Overall, this provides a new therapeutic approach for the treatment of Parkinson’s disease and other synucleinopathies.
Introduction
Parkinson’s disease (PD) is characterized pathologically by degeneration of discrete groups of neurons in the central, autonomic, and enteric nervous systems. Particularly, the loss of substantia nigra dopamine (DA) neurons causes the cardinal motor signs of PD (tremor, rigidity, and bradykinesia).1 Moreover, monoamine and non-monoamine neurons show intracellular aggregates, Lewy bodies and Lewy neurites, containing deposits of insoluble α-synuclein.2, 3, 4 Variants in the Snca gene, which encodes the α-synuclein protein, modulate both PD risk5, 6, 7 and α-synuclein expression levels,8 suggesting that α-synuclein may be an early and integral mediator of the pathological cascade that ultimately results in neurodegeneration. The identification of families with duplication or triplication of the Snca gene (PARK4 locus, PD autosomal dominant Lewy body 4 [Homo sapiens (human)]) strengthened the link between α-synuclein and PD, and indicated that increased concentrations of the wild-type protein alone can cause the disease.9, 10 In addition, growing evidence implicates cell-to-cell transmission of misfolded α-synuclein as a common pathogenic mechanism in synucleinopathies.11, 12, 13, 14, 15, 16 Overall, these data point to α-synuclein as a potential primary target for therapeutic intervention in PD.
Although the physiological function of α-synuclein remains largely unknown, it may play a significant role in the regulation of neurotransmitter release, synaptic function, and neuroplasticity. Indeed, it is abundantly localized in presynaptic terminals17, 18 and is associated with the distal reserve pool of synaptic vesicles.19, 20, 21, 22 Further, alterations in synaptic transmission have been found in mice overexpressing or down-expressing α-synuclein.23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35
Despite this, it is still unclear how α-synuclein contributes to early functional changes and cell death in PD, and potential strategies to either reduce the basal levels of α-synuclein or to inhibit its process of aggregation are being explored. Preclinical studies have successfully shown α-synuclein downregulation both in cell culture and animal models using antisense oligonucleotides (ASOs), ribozymes, small interfering RNAs (siRNAs), or microRNA without neurotoxic effects.36, 37, 38, 39, 40, 41, 42 Yet, some studies reported DA neurodegeneration after α-synuclein silencing using a viral delivery of short hairpin RNA (shRNA.)43, 44, 45 Despite these exciting prospects, the utility of oligonucleotide-based silencing strategies is severely compromised by the extreme difficulty to deliver oligonucleotides to specific neuronal populations because of the need to cross several biological barriers after administration and the enormous complexity of the mammalian brain.
We previously developed conjugated siRNA sequences that selectively target genes in the serotonin (5-HT) or norepinephrine (NE) neurons after intranasal delivery.46, 47, 48, 49 The covalent binding of the 5-HT or NE transporter (NET) blockers sertraline or reboxetine, respectively, to siRNA molecules allows their selective accumulation in 5-HT or NE neurons after crossing the semi-permeable nasal blood-brain barrier and being transported rapidly to the brain. Here, we assessed the cellular selectivity and efficacy of different oligonucleotide sequences to downregulate α-synuclein expression in vitro and in vivo. We developed siRNA and ASO molecules conjugated with indatraline (IND; triple monoamine transporter blocker) to selectively knock down α-synuclein expression in tyrosine hydroxylase (TH)+ and tryptophan-hydroxylase2 (TPH2)+ neurons of mice after intranasal administration (i.n.). In addition, we evaluated the effects of α-synuclein knockdown on dopaminergic and serotonergic function.
Results
499-siRNA and 1233-ASO Molecules Selectively Reduce α-Synuclein Expression In Vitro
Three siRNA sequences (499-siRNA, MAYO2-siRNA, SNCA2-siRNA) were designed and synthesized to target regions of the mouse and human Snca mRNA encoding α-synuclein protein. The target region was selected based on its minimal homology to Sncb or Sncg (encoding β- and γ-synuclein, respectively) (Figure 1A; Table S1). Sequences showing significant homology to other genes by BLAST search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) were rejected.
Figure 1.

499-siRNA and 1233-ASO Molecules Selectively Reduce α-Synuclein Expression in Cultured Cells
(A and G) Comparative alignment showing the oligonucleotide sequence of human Snca, Sncb, and Sncg. Green letters indicate nucleotide differences compared with Snca sequence. Red letters indicate the specific sequence targeted by (A) 499-siRNA or (G) 1233-ASO. (B, C, and E) qRT-PCR quantification of Snca (B), Sncb (C), and Sncg (E) mRNA expression in M17-EV 24 hr after transfection with 200 nM 499-siRNA, MAYO2-siRNA, or SNCA2-siRNA. Cells transfected with nonsense siRNA (NS) were used as control. Target gene expression was normalized to two different housekeeping genes: GAPDHD and RPLPO. (D) Top: immunofluorescence images showing α-synuclein expression in M17-EV and M17-Syn. Blue: Hoechst staining; green: α-synuclein. Scale bar: 20 μm. Bottom: α-synuclein immunoblot image and quantification confirming the overexpression of α-synuclein protein levels in M17-Syn (n = 3). *p < 0.05, compared with M17-EV cells (one-way ANOVA followed by Tukey’s post hoc test). (F) qRT-PCR quantification of Snca expression in M17-Syn 24 hr after transfection with 499-siRNA, MAYO2-siRNA, and SNCA2-siRNA. Cells transfected with NS-siRNA were used as a control. (H and I) qRT-PCR quantification of α-synuclein expression in (H) M17-EV or (I) M17-Syn cells transfected with 300 nM nonsense 1227-ASO or 1233-ASO for 24 hr. In all graphs, histograms represent average ± SEM. ˆp < 0.05, ˆˆˆp < 0.001, compared with cells treated with lipofectamine alone; +p < 0.05, ++p < 0.01, +++p < 0.001, compared with cells treated with NS-siRNA (one-way ANOVA followed by Tukey’s post hoc test). 499-siRNA, MAYO2-siRNA, or SNCA2-siRNA are siRNA sequences designed to target regions of the Snca mRNA encoding α-synuclein protein (Table S1 shows siRNA sequences). M17-EV, M17 human neuroblastoma cells expressing empty vector; M17-Syn, M17 human neuroblastoma cells overexpressing α-synuclein.
First, the three α-synuclein siRNA sequences (499-siRNA, MAYO2-siRNA, SNCA2-siRNA) were screened in vitro to determine their efficacy and detect off-target effects. The ability of siRNA sequences to downregulate endogenous α-synuclein expression was studied in M17 human neuroblastoma cells containing a control empty vector (M17-EV) by qRT-PCR at 24 hr after transfection. One-way ANOVA followed by Tukey’s post hoc test indicated reductions of α-synuclein expression of 78% ± 8% (p < 0.001) for 499-siRNA, 86% ± 1% (p < 0.001) for MAYO2-siRNA, and 72% ± 5% (p < 0.05) for SNCA2-siRNA, respectively (Figure 1B). In contrast, M17-EV cells transfected with nonsense siRNA (NS; control group) did not show alterations of α-synuclein mRNA levels. To confirm the selectivity of siRNAs, we also evaluated β- and γ-synuclein mRNA levels. MAYO2-siRNA significantly suppressed β- and γ-synuclein expression in M17-EV cells (66% ± 4%, p < 0.05; and 59% ± 3%, p < 0.001, respectively). However, 499-siRNA and SNCA2-siRNA did not decrease either β- or γ-synuclein expression (Figures 1C and 1E).
Next, we evaluated the efficacy of siRNAs targeting α-synuclein in an in vitro PD-like model consisting of M17 cells overexpressing wild-type human α-synuclein (M17-Syn; Figure 1D). One-way ANOVA analyses followed by Tukey’s post hoc test revealed that all siRNA sequences reduced to the same extent the increased α-synuclein levels in M17-Syn cells 24 hr after transfection (74% ± 2%, p < 0.001 for 499-siRNA; 75% ± 3%, p < 0.05 for MAYO2-siRNA; and 67% ± 3%, p < 0.001 for SNCA2-siRNA) (Figure 1F). Like in M17-EV cells, MAYO2-siRNA, but not 499-siRNA and SNC2-siRNA, significantly decreased β- and γ-synuclein levels in M17-Syn cells (35% ± 4%, p < 0.05; and 68% ± 5%, p < 0.05, respectively) (Figure S1).
In addition, an ASO molecule (1233-ASO), encompassing the same gene sequence as 499-siRNA, was also validated in vitro prior to in vivo studies (Figure 1G). As described above, M17-EV and M17-Syn cells were transfected with 300 nM nonsense ASO (1227-ASO) or 1233-ASO targeting α-synuclein. Although no statistically significant differences were detected in M17-EV cells (Figure 1H), a significant effect of 1233-ASO was observed in M17-Syn cells (61 ± 6; p < 0.05) (Figure 1I). Both M17-EV and M17-Syn cells treated with 1227-ASO showed no alterations in α-synuclein levels (Figure 1I). Based on these results, both 499-siRNA and 1233-ASO were used for subsequent studies in vivo.
Selective Delivery of Conjugated ASO Molecules to Monoamine Neurons after i.n.
As summarized in the introductory section, the covalent binding of 5-HT or NE reuptake inhibitors to siRNA sequences allows their selective enrichment into raphe 5-HT neurons or locus coeruleus (LC) NE neurons after i.n.46, 47, 48 We previously demonstrated that the main factor conferring the appropriate neuronal selectivity was the presence of covalently bound ligand rather than the oligonucleotide sequence.46, 47 Given the efficacy of 499-siRNA and 1233-ASO to suppress α-synuclein expression in vitro, we next conjugated these sequences to the triple monoamine reuptake inhibitor IND to attain cell-type-selective targeting and to enhance the uptake of oligonucleotide based on in vitro and in vivo pharmacological profile of IND. Indeed, in vitro studies indicated that this agent is a highly potent inhibitor at all three uptake sites of monoamine transporters (5-HT transporter [SERT] > NET > DA transporter [DAT]) with half maximal inhibitory concentration (IC50) values around 1–4 nM, lacking affinity for DA, 5-HT, NE, histamine, and cholinergic receptors. This in vitro profile results in a maximal in vivo occupancy and efficacy for the monoamine transporters.50
To examine the brain distribution of conjugated oligonucleotide following i.n., we first used Alexa 488-labeled IND-conjugated nonsense ASO (A488-IND-1227-ASO) or IND-conjugated 1233-ASO (A488-IND-1233-ASO) molecules. Confocal fluorescence microscopy revealed that both A488-IND-1227-ASO and A488-IND-1233-ASO were intracellularly detected specifically in TPH2+ raphe 5-HT neurons and in TH+ DA and NE neurons of substantia nigra pars compacta/ventral tegmental area (SNc/VTA) and LC, respectively (Figure 2A; Figure S2). In contrast, IND-conjugated ASO was absent in cells of brain areas close to the application site (olfactory bulbs) or to brain ventricles (hippocampus, hypothalamus, and striatum) (Figure S3), indicating that functional monoamine transporters (DAT, SERT, and NET) are a requirement for oligonucleotide uptake and internalization. Indeed, pre-treatment with selective monoamine transporter inhibitors (sertraline for SERT, nomifensine for DAT/NET, and reboxetine for NET) prior to intranasal treatment with IND-conjugated ASO prevented the incorporation of oligonucleotide in each monoamine neuronal group expressing corresponding transporter (Figures S4–S6). In addition, IND-conjugated ASO co-localized with Rab5 (early endosome marker) and Rab7 (late endosome marker) (Figures S7–S9), which suggests the involvement of complex trafficking mechanisms after internalization in monoamine neurons as previously described for sertraline-conjugated siRNA.48
Figure 2.

Preferential Accumulation of Indatraline-Conjugated ASO Molecules in Monoamine Neurons after Intranasal Administration
(A) Mice were intranasally administered with Alexa 488-labeled indatraline-conjugated 1233-ASO targeting α-synuclein (A488-IND-1233-ASO) or Alexa 488-labeled indatraline-conjugated nonsense 1227-ASO (A488-IND-1227-ASO) at 30 μg/day for 4 days and sacrificed 6 hr after last administration (n = 3 mice/group). Confocal images showing the co-localization of A488-IND-ASOs (yellow) with TH+ neurons (red) in the SNc/VTA and LC or with TPH2+ neurons (red) in the DR identified with white arrowheads. Cell nuclei were stained with DAPI (blue). Scale bars: 10 μm. (B and C) Bars show the extracellular concentration of indatraline-conjugated 1233-ASO targeting α-synuclein (IND-1233-ASO) (B) and 1233-ASO molecules (C) (expressed as 109 molecules/μL) in DR, LC, SNc/VTA, and CPu. Mice received two consecutive ASO doses (1000 μM to 60 μg and 3000 μM to 180 μg) intranasally administered with time intervals of 1 hr. Note the higher extracellular IND-1233-ASO concentration in the DR compared with all brain areas. Data are mean ± SEM. ***p < 0.001 versus brain areas (two-way ANOVA followed by Tukey’s post hoc test). (D) Proposed mechanisms for transport of indatraline-conjugated oligonucleotides to brain following intranasal administration. Three potential pathways have been indicated for compound transport to the olfactory bulb or olfactory subarachnoid space following intranasal administration: (1) receptor-mediated endocytosis into olfactory sensory neurons followed by slow intracellular transport (from hours to days) to the olfactory bulb, (2) non-specific endocytosis into olfactory sensory neurons followed by intracellular transport to the olfactory bulb, and (3) extracellular diffusion into the olfactory submucosa along open intercellular clefts in the olfactory epithelium with a rapid transport (∼30 min) directly to the olfactory bulb or the olfactory subarachnoid space and entrance to the cerebrospinal fluid (CSF) circulation.58 IND-1233-ASO could be rapidly transported to the brain following intranasal administration mediated by the pulsatile flow through CSF leading to subsequent uptake into monoaminergic neurons via functional monoamine transporters. CPu, caudate putamen; DR, dorsal raphe nucleus; LC, locus coeruleus; SNc/VTA, substantia nigra compacta/ventral tegmental area.
In order to assess whether the presence of IND led to a selective enrichment of conjugated ASO in monoamine nuclei, we determined the concentration of ASO molecules targeting α-synuclein (unmodified and IND-conjugated 1233-ASO) in the extracellular space of different brain areas using a modified microdialysis procedure and stem-loop qRT-PCR.51, 52 Mice received two consecutive intranasal ASO doses (1000 μM to 60 μg and 3000 μM to 180 μg) with a time interval of 1 hr. The oligonucleotides were detected in the dialysate samples at 10–20 min post-administration. The concentration of IND-1233-ASO was found to be greater in the dorsal raphe nucleus (DR) and LC than in SNc or VTA (Figure 2B), perhaps due to the higher affinity of IND for SERT and NET than for DAT.50 In addition, IND-1233-ASO concentration in the extracellular space of caudate putamen (CPu) was much lower than in brainstem nuclei, and similar to those found in all brain areas examined after the i.n. of the non-conjugated 1233-ASO (i.e., non-specific signal) (Figures 2B and 2C; Figure S3). Two-way ANOVA analysis followed by Tukey’s post hoc test indicated an effect of brain area F(4,52) = 6.153, p < 0.0004, but not of ASO concentration F(1,52) = 0.3864, p = 0.763, nor area-by-concentration interaction F(4,52) = 0.1057, p = 0.98.
A lower concentration of the non-conjugated 1233-ASO molecule was detected in the analyzed brain areas, likely owing to the absence of IND, which is required for the specific monoaminergic delivery. Two-way ANOVA analysis showed no significant effects between the brain areas and ASO concentration (Figure 2C).
IND-Conjugated 499-siRNA and 1233-ASO Induce Selective and Safe Knockdown of α-Synuclein In Vivo
Next, we examined the temporal pattern of α-synuclein expression in the monoaminergic nuclei after treatment with IND-conjugated 499-siRNA (IND-499-siRNA) or IND-1233-ASO. For this purpose, mice were intranasally administered with IND-499-siRNA or IND-1233-ASO at 30 μg/day for 4 consecutive days and euthanized at different times after the last administration (Figures 3A and 4A). α-Synuclein mRNA levels in the SNc/VTA, DR, and LC, assessed by in situ hybridization, were significantly lower in IND-499-siRNA-treated mice than in control groups at 1 day post-administration (81% ± 2%, 69 ± 07%, and 76% ± 2%, respectively, versus vehicle-treated mice), with a recovery of α-synuclein expression to control values at day 3 post-administration (Figures 3B and 3C). α-Synuclein mRNA levels were unchanged in cortical and subcortical brain regions (data not shown). Reduction of α-synuclein mRNA expression in the SNc/VTA was confirmed by western blot procedures for murine α-synuclein protein (Figures 3D and 3E). We found that IND-499-siRNA (30 μg/day for 4 days) decreased α-synuclein protein levels in the SNc/VTA at 1 day post-administration (70% ± 3% versus vehicle-treated mice), but not in projection areas, such as CPu (Figures 3D and 3E; Figures S10A and S10B). Importantly, IND-499-siRNA (30 μg/day, 4 days, i.n.) did not induce any loss of SNc TH+ DA neurons nor striatal TH+ fibers (Figure S11). In addition, mRNA levels of β- and γ-synuclein, as well as of DAT, SERT, and NET, were unmodified by IND-499-siRNA in the SNc/VTA, DR, and LC, respectively (Figure S12). Altogether, these data support the specificity and safety of IND-499-siRNA effects.
Figure 3.

Intranasal Indatraline-Conjugated 499-siRNA Treatment Downregulates α-Synuclein Expression
(A) Schematic representation of the treatment. Mice were intranasally administered with PBS, indatraline-conjugated nonsense siRNA (IND-NS-siRNA), or indatraline-conjugated 499-siRNA (IND-499-siRNA) at 30 μg/day for 4 days and were sacrificed at 1, 3, or 7 days after last administration (1, 3, or 7 days, respectively; n = 5–10 mice/group). (B) Coronal brain sections showing reduced α-synuclein mRNA levels in the SNc, VTA, DR, and LC of mice treated with IND-499-siRNA (4 days) and sacrificed at 1 day post-administration were assessed by in situ hybridization (ISH). Signal represents the relative optical density (ROD) of autoradiograms as indicated at the right-hand side of the image. White arrowheads show α-synuclein mRNA expression in SNc, VTA, DR and LC. Scale bar: 500 μm. (C) Time course of α-synuclein mRNA suppression in the monoaminergic nuclei after intranasal IND-499-siRNA administration. Bar graphs showing a significant reduction of α-synuclein mRNA level compared with their respective controls at day 1 post-administration. Conversely, no difference was detected at days 3 and 7 post-administration. **p < 0.01, ***p < 0.001, versus PBS-treated mice; +p < 0.05, ++p < 0.01, +++p < 0.001 versus IND-NS-siRNA (two-way ANOVA followed by Tukey’s post hoc test). (D) Image of immunoblot of α-synuclein, β-actin, and tyrosine hydroxylase (TH) in SNc/VTA of mice treated with PBS or IND-conjugated siRNAs. (E) Bar graphs showing α-synuclein protein levels in SNc/VTA normalized against β-actin or TH, and TH protein levels normalized against β-actin. IND-499-siRNA reduced α-synuclein protein level in SNc/VTA 24 hr after last administration; then α-synuclein level was recovered 3 days later. *p < 0.05 versus control groups (two-way ANOVA followed by Tukey’s post hoc test). Data are mean ± SEM. DR, dorsal raphe nucleus; LC, locus coeruleus; SNc/VTA, substantia nigra compacta/ventral tegmental area.
Figure 4.

Intranasal Indatraline-Conjugated 1233-ASO Treatment Downregulates α-Synuclein Expression
(A) Schematic representation of the treatment. Mice were intranasally administered with PBS, indatraline-conjugated nonsense ASO (IND-1227-ASO), or indatraline-conjugated 1233-ASO (IND-1233-ASO) at 30 μg/day for 4 days and were sacrificed at 1, 3, or 7 days after last administration (1, 3, or 7 days, respectively; n = 5–10 mice/group). (B) Coronal brain sections showing reduced α-synuclein mRNA levels in the SNc, VTA, DR, and LC of mice treated with IND-1233-ASO (4 days) and sacrificed at 1 day post-administration assessed by in situ hybridization (ISH). Signal represents the relative optical density (ROD) of autoradiograms as indicated at the right-hand side of the image. White arrowheads show α-synuclein mRNA expression in SNc, VTA, DR and LC. Scale bar: 500 μm. (C) Time course of α-synuclein mRNA suppression in the monoaminergic nuclei after intranasal IND-1233-ASO administration. Bar graphs showing a significant reduction of α-synuclein mRNA level compared with their respective controls at day 1, but not at 3 and 7 days post-administration. *p < 0.05, **p < 0.01 versus PBS-treated mice; +p < 0.05, ++p < 0.01, +++p < 0.001 versus IND-1227-ASO (two-way ANOVA followed by Tukey’s post hoc test). (D) Image of immunoblot of α-synuclein, β-actin, and tyrosine hydroxylase (TH) in SNc/VTA of mice treated with PBS or IND-conjugated ASOs. (E) Bar graphs showing α-synuclein protein levels in SNc/VTA normalized against β-actin or TH, and TH protein levels normalized against β-actin. IND-1233-ASO decreased α-synuclein protein levels in SNc/VTA up to 3 days post-treatment, with a subsequent recovery to basal values at 7 days. *p < 0.05 versus control groups (two-way ANOVA followed by Tukey’s post hoc test). Data are mean ± SEM. DR, dorsal raphe nucleus; LC, locus coeruleus; SNc/VTA, substantia nigra compacta/ventral tegmental area.
Likewise, intranasal IND-1233-ASO treatment (30 μg/day for 4 days) selectively reduced endogenous α-synuclein mRNA expression in monoaminergic nuclei at 1 day post-administration (SNc/VTA: 84% ± 2%, DR: 70% ± 5%, and LC: 79% ± 2%, respectively, versus vehicle-treated mice) (Figures 4B and 4C). However, unlike IND-499-siRNA, IND-1233-ASO effect on α-synuclein protein levels in the SNc/VTA and CPu was time dependent, with protein levels decreasing up to 3 days post-treatment and a recovery to basal levels at day 7 post-treatment (SNc/VTA: 68% ± 5%, 44% ± 3%, and 95% ± 20%; and CPu: 67% ± 7%, 97% ± 5%, and 99% ± 5% at 1, 3, and 7 days post-administration, respectively, versus vehicle-treated mice) (Figures 4D and 4E; Figures S10C and S10D). IND-1233-ASO treatment did not induce any DA neurotoxicity (Figure S11). Furthermore, β-synuclein, γ-synuclein, DAT, SERT, and NET mRNA densities were unchanged by IND-1233-ASO in the monoaminergic nuclei (Figure S13). Overall, these results indicate that intranasal treatment with IND-1233-ASO would be a better choice for decreasing α-synuclein levels specifically in monoaminergic nuclei than IND-499-siRNA, because it provides a more sustained knockdown of α-synuclein expression.
ASO-Induced α-Synuclein Knockdown Enhances Forebrain DA Neurotransmission
To evaluate whether IND-1233-ASO-induced α-synuclein silencing in SNc/VTA can modulate forebrain DA function, we performed a series of microdialysis experiments in freely moving mice. No difference was observed in baseline DA concentration in the CPu and medial prefrontal cortex (mPFC) among knockdown mice and control groups treated with PBS or IND-1227-ASO (Table S2). However, the infusion of the depolarizing agent veratridine (50 μM) by reverse dialysis increased ∼5-fold striatal DA release in IND-1233-ASO-treated mice versus ∼2-fold in the control groups treated with PBS or IND-1227-ASO (Figure 5A). Two-way ANOVA followed by Tukey’s post hoc test analysis showed an effect of group F(2,21) = 3.58 (p < 0.05), time F(15,315) = 8.94 (p < 0.0001), and group-by-time interaction F(30,315) = 1.761 (p < 0.01). A similar effect of veratridine on DA release was detected in the mouse mPFC (Figure 5F). Two-way ANOVA showed effect of group F(1,10) = 7.26 (p < 0.05), time F(15,150) = 8.12 (p < 0.0001), but not group-by-time interaction.
Figure 5.

Neurochemical Effects on Forebrain DA Neurotransmission of IND-1233-ASO-Induced α-Synuclein Knockdown
Mice were intranasally administered with PBS, indatraline-conjugated nonsense ASO (IND-1227-ASO), or indatraline-conjugated 1233-ASO (IND-1233-ASO) at 30 μg/day for 4 days. Microdialysis experiments were conducted between 1 and 3 days after last administration. (A and F) Local veratridine infusion (depolarizing agent, 50 μM) by reverse dialysis increased extracellular DA release in CPu (A) and mPFC (F). This effect was more significant in IND-1233-ASO-treated mice than in control groups. (B and G) Nomifensine (DAT inhibitor, 1-10-50 μM) increased more significantly extracellular DA levels in CPu (B) and mPFC (G) of α-synuclein knockdown mice than in control groups. (C and H) Similarly, local infusion of amphetamine (DA releaser, 1-10-100 μM) induced a higher DA release in CPu (C) and mPFC (H) of IND-1233-ASO-treated mice versus control mice. (D and I) Local tetrabenazine (VMAT2 inhibitor, 100 μM) application significantly reduced DA release in both brain areas: CPu (D) and mPFC (I). Only in the CPu (D) was this effect faster in control mice than in α-synuclein knockdown mice. (E and J) Local activation of DA D2/3 receptor in CPu (E) and mPFC (J) by the infusion of 10 μM quinpirole by reverse dialysis reduced DA release in all treatments. However, after quinpirole was removed, control groups recovered baseline DA levels faster than α-synuclein knockdown mice. The number of mice used in each experiment is shown in parentheses. *p < 0.05, **p < 0.01, ***p < 0.001 compared with control groups (two-way ANOVA followed by Tukey’s post hoc test). Data are mean ± SEM. CPu, caudate putamen; mPFC, medial prefrontal cortex.
α-Synuclein modulates the function of DAT and vesicular monoamine transporter (VMAT2) in DA terminals.53, 54 Local application of DAT/NET inhibitor, nomifensine (1-10-50 μM) increased dose-dependently the extracellular DA concentration in the CPu. This effect was more pronounced in α-synuclein knockdown mice than in control groups (Figure 5B). Two-way ANOVA showed the effect of group F(2,16) = 8.38 (p < 0.01), time F(19,304) = 54.19 (p < 0.0001), and group-by-time interaction F(38,304) = 6.463 (p < 0.0001). Similarly, amphetamine (DA releaser and DAT inhibitor, 1-10-100 μM) dose-dependently elevated dialysate DA concentration in the striatum, being significantly higher in IND-1233-ASO-treated mice [effect of group F(2,14) = 17.18, p < 0.001; time F(19,266) = 49.19, p < 0.0001; and group-by-time interaction F(38,266) = 8.62, p < 0.0001] (Figure 5C). Likewise, local nomifensine or amphetamine administration produced a more robust increase of extracellular DA levels in the mPFC of α-synuclein knockdown mice than in control group (Figures 5G and 5H). Two-way ANOVA showed an effect of time F(19,171) = 9.36 (p < 0.0001) and group-by-time interaction F(19,171) = 2.23 (p < 0.01), but not group for nomifensine, and an effect of group F(1,7) = 5.68 (p < 0.05), time F(17,119) = 13.31 (p < 0.0001), and group-by-time interaction F(17.119) = 3.58 (p < 0.0001) for amphetamine. In addition, local tetrabenazine application (VMAT2 inhibitor, 100 μM) decreased dialysate DA concentration to a minimum of ∼28% (CPu) and ∼34% (mPFC) of baseline in all experimental groups at 30–40 min post-infusion (Figures 5D and 5I). However, in the CPu (but not mPFC), prior to the massive DA depletion, tetrabenazine produced a significantly lesser decline in IND-1233-ASO-treated mice versus control groups.
To obtain more information on the mechanisms controlling DA neurotransmission in SNc/VTA α-synuclein knockdown mice, we examined the effect of the DA D2 receptor agonist quinpirole on DA release. Local quinpirole infusion (10 μM) markedly reduced DA release in CPu and mPFC of all experimental groups (Figures 5E and 5J). After the replacement of the artificial cerebrospinal fluid (aCSF) containing quinpirole by standard aCSF, DA release returned to baseline values in control groups (PBS- and IND-1227-ASO-treated mice), but remained reduced in IND-1233-ASO-treated mice (Figure 5E). Two-way ANOVA showed a significant effect of the group F(2,19) = 6.22 (p < 0.01), time F(17, 323) = 16.19 (p < 0.0001), and group-by-time interaction F(34,323) = 1.71 (p < 0.01). Similar effects were detected in the mPFC of IND-1233-ASO-treated compared with PBS-treated mice (Figure 5J). Two-way ANOVA analysis showed an effect of time F(17, 136) = 22.89 (p < 0.0001) and group-by-time interaction F(17,136) = 2.01 (p < 0.01), but not of group.
ASO-Induced α-Synuclein Knockdown Enhances Forebrain 5-HT Neurotransmission
Given the involvement of α-synuclein in the regulation of synaptic transmission in general and not only of the DA system,29 we also examined whether the knockdown of α-synuclein expression in DR serotonergic neurons may affect forebrain 5-HT function. As previously observed for DA (see above), there were no differences in basal 5-HT concentration in CPu and mPFC between the different groups (Table S2). We first evaluated the effect of veratridine (50 μM) on striatal or cortical 5-HT release (Figures 6A and 6F). Compared with PBS-treated mice, veratridine-stimulated 5-HT levels in CPu (but not in mPFC) were higher in IND-1233-ASO-treated mice [effect of time F(15,135) = 69.05, p < 0.0001, and group-by-time interaction F(15,135) = 10.71, p < 0.0001, but not of group]. Furthermore, local application of the SERT inhibitor citalopram (1-10-50 μM) dose-dependently increased the extracellular 5-HT concentration in the CPu and mPFC. This effect was significantly higher in IND-1233-ASO-treated mice than in PBS-treated mice (Figures 6B and 6G). Two-way ANOVA showed an effect of time F(19,152) = 18.84 (p < 0.0001) and group-by-time interaction F(19,152) = 2.092 (p < 0.001), but not of group in CPu, and an effect time F(19,133) = 14.82 (p < 0.0001) and group-by-time interaction F(19,133) = 2.07 (p < 0.01), but not group in the mPFC. Likewise, we found that local tetrabenazine infusion (100 μM) decreased 5-HT release in the striatum, but not in mPFC, more markedly in control mice than in IND-1233-ASO-treated mice [marginal effect of group F(1,9) = 4.92, p = 0.053; effect of time F(14,126) = 56.25, p < 0.0001; and group-by-time interaction F(14,126) = 3.41, p < 0.0001] (Figures 6C and 6H).
Figure 6.

Neurochemical Effects on Forebrain 5-HT Neurotransmission of IND-1233-ASO-Induced α-Synuclein Knockdown
Mice were intranasally administered with PBS or indatraline-conjugated 1233-ASO (IND-1233-ASO) at 30 μg/day for 4 days. Microdialysis experiments were conducted between 1 and 3 days after last administration. (A and F) Local veratridine infusion (depolarizing agent, 50 μM) by reverse dialysis increased extracellular 5-HT release in CPu (A), but not in mPFC (F). This effect was more significant in IND-1233-ASO-treated mice than in PBS-treated mice. (B and G) Citalopram (SERT inhibitor, 1-10-50 μM) increased more significantly extracellular 5-HT levels in CPu (B) and mPFC (G) of α-synuclein knockdown mice than in PBS mice. (C and H) Local tetrabenazine (VMAT2 inhibitor, 100 μM) application significantly reduced 5-HT release in CPu (C) and mPFC (H). However, only CPu reached a marginal statistical difference between phenotypes (p = 0.053), with a greater effect of tetrabenazine in control than in α-synuclein knockdown mice. (D and I) Local activation of 5-HT1B receptor in CPu (D) and mPFC (I) by the infusion of 300 μM CP93129 by reverse dialysis reduced 5-HT release in PBS and α-synuclein knockdown mice. However, after CP93129 was removed, PBS mice recovered baseline 5-HT levels more rapidly than α-synuclein knockdown mice. (E and J) Systemic 8-OH-DPAT (5-HT1A receptor agonist, 1 mg/kg i.p.) administration reduced 5-HT release similarly in CPu (E) and mPFC (J) of PBS and α-synuclein knockdown mice. The number of mice used in each experiment is shown in parentheses. *p < 0.05, **p < 0.01, ***p < 0.001 compared with PBS mice (two-way ANOVA followed by Tukey’s post hoc test). Data are mean ± SEM. CPu, caudate putamen; mPFC, medial prefrontal cortex.
Systemic administration of 5-HT1A receptor agonist 8-OH-DPAT (1 mg/kg intraperitoneally [i.p.]) comparably reduced the extracellular 5-HT level in control and IND-1233-ASO mice [effect of time F(15,195) = 15.77, p < 0.0001, and F(15,120) = 5.37, p < 0.0001, but not of group and group-by-time interaction in CPu and mPFC, respectively] (Figures 6E and 6J). However, the local perfusion of 5-HT1B receptor agonist CP93129 (300 μM) reduced more markedly 5-HT release in CPu and mPFC of IND-1233-ASO-treated mice than in control mice (Figures 6D and 6I). Two-way ANOVA showed an effect of group F(1,14) = 13.47 (p < 0.001) and time F(17,144) = 4.19 (p < 0.0001), but not group-by-time interaction in CPu, and an effect of group F(1,12) = 15.62 (p < 0.001) and time F(17,119) = 6.46 (p < 0.0001), but not group-by-time interaction in mPFC. As observed for the effect of quinpirole on DA release, the effect of CP93129 on 5-HT release persisted after the end of reverse dialysis only in IND-1233-ASO-treated mice. Overall, these data support that α-synuclein knockdown in the cell bodies of DA and 5-HT neurons facilitates the terminal release of the respective neurotransmitters in projection areas of the forebrain, also altering the function of axon terminal autoreceptors (DA D2 and 5-HT 5-HT1B).
α-Synuclein Overexpression in TH+ Neurons Impairs DA Release in the Nigrostriatal Pathway
To obtain more information about the role of α-synuclein on the in vivo DA release and reuptake in the CPu, we used a mouse transgenic model overexpressing human wild-type α-synuclein selectively in TH+ neurons (referred as TG+ onward). Immunoblot with an antibody that recognizes both rodent and human α-synuclein showed that α-synuclein expression in TG+ mice is 2- to 3-fold greater than murine endogenous levels. This range not only reduces the likelihood of toxicity due to massive protein expression, but also reproduces the levels of α-synuclein accumulation in patients with duplication or triplication of the gene.29, 55, 56 Indeed, TG+ mice showed a normal development, maintenance of TH-immunoreactive nigral neurons and locomotor activity, being an appropriate model to evaluate the biological, but not pathological, function of α-synuclein on synaptic DA neurotransmission.55 Like α-synuclein knockdown mice (see above), TG+ mice showed no differences in baseline DA levels in CPu as compared with TG− mice (Table S2). In contrast, veratridine increased extracellular DA in CPu much less in TG+ mice than in the TG− mice [marginal effect of group F(1,8) = 4.45, p = 0.06; time F(15,120) = 10.17, p < 0.0001; and group-by-time interaction F(15,120) = 2.96, p < 0.001] (Figure 7A). Moreover, local nomifensine application (1-10-50 μM) evoked a significantly lesser increase of extracellular DA concentration in the CPu of TG+ compared with TG− mice [group F(1,8) = 5.93, p < 0.05; time F(19,152) = 10.45, p < 0.0001; and group-by-time interaction F(19,152) = 1.85, p < 0.05] (Figure 7B). No significant differences on DA release were seen between both phenotypes after local amphetamine or tetrabenazine infusion by reverse dialysis (Figures 7C and 7D). However, the reduction of striatal DA release after local quinpirole infusion in TG− mice was absent in TG+ mice, suggesting that the inhibitory feedback mechanism mediated by DA D2 receptor activation is attenuated by overexpressed α-synuclein (Figure 7E). Two-way ANOVA analysis showed an effect of time F(17,221) = 4.961 (p < 0.001) and of group-by-time interaction F(17,221) = 2,402 (p < 0.01), but not of group. These results concur with previous findings suggesting that increased expression of α-synuclein reduces neurotransmitter release.29
Figure 7.
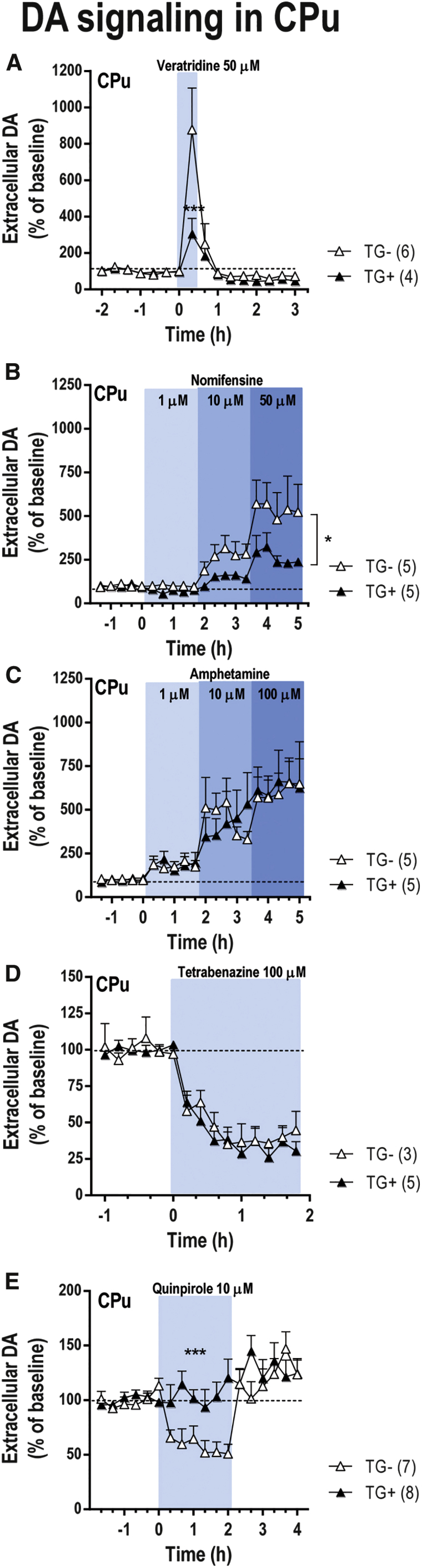
Neurochemical Effects on Nigrostriatal DA Neurotransmission in Transgenic Mice Overexpressing α-Synuclein
Transgenic mice expressing human wild-type α-synuclein cDNA under the control of TH promotor (TG+) and their respective controls (TG−) were implanted with a microdialysis probe into CPu. Microdialysis experiments were conducted between 1 and 3 days post-implantation. (A) Local veratridine infusion (depolarizing agent, 50 μM) by reverse dialysis increased extracellular DA release in CPu, but this effect was more significant in TG- mice than in TG+ mice. (B) Local nomifensine (DAT inhibitor, 1-10-50 μM) application increased more significantly extracellular DA levels in CPu of TG− mice than in TG+ mice. (C) However, local infusion of amphetamine (DA releaser, 1-10-100 μM) increased striatal DA levels similarly in both phenotypes. (D) In addition, tetrabenazine (VMAT2 inhibitor, 100 μM) induced a comparable effect on striatal DA release in TG− and TG+ mice. (E) Local activation of DA D2/3 receptor in CPu by the infusion of 10 μM quinpirole by reverse dialysis only reduced DA release in TG− mice, but not in TG+ mice. The number of mice used in each experiment is shown in parentheses. *p < 0.05, ***p < 0.001, compared with TG− mice (two-way ANOVA followed by Tukey’s post hoc test). Data are mean ± SEM. CPu, caudate putamen.
Discussion
The present study confirms and extends previous studies by our group on the use of oligonucleotides (siRNA and ASO) conjugated with monoamine transporter inhibitors to selectively silence genes expressed by monoaminergic neurons in vivo.46, 47, 48, 49 The design of IND-conjugated oligonucleotides allowed us to selectively enrich them in DA, 5-HT, and NE neurons after i.n. Using this strategy, we were able to reduce the expression of α-synuclein only in the monoaminergic neurons vulnerable to PD, with no signs of neuronal toxicity or compensatory mechanisms involving the expression of other members of the synuclein family. α-Synuclein knockdown in monoaminergic nuclei was followed by increased DA and 5-HT neurotransmission in the forebrain areas examined: CPu and mPFC. These findings improve the current knowledge of the physiological role of α-synuclein to regulate monoaminergic synaptic plasticity. But, more importantly, represent a major advance in the development of new therapies using non-viral inhibitory oligonucleotides to treat PD and other neurological diseases involving α-synuclein accumulation.
The potential use of siRNA and ASO oligonucleotides as therapeutic tools has aroused a great deal of interest. However, this interest is greatly dampened by the difficulties into delivering these oligonucleotide molecules to the desired neurons/circuits in the mammalian brain. As previously reported,46, 47, 48, 49 our strategy was to develop conjugates of siRNA or ASO in which the oligonucleotide sequence was covalently bound to a triple inhibitor of monoamine transporters IND to selectively accumulate it in monoaminergic cells via monoamine transporters. The precise mechanism(s) by which IND-conjugated siRNA/ASO reaches DA, 5-HT, and NE neurons after i.n. are not fully understood. At least three potential pathways have been proposed for oligonucleotide transport to the brain following i.n.: (1) receptor-mediated endocytosis into olfactory sensory neurons followed by slow intracellular transport (from hours to days) to the olfactory bulb, (2) non-specific fluid-phase endocytosis into olfactory sensory neurons followed by intracellular transport to the olfactory bulb, and (3) extracellular diffusion into the olfactory submucosa along open intercellular clefts in the olfactory epithelium with rapid transport (∼30 min) directly to the olfactory bulb or the olfactory subarachnoid space and CSF circulation57, 58, 59 (Figure 2D). Therefore, conjugated siRNA/ASO molecules may use the extracellular pathway to be rapidly transported over large distances via CSF by pulsatile flow and then be taken up by the dense network of axons emerging from monoamine cell bodies that contain the greatest densities of SERT, DAT, and NET.60, 61 This possibility is supported by: (1) the absence of fluorescence-linked conjugated oligonucleotides in the projection brain areas closer to the administration site, as shown by previous46, 48 and present observations; (2) the short time required for IND-conjugated ASO to reach brainstem monoamine nuclei after i.n. (10–20 min); (3) the preferential extracellular concentration of conjugated ASO in the monoaminergic nuclei in a concentration gradient (DR > LC > SNc/VTA) that is consistent with in vitro affinity and in vivo occupancy of IND by the respective transporters (Ki = 0.6, 2, and 4 nM for SERT, NET, and DAT, respectively);50 and (4) the association of conjugated siRNA47, 48 or ASO (present study) with Rab5 and Rab7 in the monoamine neurons supporting endocytosis and intracellular traffic processes via early and late endomembrane compartments. Overall, our results demonstrate a selective and active accumulation of conjugated oligonucleotides in DA, 5-HT, and NE neurons.
After i.n., both 499-siRNA and 1233-ASO conjugated with IND and targeting a similar region of mRNA induced comparable α-synuclein mRNA downregulation in the monoaminergic brainstem nuclei. However, the effect of IND-1233-ASO on α-synuclein protein levels in the SNc/VTA was more sustained than that produced by IND-499-siRNA (72 versus 24 hr post-administration). Likewise, a decrease of striatal α-synuclein protein density was evident after treatment with IND-1233-ASO, but not with IND-499-siRNA. Reduced α-synuclein protein levels for 3 or more days are consistent with its half-life (48–54 hr) and indicate that stable and long-lasting reductions can be achieved.62, 63 Hence, in our in vivo model, the use of ASO may be more advantageous than that of siRNA in terms of a longer-lasting effect of the former on α-synuclein knockdown. Previously, different silencing efficacies have been reported using siRNA and ASO in vitro and in vivo.64, 65, 66 Both strategies powerfully knock down genes, and there is no clear evidence that the silencing is substantially stronger for either siRNA or ASO. The choice of the optimal mode will probably depend on the molecular mechanisms by which oligonucleotides modulate RNA expression/function in brain cells. Indeed, ASO molecules have inherent advantages: (1) they are half the molecular weight of siRNAs; (2) they do not require hybridization, thus large-scale preparation of drugs is more straightforward; and (3) because endogenous duplex RNAs control important physiological processes, introducing synthetic siRNA may result in competition for binding to the RNA-induced silencing complex (RISC) and cause toxicity in vivo.67, 68 To the best of our knowledge, similar studies of ASOs showing competition for the RNase H enzyme and in vivo toxicity have not been reported.69 In particular, three observations of the present study indicate that intranasal IND-1233-ASO treatment is safe and highly specific: (1) selective α-synuclein silencing occurred only in monoamine neurons, leaving other genes expressed by these neuronal populations, including β- and γ-synuclein, unaffected; (2) it was functionally effective for at least 3 days post-treatment; and (3) there were no signs of cellular toxicity in DA neurons in the animals treated.
Interestingly, 1233-ASO was more effective in vivo than in vitro at reducing α-synuclein expression; a difference that may be because of the experimental conditions used, such as the requirement for a delivery agent (e.g., lipofectamine) in the in vitro, but not in in vivo, studies.70 Moreover, cultured cells were only briefly exposed to the 1233-ASO oligonucleotide, whereas in vivo experiments involved multiple doses of conjugated 1233-ASO, thereby allowing gradual intracellular accumulation of the oligonucleotides. Likewise, it was reported that cell systems with high gene expression are more susceptible to oligonucleotide-mediated silencing than low-expression cell systems,71 as we also showed using M17-Syn cells overexpressing α-synuclein (but not in M17-EV cells).
Downregulation of α-synuclein in mouse monoamine neurons induced by intranasal IND-1233-ASO treatment enhanced DA and 5-HT neurotransmission in projection brain areas, such as CPu and mPFC (the effects on NE release were not assessed because of lack of adequate high performance liquid chromatography (HPLC) methods in our laboratory). Previous findings indicated that nigro-striatal terminals of α-synuclein-deficient mice display a standard tonic DA activity after electrical stimulation with single pulses; however, they also exhibited increased DA release with paired stimuli that elevated Ca2+ levels.23 The present data are in full agreement with those observations. Although α-synuclein knockdown mice exhibited unchanged DA and 5-HT baseline values, the release of these neurotransmitters was increased by the depolarizing agent veratridine mainly in the striatum. An inverse effect on stimulated DA release was observed in the CPu of transgenic mice α-synuclein overexpressing.
Moreover, recent studies showed that α-synuclein inhibits exocytosis and the refilling of releasable synaptic vesicles.29, 72 α-Synuclein modulates the SNARE-complex assembly in glutamatergic presynaptic terminals.28 Although it is so far unclear whether monoaminergic terminals share the same mechanism of action, α-synuclein probably plays a dynamic role in regulating synaptic vesicle release or reuptake at the DA and 5-HT terminals. In vitro studies showed that α-synuclein and γ-synuclein regulate the expression, trafficking, and function of monoamine transporters (DAT, SERT, and NET) at the cell surface.53, 73 In the present in vivo study, nomifensine (DAT inhibitor) and amphetamine (DA releaser and DAT inhibitor), as well as citalopram (SERT inhibitor), produced robust increases of DA and 5-HT levels, respectively, in the CPu and mPFC of α-synuclein knockdown mice. Similarly, a decrease of striatal DA reuptake with a concomitant increase of extracellular DA levels was reported in α-synuclein-deficient mice.74 In contrast, striatal nomifensine application reduced extracellular DA levels by approximately 50% in mice overexpressing α-synuclein, thereby suggesting that DAT function was markedly impaired in these animals. Together, this evidence confirms that α-synuclein plays a key role in the regulation of monoamine reuptake function.
Early studies reported that the overexpression of mutant α-synuclein variants also decreases VMAT2 expression/function leading to increased cytosolic DA levels at the presynaptic terminals and resulting in DA neurotoxicity.53 Our data showed that a moderate wild-type α-synuclein downregulation (∼20%–40% reduction) or overexpression (∼2- to 3-fold increase) in monoamine neurons does not cause significant alteration of VMAT2 function, as evaluated with tetrabenazine, and thereby that the physiological intracellular DA and 5-HT concentrations in projection areas of the brain are maintained.
A key observation in the present study is that the selective downregulation or upregulation of α-synuclein expression evoked opposite changes in terminal autoreceptors controlling DA and 5-HT release, a crucial mechanism in the regulation of monoamine homeostasis.75, 76, 77 The local administration of quinpirole (DA D2 receptor agonist) or CP93139 (5-HT1B receptor agonist) induced a long-lasting reduction of DA and 5-HT release, respectively, in the CPu and mPFC of α-synuclein knockdown mice. In contrast, quinpirole had no effect on striatal DA release in α-synuclein-overexpressing transgenic mice. This reveals an interesting compensatory mechanism regulated by α-synuclein, by which monoaminergic terminals can maintain functional extracellular DA and 5-HT activation of postsynaptic receptors in conditions of presynaptic deficit, such as that occurring in early PD stages when DA release is impaired.34, 35 So far, the cellular mechanism by which α-synuclein affects the downstream signaling of monoaminergic receptors remains unclear. Notwithstanding, using whole-cell patch-clamp recordings from medium-sized striatal spiny neurons in slices of wild-type and α-synuclein-overexpressing mice, it has been reported that quinpirole produces opposing effects on spontaneous excitatory postsynaptic currents (sEPSCs) mediated by the DA D2 receptor leading to an altered DA efflux.78 Likewise, α-synuclein modified DA D2-agonist-mediated inhibition of adenylate cyclase, which consequently affected its downstream cAMP-responsive element (CRE)-mediated gene transcription in CHO cells transfected with DA D2 receptors.79 Altogether, this evidence confirms that α-synuclein plays an essential physiological role as a fine-tuning regulator for the maintenance of monoaminergic plasticity.
In conclusion, the cellular selectivity and efficacy obtained with the intranasal IND-1233-ASO administration indicate that this may be a new approach to reduce α-synuclein expression specifically in monoamine neurons, with a high translational value in the treatment of PD. In addition, α-synuclein silencing would improve a deficit in DA and 5-HT neurotransmission in PD by enhancing monoamine release and/or reducing uptake. Notably, preliminary data showed that IND-1233-ASO induces a functional recovery of the nigro-striatal DA pathway in an animal PD model.80 Overall, these effects would potentially delay the progression of PD symptomatology without serious deleterious effects.
Materials and Methods
Small Interfering RNA and ASO Molecules
Three unmodified siRNA and ASO molecules targeting α-synuclein were chosen for in vitro and in vivo studies. In addition, an unrelated siRNA duplex (nonsense siRNA [NS-siRNA]) or ASO (1227-ASO) with no homology to mouse genome were used as negative controls. Sequences of different oligonucleotides are shown in Table S1. IND-conjugated siRNA or ASO targeting α-synuclein or nonsense were synthesized by nLife Therapeutics (Granada, Spain) as previously reported.46, 48 Moreover, to study in vivo brain distribution and intracellular incorporation of IND-conjugated ASO into DA, 5-HT, and NE neurons, IND-1227-ASO and IND-1233-ASO were additionally bound to fluorophore Alexa 488 as previously reported by siRNA molecule.48 Stock solutions of all siRNAs and ASOs were prepared in RNase-free water and stored at −20°C until use. Details are shown in the Supplementary Information.
In Vitro Experiments
Cell Culture
M17 human neuroblastoma cells overexpressing α-synuclein (M17-Syn) or the corresponding empty vector (M17-EV) were grown in Opti-MEM (GIBCO) medium supplemented with 10% fetal calf serum (FCS) and 0.5 mg/mL active Geneticin (GIBCO). Transfections were performed with 200 nM siRNAs or 300 nM ASOs in 24-well plates using lipofectamine RNAiMax (Invitrogen). Equal transfection efficiency in independent experiments was controlled by using BLOCK-IG Fluorescent Oligo (Invitrogen).
mRNA Extraction and RT-PCR
Isolation of mRNA was performed 24 hr post-transfection by the TRIzol (Invitrogen) following the manufacturer’s protocol. In brief, cell pellets were lysed in 1 mL TRIZOLTM Reagent (Invitrogen) and frozen overnight to help homogenization. Then 0.2 mL chloroform was added and samples were centrifuged for 15 min at 10,000 rpm at 4°C. The aqueous supernatant was mixed with 0.5 mL isopropanol and centrifuged again for 20 min at 15,000 rpm at 4°C. Finally, isopropanol was replaced by 0.9 mL of cold 75% ethanol, and the samples were centrifuged for 5 min at 10,000 rpm at 4°C. Air-dried samples were re-suspended in RNase-free water, and genomic DNA was removed by digestion with DNase I (QIAGEN). Total mRNA concentration was measured on a NanoDrop 200 (Thermo Fischer Scientific). One microgram of total mRNA was reverse-transcribed with Oligo dT by using SuperScript III first-strand synthesis system for RT-PCR (Invitrogen). After RNase treatment, cDNA was diluted to 8 ng/μL in 10 mM Tris-HCl (pH 8.0).
qRT-PCR
qRT-PCR was performed on a 7900HT SDS (Applied Biosystems) with TaqMan Universal Master Mix II with UNG (Roche Applied Biosystems) for detection using 20 ng of cDNA per reaction in a total volume of 10 μL. The cycling conditions were as follows: 2 min at 50°C and 10 min at 95°C, followed by 40 cycles, each consisting of 15 s at 95°C and 1 min at 60°C. Fluorescence-labeled specific probes were used to detect the expression levels of the target genes α- (SNCA-FAM, Hs00240907-m1), β- (SNCB-FAM, Hs00608185-m1), or γ-synuclein (SNCG-FAM, Hs00268306-m1), normalized against the housekeeping genes β-actin (FAM, 4333762F), RPLPO (VIC, 4326314E), and GADPH (FAM, 4333764F), all from Applied Biosystems. Threshold cycles (Ct) were calculated using the software ABI PRISM 7900HT SDS version 2.2 (Applied Biosystems). Relative quantification using the comparative Ct method was used to analyze the data output. Values were expressed as fold change over corresponding values for the control by the 2−ΔΔCt method.
Western Blot Analysis
M17-EV and M17-Syn cells grown on culture plates for 48 hr were harvested and centrifuged at 1500 rpm for 5 min. Pellets were washed with PBS three times. Lysis buffer (Tris-HCl 50 mM [pH 7.4], NaCl 150 mM, EDTA 1 mM, Triton X-100 1%) was added to the pellet and then incubated on ice for 30 min. Total protein concentration was determined using bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Thirty micrograms of protein was resolved by SDS-PAGE on 12% polyacrylamide gels and electrotransferred onto nitrocellulose membranes (GE Healthcare), which were blocked in 5% non-fat milk powder in PBS for 1 hr at room temperature and incubated overnight at 4°C with human α-synuclein primary antibody (mouse monoclonal anti-α-synuclein [BD Biosciences], 610786, dilution 1:1,000). Incubation with the anti-mouse secondary antibody coupled to horseradish peroxidase (dilution 1:5,000; Amersham Bioscience) was performed at room temperature for 1 hr, followed by repeat washing with PBS. Immunoreactive bands were visualized using SuperSignal Femto Chemiluminescent Substrate (Pierce) according to manufacturer’s instructions on an ImageQuant RT ECL imaging system (GE Healthcare).
In Vivo Experiments
Animals
Wild-type male C57BL/6J mice (10–14 weeks; Charles River, Lyon, France) were housed under controlled conditions (22 ± 1°C; 12 hr light/dark cycle) with food and water available ad libitum. Animal procedures were conducted in accordance with standard ethical guidelines (EU directive 2010/63 of September 22, 2010) and approved by the local ethical committee. In addition, transgenic mice containing human wild-type α-synuclein cDNA under the control of TH promotor (TG+) and their respective controls (TG−) on a C57BL/6 background were also used in microdialysis studies.55
Treatments
For i.n., mice were slightly anesthetized by 2% isoflurane inhalation and placed in a supine position.46, 47, 48 A 5-μL drop of PBS or IND-conjugated siRNA or ASO molecules was applied alternatively to each nostril once daily. A total of 10 μL of solution containing 30 μg (2.3 nmol/day siRNA or 4.6 nmol/day ASO) of conjugated oligonucleotides was delivered for 4 days, and mice were killed at 1, 3, or 7 days after last administration.
Western Blot Analysis
Mice (n = 8–10 per group) were sacrificed at all time points, brains were rapidly removed, and CPu and ventral midbrain were dissected and snap-frozen on dry ice. Tissues were homogenized in lysis buffer (Tris-EDTA-SDS-NP40 buffer) with protease inhibitors, and protein was quantified using a Pierce BCA protein assay kit (Thermo Fisher Scientific). A total of 10 μg of protein per well in the presence of Laemmli buffer was loaded in a Mini-Protean TGX precast gel (Bio-Rad, Madrid, Spain). Electrophoresis was run 5 min at 80 V followed by 40 min at 120 V. Protein transfer was done using the Trans-blot turbo system (Bio-Rad) in 0.2-μm polyvinylidene fluoride (PVDF) membranes. After transfer, membranes were blocked and incubated in primary antibodies (anti-α-synuclein, 1 μg/mL, ab6162 [Abcam, Cambridge, UK]; anti-β-actin, 0.2 μg/mL, A5316 [Sigma-Aldrich, Madrid, Spain]; and anti-TH, 0.5 μg/mL, AB152 [Sigma-Aldrich, Madrid, Spain]) overnight at 4°C. Secondary antibody incubation was done at room temperature during 1 hr in blocking solution (4% BSA in PBS-Tween 0.05%). Detection was done by chemiluminescence using SuperSignal Chemiluminescence ECL substrate kit (Thermo Fisher Scientific), and pictures were taken using ImageQuant LAS500 (GE Healthcare, Madrid, Spain). Images were analyzed using ImageJ software (https://imagej.nih.gov/ij/).
In Situ Hybridization
Mice were killed by pentobarbital overdose, and brains were rapidly removed, frozen on dry ice, and stored at −80°C. Coronal tissue sections (14 μm thick) were cut using a microtome-cryostat (HM500-OM; Microm, Walldorf, Germany), thaw-mounted onto 3-aminopropyltriethoxysilane (Sigma-Aldrich)-coated slides, and kept at −20°C until use. Antisense oligoprobes were complementary to bases: α-synuclein/411-447 (GenBank: NM_001042451), β-synuclein/1161-1194 (GenBank: NM_033610.2), γ-synuclein/366-416 (GenBank: NM_011430), DAT-DAT/564-614 (GenBank: NM_010020), SERT-SERT/820-863 (GenBank: NM_010484.1), and NET-NE/1210-1260 (GenBank: NM_009209), respectively (Göttingen, Germany). Each oligoprobe was labeled (2 pmol) at the 3′ end with [33P]-dATP (>2,500 Ci/mmol; DuPont-NEN, Boston, MA) using terminal deoxynucleotidyltransferase (TdT; Calbiochem, La Jolla, CA). Sections were hybridized as previously described.46, 48 Details are shown in the Supplementary Information.
Quantitative Image Analysis of Film Autoradiograms
Autoradiograms were analyzed and relative optical densities (RODs) were obtained using a computer-assisted image analyzer (MCID, Mering, Germany). For binding experiments, the system was calibrated with 3H-microscales standards to obtain fmol/mg protein equivalents from ROD data. The slide background and non-specific densities were subtracted. ROD was evaluated in two to three duplicate adjacent sections from each mouse and averaged to obtain individual values. MCID system was also used to acquire pseudocolor images. Black-and-white photographs were taken from autoradiograms using a Wild 420 microscope (Leica, Heerbrugg, Germany) equipped with Nikon DXM1200F digital camera and ACT-1 Nikon software (Soft Imaging System, Münster, Germany). Figures were prepared for publication using Adobe Photoshop software (Adobe Software, San Jose, CA, USA). Contrast and brightness of images were the only variables we adjusted digitally.
Immunohistochemistry
Mice were anesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde in sodium-phosphate buffer (pH 7.4). Brains were collected, post-fixed 24 hr at 4°C in the same solution, and then placed in gradient sucrose 10%–30% for 3 days at 4°C. For immunohistochemical procedures, 20- or 30-μm-thick brain sections were inactivated with 1× PBS containing 48% methanol and 1.5% H2O2 for 25 min. After several 1× PBS washes, tissues were blocked with normal serum from secondary antibody host in a 1× PBS/0.2% Triton for 120 min. Then sections were rinsed with 1× PBS/0.2% Triton and incubated at 4°C overnight with primary antibodies: anti-human α-synuclein (1:20 for CPu, SNc/VTA, and LC; ab75305; Abcam, Cambridge, UK), anti-α-synuclein (1:2,500 for CPu, SNc/VTA, and LC; 610786; BD Biosciences, NJ, Franklin Lakes, USA; EEUU) or anti-TH (1:5,000; ab112; Abcam). Once washed with 1× PBS/0.2% Triton, sections were incubated with the following biotinylated secondary antibodies: anti-mouse IgG1 (1:200; A-10519; Life Technologies, Carlsbad, CA, USA) for anti-human α-synuclein and anti-α-synuclein, and anti-rabbit (1:200; BA-1000, Vector Laboratories, Burlingame, CA, USA) for anti-TH. Then tissues were incubated with 1% avidin/biotin complex (Vectastain Elite ABC Kit; Vector Laboratories) for 60 min. Finally, sections were washed and reacted for visualization using diaminobenzidine tetrahydrochloride (DAB; D5905-50TAB; Sigma-Aldrich) solution in a peroxidase reaction to produce a brown reaction product. Sections were mounted and embedded in Entellan (Electron Microscopy Sciences).
For quantitative morphology, the total number of TH+ SNc neurons was assessed by stereology in regularly spaced 20-μm-thick sections spanning the entire SNc using StereoInvestigator software (MBF Bioscience). In brief, SNc was delineated for each slide and probes for stereological counting were applied to the map obtained (size of counting frame was 100 × 80 μm spaced by 600 × 400 μm). Each TH+ cell with its nucleus included within the counting frame was counted. Striatal TH innervation was assessed by ROD in regularly spaced 20-μm-thick sections corresponding to different striatal anatomical levels using Sigma Scan.81 Sections were scanned using an Epson high-resolution scanner, and Sigma Scan software was used to compare the optical density in each region of interest.
Confocal Fluorescence Microscopy
Intracellular IND-1227-ASO distribution in monoamine neurons was examined by confocal microscopy using a Leica TCS SP5 laser scanning confocal microscope (Leica Microsystems Heidelberg, Manheim, Germany) equipped with a DMI6000 inverted microscope, blue diode (405 nm), argon (458/476/488/496/514), diode pumped solid-state (561 nm), and HeNe (594/633 nm) lasers. After i.n. administration with Alexa 488-labeled IND-1227-ASO at 30 μg/day for 4 days, mice were sacrificed at 30 min or 6 hr, and their brain was extracted and processed for immunofluorescence. Details are shown in the Supplemental Information.
Drug and Reagents
All reagents used were of analytical grade and were obtained from Merck (Darmstadt, Germany). 5-HT oxalate, (±)-8-hydroxi-2(dipropylamino)tetralin hydrobromide (8-OH-DPAT), DA hydrochloride, nomifensine maleate, (-)-quinpirole hydrochloride, and tetrabenazine were from Sigma-Aldrich-RBI (Madrid, Spain). Moreover, D-amphetamine sulfate, 1,4-dihydro-3-(1,2,3,6-tetrahydro-4-pyridinyl-5H-pyrrol[3,2-b] pyridine-5-one)-dihydro chloride (CP93129), citalopram hydrobromide, and veratridine were purchased from Tocris (Madrid, Spain). To assess local effects in microdialysis experiments, we dissolved drugs in aCSF (in mM: NaCl, 125; KCl, 2.5; CaCl2, 1.26; and MgCl2, 1.18) and administered by reverse dialysis at the stated concentrations (uncorrected for membrane recovery). Stock solutions of tetrabenazine and veratridine were made in DMSO and were diluted to appropriate concentrations in aCSF to reach 1% DMSO. All other drugs were dissolved in saline or aCSF, as required. Concentrated solutions (1 mM; pH adjusted to 6.5–7 with NaHCO3 when necessary) were stored at −80°C, and working solutions were prepared daily by dilution in aCSF.
Microdialysis Procedures
Extracellular DA and 5-HT concentration were measured by in vivo microdialysis as previously described.46, 82, 83 In brief, one concentric dialysis probe (Cuprophan membrane; 6,000 Da molecular weight cutoff; 1.5 mm long) was implanted in the CPu (coordinates in mm: anteroposterior [AP], +0.5; mediolateral [ML], −1.7; dorsoventral [DV], −4.5) or mPFC (AP, +2.2; ML, −0.2; DV, −3.4) of pentobarbital-anaesthetized mice.84 Experiments were performed 24–48 hr after surgery in freely moving mice. Nomifensine (10–50 μM) or citalopram (1–50 μM), respectively, was added to aCSF to assess quinpirole, 8-OH-DPAT, CP93129, or tetrabenazine effects on extracellular DA or 5-HT levels. The aCSF was pumped (WPI model, SP220i) at 1.5 μL/min, and 20-min samples were collected (except for the experiment using tetrabenazine, where aCSF was perfused at 3 μL/min and collected 10-min samples). DA and 5-HT concentrations were analyzed by HPLC-amperometric detection (+0.75 and +0.6 V, respectively; Hewlett Packard 1049, Palo Alto, CA, USA) with 3-fmol detection limits. Baseline DA and 5-HT levels were calculated as the average of the five to six pre-drug samples.
In addition, we evaluated extracellular 1233-ASO concentration in a different mouse cohort after i.n. using in vivo microdialysis and stem-loop qRT-PCR detection. One concentric dialysis probe (Cuprophan membrane as above described; 1 or 1.5 mm long) was implanted in the CPu, SNc/VTA (AP, −3.0; ML, −0.75; DV, −5.0), DR (AP, −4.5; ML, −1.0; DV, −4,2 with a lateral angle of 20°), or LC (AP, −5.4; ML, −0.9; DV, −3.5).84 Experiments were performed 24 hr after surgery. The aCSF was perfused at 3 μL/min, and 10-min samples were collected. 1233-ASO concentration was quantified using stem-loop qPCR. Reverse transcription (RT) was performed directly on the samples using the SuperScript II Enzyme at a final concentration of 6.67 U/μL (18064-014; Invitrogen, Thermo-Fisher) and the antisense RT stem loop primer at a final concentration of 0.05 μM (IDT, Coralville, IA, USA) on a Bio-Rad Thermocycler C1000 (Bio-Rad, Madrid, Spain). After the RT, 2 μL of the reaction was mixed with Immolase DNA polymerase (final concentration of 0.04 U/μL) and reaction buffer (BIO-21046; Bioline, London, UK) with the forward and reverse primers and probe (final concentrations of 1.5, 0.7, and 0.2 μM, respectively) for the qPCR. The qPCR was performed in a LightCycler 480 System (Roche Molecular Systems, CA, USA).
Statistical Analyses
All results are given as mean ± SEM. Data were analyzed using GraphPad Prism 7.01 (San Diego, CA, USA). Statistical analyses were performed by two-tailed Student’s t test and one-way or two-way ANOVA followed by Tukey’s post hoc test as appropriate. Differences were considered significant when p < 0.05.
Author Contributions
R.R., M.V., and A.B. designed experiments and supervised the research. R.R., A.M., F.A., M.V., and A.B. designed the conjugated oligonucleotides. A.R. and I.C.-C. performed in vitro experiments. D.A.-A., M.G., A.F.-C., R.P.-C., N.Z., E.R.-B., R.C., and M.S. performed in vivo experiments. D.A.-A. and N.Z. performed microdialysis experiments. M.G. and E.R.-B. performed in situ hybridization experiments. A.F.-C. and R.P.-C. performed confocal microscopy experiments. R.C. and M.S. performed western blot experiments. I.F. provided the α-synuclein transgenic mice. F.A., M.V., and A.B. wrote the manuscript, with all 13 authors providing input.
Conflicts of Interest
F.A., M.V., R.R., A.M., and A.B. are authors of the patent WO/2011/131693 for the siRNA and ASO (antisense oligonucleotides) molecules and the targeting approach related to this work. N.Z., R.C., M.S., R.R., and A.M. are board members of nLife Therapeutics S.L. The rest of authors declare no competing financial interest.
Acknowledgments
We thank María Calvo, Elisenda Coll, and Anna Bosch for outstanding technical support in the Confocal microscopy unit (CCiT-UB), and María C. Carmona for advice on design of small RNA and oligonucleotide molecules. We thank Letizia Campa, Verónica Paz, and Lluis Miquel for their excellent technical support. We would like to thank Theresa Branchek for her valuable comments on the manuscript. This work was supported by grant SAF2016-75797-R (to A.B.), INNPACTO Subprogram IPT-2012-1208-300000 (to A.B.), and Retos-Colaboración Subprogram RTC-2014-2812-1 (to M.V. and A.B.); Ministry of Economy and Competitiveness (MINECO) and European Regional Development Fund (ERDF), UE; grants PI13/01897 (to M.V.) and PI13/01390 (to A.B.), Fondo de Investigación Sanitaria-Instituto de Salud Carlos III (Spain), co-financed by ERDF; and Centro de Investigación Biomédica en Red de Salud Mental (CIBERSAM) and Centro de Investigación Biomédica en Red Enfermedades Neurodegenerativas (CIBERNED). CERCA Programme/Generalitat de Catalunya is also acknowledged.
Footnotes
Supplemental Information includes Supplemental Materials and Methods, 13 figures and two tables and can be found with this article online at https://doi.org/10.1016/j.ymthe.2017.11.015.
Contributor Information
Miquel Vila, Email: miquel.vila@vhir.org.
Analia Bortolozzi, Email: analia.bortolozzi@iibb.csic.es.
Supplemental Information
References
- 1.Braak H., Braak E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000;247(Suppl 2):II3–II10. doi: 10.1007/PL00007758. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini M.G., Crowther R.A., Jakes R., Hasegawa M., Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba M., Nakajo S., Tu P.H., Tomita T., Nakaya K., Lee V.M., Trojanowski J.Q., Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 5.Satake W., Nakabayashi Y., Mizuta I., Hirota Y., Ito C., Kubo M., Kawaguchi T., Tsunoda T., Watanabe M., Takeda A. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 6.Simón-Sánchez J., Schulte C., Bras J.M., Sharma M., Gibbs J.R., Berg D., Paisan-Ruiz C., Lichtner P., Scholz S.W., Hernandez D.G. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards T.L., Scott W.K., Almonte C., Burt A., Powell E.H., Beecham G.W., Wang L., Züchner S., Konidari I., Wang G. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fuchs J., Tichopad A., Golub Y., Munz M., Schweitzer K.J., Wolf B., Berg D., Mueller J.C., Gasser T. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB J. 2008;22:1327–1334. doi: 10.1096/fj.07-9348com. [DOI] [PubMed] [Google Scholar]
- 9.Chartier-Harlin M.C., Kachergus J., Roumier C., Mouroux V., Douay X., Lincoln S., Levecque C., Larvor L., Andrieux J., Hulihan M. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 10.Singleton A.B., Farrer M., Johnson J., Singleton A., Hague S., Kachergus J., Hulihan M., Peuralinna T., Dutra A., Nussbaum R. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 11.Braak H., Rüb U., Gai W.P., Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. (Vienna) 2003;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- 12.Li J.Y., Englund E., Holton J.L., Soulet D., Hagell P., Lees A.J., Lashley T., Quinn N.P., Rehncrona S., Björklund A. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 13.Desplats P., Lee H.J., Bae E.J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kordower J.H., Brundin P. Propagation of host disease to grafted neurons: accumulating evidence. Exp. Neurol. 2009;220:224–225. doi: 10.1016/j.expneurol.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 15.Dehay B., Vila M., Bezard E., Brundin P., Kordower J.H. Alpha-synuclein propagation: new insights from animal models. Mov. Disord. 2016;31:161–168. doi: 10.1002/mds.26370. [DOI] [PubMed] [Google Scholar]
- 16.Stuendl A., Kunadt M., Kruse N., Bartels C., Moebius W., Danzer K.M., Mollenhauer B., Schneider A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain. 2016;139:481–494. doi: 10.1093/brain/awv346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H.A., Kittel A., Saitoh T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 18.George J.M., Jin H., Woods W.S., Clayton D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron. 1995;15:361–372. doi: 10.1016/0896-6273(95)90040-3. [DOI] [PubMed] [Google Scholar]
- 19.Murphy D.D., Rueter S.M., Trojanowski J.Q., Lee V.M. Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yavich L., Tanila H., Vepsäläinen S., Jäkälä P. Role of α-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004;24:11165–11170. doi: 10.1523/JNEUROSCI.2559-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott D., Roy S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012;32:10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lashuel H.A., Overk C.R., Oueslati A., Masliah E. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abeliovich A., Schmitz Y., Fariñas I., Choi-Lundberg D., Ho W.H., Castillo P.E., Shinsky N., Verdugo J.M., Armanini M., Ryan A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 24.Cabin D.E., Shimazu K., Murphy D., Cole N.B., Gottschalk W., McIlwain K.L., Orrison B., Chen A., Ellis C.E., Paylor R. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002;22:8797–8807. doi: 10.1523/JNEUROSCI.22-20-08797.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dauer W., Kholodilov N., Vila M., Trillat A.C., Goodchild R., Larsen K.E., Staal R., Tieu K., Schmitz Y., Yuan C.A. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA. 2002;99:14524–14529. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra S., Fornai F., Kwon H.B., Yazdani U., Atasoy D., Liu X., Hammer R.E., Battaglia G., German D.C., Castillo P.E., Südhof T.C. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. USA. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson D.C., Schmidt O., Ninkina N., Jones P.A., Sharkey J., Buchman V.L. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem. 2004;89:1126–1136. doi: 10.1111/j.1471-4159.2004.02378.x. [DOI] [PubMed] [Google Scholar]
- 28.Burré J., Sharma M., Tsetsenis T., Buchman V., Etherton M.R., Südhof T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nemani V.M., Lu W., Berge V., Nakamura K., Onoa B., Lee M.K., Chaudhry F.A., Nicoll R.A., Edwards R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anwar S., Peters O., Millership S., Ninkina N., Doig N., Connor-Robson N., Threlfell S., Kooner G., Deacon R.M., Bannerman D.M. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci. 2011;31:7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chadchankar H., Yavich L. Sub-regional differences and mechanisms of the short-term plasticity of dopamine overflow in striatum in mice lacking alpha-synuclein. Brain Res. 2011;1423:67–76. doi: 10.1016/j.brainres.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 32.Al-Wandi A., Ninkina N., Millership S., Williamson S.J., Jones P.A., Buchman V.L. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging. 2010;31:796–804. doi: 10.1016/j.neurobiolaging.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin X., Parisiadou L., Sgobio C., Liu G., Yu J., Sun L., Shim H., Gu X.L., Luo J., Long C.X. Conditional expression of Parkinson’s disease-related mutant α-synuclein in the midbrain dopaminergic neurons causes progressive neurodegeneration and degradation of transcription factor nuclear receptor related 1. J. Neurosci. 2012;32:9248–9264. doi: 10.1523/JNEUROSCI.1731-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundblad M., Decressac M., Mattsson B., Björklund A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA. 2012;109:3213–3219. doi: 10.1073/pnas.1200575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janezic S., Threlfell S., Dodson P.D., Dowie M.J., Taylor T.N., Potgieter D., Parkkinen L., Senior S.L., Anwar S., Ryan B. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA. 2013;110:E4016–E4025. doi: 10.1073/pnas.1309143110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayashita-Kinoh H., Yamada M., Yokota T., Mizuno Y., Mochizuki H. Down-regulation of alpha-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 2006;341:1088–1095. doi: 10.1016/j.bbrc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 37.Junn E., Lee K.W., Jeong B.S., Chan T.W., Im J.Y., Mouradian M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA. 2009;106:13052–13057. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis J., Melrose H., Bumcrot D., Hope A., Zehr C., Lincoln S., Braithwaite A., He Z., Ogholikhan S., Hinkle K. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 2008;3:19. doi: 10.1186/1750-1326-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sapru M.K., Yates J.W., Hogan S., Jiang L., Halter J., Bohn M.C. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 2006;198:382–390. doi: 10.1016/j.expneurol.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 40.Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010;285:12726–12734. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zharikov A.D., Cannon J.R., Tapias V., Bai Q., Horowitz M.P., Shah V., El Ayadi A., Hastings T.G., Greenamyre J.T., Burton E.A. shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 2015;125:2721–2735. doi: 10.1172/JCI64502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Recasens A., Perier C., Sue C.M. Role of microRNAs in the regulation of α-synuclein expression: a systematic review. Front. Mol. Neurosci. 2016;9:128. doi: 10.3389/fnmol.2016.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gorbatyuk O.S., Li S., Nash K., Gorbatyuk M., Lewin A.S., Sullivan L.F., Mandel R.J., Chen W., Meyers C., Manfredsson F.P., Muzyczka N. In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010;18:1450–1457. doi: 10.1038/mt.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khodr C.E., Sapru M.K., Pedapati J., Han Y., West N.C., Kells A.P., Bankiewicz K.S., Bohn M.C. An α-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res. 2011;1395:94–107. doi: 10.1016/j.brainres.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collier T.J., Redmond D.E., Jr., Steece-Collier K., Lipton J.W., Manfredsson F.P. Is alpha-synuclein loss-of-function a contributor to Parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 2016;10:12. doi: 10.3389/fnins.2016.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bortolozzi A., Castañé A., Semakova J., Santana N., Alvarado G., Cortés R., Ferrés-Coy A., Fernández G., Carmona M.C., Toth M. Selective siRNA-mediated suppression of 5-HT1A autoreceptors evokes strong anti-depressant-like effects. Mol. Psychiatry. 2012;17:612–623. doi: 10.1038/mp.2011.92. [DOI] [PubMed] [Google Scholar]
- 47.Ferrés-Coy, A., Ruiz-Bronchal, E., Paz, V., Galofré, M., Artigas, F., and Bortolozzi, A. (2014). Selective siRNA-mediated suppression of TASK3 channels in monoaminergic neurons: a new antidepressant target. Neuroscience 2014 Annual Meeting, Poster 322.04/Y21, Society for Neuroscience, November 15–19, 2014, Washington, DC.
- 48.Ferrés-Coy A., Galofré M., Pilar-Cuéllar F., Vidal R., Paz V., Ruiz-Bronchal E., Campa L., Pazos Á., Caso J.R., Leza J.C. Therapeutic antidepressant potential of a conjugated siRNA silencing the serotonin transporter after intranasal administration. Mol. Psychiatry. 2016;21:328–338. doi: 10.1038/mp.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Artigas F., Bortolozzi A. Therapeutic potential of conjugated siRNAs for the treatment of major depressive disorder. Neuropsychopharmacology. 2017;42:371. doi: 10.1038/npp.2016.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lengyel K., Pieschl R., Strong T., Molski T., Mattson G., Lodge N.J., Li Y.W. Ex vivo assessment of binding site occupancy of monoamine reuptake inhibitors: methodology and biological significance. Neuropharmacology. 2008;55:63–70. doi: 10.1016/j.neuropharm.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 51.Krupp J.L., Bernards C.M. Pharmacokinetics of intrathecal oligodeoxynucleotides. Anesthesiology. 2004;100:315–322. doi: 10.1097/00000542-200402000-00021. [DOI] [PubMed] [Google Scholar]
- 52.Hurley J., Roberts D., Bond A., Keys D., Chen C. Stem-loop RT-qPCR for microRNA expression profiling. Methods Mol. Biol. 2012;822:33–52. doi: 10.1007/978-1-61779-427-8_3. [DOI] [PubMed] [Google Scholar]
- 53.Sidhu A., Wersinger C., Vernier P. Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? FASEB J. 2004;18:637–647. doi: 10.1096/fj.03-1112rev. [DOI] [PubMed] [Google Scholar]
- 54.Zhou Z., Kim J., Insolera R., Peng X., Fink D.J., Mata M. Rho GTPase regulation of α-synuclein and VMAT2: implications for pathogenesis of Parkinson’s disease. Mol. Cell. Neurosci. 2011;48:29–37. doi: 10.1016/j.mcn.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pérez-Sánchez F., Milán M., Buendía P., Cano-Jaimez M., Ambrosio S., Rosenthal A., Fariñas I. Prosurvival effect of human wild-type alpha-synuclein on MPTP-induced toxicity to central but not peripheral catecholaminergic neurons isolated from transgenic mice. Neuroscience. 2010;167:261–276. doi: 10.1016/j.neuroscience.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 56.Sánchez-Danés A., Richaud-Patin Y., Carballo-Carbajal I., Jiménez-Delgado S., Caig C., Mora S., Di Guglielmo C., Ezquerra M., Patel B., Giralt A. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dhuria S.V., Hanson L.R., Frey W.H., 2nd Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J. Pharm. Sci. 2010;99:1654–1673. doi: 10.1002/jps.21924. [DOI] [PubMed] [Google Scholar]
- 58.Lochhead J.J., Thorne R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 59.Renner D.B., Frey W.H., 2nd, Hanson L.R. Intranasal delivery of siRNA to the olfactory bulbs of mice via the olfactory nerve pathway. Neurosci. Lett. 2012;513:193–197. doi: 10.1016/j.neulet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 60.Javitch J.A., Strittmatter S.M., Snyder S.H. Differential visualization of dopamine and norepinephrine uptake sites in rat brain using [3H]mazindol autoradiography. J. Neurosci. 1985;5:1513–1521. doi: 10.1523/JNEUROSCI.05-06-01513.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cortés R., Soriano E., Pazos A., Probst A., Palacios J.M. Autoradiography of antidepressant binding sites in the human brain: localization using [3H]imipramine and [3H]paroxetine. Neuroscience. 1988;27:473–496. doi: 10.1016/0306-4522(88)90282-5. [DOI] [PubMed] [Google Scholar]
- 62.Okochi M., Walter J., Koyama A., Nakajo S., Baba M., Iwatsubo T., Meijer L., Kahle P.J., Haass C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J. Biol. Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- 63.Tofaris G.K., Layfield R., Spillantini M.G. alpha-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 64.Bertrand J.R., Pottier M., Vekris A., Opolon P., Maksimenko A., Malvy C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem. Biophys. Res. Commun. 2002;296:1000–1004. doi: 10.1016/s0006-291x(02)02013-2. [DOI] [PubMed] [Google Scholar]
- 65.Senn C., Hangartner C., Moes S., Guerini D., Hofbauer K.G. Central administration of small interfering RNAs in rats: a comparison with antisense oligonucleotides. Eur. J. Pharmacol. 2005;522:30–37. doi: 10.1016/j.ejphar.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 66.Corey D.R. RNA learns from antisense. Nat. Chem. Biol. 2007;3:8–11. doi: 10.1038/nchembio0107-8. [DOI] [PubMed] [Google Scholar]
- 67.Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K., Davis C.R., Marion P., Salazar F., Kay M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 68.Koller E., Propp S., Murray H., Lima W., Bhat B., Prakash T.P., Allerson C.R., Swayze E.E., Marcusson E.G., Dean N.M. Competition for RISC binding predicts in vitro potency of siRNA. Nucleic Acids Res. 2006;34:4467–4476. doi: 10.1093/nar/gkl589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Southwell A.L., Skotte N.H., Bennett C.F., Hayden M.R. Antisense oligonucleotide therapeutics for inherited neurodegenerative diseases. Trends Mol. Med. 2012;18:634–643. doi: 10.1016/j.molmed.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 70.Juliano R., Alam M.R., Dixit V., Kang H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008;36:4158–4171. doi: 10.1093/nar/gkn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hong S.W., Jiang Y., Kim S., Li C.J., Lee D.K. Target gene abundance contributes to the efficiency of siRNA-mediated gene silencing. Nucleic Acid Ther. 2014;24:192–198. doi: 10.1089/nat.2013.0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sulzer D. Clues to how alpha-synuclein damages neurons in Parkinson’s disease. Mov. Disord. 2010;25(Suppl 1):S27–S31. doi: 10.1002/mds.22639. [DOI] [PubMed] [Google Scholar]
- 73.Oaks A.W., Sidhu A. Synuclein modulation of monoamine transporters. FEBS Lett. 2011;585:1001–1006. doi: 10.1016/j.febslet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chadchankar H., Ihalainen J., Tanila H., Yavich L. Decreased reuptake of dopamine in the dorsal striatum in the absence of α-synuclein. Brain Res. 2011;1382:37–44. doi: 10.1016/j.brainres.2011.01.064. [DOI] [PubMed] [Google Scholar]
- 75.Adell A., Celada P., Artigas F. The role of 5-HT1B receptors in the regulation of serotonin cell firing and release in the rat brain. J. Neurochem. 2001;79:172–182. doi: 10.1046/j.1471-4159.2001.00550.x. [DOI] [PubMed] [Google Scholar]
- 76.Sokoloff P., Guillin O., Diaz J., Carroll P., Griffon N. Brain-derived neurotrophic factor controls dopamine D3 receptor expression: implications for neurodevelopmental psychiatric disorders. Neurotox. Res. 2002;4:671–678. doi: 10.1080/1029842021000045499. [DOI] [PubMed] [Google Scholar]
- 77.Bortolozzi A., Amargós-Bosch M., Toth M., Artigas F., Adell A. In vivo efflux of serotonin in the dorsal raphe nucleus of 5-HT1A receptor knockout mice. J. Neurochem. 2004;88:1373–1379. doi: 10.1046/j.1471-4159.2003.02267.x. [DOI] [PubMed] [Google Scholar]
- 78.Lam H.A., Wu N., Cely I., Kelly R.L., Hean S., Richter F., Magen I., Cepeda C., Ackerson L.C., Walwyn W. Elevated tonic extracellular dopamine concentration and altered dopamine modulation of synaptic activity precede dopamine loss in the striatum of mice overexpressing human α-synuclein. J. Neurosci. Res. 2011;89:1091–1102. doi: 10.1002/jnr.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim S.J., Kim S.Y., Na Y.S., Lee H.J., Chung K.C., Baik J.H. Alpha-synuclein enhances dopamine D2 receptor signaling. Brain Res. 2006;1124:5–9. doi: 10.1016/j.brainres.2006.09.079. [DOI] [PubMed] [Google Scholar]
- 80.Alarcón-Arís, D., Ruiz-Bronchal, E., Montefeltro, A., Artigas, F., and Bortolozzi, A. (2016). Antisense oligonucleotide-induced reduction of human alpha-synuclein accumulation in dopamine and serotonin neurons prevents early dysfunctions in a mouse model of Parkinson’s disease. Neuroscience 2016 Annual Meeting, Poster 602.03/T12, Society for Neuroscience, November 12–16, 2016, San Diego, CA.
- 81.Dehay B., Bové J., Rodríguez-Muela N., Perier C., Recasens A., Boya P., Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Díaz-Mataix L., Scorza M.C., Bortolozzi A., Toth M., Celada P., Artigas F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: role in atypical antipsychotic action. J. Neurosci. 2005;25:10831–10843. doi: 10.1523/JNEUROSCI.2999-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bortolozzi A., Masana M., Díaz-Mataix L., Cortés R., Scorza M.C., Gingrich J.A., Toth M., Artigas F. Dopamine release induced by atypical antipsychotics in prefrontal cortex requires 5-HT(1A) receptors but not 5-HT(2A) receptors. Int. J. Neuropsychopharmacol. 2010;13:1299–1314. doi: 10.1017/S146114571000009X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Franklin K.B.J., Paxinos G. Academic Press; 2008. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.